

Abstract
Chimeric antigen receptor (CAR) engineering is a branch of cancer immunotherapy that equips immune cells to target tumor antigens expressed on the cell surface using antibody-derived single-chain variable fragments (scFvs). However, other antibody mimetics, such as designed ankyrin repeat proteins (DARPins), can also serve as antigen-binding domains in CARs. This study shows that CAR-engineered T (CAR-T) cells utilizing Her2-targeting DARPins G3 and 929 can target human epidermal growth factor receptor 2 (Her2)-overexpressing cancer cells as effectively as CAR-T cells with the scFv 4D5 in vitro, and G3 CAR-T cells can slow or eliminate tumor growth in vivo as effectively as 4D5 CAR-T cells. Some DARPins may offer an attractive alternative to scFv usage in CARs, as they are smaller, thermodynamically stable, poorly immunogenic, and can be generated with different binding properties from DARPin libraries.
Keywords: : cancer immunotherapy, chimeric antigen receptors, designed ankyrin repeat proteins, antibody mimetic proteins
Introduction
Cancer immunotherapy has the potential to achieve long-term remission by harnessing the patient's own immune system to attack cancer cells. Chimeric antigen receptors (CARs) are engineered proteins that offer a method of reprogramming autologous immune cells to recognize and target cancer cells.1 CAR-engineered T (CAR-T) cells are not major histocompatibility complex (MHC)-restricted and can be engineered to recognize specific tumor-associated surface antigens (TAA). Typically, this recognition is due to a single-chain variable fragment (scFv), which is derived from the desired antibody and fused to intracellular T-cell signaling and costimulatory domains. Antigen binding to the scFv triggers a signaling cascade that activates the CAR-T cell against the TAA-expressing cancer cell.2
CAR-T therapy has been largely successful in hematological cancers3–6 but has faced many setbacks in treating solid tumors.7–10 Often this is because CAR-T cells become exhausted and lose their cytotoxic capabilities after long exposures to TAA.11 Many factors are thought to contribute to CAR-T exhaustion, and problems can stem from both the immunosuppressive tumor microenvironment and the CARs themselves. A growing body of evidence suggests that the milieu surrounding the tumor suppresses both native and adoptively transferred immune cells that would normally attack the tumor cells.12,13 One problem with the CARs themselves is that scFvs tend to aggregate, and scFv crosslinking on the cell surface can stimulate the CAR-T cell, even in the absence of the appropriate antigen.14 Prolonged stimulation leads to eventual exhaustion and compromised function, including diminished cytokine release and decreased cytotoxicity. Additionally, many scFvs are of mouse origin, and even humanized scFvs, such as 4D5, carry the risk of immunogenicity and adverse side effects.15
To date, many CARs, including those targeting the TAA human epidermal growth factor receptor 2 (Her2),16,17 recognize their target antigens by using scFvs. However, other proteins potentially can be used in place of the scFv to recognize the cognate antigen. Like scFvs, antibody mimetic proteins also are capable of binding to specific antigens. One such protein family is designed ankyrin repeat proteins (DARPins), which are based off naturally occurring ankyrin proteins. In general, these proteins are smaller than scFvs and consist of two or three repeat units of 33 amino acids each, which are flanked by N and C caps.18 Constructs with specific binding affinities, sizes, or other desired qualities can be generated from DARPin libraries.19 DARPins have been shown to be very stable and are not prone to aggregation,15 unlike scFvs. DARPins are being explored as an alternative to scFv use in CARs.
This study explored CARs capable of targeting Her2, which is commonly overexpressed on a variety of cancers, including breast and ovarian.20 Two Her2-binding DARPin CARs were compared to a traditional anti-Her2 scFv CAR. Two previously reported DARPins, G3 and 929, were selected to compare to the scFv 4D5. G3 is the smallest of the three Her2 targeting proteins and has the highest binding affinity (Kd = 90 pM).21 DARPin 929 binds to a different Her2 domain than either 4D5 or G3. In fact, many anti-Her2 scFvs, including trastuzumab-derived 4D5,16 bind to membrane proximal Her2 domain IV,22 but 929 binds to membrane distal domain I.23 The activation and tumor-killing efficacy of these CARs in vitro and in vivo is reported.
Materials and Methods
Cell culture
4T1 (ATCC CRL-2539), MDA.MB.231 (ATCC HTB-26), MDA.MB.468 (ATCC HTB-132), and SKOV3 (ATCC HTB-77) tumor cell lines were maintained in a 5% CO2 environment in RPMI 1640 (Gibco) media supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (pen-strep), and 2 mM of L-glutamine.
DARPin sequences
Gene fragments (gBlocks IDT) were created based on previously published DARPin sequences21 and are listed below.
G3: ATGTCCGATCTCGGGAAGAAGCTTCTGGAGGCAGCAAGGGCCGGTCAGGATGATGAAGTGCGAATCCTCATGGCGAACGGAGCCGATGTGAACGCTAAGGACGAGTATGGACTGACCCCGCTCTATCTTGCAACCGCACACGGTCACCTGGAAATAGTTGAAGTACTCCTCAAAAACGGAGCCGACGTGAACGCCGTCGACGCAATCGGCTTCACACCTCTGCACCTGGCAGCCTTCATCGGGCATCTCGAAATAGCAGAGGTTTTGCTGAAGCATGGCGCCGATGTTAACGCCCAGGACAAATTCGGCAAAACTGCTTTCGACATTAGCATAGGAAACGGCAACGAGGACCTCGCAGAGATTCTGCAAAAGCTGAAC
929: ATGAGCGATCTGGGGAAGAAGCTGCTCGAAGCGGCCAGAGCGGGTCAAGATGACGAAGTGCGCATACTGATGGCCAACGGAGCTGACGTGAACGCGCACGACTTCTACGGAATCACTCCACTGCATCTCGCAGCCAACTTTGGCCATCTCGAGATCGTGGAGGTTCTTCTCAAGCATGGCGCCGACGTTAACGCCTTTGATTATGACAACACACCTCTCCATTTGGCAGCTGACGCAGGGCACTTGGAGATCGTTGAGGTGTTGCTTAAATACGGCGCAGATGTAAATGCCTCCGATAGGGACGGTCACACGCCTCTGCACCTGGCCGCTCGGGAAGGCCACCTTGAAATCGTGGAGGTGCTGCTTAAGAATGGAGCCGATGTCAACGCCCAGGATAAATTTGGAAAAACTGCATTCGACATCAGTATCGATAACGGCAATGAGGATTTGGCTGAGATCTTGCAGAAGCTCAAC
Lentiviral production
Antigen-binding domains in the third-generation CAR constructs consisted of either the scFv 4D5 or the DARPins G3 or 929. These constructs were cloned into a lentiviral pCCW vector (restriction sites EcoR1 and BamH1), which is based on the pCCL vector24,25 with an added WRE post-transcriptional regulatory region. The CARs consisted of an antigen-binding domain, CD8 hinge and transmembrane region, and CD28, 4-1BB, and CD3ζ cytoplasmic regions, and were cloned into the pCCW vector using Gibson assembly (New England Biolabs). These plasmids were used to transfect HEK 293T cells in 30 mL plates using CaCl2 precipitation methods. Fresh media (high glucose Dulbecco's modified Eagle's medium supplemented with 10% FBS and 1% pen-strep) was plated onto the cells 4 h after initial transfection. Supernatants were harvested and filtered (0.45 μm) 48 h later and concentrated 30 × (106,559 g for 90 min at 4°C). Viral stocks were re-suspended in Hank's balanced salt solution and frozen at −80°C until later use.
Transduction of peripheral blood mononuclear cell–derived human T cells and MDA.MB.468 cells
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were thawed and cultured in T-cell media. Twenty-four hours after thawing, 1 × 106 cells were activated with anti-CD3/CD28 beads at a 3:1 ratio (Life Technologies) in a 24-well plate with 50 IU/mL of recombinant human IL-2 (rhIL2; Peprotech). Forty-eight hours after activation, T cells were transduced with concentrated lentivirus at a multiplicity of infection (MOI) of 40; the titer was based on transduction of 293T cells. CAR-T cell cultures were expanded for up to 10 days in fresh media supplemented with 100 IU/mL of rhIL-2.
MDA.MB.468 cells were similarly transduced to express human Her2 via a lentiviral vector. One million cells were added to 2 mL of fresh viral supernatant and centrifuged at 1,050 g for 90 min at room temperature. Her2+ cells were sorted using fluorescence-activated cell sorting and cultured as described for MDA.MB.468 cells.
CAR detection on T-cell surface
Eight days after transduction, T cells (1 × 105) were incubated with rhHer2-Fc chimera (Peprotech) at a volume ratio of 1:50 (2 μg/mL) in phosphate-buffered saline (PBS) at 4°C for 30 min and rinsed with PBS. The cells were subsequently incubated with phycoerythrin (PE)-labeled goat anti-human Fc (Jackson ImmunoResearch) at a volume ratio of 1:150 in PBS at 4°C for 10 min, rinsed, and read using flow cytometry. Nontransduced (NT) T cells served as a negative control.
Her2 binding assay
Frozen stocks of CAR-T cells were thawed and rested overnight before staining with rhHer2-Fc and PE-labeled goat anti-human Fc, as mentioned previously. rhHer2 concentrations varied from 0 to 10 μg/mL. Results were read using flow cytometry, and the data were determined in triplicate and presented as the mean ± standard error of the mean (SEM).
Cytokine release assay
T cells (1 × 105 cells/well) were co-incubated with target cells in 96-well plates at a 1:1 ratio for 6 h at 37°C. Brefeldin-A (1 μg; Sigma–Aldrich) was added to each well to prevent protein transport. At the end of the incubation, cells were permeabilized using the CytoFix/CytoPerm kit (BD Biosciences) and stained for CD8 and interferon gamma (IFN-γ) using Pacific Blue-conjugated anti-human CD8 (Biolegend) and PE-conjugated anti-human IFN-γ (Biolegend). Cells stimulated with anti-human CD3/anti-human CD28 were used as a positive control. Results were read using flow cytometry. The data were determined in triplicate and presented as the mean ± SEM.
Cytotoxicity assay
Target cells (1 × 104 cells/well) were labeled with 5 μM of carboxyfluorescein succinimidyl ester (CFSE; Life Technologies), as previously described,26 and co-incubated with T cells at various ratios in 96-well plates for 24 h at 37°C. The cells were then incubated in 7-AAD (Life Technologies) in PBS (1:1,000 dilution) for 10 min at room temperature and analyzed via flow cytometry. Percentages of killed cells were calculated as (CFSE+7-AAD+ cells/[CFSE+7-AAD− + CFSE+7-AAD+]) cells, with live/dead gates based on control wells of target cells only to account for spontaneous cell death. The cytotoxicity was determined in triplicate and presented as the mean ± SEM.
Xenograft tumor model
All animal experiments were conducted according to the animal protocol approved by the University of Southern California Institutional Animal Care and Use Committee (IACUC). Ten-week-old female NOD.Cg-PrkdcscidIL2Rγtm1Wj1/SZ (NSG) mice (Jackson Laboratories) were inoculated subcutaneously with 3.5 × 106 SKOV3 cells, and tumors were grown to 70–100 mm3. On day 14, the mice were injected intravenously through the tail vein with 10 × 106 CAR-T cells. The solutions were adjusted with NT T cells to ensure that all CAR-T groups had 50% CAR expression (5 × 106 CAR-T cells per mouse). One week after CAR-T injection (day 21), a subset of mice was sacrificed for ex vivo staining. Tumor growth and the body weight of the remaining mice were recorded until the end of the experiment. The tumor length and width were measured with a fine caliper three times a week, and tumor volume was calculated as 0.5 × (length) × (width)2. The survival endpoint was determined to be when the tumor reached 1,000 mm3.
Ex vivo T-cell purification and flow cytometry
Seven days after CAR-T injection, a subset of mice was sacrificed for analysis of T cells from the tumor, blood, and spleen. Briefly, a 50% Percoll (GE Healthcare) solution was made with 100% Percoll and PBS. Each tissue sample was made into a single-cell suspension through a 70 μm strainer and re-suspended in 15 mL of 50% Percoll solution. The samples were centrifuged with no braking at 800 g for 30 min at room temperature, and the pellet was re-suspended in 10 mL of TAC buffer for 10 min at room temperature. The samples were then washed and re-suspended with PBS. Fluorescent-conjugated antibodies (Biolegend) were used to stain the samples in 100 μL of PBS at a 1:100 volume ratio for 15 min at 4°C in a 96-well plate. Samples were analyzed using flow cytometry in triplicate and presented as the mean ± SEM.
Quantitative real-time polymerase chain reaction
Total mRNA was extracted from the tumor and blood samples using an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Quantitative real-time polymerase chain reaction (PCR) was performed with SYBR Green reagents (Thermo Fisher) using a 100 ng template and 150 nM of forward and reverse primers. Quantitative PCR was performed for 40 cycles (denaturation at 95°C for 15 s; annealing/extension at 60°C for 1 min) using an Applied Biosystems 7300 real-time PCR system. Standard curves were prepared from serial dilutions of known percentages of CAR+ T cells. The percentages of CAR+ T cells from unknown ex vivo samples were determined using the standard curves. GAPDH was used as a housekeeping gene. Each sample was performed in triplicate and presented as the mean ± SEM. Primers are listed below.
GAPDH: 5′-ACCAGCCCCAGCAAGAGCACAAG-3′ (forward); 5′-TTCAAGGGGTCTACA TGGCAACTG-3′ (reverse). 4D5: 5′-CATTGTCAGCAAGTGTCGGCG-3′ (forward); 5′-CGTGCCTTGCCCGAACG-3′; (reverse). G3: 5′-CGGAGCCGATGTGAACGCTAAG-3′ (forward); 5′-CTCTGCGAGGTCCTCGTTGC-3′ (reverse). 929: 5′-GGGTCAAGATGACGAAGTGCGC-3′ (forward); 5′-CTGGGCGTTGACATCGGCT-3′ (reverse).
Statistical analysis
Statistical analysis was performed in GraphPad Prism v6. One-way analysis of variance with Tukey's multiple comparison was used to assess the data between different groups. The mouse survival curve was analyzed using Kaplan–Meier analysis (log-rank test with Bonferroni correction). p-Values of <0.05 were considered significant and denoted as follows: NS, not significant (p > 0.05); *p < 0.05; **p < 0.01; ***p < 0.001.
Results
DARPin and scFv CARs are both efficiently transduced in human T cells
The lentiviral vectors described in Materials and Methods were used to transduce the CAR constructs (Fig. 1A and B) into human PBMC-derived T cells. All viral stocks were made simultaneously, and all CAR-T groups were transduced simultaneously with the same MOI to minimize differences in experimental methods. All three CAR-T groups were stained for CAR expression on day 8, using non-transduced (NT) T cells as a negative control. All CAR groups were >50% CAR+ (representative data shown in Fig. 1C), with mean fluorescence intensity (MFI) values of 505 for NT cells, 2,876 for 4D5, 3,773 for G3, and 2,712 for 929. This indicates that like scFvs, DARPins are capable of being efficiently expressed in human T cells using previously established lentiviral transduction methods.
Figure 1.

Designed ankyrin repeat protein (DARPin) chimeric antigen receptors (CARs) are efficiently expressed on human T cells. (A) Schematic diagram of single-chain variable fragments (scFv) and DARPin CARs shows that either an antibody-derived scFv (left) or DARPin (right) is added to costimulatory domains and CD3ζ signaling domains derived from T-cell receptors. The CARs are then expressed on T-cell surfaces. (B) Schematic diagram shows that different third-generation CAR constructs contain the antigen-binding domain, two costimulatory domains (CD28 and 4-1BB), and the main CD3ζ signaling domain. (C) On day 8 of ex vivo expansion, CAR expression in transduced human T cells was detected using soluble Her2-Fc chimera followed by a PE-conjugated anti-human Fc antibody.
Higher percentages of DARPin CAR-T cells bind to soluble Her2 than scFv CAR-T cells at intermediate concentrations
The ability of different CAR-T cells to bind to a range of soluble Her2 concentrations was tested (Fig. 2A). For intermediate concentrations (1 and 2 μg/mL), the DARPin CAR groups had a significantly higher percentage of cells bound to soluble Her2 compared to the 4D5 CAR group. Previously published data analyzing DARPins alone show that G3 has a very high binding affinity (Kd = 90 pM), while 4D5 has a Kd of 190 pM and 929 has a Kd of 3.9 nM.21 The data show that both G3 and 929 CAR-T cells are overall more effective at binding to Her2 than 4D5 CAR-T cells are.
Figure 2.

DARPin CAR-T cells bind to Her2 and release cytokines in response to Her2-expressing target cells in vitro. (A) CAR-T cells were bound to soluble Her2 in triplicate by incubating with various concentrations of soluble Her2-Fc chimera followed by a PE-conjugated anti-human Fc antibody (n = 3, mean ± standard error of the mean [SEM]; NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001). (B) Her2 expression on target cells was detected by labeling with PE-conjugated anti-human Her2 antibody. (C) On day 8 of ex vivo expansion, CAR-T cells were co-cultured with MDA.MB.468, MDA.MB.231, SKOV3, or MDA.MB.468.Her2 cells with Brefeldin A protein transport inhibitor for 6 h to detect interferon gamma (IFN-γ) release. Unstimulated CAR-T cells served as a negative control, while CAR-T cells stimulated with anti-human CD3/CD28 were used as positive controls. IFN-γ was measured with intracellular staining. CD8+IFN-γ+ cells are shown in each gate. Representative data from one donor are shown herein. (D) Summarized data from IFN-γ assay performed in triplicate are displayed in a bar graph (n = 3, mean ± SEM; NS, not significant; *p < 0.05; **p < 0.01.
Exposure to Her2+ target cells induces more IFN-γ release in DARPin CAR-T cells than scFv CAR-T cells
To detect T-cell activation upon antigen binding, an IFN-γ release assay was performed with the CAR-T cells. First, various target cell lines were stained for Her2 expression, using the murine breast cancer line 4T1 as a negative control. Human breast cancer line MDA.MB.468 was also a Her2− line, while human breast cancer line MDA.MB.231 and human ovarian line SKOV3 were Her2+. MDA.MB.468.Her2 cells, which were transduced to overexpress Her2, were also tested (Fig. 2B).
The functionality of the CAR-T cells was tested using flow cytometry to analyze the IFN-γ+ populations after co-incubation with various target cell lines, using NT T cells as a negative control. In all cases, none of the CAR-T cells reacted to Her2− lines, but Her2+ lines induced IFN-γ release from all CAR-T groups. Both DARPin CARs yielded a greater percentage of IFN-γ+ cells than did the 4D5 CAR when co-incubated with MDA.MB.231 (p < 0.05). When exposed to SKOV3 cells, 929 had the greatest percentage of IFN-γ+ cells (p < 0.01), followed by G3 (p < 0.05), when compared to 4D5 cells. Both DARPin CARs yielded a greater percentage of IFN-γ+ cells than did the 4D5 CAR when co-incubated with MDA.MB.468.Her2 (p < 0.01). None of the CAR-T groups released IFN-γ when alone or when co-incubated with Her2− lines, demonstrating specificity toward the appropriate TAA (Fig. 2C and D). These data indicate that DARPin CARs exhibit higher activation and induce greater IFN-γ release compared to the 4D5 CAR when in the presence of cells expressing the cognate antigen.
G3 CAR-T cells exhibit the greatest cytotoxicity against Her2+ target cells
The ability of the different CARs to trigger cytotoxic effects against target cells was assessed by co-incubating the CAR-T cells with various target cell lines and reading the results with flow cytometry. Both DARPin groups demonstrated significantly greater cytotoxicity against MDA.MB.231 compared to 4D5 at lower effector-to-target ratios (p < 0.01 for 1:1 and p < 0.05 for 3:1) but were similar to 4D5 at higher ratios. With SKOV3 target cells, G3 exhibited the greatest killing ability (p < 0.001 for 5:1 and p < 0.05 for all other ratios), followed by 929 and then 4D5. With MDA.MB.468.Her2 cells, both G3 and 929 had greater cytotoxicity than 4D5 at higher ratios (p < 0.05). None of the CAR-T cells showed cytotoxicity toward Her2− cells, again confirming CAR-specific activation (Fig. 3). These assays demonstrate that all CAR-T groups were cytotoxic against Her2+ cells, with DARPin CARs generally having higher killing ability than the scFv CAR.
Figure 3.

DARPin CAR-T cells are cytotoxic to Her2-expressing target cells in vitro. CAR-T cells were co-cultured for 24 h with MDA.MB.468, MDA.MB.231, SKOV3, or MDA.MB.468.Her2 target cells at 1:1, 3:1, 5:1, or 10:1 effector-to-target ratios. Cytotoxicity against each target cell type was measured in triplicate and shown in the figure (n = 3, mean ± SEM; NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001).
G3 and 4D5 CAR-T cells inhibit tumor growth and enhance survival in a mouse xenograft model
After the functionality of the CARs was confirmed in vitro, a mouse xenograft model was established to observe the effects in vivo. When subcutaneously injected with SKOV3 cells, all mice developed tumors within 1 week, which were monitored and measured until the end of the study. At 2 weeks, they were injected with either CAR-T cells or NT T cells. Mice treated with 929 CAR-T cells had somewhat delayed tumor growth, but this was not statistically different from the NT group. In contrast, mice treated with either 4D5 or G3 CAR-T cells had dramatically slowed tumor growth (p < 0.01; Fig. 4A), including a few mice with complete tumor regression. Mice were euthanized once the tumor size reached 1,000 mm3. The survival curve also shows slightly extended survival in the 929 group (median survival time of 32 days) over the NT group (median survival time of 30 days). There was significantly enhanced survival in the 4D5 group (median survival time of 38 days), and the longest-surviving mice were in the G3 group (median survival time of 35 days), although differences between the 4D5 and G3 groups were not statistically significant (Fig. 4B). The differences in survival for all CAR-T groups compared to the NT group were statistically significant (p < 0.01 for 4D5 and G3; p < 0.05 for 929). In summary, 4D5 and G3 CAR-T cells effectively slowed overall tumor growth, while 929 CAR-T cells modestly reduced the tumor growth but were not as effective as the other CAR types. Mice treated with 4D5 and G3 CAR-T cells had the most significantly enhanced survival.
Figure 4.
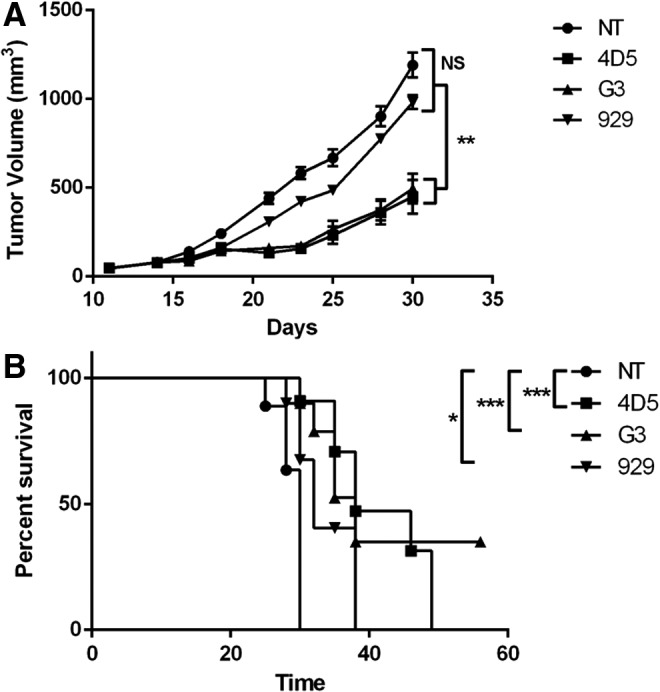
G3 and 4D5 CAR-T cells inhibit tumor growth in vivo. (A) SKOV3 cells were injected subcutaneously into the right flank of NSG mice on day 0. Mice were randomized into four groups of seven mice each and treated with 10 million NT, 4D5, G3, or 929 CAR-T cells on day 14 via tail-vein injection. Tumor size was measured with a fine caliper three times per week and is shown here (n = 7, mean ± SEM; NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001). (B) Mouse survival curve was calculated using the Kaplan–Meier method.
G3 and 4D5 CAR-T cells have greater T-cell infiltration into the tumor
T-cell infiltration into the tumors was compared for the different CAR-T groups. Seven days after CAR-T injection, a subset of mice was sacrificed, and cells from the blood, spleen, and tumor were analyzed with flow cytometry. Representative plots of CD45+CD3+ T cells and CD45+CD8+ T cells from tumor samples are shown in Fig. 5A and B. Significantly higher percentages of CD45+CD3+ T cells were observed in the tumors of mice in the 4D5 and G3 groups compared to the NT group (p < 0.05; Fig. 5C). Interestingly, but in accordance with the tumor growth curve, the infiltration of the 929 CAR-T group was not significantly different from the NT control group. CD45+CD8+ cells were also stained for in the tumor, and similar results were found, with the 4D5 and G3 groups having greater percentages than 929 or NT groups (p < 0.05; Fig. 5D). Percentages of CD45+CD3+ or CD45+CD8+ T cells in the blood and spleen were not statistically different between treatment groups.
Figure 5.

G3 and 4D5 CAR-T cells exhibit better tumor infiltration. Tumors and spleens were made into single-cell suspensions with a 70 μm strainer and were stained with the appropriate fluorescent antibodies. (A and B) Representative plots of CD45+CD3+ and CD45+CD8+ cells isolated from tumor tissue were stained using PacificBlue-conjugated anti-human CD45 and FITC-conjugated anti-human CD3 or CD8, respectively. (C and D) Summarized data from CD45+CD3+ and CD45+CD8+ cells isolated from tumor, blood, and spleen is displayed herein. (E) Quantiative polymerase chain reaction (qPCR) data detecting CAR+ T cells in the tumor. Total mRNA was extracted from tumor samples and was used for qPCR in triplicate, and percentages of CAR+ T cells subsequently were obtained from a standard curve. For all graphs, cells were harvested 7 days after CAR-T cell injection (n = 3, mean ± SEM; NS, not significant; *p < 0.05.
Finally, quantitative PCR was used to detect CAR+ T cells in the tumor as well, and the percentages of CAR+ T cells were significantly greater in all three CAR groups compared to the NT control (p < 0.05; Fig. 5E), with the G3 group having the largest percentage. In conclusion, the ex vivo results were overall in accordance with the tumor growth curve, with 4D5 and G3 CAR-T groups having better T-cell infiltration into the tumor.
Discussion
These findings demonstrate that DARPins offer an alternative to scFvs for the recognition and binding of TAAs in the context of CAR-T therapy. In vitro, both DARPin CAR-T groups performed as well or better than the scFv 4D5 CAR-T in soluble Her2 binding, cytokine release, and cytotoxicity assays. To the authors' knowledge, this is the first study to report their function in vivo. Somewhat unexpectedly, in vivo, only G3 CAR-T cells were as effective as 4D5 CAR-T cells at infiltrating the tumor to slow or reverse growth in an ovarian cancer xenograft model. This is surprising, given that the 929 CAR performed better than the 4D5 CAR in vitro. The most notable difference between 929 and the other Her2-targeting domains is that 929 targets membrane distal domain I on Her2, while G3 and 4D5 target membrane proximal domain IV. Perhaps this is part of the reason why 929 CARs did not perform as well as the other CARs. Binding to membrane proximal Her2 domain IV physically blocks the Her2 receptor from dimerizing with either itself27 or with Her3,28 so targeting domain IV may be advantageous. In addition, many research groups have reported that binding to membrane proximal regions of various TAAs,29 including carcinoembryonic antigen,30,31 CD22,32,33 and ROR1,34 yielded better T-cell activation than those binding to membrane distal regions. One hypothesis is that CAR binding to membrane distal domains does not allow the T cell to form an immunological synapse effectively with the target cell. Further studies are needed to elucidate the combination of properties that make the G3 DARPin a suitable replacement for scFvs in CARs while the 929 DARPin is not as effective.
Although some cancer immunotherapies have shown promising results, one major issue of any protein-based therapeutic is its potential immunogenicity. For example, many scFvs are based on chimeric or humanized antibodies of nonhuman origin, and even fully humanized antibodies can be immunogenic.15 Various antibody mimetic proteins, including adnectins, affibodies, anticalins, and DARPins, have been researched in the hope of circumventing the issue of immunogenicity.35 DARPins are highly stable and do not aggregate, which are characteristics of poorly immunogenic proteins.36 While DARPins have not yet been extensively tested in the clinic, anti-VEGF DARPins were shown to be safe and well-tolerated in patients with age-related macular degeneration in a Phase I/II clinical trial,37 which is encouraging and points to low immunogenicity.
DARPins have been used in a variety of immunotherapy platforms, both as diagnostic and therapeutic tools. Certain anti-Her2 DARPins have been shown to bind to Her2 in paraffin-embedded tissue sections with more specificity than Food and Drug Administration–approved anti-Her2 antibodies, demonstrating their worth in immunohistochemistry and pathology diagnostics.38 DARPins are being investigated as antitumor therapeutics, both on their own and in combination with cytotoxic agents. Bispecific DARPins that bind to two different Her2 domains have been shown to “lock” Her2 into place and prevent phosphorylation, killing Her2-dependent tumor cells with minimal cardiac toxicity.15 Epithelial cell adhesion molecule (EpCAM)-targeting DARPins fused with Pseudomonas aeruginosa exotoxin A efficiently trafficked to and killed EpCAM+ tumor cells.39 Anti-EpCAM DARPins also have delivered small interfering RNA, targeting the anti-apoptotic factor Bcl, to tumor sites.40 DARPins have also been used to improve viral delivery; anti-Her2 DARPins incorporated into a lentiviral vector enhanced transduction of Her2+ tumor tissue.41 It is believed that the use of DARPins can develop further in the field of cancer immunotherapy, specifically as TAA-targeting domains in CAR-engineered immune cells.
One previous study demonstrated that G3 was an effective replacement of the anti-Her2 scFv FRP5 in CARs in vitro.42 They showed enhanced cytokine release and cytotoxicity against Her2-overexpressing cell lines, in accordance with the present in vitro data for G3. This study has expanded the testing of DARPin CARs to include 929, and their in vivo activity in a mouse xenograft model has also been shown. These findings support the additional investigation of the G3 DARPin CAR. There are multiple possible benefits to using DARPins in place of scFvs in CARs. In particular, G3 is about half the size of the scFv 4D5 (G3 has 126 amino acids, while 4D5 has 244), making it easier to insert in lentiviral vectors and leaving more space available for other CAR components, such as additional costimulatory domains. scFvs require both heavy and light chains connected by a linker in order to function in CARs, necessitating a complicated cloning process. DARPins are more streamlined, as they contain a single component necessary for antigen recognition, and are resistant to aggregation, unlike scFvs. As mentioned previously, DARPins are also thought to be less immunogenic than scFvs, potentially increasing the safety of CAR-T therapy in the clinic. Finally, the variety of target molecules to which DARPins can bind is growing rapidly, and DARPins can also be linked to form multivalent adapters,43 which could be used to target multiple TAAs and reduce on-target off-tumor effects of CAR-T therapy. This study tested two Her2-binding DARPins, but many more have been developed, and countless others can be generated from libraries44 to have different properties that may boost their antitumor effects in the context of CAR therapy.
Acknowledgments
This work was supported by National Institutes of Health grants (R01AI068978, R01CA170820, R01EB017206, and P01CA132681) and a translational acceleration grant from the Joint Center for Translational Medicine. Portions of the introduction section of this manuscript are adapted from a poster presented by E.S. and P.W. at the American Institute of Chemical Engineers 2016 annual meeting.
Author Disclosure
The authors declare no competing financial interests.
References
- 1.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov 2013;3:388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Srivistava S, Riddell S. Engineering CAR-T cells: design concepts. Trends Immunol 2015;36:494–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee D, Kochenderfer J, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a Phase 1 dose-escalation trial. Lancet 2015;385:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brentjens R, Davila M, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013;5:177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer J, Dudley M, Kassim S, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015;33:540–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude S, Frey N, Shaw P, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J, Digiusto D, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 2007;4:825–833 [DOI] [PubMed] [Google Scholar]
- 8.Lamers C, Sleijfer S, Vulto A, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006;24:e20–e22 [DOI] [PubMed] [Google Scholar]
- 9.Brown C, Badie B, Barish M, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015;21:4062–4072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katz S, Burga R, McCormack E, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res 2015;21:3149–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pauken K, Wherry E. Overcoming T cell exhaustion in infection and cancer. Trends Immunol 2015;36:265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gargett T, Yu W, Dotti G, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther 2016;24:1135–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai H, Wang Y, Lu X, et al. Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst 2016;108:djv439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long A, Haso W, Shern J, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015;21:581–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pluckthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol 2015;55:489–511 [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Wang Q, Yang S, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol 2009;183:5563–5574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun M, Shi H, Liu C, et al. Construction and evaluation of a novel humanized Her2-specific chimeric receptor. Breast Cancer Res 2014;16:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boersma Y, Pluckthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol 2011;22:849–857 [DOI] [PubMed] [Google Scholar]
- 19.Seeger M, Zbinden R, Flütsch A, et al. Design, construction, and characterization of a second-generation DARP in library with reduced hydrophobicity. Protein Sci 2013;22:1239–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int 2014;2014:852748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Münch R, Mühlebach D, Schaser T, et al. DARPins: an efficient targeting domain for lentiviral vectors. Mol Ther 2011;19:686–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epa V, Dolezal O, Doughty L, et al. Structural model for the interaction of a designed ankyrin repeat protein with the human epidermal growth factor receptor 2. PLoS One 2013;8:e59163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jost C, Schilling J, Tamaskovic R, et al. Structural basis for eliciting a cytotoxic effect in HER2-overexpressing cancer cells via binding to the extracellular domain of HER2. Structure 2013;21:1971–1991 [DOI] [PubMed] [Google Scholar]
- 24.Haas D, Lutzko C, Logan A, et al. The moloney murine leukemia virus repressor binding site represses expression in murine and human hematopoietic stem cells. J Virol 2003;77:9439–9450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dull T, Zuffery R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol 1998;72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han X, Bryson P, Zhao Y, et al. Masked chimeric antigen receptor for tumor-specific activation. Mol Ther 2017;25:274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh R, Narasanna A, Wang S, et al. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res 2011;71:1871–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Junttila T, Akita R, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009;15:429–440 [DOI] [PubMed] [Google Scholar]
- 29.Zahnd C, Kawe M, Stumpp M, et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res 2010;70:1595–1605 [DOI] [PubMed] [Google Scholar]
- 30.Guest R, Hawkins R, Kirillova N, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother 2005;28:203–211 [DOI] [PubMed] [Google Scholar]
- 31.Hombach A, Schildgen V, Heuser C, et al. T cell activation by antibody-like immunoreceptors: the position of the binding epitope within the target molecule determines the efficiency of activation of redirected T cells. J Immunol 2007;178:4650–4657 [DOI] [PubMed] [Google Scholar]
- 32.Haso W, Lee D, Shah N, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013;121:1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James S, Greenberg P, Jensen M, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol 2008;180:7028–7038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hudecek M, Lupo-Stanghellini M, Kosasih P, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res 2013;19:3153–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skerra A. Alternative non-antibody scaffolds for molecular recognition. Curr Opin Biotechnol 2007;18:295–304 [DOI] [PubMed] [Google Scholar]
- 36.Stumpp M, Binz H, Amstutz P. DARPins: a new generation of protein therapeutics. Drug Discov Today 2008;13:695–701 [DOI] [PubMed] [Google Scholar]
- 37.Campochiaro P, Channa R, Berger B, et al. Treatment of diabetic macular edema with a designed ankyrin repeat protein that binds vascular endothelial growth factor: a Phase I/II study. Am J Opthalmol 2013;155:697–704.e2 [DOI] [PubMed] [Google Scholar]
- 38.Theurillat J, Dreier B, Nagy-Davidescu G, et al. Designed ankyrin repeat proteins: a novel tool for testing epidermal growth factor receptor 2 expression in breast cancer. Mod Pathol 2010;23:1289–1297 [DOI] [PubMed] [Google Scholar]
- 39.Martin-Killias P, Stefan N, Rothschild S, et al. A novel fusion toxin derived from an EpCAM-specific designed ankyrin repeat protein has potent antitumor activity. Clin Cancer Res 2011;17:100–110 [DOI] [PubMed] [Google Scholar]
- 40.Winkler J, Martin-Killias P, Pluckthun A, et al. EpCAM-targeted delivery of nanocomplexed siRNA to tumor cells with designed ankyrin repeat proteins. Mol Cancer Ther 2009;9:2674–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Münch R, Janicki H, Völker I, et al. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol Ther 2013;21:109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammill J, VanSeggelen H, Helsen C, et al. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J Immunother Cancer 2015;3:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dreier B, Mikheeva G, Belousova N, et al. Her2-specific multivalent adapters confer designed tropism to adenovirus for gene targeting. J Mol Biol 2011;405:410–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munch R, Muth A, Muik A, et al. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat Commun 2015;6:6246. [DOI] [PubMed] [Google Scholar]