

Abstract
Primary pulmonary myxoid sarcoma (PPMS), classified as low to intermediate grade malignant myxoid endobronchial tumor, is rarely reported. Most reported cases occurred in lung parenchyme with an endobronchial component. Herein, we report a case of PPMS in a 29‐year‐old woman that developed in a major fissure of the left lung without parenchymal invasion. Histopathologically, the diagnosis was compatible to PPMS with EWSR1‐CREB1 translocation.
Keywords: Myxoid sarcoma, pulmonary, thoracoscopy/VATS
Introduction
In 1999, Nicholson et al. were the first to report two cases of what was then classified as a novel low‐grade, malignant, myxoid endobronchial tumor.1 Jeon et al. subsequently performed histopathological analysis of 17 primary pulmonary myxoid sarcoma (PPMS) cases and concluded that the sarcoma was of intermediate malignancy and may originate from mesenchymal cells undergoing fibroblastic or myofibroblastic differentiation.2 Most cases underwent pulmonary resection and experienced relatively good prognoses (no local recurrence despite the presence of distant metastases).2 We encountered a PPMS case in a location different from previously reported cases. The mass lay in the interlobar fissure of the left lung and lacked any definite parenchymal invasion.
Case report
A 29‐year‐old woman was referred to our hospital with a mass shadow in her left mid‐lung field detected by chest X‐ray during a routine health screen. Chest computed tomography (CT) revealed a slightly attenuating 3 × 3 cm mass in the interlobar fissure, in contact with the interlobar pulmonary artery (Fig 1a,b). The mass exhibited mild heterogeneous enhancement (Fig 1c). We considered the possibility of primary lung cancer because the patient was young, female, with no relevant family history. No abnormality was evident on gross bronchoscopic and cytological examinations. Blood test data and tumor marker levels were normal. We considered percutaneous needle biopsy, but decided that such a procedure would be dangerous as the tumor was located centrally. Although chest CT radiologic findings suggested sclerosing hemangioma or carcinoid tumors, we planned surgical resection because we could not rule out malignancy. During surgery, a mass was evident in the interlobar fissure above the interlobar pulmonary artery. The mass was covered by visceral pleura and was well encapsulated; no invasion of the adjacent lung parenchyma was evident. Internally, the mass was composed of myxoid materials that were easily removed by surgical suction. The internal surface of the capsule was smooth, but vascularity was well developed and the bleeding tendency was high. We completely enucleated the mass via video‐assisted thoracoscopic surgery. There was no need for pulmonary parenchymal resection. The postoperative course was uneventful.
Figure 1.
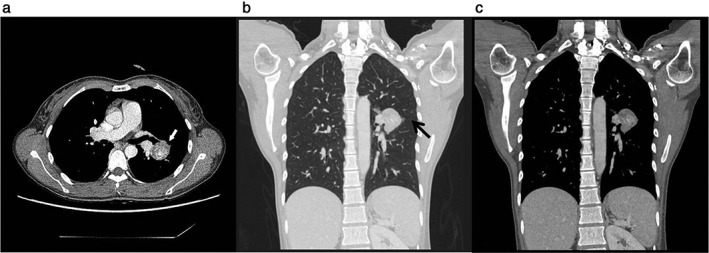
Radiologic findings. Chest computed tomography showing (a) a 3 × 3 cm round mass exhibiting mild, heterogeneous internal enhancement (white arrow), (b) coronal reconstruction (chest setting) of a lesion in the left pulmonary interlobar fissure (black arrow), and (c) coronal reconstruction (mediastinal setting) of a heterogenous enhancement beside the interlobar pulmonary artery.
Microscopically, the tumor was composed of short spindle‐shaped to ovoid and stellate cells embedded in the reticular network of a myxoid stroma. Lymphoplasmacytic cell infiltration was also evident. The extent of cellular atypia was mild and mitosis was uncommon. Alveolar cells of normal appearance were observed in the peripheral region of the tumor (Fig 2a–d). Although the pathologic findings did not reveal mesothelial cells, we concluded that the tumor did not originate from the pleura because the tumor was well encapsulated, covered by visceral pleura and was located in the interlobar fissure, as previously described. On immunohistochemical staining, the tumor cells were positive for vimentin and epithelial membrane antigen (EMA), but negative for rhabdoid cells (Fig 2e), smooth muscle actin, smooth muscle myosin heavy chain (SM‐MHC), calretinin, thyroid transcription factor‐1 (TTF‐1), cytokeratin 7 (CK7), p63, S‐100, CD34, and CD56. The Ki‐67 labeling index was ca. 1–2%. The overall histological profile suggested PPMS with an EWSR1‐CREB1 translocation, as described in the 4th edition of the World Health Organization lung tumor classification.3 We performed fluorescence in situ hybridization on formalin‐fixed, paraffin‐embedded tumor tissue. The EWSR1 break‐apart probe revealed split red and green signals suggestive of an EWSR1 translocation (Fig 2f). During 17 months of follow‐up, the patient has remained well and has not reported any medical issues.
Figure 2.
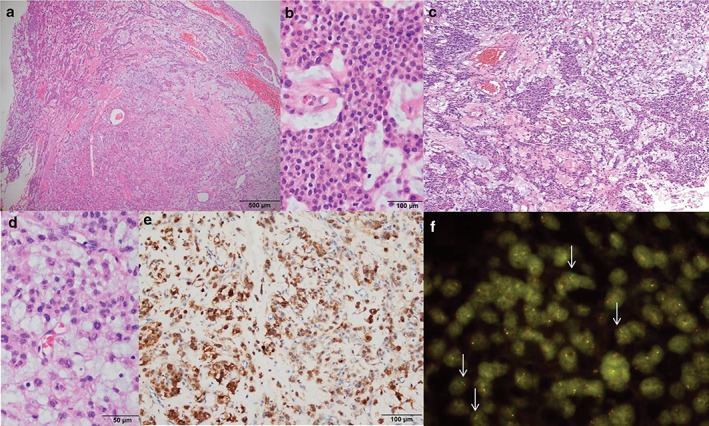
Pathological features. (a) A well‐demarcated cellular tumor in the myxoid stroma (hematoxylin end eosin [H&E] x 20). (b) Uniform tumor cells with a solid reticular growth pattern (H&E x 200). (c) Typical reticular features resembling extraskeletal myxoid chondrosarcoma (H&E x 100). (d) Mild nuclear atypia with multinucleated cells (H&E x 200). (e) Epithelial membrane antigen immunoreactivity (immunohistochemistry x 10). (f) Fluorescence in situ hybridization using the EWSR1 break‐apart probe revealed split red and green signals (arrows) suggestive of EWSR1 translocation.
Discussion
Primary pulmonary myxoid sarcoma is an extremely rare intrapulmonary sarcoma. Histologically, PPMS is characterized by an extensive myxoid stroma with a multi‐lobulated architecture, and contains spindle‐shaped or stellate to polygonal tumor cells arranged in a cord‐like or reticulated pattern, with mild to moderate atypia.2 The reticular network consists of delicate lacelike strands and cords within a prominent myxoid stroma that may be lightly basophilic, although more solid areas may be evident.3 Such tumors have recently been classified as PPMSs and harbor gene translocations involving EWSR1‐CREB1 fusions.4 EWSR1–CREB1 is also seen in tumors such as angiomatoid fibrous histiocytoma and clear cell sarcomas, but PPMS are morphologically differentiated from these entities.5 EWSR1 is a well‐known indiscriminate gene involved in many types of chromosomal translocation in a variety of sarcomas, myoepithelial and epithelial carcinomas, and acute leukemias.2 cAMP response element‐binding protein (CREB) is a transcription factor that performs an oncogenic function in several types of cancer and CREB1 is part of the CREB family of molecules along with cAMP‐dependent transcription factor 1 (ATF1).2 EWSR1‐CREB1 fusions have been found in tumors, including angiomatoid fibrous histiocytoma, clear cell sarcoma of soft tissue, clear cell sarcoma‐like tumor of the gastrointestinal tract, and hyalinizing clear cell carcinoma of the salivary gland.2
All patients in the cases reported to date underwent pulmonary resection, including wedge resection, segmentectomy, lobectomy, and pneumonectomy. Most cases involved the pulmonary parenchyma and had definite endobronchial components. Notably, in our case, the tumor was located in the interlobar fissure over the interlobar pulmonary artery and lacked gross parenchymal invasion. Histologically, however, alveolar cells of normal appearance were observed in the peripheral portion of the tumor. We consider that these may have been incidentally collected during surgery.
The male:female ratio of PPMS patients diagnosed in previous reports was 6:11. Patient age varied (26–68 years) and the most common symptom was a cough. However, half of the patients were incidentally found to have lung masses. Postoperative recurrence (distant metastases) developed in three patients.1, 2, 4 The histological features of PPMS overlap somewhat with those of other myxoid soft‐tissue and salivary gland‐type tumors, including extraskeletal myxoid chondrosarcoma (EMC), myoepithelial neoplasms, epithelioid hemangioendothelioma, and inflammatory myofibroblastic tumor.2 Of these tumors, EMC is morphologically similar to PPMS. The various tumors have distinct genetic features. EWSR1‐CREB1 fusion is characteristic of PPMS, whereas EMC harbors chromosomal translocations involving other genes.6
In summary, we successfully enucleated a PPMS with an EWSR1‐CREB1 translocation located in the interlobar fissure in a young woman; pulmonary resection was not required.
Disclosure
No authors report any conflict of interest.
References
- 1. Nicholson AG, Baandrup U, Florio R, Sheppard MN, Fisher C. Malignant myxoid endobronchial tumour: A report of two cases with a unique histological pattern. Histopathology 1999; 35: 313–8. [DOI] [PubMed] [Google Scholar]
- 2. Jeon YK, Moon KC, Park SH, Chung DH. Primary pulmonary myxoid sarcomas with EWSR1‐CREB1 translocation might originate from primitive peribronchial mesenchymal cells undergoing (myo)fibroblastic differentiation. Virchows Arch 2014; 465: 453–61. [DOI] [PubMed] [Google Scholar]
- 3. Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart, 4th edn. IARC, Lyon: 2015. [DOI] [PubMed] [Google Scholar]
- 4. Thway K, Nicholson AG, Lawson K et al. Primary pulmonary myxoid sarcoma with EWSR1‐CREB1 fusion: A new tumor entity. Am J Surg Pathol 2011; 35: 1722–32. [DOI] [PubMed] [Google Scholar]
- 5. Travis WD, Brambilla E, Nicholson AG et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol 2015; 10: 1243–60. [DOI] [PubMed] [Google Scholar]
- 6. Inayama Y, Hayashi H, Ogawa N, Mitsui H, Nakatani Y. Low‐grade pulmonary myxoid sarcoma of uncertain histogenesis. Pathol Int 2001; 51: 204–10. [DOI] [PubMed] [Google Scholar]