

Abstract
The aim of the present study was to investigate the protective effect of the NADPH oxidase inhibitor, diphenyleneiodonium (DPI) against necroptosis in renal tubular epithelial cells. A necroptosis model of HK-2 cells was established using tumor necrosis factor-α, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone and antimycin A (collectively termed TZA), as in our previous research. The necroptosis inhibitor, necrostatin-1 (Nec-1) or the NADPH oxidase inhibitor, DPI were administered to the necroptosis model. Production of reactive oxygen species (ROS) was detected by dichlorodihydrofluorescein diacetate in the different groups, and the manner of cell death was identified by flow cytometry. Western blot analysis was used to determine the levels of phosphorylation of receptor-interacting protein kinase 3 (RIP-3) and mixed lineage kinase domain-like (MLKL), which are essential to necroptosis. The results revealed that TZA increased the percentages of propidium iodide-positive HK-2 cells from 1.22±0.69 to 8.98±0.73% (P<0.001), and augmented the phosphorylation of RIP-3 and MLKL. ROS levels were increased in the TZA group compared with the control group (27.74±1.60×104 vs. 18.51±1.10×104, respectively; P<0.001), and could be inhibited by Nec-1 (TZA + Nec-1 group, 22.90±2.22×104 vs. TZA group, 27.74±1.60×104; P=0.01). DPI decreased ROS production (TZA + DPI group, 22.13±1.86×104 vs. TZA group, 27.74±1.60×104; P<0.001) and also reduced the proportions of necrosis in the necroptosis model (TZA + DPI group, 4.40±1.51% vs. TZA group, 8.98±0.73%; P<0.001). Phosphorylated RIP-3 and MLKL were also decreased by DPI treatment. The results indicate that ROS production increases in HK-2 cells undergoing necroptosis, and that the NADPH oxidase inhibitor, DPI may protect HK-2 cells from necroptosis via inhibition of ROS production.
Keywords: necroptosis, renal tubular epithelial cell, acute kidney injury, reactive oxygen species, diphenyleneiodonium
Introduction
Acute kidney injury (AKI) is a common clinical syndrome that is characterized by the rapid loss of kidney function and abrupt kidney damage. It is associated with high morbidity and mortality worldwide (1). Acute tubular necrosis (ATN) is the most common and severe pathological manifestation of AKI. Although necrosis has long been considered to be unregulated, previous studies have revealed various types of regulated necrosis that are independent of caspase activities (2). Necroptosis is an important type of regulated necrosis and is involved in various pathological conditions (3–7). Receptor-interacting protein kinase (RIP)-1 receives signals from various death stimuli, and subsequently recruits RIP-3 via RIP homotypic interaction motif domain-mediated interactions and promotes RIP-3 phosphorylation (8,9). Phosphorylated RIP-3 mediates the phosphorylation of mixed-lineage kinase domain-like (MLKL), ultimately leading to rupture of the plasma membrane (7). Previous studies have identified necroptosis in ATN induced by various stimuli, including ischemia/reperfusion, contrast medium and cisplatin (10–12). RIP-1 inhibition by necrostatin-1 (Nec-1), or knockout of RIP-3 or MLKL all prevent ATN (10–12).
Reactive oxygen species (ROS) promote necroptosis in cardiomyocytes, liver cells, Jurkat cells and lung adenoma cells (13–16). However, the effect of ROS on the necroptosis of renal tubular epithelium remains unknown. The present study hypothesized that excessive ROS production may promote necroptosis in HK-2 human kidney cells. Diphenyleneiodonium (DPI), an NADPH oxidase inhibitor, has been demonstrated to reduce ROS production and exert a protective role in numerous cell types (17–19). The present study investigated the protective effect of DPI in a renal tubular epithelium necroptosis model, which was established in our previous study (20).
Materials and methods
Cell culture
The HK-2 human kidney proximal tubular cell line (American Type Culture Collection, Rockville, MD, USA) was maintained in a mixture of Gibco Ham's F12 and Dulbecco's modified Eagle's medium (DMEM/F12; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere of 5% CO2 at 37°C. The cells were seeded at an appropriate cell density for different assays and allowed to grow to 80% confluence. Cell synchronization was routinely performed by incubating cells in serum-free medium at 37°C for 24 h prior to each experiment. Finally, the cells were exposed to different experimental conditions.
Cell treatment
Cells were divided into four groups as follows: i) Control group, cells were maintained in DMEM/F12 supplemented with 10% FBS for 2 h; ii) tumor necrosis factor-α (TNF-α), benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (z-VAD-fmk) and antimycin A group [TZA group; HK-2 cells were treated with 10 ng/ml TNF-α (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and 50 µM z-VAD-fmk (Sigma-Aldrich; Merck KGaA) for 2 h, and with 10 µM antimycin A (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 1 h], as described in our previous study (20); iii) TNF-α, z-VAD-fmk, antimycin A and Nec-1 group [TZA + Nec-1 group; cells were pretreated with 50 µM of Nec-1 (Sigma-Aldrich; Merck KGaA) for 6 h, and continuously treated with 10 ng/ml TNF-α and 50 µM z-VAD-fmk for 2 h, and with 10 μM antimycin A for 1 h]; and iv) TNF-α, z-VAD-fmk, antimycin A and DPI group [TZA + DPI group; cells were pretreated with 500 µM of DPI (Santa Cruz Biotechnology, Inc.) for 6 h, and continuously treated with 10 ng/ml TNF-α and 50 µM z-VAD-fmk for 2 h, and with 10 µM antimycin A for 1 h].
ROS measurements
ROS content was quantified using an ROS assay kit (cat. no. S0033; Beyotime Institute of Biotechnology, Shanghai, China). The cells were harvested using trypsin (cat. no. 25300054; Thermo Fisher Scientific, Inc.) and washed with PBS. The harvested cells were exposed to 10 µM 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) for 20 min at 37°C, and the labeled cells were washed twice in PBS and subjected to flow cytometry (Beckman Coulter, Inc., Brea, CA, USA) for ROS detection using the 488 nm laser for excitation and detected at 535 nm. The ROS level was represented by the median fluorescence intensity.
Western blot analysis
HK-2 cells subjected to the different experimental conditions were lysed in radioimmunoprecipitation assay lysis buffer (cat. no. P0013B; Beyotime Institute of Biotechnology). The total protein concentration from the resultant supernatant was determined using a Pierce™ BCA Protein Assay kit (Thermo Fisher Scientific, Inc.). Samples of equal protein content (35 µg/lane) were separated by SDS-PAGE using 8% gels (130 V for 90 min) and transferred to polyvinylidene difluoride membranes (90 V for 60 min). Membranes were blocked with 3% bovine serum albumin in TBS-Tween-20 (cat. no. ST825; Beyotime Institute of Biotechnology) and incubated overnight at 4°C with antibodies against RIP-3 phospho-S227 (cat. no. ab209384 at 1:2,000; Abcam, Shanghai, China) and MLKL phospho-S358 (cat. no. ab187091 at 1:1,000; Abcam). β-actin (cat. no. 8457; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA) was used as loading control. After being washed in TBS, blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit antibodies (cat. no. 7077; 1:2,000; Cell Signaling Technology, Inc.) for 1 h at room temperature. After a final wash as described above, the corresponding secondary antibodies were visualized using enhanced Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific, Inc.). Signals were quantified with a chemiluminescence detector (Tanon 4200; Tanon Science & Technology Co., Ltd., Shanghai, China) and the accompanying densitometry software.
Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) staining
Annexin V and PI staining were performed using an Annexin V-FITC Apoptosis Detection kit (cat. no. PF032; EMD Millipore, Billerica, MA, USA). Cells were trypsinized, washed twice with ice-cold PBS and resuspended in 1X binding buffer The cell concentration was adjusted to 1×106/ml, and 400 µl cell suspensions were added to 5-ml centrifuge tubes, followed by the addition of 1.25 µl Annexin V-FITC. Cells were incubated for 30 min at room temperature and centrifuged at 1,000 × g for 3 min at room temperature. Following resuspension in 400 µl 1X binding buffer, 5 ml PI was added to each cell suspension and incubated for 5 min in the dark at 2–8°C. The rates of necrosis and apoptosis of the HK-2 cells were analyzed by flow cytometry (Beckman Coulter, Inc., Brea, CA, USA).
Statistical analysis
All values are expressed as the mean ± standard deviation. Multiple comparisons among the groups were conducted by one-way ANOVA followed by a least significant difference multiple comparison test. All statistical analyses were performed using SPSS software (version 21.0; IBM SPSS, Armonk, NY, USA), with P<0.05 considered to indicate a statistically significant difference.
Results
Construction and validation of the necroptosis model in HK-2 cells
Following the treatment of HK-2 cells with TNF-α, zVAD-fmK and antimycin A, the percentages of PI-positive cells increased from 1.22±0.69 to 8.98±0.73% (P<0.001; Fig. 1). However, TZA treatment did not increase the percentage of Annexin V-positive cells (control group, 0.87±0.15% vs. TZA group, 0.89±0.13%; P=0.098). Administration of Nec-1 was observed to reduce the rate of TZA-induced necrosis markedly (PI-positive cells, 2.74±0.62%; P<0.001 vs. TZA group). This result indicates that TZA-induced necrosis may be regulated by Nec-1.
Figure 1.
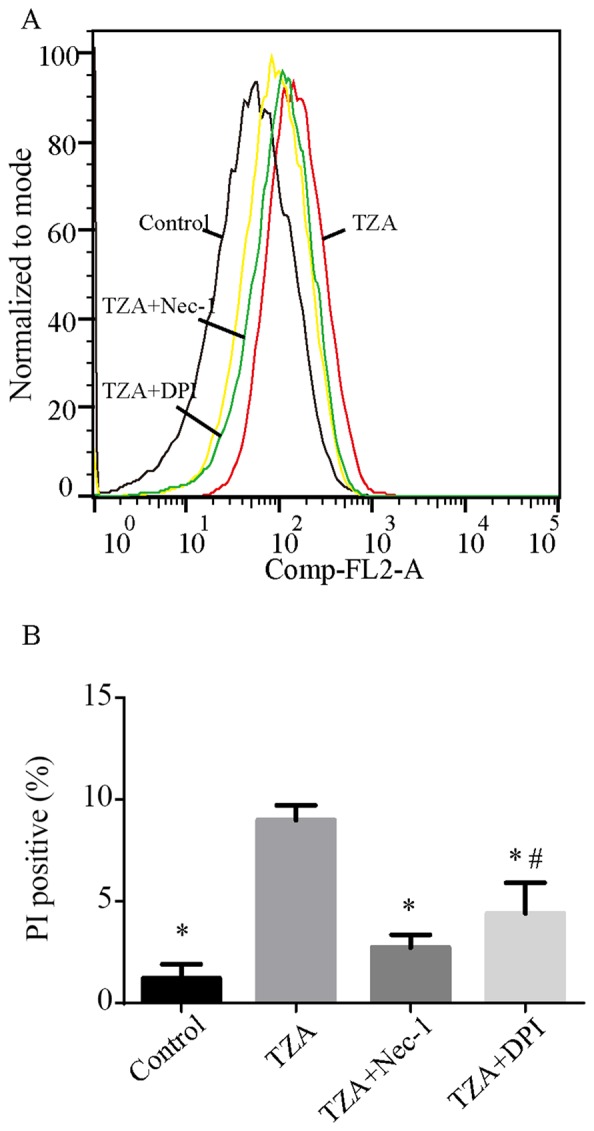
PI+ HK-2 cells detected by flow cytometry in different groups. (A) TZA increased the percentage of PI+ HK-2 cells. Nec-1 and DPI exposure reduced the percentages of PI+ HK-2 cells induced by TZA. (B) The amount of the dead cells are presented as mean ± standard deviation. *P<0.05 vs. TZA group; #P<0.05 vs. control group. PI, propidium iodide; TZA, tumor necrosis factor-α + z-VAD-fmk + antimycin A; Nec-1, necrostatin-1; DPI, diphenyleneiodonium.
As RIP-3 and MLKL are essential for the necroptosis pathway, the phosphorylation of RIP-3 (p-RIP-3) and MLKL (p-MLKL) were assessed by western blotting to confirm that the observed cell death was by necroptosis. p-RIP-3 and p-MLKL expression levels were elevated in the TZA group, and Nec-1 reversed the increased p-RIP-3 and p-MLKL expression levels (Fig. 2). These results indicate that the cell death induced by TZA is necroptosis.
Figure 2.

Essential necroptosis proteins detected by western blotting. (A and B) TZA increased the expression levels of p-RIP-3 and p-MLKL, and Nec-1 and DPI exposure reversed this effect. (C and D) Quantification of p-RIP-3 and p-MLKL are presented as mean ± standard deviation. *P<0.05 vs. TZA group; #P<0.05 vs. control group. TZA, tumor necrosis factor-α + z-VAD-fmk + antimycin A; p-RIP-3, phosphorylated receptor-interacting protein kinase 3; p-MLKL, phosphorylated mixed-lineage kinase domain-like; Nec-1, necrostatin-1; DPI, diphenyleneiodonium.
ROS production is augmented in TZA-induced HK-2 necroptosis
Total ROS production, as measured by DCFH-DA fluorescence intensity (Fig. 3), was increased in the TZA group compared with the control group (29.4×104 vs. 17.5×104). Additionally, the increase in ROS generation could be reversed by Nec-1 treatment (22.6×104). Thus, it was concluded that ROS production increases in HK-2 cells undergoing TZA-induced necroptosis.
Figure 3.

ROS content detected by flow cytometry in different groups. TZA increased ROS production in the HK-2 cells, whereas Nec-1 and DPI each inhibited ROS production. ROS, reactive oxygen species; TZA, tumor necrosis factor-α + z-VAD-fmk + antimycin A; Nec-1, necrostatin-1; DPI, diphenyleneiodonium; DCFH-DA, dichlorodihydrofluorescein diacetate.
NADPH oxidase inhibitor, DPI protects against necroptosis via inhibition of ROS production
To determine the effect of the NADPH oxidase inhibitor, DPI on necroptosis in HK-2 cells, DPI was administered to the TZA-induced necroptosis cell model. DPI inhibited ROS generation in the TZA-induced necroptosis (TZA + DPI group, 21.1×104 vs. TZA group, 29.4×104; Fig. 3) and ameliorated TZA-induced necrosis (PI-positive cells: TZA + DPI group, 4.40±1.51% vs. TZA group, 8.98±0.73%; P<0.001; Fig. 1). In addition, p-RIP-3 and p-MLKL levels were decreased by DPI pre-treatment (Fig. 2). These results indicate that the NADPH oxidase inhibitor, DPI may reduce ROS production in HK-2 cells and prevent HK-2 cell necroptosis.
Discussion
AKI caused by ATN is a common clinical syndrome with high rates of morbidity and mortality (1). Although necrosis used to be considered as an unregulated form of cell death, necroptosis, a type of regulated necrosis, has been identified in ATN, as well as in drug-induced liver injury and ischemic brain injury (10–12). Mechanistically, RIP-1 phosphorylates RIP-3 and forms a necrosome (8,9). Subsequently, the necrosome phosphorylates MLKL, resulting in necroptosis in various types of cells (7,21). Inhibition of RIP-1 by Nec-1 and knockout of RIP-3 or MLKL prevents ATN in response to various stimuli, such as cisplatin, sepsis and ischemia/reperfusion injury (10–12).
ROS are chemically reactive species containing oxygen, including peroxides, superoxides, hydroxyl radicals and singlet oxygen. Although ROS are formed as a natural byproduct of the normal metabolism of oxygen, and have important roles in cell signaling and homeostasis in biological conditions, excessive ROS are detrimental. Various studies have demonstrated that excessive ROS promotes necroptosis in various cell types, whereas inhibiting ROS production reduces necroptosis (13–16). In addition, previous studies revealed that ROS production is augmented in AKI, and that ROS lead to renal tubular epithelium injury via inflammasome activation, mitochondrion damage and tubular epithelium apoptosis (22–25). To the best of our knowledge, there are no studies that have investigated the effect of ROS on necroptosis in renal tubular epithelium. The current study hypothesized that excessive ROS may promote necroptosis in HK-2 cells, and that DPI may reduce ROS production and thus exert a protective role against necroptosis in HK-2 cells.
A necroptosis model in HK-2 cells, established in our previous study, was validated in the present study, and revealed that ROS production increased in HK-2 cells undergoing necroptosis induced by TZA. Nec-1, a necroptosis inhibitor decreased ROS production in necroptosis. Similarly, pre-treatment of HK-2 cells with DPI, an NADPH oxidase inhibitor, decreased ROS generation, and ameliorated TZA-induced necroptosis, and attenuated p-RIP-3 and p-MLKL expression levels.
In conclusion, the results of the current study indicate that excessive ROS levels augment HK-2 cell necroptosis and that DPI may protect HK-2 cells against necroptosis by inhibiting ROS generation. Currently, the mechanism underlying precisely how ROS participates in necroptosis remains unclear. The molecular mechanism of ROS-mediated necroptosis was not investigated in the present study and further investigations of this in the renal tubular epithelium are required. Further research may provide a novel therapeutic strategy for the treatment of AKI.
Acknowledgements
The present study was supported by the National Natural Science Foundation (grant no. 81570609), the Natural Science Foundation of Guangdong Province (grant no. 2014A030313545) and the National Clinical Key Specialty Construction Preparatory Projects.
References
- 1.Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, Jaber BL. Acute Kidney Injury Advisory Group of the American Society of Nephrology: World incidence of AKI: A meta-analysis. Clin J Am Soc Nephrol. 2013;8:1482–1493. doi: 10.2215/CJN.00710113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, Sluijter JP. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107:270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 4.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 9.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 10.Linkermann A, Bräsen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–761. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 11.Linkermann A, Heller JO, Prókai A, Weinberg JM, De Zen F, Himmerkus N, Szabó AJ, Bräsen JH, Kunzendorf U, Krautwald S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol. 2013;24:1545–1557. doi: 10.1681/ASN.2012121169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol. 2015;26:2647–2658. doi: 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takemoto K, Hatano E, Iwaisako K, Takeiri M, Noma N, Ohmae S, Toriguchi K, Tanabe K, Tanaka H, Seo S, et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 2014;4:777–787. doi: 10.1016/j.fob.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song KJ, Jang YS, Lee YA, Kim KA, Lee SK, Shin MH. Reactive oxygen species-dependent necroptosis in Jurkat T cells induced by pathogenic free-living Naegleria fowleri. Parasite Immunol. 2011;33:390–400. doi: 10.1111/j.1365-3024.2011.01297.x. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Y, Shan S, Chi L, Zhang G, Gao X, Li H, Zhu X, Yang J. Methyl methanesulfonate induces necroptosis in human lung adenoma A549 cells through the PIG-3-reactive oxygen species pathway. Tumour Biol. 2016;37:3785–3795. doi: 10.1007/s13277-015-3531-y. [DOI] [PubMed] [Google Scholar]
- 16.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 17.Farrow MA, Chumbler NM, Lapierre LA, Franklin JL, Rutherford SA, Goldenring JR, Lacy DB. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci USA. 2013;110:18674–18679. doi: 10.1073/pnas.1313658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostergaard M, Christensen M, Nilsson L, Carlsen I, Frøkiær J, Nørregaard R. ROS dependence of cyclooxygenase-2 induction in rats subjected to unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2014;306:F259–F270. doi: 10.1152/ajprenal.00352.2013. [DOI] [PubMed] [Google Scholar]
- 19.Das R, Xu S, Quan X, Nguyen TT, Kong ID, Chung CH, Lee EY, Cha SK, Park KS. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am J Physiol Renal Physiol. 2014;306:F155–F167. doi: 10.1152/ajprenal.00438.2013. [DOI] [PubMed] [Google Scholar]
- 20.Liang X, Chen Y, Zhang L, Jiang F, Wang W, Ye Z, Liu S, Yu C, Shi W. Necroptosis, a novel form of caspase-independent cell death, contributes to renal epithelial cell damage in an ATP-depleted renal ischemia model. Mol Med Rep. 2014;10:719–724. doi: 10.3892/mmr.2014.2234. [DOI] [PubMed] [Google Scholar]
- 21.Moujalled DM, Cook WD, Murphy JM, Vaux DL. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014;5:e1086. doi: 10.1038/cddis.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintavalle C, Brenca M, De Micco F, Fiore D, Romano S, Romano MF, Apone F, Bianco A, Zabatta MA, Troncone G, et al. In vivo and in vitro assessment of pathways involved in contrast media-induced renal cells apoptosis. Cell Death Dis. 2011;2:e155. doi: 10.1038/cddis.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winterberg PD, Wang Y, Lin KM, Hartono JR, Nagami GT, Zhou XJ, Shelton JM, Richardson JA, Lu CY. Reactive oxygen species and IRF1 stimulate IFNα production by proximal tubules during ischemic AKI. Am J Physiol Renal Physiol. 2013;305:F164–F172. doi: 10.1152/ajprenal.00487.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn JM, You SJ, Lee YM, Oh SW, Ahn SY, Kim S, Chin HJ, Chae DW, Na KY. Hypoxia-inducible factor activation protects the kidney from gentamicin-induced acute injury. PLoS One. 2012;7:e48952. doi: 10.1371/journal.pone.0048952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao WY, Zhang L, Sui MX, Zhu YH, Zeng L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute. doi: 10.1038/srep33201. [DOI] [PMC free article] [PubMed] [Google Scholar]