

Abstract
In higher eukaryotes, the integration of signals triggered in response to certain types of stress can result in programmed cell death. Central to these events is the sequential activation of a cascade of proteinases known as caspases. The final activated effector caspases of this cascade digest a number of cellular proteins, in some cases increasing their enzymatic activity, in others destroying their function. Of the proteins shown to be targets for caspase-mediated proteolysis, a surprisingly large proportion are proteins involved in the signalling or repair of DNA damage. Here we investigate whether BLM, the product of the gene mutated in Bloom’s syndrome, a human autosomal disease characterised by cancer predisposition and sunlight sensitivity, is cleaved during apoptosis. BLM interacts with topoisomerase IIIα and has been proposed to play an important role in maintaining genomic integrity through its roles in DNA repair and replication. We show that BLM is cleaved during apoptosis by caspase-3 and reveal that the main cleavage site is located at the junction between the N-terminal and central helicase domains of BLM. Proteolytic cleavage by caspase-3 produces a 120 kDa fragment, which contains the intact helicase domain and three smaller fragments, the relative amounts of which depend on time of incubation with caspase-3. The 120 kDa fragment retains the helicase activity of the intact BLM protein. However, its interaction with topoisomerase IIIα is severely impaired. Since the BLM–topoisomerase interaction is believed to be necessary for many of the replication and recombination functions of BLM, we suggest that caspase-3 cleavage of BLM could alter the localisation and/or function of BLM and that these changes may be important in the process of apoptosis.
INTRODUCTION
Apoptosis, or programmed cell death, is an important biological process by which individual cells or groups of cells are destroyed in a regulated manner. The process is of great importance both during development and adult life (1,2). A variety of signals can induce apoptosis. These signals can be extracellular, for example produced by tumour necrosis factor α (TNF-α) during inflammatory responses, or intracellular, for example DNA damage. Although a multitude of signals can initiate the process of apoptosis, all the signalling pathways finally converge on the central executioners of apoptosis: the caspases, a class of cysteine proteases that act C-terminal to an aspartate residue.
Caspases are present in normal cells as inactive zymogens that become active only upon proteolysis by other caspases. Thus, reminiscent of the complement system, a series of caspases form a proteolytic cascade that serves to transmit and amplify death signals. Upstream activator caspases, such as caspase-8 and caspase-9, integrate the various different apoptotic signals (3,4). Downstream effector caspases (for example, caspase-3, caspase-6 and caspase-7) are cleaved by activator caspases and in turn cleave a series of cellular proteins. It is these latter proteolytic cleavages that are believed to result in the disruption of normal cell function and, ultimately, cell death.
An important characteristic of the caspases is that they perform proteolysis at only a limited number of sites within their targets and thus do not totally degrade the protein substrate (3,5). Indeed, in several cases, caspase cleavage activates the target. For example, caspase-activated deoxyribonucleose (CAD) is responsible for DNA degradation during apoptosis (6,7) and the kinase MEKK-1, which is activated by caspase cleavage, is involved in the JNK signal transduction pathway (8), an important pathway involved in the general stress response. Other proteins are inactivated by the action of caspases, for example structural proteins such as gelsolin (9). The process of apoptosis, then, is a complex collection of events that ultimately results in cellular destruction. To understand fully how and why a cell dies, we therefore need to identify and understand the functions of the downstream targets of the caspases.
In recent years, several proteins involved in the detection, signalling and/or repair of DNA damage have been identified as caspase targets. These include poly(ADP-ribose) polymerase (PARP) (10,11); poly(ADP-ribose) glycohydrolase (PARG) (12); the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) (13,14); and ATM, a kinase that is the product of the gene defective in ataxia telangiectasia (15). All these enzymes are inactivated by caspase action. This ensures that the systems that normally act to repair or signal the presence of DNA damage are not active at the same time as CAD, which damages DNA as part of the cell suicide mechanism.
We were interested in studying the effect of apoptosis on another protein involved in maintaining genomic integrity, the product of the gene deficient in Bloom’s syndrome (BS). BS is a rare autosomal recessive disease in which affected individuals have a small stature, are sub-fertile and immunodeficient, have sunlight-sensitive skin and a predisposition to cancer, both solid tumours and leukaemias. The high incidence of malignancies has been associated with genomic instability in BS cells. This instability is manifested by a high frequency of chromosome breaks and exchanges as well as frequent sister chromatid exchanges (16). The gene mutated in BS, BLM, encodes a 160 kDa protein containing a central RecQ-like helicase domain (17). The helicase region contains seven motifs conserved in other members of the helicase family and a DEXH box. The helicase domain is flanked by extended N- and C-terminal regions, which are poorly conserved but are also present in other members of the same subfamily of helicases. These include the genes mutated in two other human autosomal diseases, WRN, which is mutated in Werner’s syndrome (18), and RECQ4, which is mutated in Rothmund–Thomson syndrome (19). Also in the RecQ helicase family are two yeast genes, Saccharomyces cerevisiae SGS1 (20,21) and Schizosaccharomyces pombe rqh1+ (22,23). Individuals with Werner’s syndrome and Rothmund–Thomson syndrome, like those with BS, show chromosomal instability and have a high incidence of certain types of cancer.
Genetic studies of SGS1 and rqh1+ have shed some light on the in vivo roles of the RecQ family of helicases. These include functions in recombination, replication, ageing and checkpoint control (reviewed in 24,25). Furthermore, several models have been proposed to explain how deficiencies in BLM protein function could account for the genomic instability observed in BS cells (24,26). Importantly, the in vivo function of BLM and of its yeast homologue Sgs1p are linked to another class of enzymes: the topoisomerases. Yeast sgs1 and topoisomerase III (top3) single mutants both have an increased frequency of homologous recombination between repetitive DNA sequences (20) and elsewhere in the genome (27) when compared with wild-type cells. top3 deletion mutants also grow slowly due to an accumulation of cells in the late S/G2 phases of the cell cycle. However, mutations in SGS1 are able to rescue the slow growth phenotype of a top3 deletion mutant, indicating a functional connection between the proteins (20). Moreover, yeast two-hybrid and biochemical data show a physical interaction between Sgs1p and several topoisomerases (20,28). In addition, interspecies cross-functionality between BLM and SGS1 has been described, indicating that they are real homologues. BLM is able to reduce the hyper-recombination phenotype of sgs1 mutants and it can restore the slow growth phenotype of a top3 sgs1 double mutant (29). Notably, BLM protein has also been shown to interact physically with human topoisomerase IIIα in normal somatic human cells (30,31) and in spermatocytes that are undergoing meiotic recombination (31).
In this paper, we study the effects of apoptosis on BLM. We show that during apoptosis BLM protein is cleaved in its N-terminal region, giving rise to an intact RecQ helicase domain with a C-terminal extension. This product retains helicase activity, indicating that the N-terminus is not essential for this activity, but its interaction with topoisomerase IIIα is severely diminished, providing a functional role for this cleavage during apoptosis.
MATERIALS AND METHODS
Reagents
All reagents were obtained from Sigma (Poole, UK) unless otherwise stated. Staurosporine was from Alexis Corp. UK (Nottingham, UK). Anti-C-terminal BLM, anti-N-terminal BLM and anti-topoisomerase IIIα antibodies have been described previously (30,32). The anti-PARP monoclonal antibody (isotype immunoglobulin G1) was obtained from Serotec (Oxford, UK). HeLa nuclear extracts were purchased from Computer Cell Culture Centre (Mons, Belgium).
Cell lines and apoptosis induction
The human lymphoblastoid cell line HL-60 was grown in RPMI 1640 medium supplemented with 15% fetal calf serum, 100 IU/ml penicilin and 10 IU/ml streptomycin at 37°C with 5% CO2. HeLa cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 100 IU/ml penicillin and 10 IU/ml streptomycin at 37°C with 5% CO2. For induction of apoptosis, HL-60 cells were treated with 68 µM etoposide for various times. Apoptosis in HeLa cells was induced with 10 ng/ml TNF-α (Peprotech EC, London, UK) and 20 µg/ml cycloheximide.
Extraction and analysis of apoptotic DNA
Approximately 6 × 106 cells were harvested at different times after etoposide treatment. DNA was extracted using DNA-zol (Life Technologies, Paisley, UK) according to the manufacturer’s instructions. Isolated DNA was electrophoresed (∼15 µg/lane) on a 1.5% agarose gel in Tris–acetate–EDTA buffer at 50 mA. Fluorescence-activated cell sorting (FACS) analysis was performed with a Becton Dickinson FACSort apparatus to determine the percentage of sub-G1 population, a measure indicative of the apoptotic population. Aproximately 106 HeLa or HL-60 cells were collected, washed twice in phosphate-buffered saline (PBS) and fixed with ice-cold 70% methanol for at least 1 h. After washing in PBS, cells were suspended in PBS containing 1% Tween, 20 µg/ml propidium iodide and 40 µg/ml RNase A and incubated for 30 min at room temperature before FACS analysis.
Cell extract preparation
Approximately 1 × 107 HL-60 cells or 5 × 106 HeLa cells were used to prepare protein extracts from control and drug-treated cells for western blot analysis. HeLa cells were trypsinised and HL-60 cells were collected directly from suspension culture. The cells were washed in PBS and lysed in 150 µl of a buffer containing 6 M urea, 20 mM Tris–HCl, pH 6.8, and 50 µg/ml ethidium bromide. Extracts were sonicated and centrifuged for 10 min at 13 000 g before western blot analysis (100 µg extracted protein/lane).
Nuclear extracts from control HL-60 cells or cells treated with 68 µM etoposide for 5 h were prepared as described (33). Briefly, ∼2 × 107 cells were harvested and washed twice with PBS. Cells were resuspended in five packed cell volumes of buffer A [10 mM HEPES–KOH, pH 7.6, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 10 nM microsystin (Alexis Corporation), 1 mM sodium orthovanadate and complete protease inhibitor mixture (Roche Diagnostics, Lewes, UK)] and were incubated on ice for 2 min. After centrifugation at 28 000 g for 10 min at 4°C, cells were resuspended in two packed cell volumes of buffer A and were disrupted by 10 strokes in a Dounce homogeniser. Nuclei were pelleted by centrifugation at 2800 g for 10 min at 4°C and a 0.11 volume of buffer B (0.3 M HEPES–KOH, pH 7.6, 1.4 M KCl, 30 mM MgCl2) was added. Nuclear extracts were obtained by adding an equal volume of buffer C (10 mM HEPES–KOH, pH 7.6, 5 mM MgCl2, 300 mM NaCl, 1 mM dithiothreitol (DTT), 0.2 mM EDTA, 50 µg/ml ethidium bromide, 10 nM microsystin, 1 mM sodium orthovanadate and complete protease inhibitor mixture) and gently stirring the solution for 30 min at 4°C on a rotating wheel. Nuclear debris was removed by centrifugation at 13 000 g for 10 min at 4°C.
In vitro cleavage of BLM protein
HeLa nuclear extracts or purified recombinant BLM (described in 34) were incubated with different amounts of caspase-3 in a final buffer composition of 20 mM HEPES, pH 7.6, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 20 mM DTT and 5 mM EDTA at 37°C for 30 min. Reactions were terminated by the addition of an equal volume of SDS-protein sample buffer to the reaction mixture.
Helicase assays
Helicase assays were performed as described (34). Briefly, purified recombinant BLM treated with caspase-3 or mock-treated was incubated with a 3′-labelled oligonucleotide annealed to single-stranded M13mp18 DNA. The reaction was carried out in a 20 µl volume containing 50 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 5 mM ATP, 100 µg/ml bovine serum albumin, 50 mM NaCl, ∼50 ng labelled DNA substrate and increasing amounts of BLM protein. Samples were incubated at 37°C for 30 min and were run on a 12% non-denaturing polyacrylamide gel at 20°C.
Inmunoprecipitations and western blot analysis
HL-60 nuclear cell extracts (200 µg protein) were mixed with anti-C-terminal BLM antibody (5–10 µl of serum) in a 200 µl volume reaction containing 10 mM HEPES–KOH, pH 7.5, 10 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100, 1 mM DTT, 50 µg/ml ethidium bromide, 10 nM microsystin, 1 mM sodium orthovanadate and complete protease inhibitor mixture (buffer D). Reactions were incubated for 1 h at 4°C on a rotating wheel. Antibody was captured by adding 25 µl of a slurry of Dynabeads M-280 Sheep Anti-Rabbit IgG (Dynal Biotech, Oslo, Norway) for 45 min at 4°C with agitation on a rotating wheel. Dynabeads were separated using the appropriate magnet (Dynal biotech) and washed four times with 300 µl of buffer D before being boiled in SDS-protein sample buffer. Solubilised samples were then subjected to electrophoresis in 8 or 12% SDS–polyacrylamide gels, transfered to nitrocellulose membranes and probed with the indicated antibodies. Western immunoblot analyses were performed as described previously (35) and were developed with the Enhanced Chemiluminescence reagent (Nycomed Amersham, Little Chalfont, UK) according to the manufacturer’s instructions.
In vitro binding assay
Different amounts of recombinant BLM, BLMΔN(212–1417) and bovine serum albumin (BSA) were blotted on a nitrocellulose membrane (MSI NitroBind; Osmonics, Minnetonka, USA). BLMΔN(212–1417) was overexpressed in S.cerevisiae and purified by the method described (34). After blocking for 1 h at room temperature using TBST with 5% milk, the membrane was incubated for 3 h at 4°C with different proteins that were 35S-labelled using a TNT T7 quick coupled transcription/translation system (Promega, Madison, WI). The constructs used in the coupled transcription/translation system were described previously (30,36). After brief washes with TBST, membranes were dried and exposed to a phosophorimager (Molecular Dynamics, Sunnyvale, CA).
Microsequencing
Recombinant purified BLM protein (5 µg) was incubated with 300 ng caspase-3 for 30 min at 37°C. Samples were resolved by SDS–PAGE on a 10% gel and then transferred to a polyvinylidine difluoride membrane. The membrane was stained with 0.1% Coomassie blue R250 in 50% methanol and 1% acetic acid. After destaining with 50% methanol, the ∼120 kDa specific band in the cleaved sample was excised and sequenced by Edman degradation using an Applied Biosystems Procise sequencer according to the manufacturer’s protocols.
RESULTS
BLM is cleaved during apoptosis
Studies in human cells and yeast indicate that BLM is a multidomain protein involved in the maintenance of genomic integrity (24,26). To investigate whether cellular levels of BLM protein change after DNA damage, we monitored BLM levels in the human lymphocyte-derived cell line HL-60 before and after treatment with radiomimetic drugs by use of an antibody raised against the most C-terminal 380 amino acid residues of the protein (32). Notably, these studies revealed that BLM underwent proteolysis at a limited number of sites (data not shown). As this was a limited proteolyis and because other DNA repair-related proteins have been shown to undergo limited degradation during apoptosis, we hypothesised that the digestion of BLM after radiomimetic drug treatment could be part of the cellular apoptotic response. To test this hypothesis, we used a model system developed by Han et al. (37,38) in which HL-60 cells are induced to enter apoptosis by treatment with the topoisomerase II inhibitor etoposide. Figure 1A (top) shows a western blot of equal amounts of HL-60 whole cell extracts taken after different periods of incubation with etoposide that were probed with the anti-C-terminal BLM antibody. In untreated cells, BLM protein ran as a single band of ∼190 kDa. As etoposide treatment time increased, the full-length protein was specifically and progressively degraded to a single ∼120 kDa band, indicating that precise cleavage of BLM rather than non-specific degradation had occurred. Moreover, after 5 h of etoposide treatment, almost all of the 190 kDa band was degraded to the lower molecular weight form. When we reprobed the blot with antibodies against PARP, a protein known to be cleaved during apoptosis (11), proteolysis of PARP occurred with similar (or possibly slightly delayed) kinetics to those for BLM (Fig. 1A, compare top and middle). Because Ku70 is not degraded during apoptosis (14), we also reprobed the blot with an anti-Ku70 antibody as a loading control (bottom panel).
Figure 1.

BLM is cleaved during apoptosis in HL-60 cells treated with etoposide. HL-60 cells were grown in the presence of 68 µM etoposide and collected at the indicated times. (A) Cell extracts were prepared and 150 µg protein were loaded/lane. After electrophoresis, the proteins were transferred to a nitrocellulose membrane and analysed by sequentially probing with anti-C-terminal BLM, anti-PARP and anti-Ku70 antibodies, as indicated. (B) Agarose gel electrophoresis (1.5%) of genomic DNA from cells treated as in (A).
To demonstrate that apoptosis was indeed occurring in these cells, we isolated genomic DNA from the control and etoposide-treated cells and analysed it by agarose gel electrophoresis. No genomic DNA degradation fragments were observed in the untreated cells, but a DNA degradation ladder typical of apoptosis became obvious as the incubation time with the drug increased (Fig. 1B). Finally, to gain a quantitative estimate of the percentage of apoptotic cells at the time points studied, we measured cellular DNA content by flow cytometry after staining the cells with propidium iodide. In untreated cells and cells treated with etoposide for 1 h, only 4.2% of the cells had a sub-G1 DNA content. This indicated that little DNA degradation had occurred at times 0 and 1 h. By 2 h, 10.9% of the cells had a sub-G1 DNA content and this percentage continued to rise such that by 5 h 47% of the cells had a sub-G1 DNA content. These results, taken with those shown in Figure 1, indicate that BLM is specifically cleaved during apoptosis.
To see whether BLM cleavage was a general feature of the apoptotic response, we treated HL-60 cells with other agents known to induce apoptosis. When cells were treated with alcohol or staurosporin, a similar 120 kDa band to that seen after treatment with etoposide was observed on western blots probed with the anti-BLM C-terminal antibody (data not shown). We also studied BLM cleavage in another cell line in response to apoptosis-inducing agents. When HeLa cells were treated with TNF-α and the protein synthesis inhibitor cycloheximide to trigger apoptosis (15), once again a 120 kDa proteolytic product appeared (Fig. 2). These results indicate that BLM proteolysis in response to apoptosis-inducing agents is not cell line or agent specific.
Figure 2.
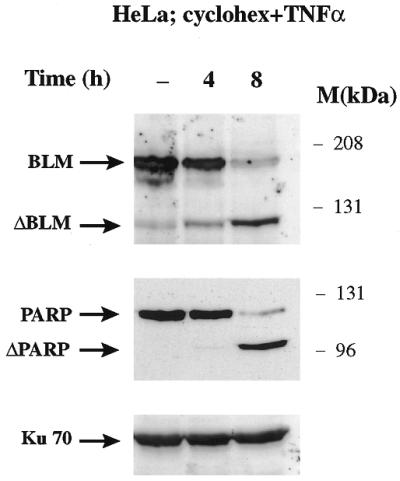
BLM is cleaved in HeLa cells induced to apoptosis. HeLa cells were treated with TNF-α (10 ng/ml) and cycloheximide (20 µM) at the indicated times. Protein extracts were prepared, electrophoresed and then analysed by western blotting using anti-C-terminal BLM, anti-PARP and anti-Ku70 antibodies, as indicated.
BLM is cleaved by caspase-3 in its N-terminal region
The above results indicate that BLM is cleaved during apoptosis. To characterise this cleavage, we next investigated whether caspase-3, one of the best-studied effector caspases and the one responsible for cleavage of other DNA repair proteins, including PARP, DNA-PKcs and ATM (10,11,13–15), was capable of producing the observed cleavage. To do this, we incubated HeLa nuclear extracts or recombinant BLM with purified recombinant caspase-3. Figure 3 shows that both the BLM present in extracts and recombinant BLM were cleaved by caspase-3. Addition of acetyl-Asp-Glu-Val-Asp-aldehyde, a specific caspase-3 inhibitor, prevented cleavage of BLM in HeLa nuclear extracts (Fig. 3) and cleavage of recombinant BLM (data not shown). Moreover, the cleavage product in HeLa nuclear extract had the same electrophoretic mobility as the band seen in HeLa cells treated with TNF-α and cycloheximide for 8 h (Fig. 3) and HL-60 cells treated with etoposide for 5 h (data not shown). The fragment derived from treating recombinant BLM with caspase-3 migrated slightly slower, most likely as a consequence of the hexahistidine tag that had been introduced at the C-terminus of BLM to facilitate its purification (34). Consistent with the idea that caspase-3 was responsible for the apoptosis-induced digestion of BLM, we found that treatment of caspase-3-deficient MCF-7 cells (15) with TNF-α and cycloheximide led to apoptosis but no discernible BLM cleavage (data not shown).
Figure 3.

BLM is cleaved by caspase-3 in vitro. Western blot analysis of control HeLa extract (lane 1), HeLa extract treated with TNF-α (10 ng/ml) and cycloheximide (20 µM) for 8 h (lane 2); HeLa nuclear extract (HNE) (50 µg total protein/lane) treated with no caspase-3 (lane 3) and with 30 ng caspase-3 in the absence (lane 4) or presence (lane 5) of the caspase inhibitor acetyl-Asp-Glu-Val-Asp-aldehyde; recombinant BLM (10 ng) mock treated (–) (lane 6) or treated (lane 7) with 30 ng caspase-3. Treatment with caspase-3 was for 30 min at 30°C. The western blot was probed with anti-C terminal BLM or anti-PARP antibodies, as indicated.
In the above experiments, the anti-BLM antibody used had been raised against the most C-terminal 380 amino acid residues of the BLM protein, meaning that it was not possible from these data alone to establish where the site(s) of BLM cleavage might be. Notably, when we probed the western blot shown in Figure 3 with an anti-histidine tag antibody, we detected both the uncleaved and cleaved versions of recombinant BLM (data not shown). Since BLM was tagged on the C-terminus, this indicated that the cleavage site(s) must be in the N-terminal part of the protein. To further map the caspase-3 cleavage site(s) in BLM, we induced HL-60 cells to undergo apoptosis by treating them with etoposide then probed the resultant western blots with an antibody raised against the N-terminal 449 amino acid residues of BLM (30). As shown in Figure 4A, this antibody recognised undigested BLM in control cells but, upon incubation with etoposide for increasing times, at least two new bands appeared (marked with asterisks): one of ∼40 kDa at intermediate times and another of ∼25 kDa after longer incubation with the drug.
Figure 4.

In vivo and in vitro analysis reveals several cleavage sites at the N-terminal end of BLM. (A) HL-60 cells were grown in the presence of 68 µM etoposide and were collected at the indicated times. Cell extracts were prepared and 150 µg protein loaded/lane. Western blot analysis was carried out by probing with anti-N-terminal BLM, anti-PARP and anti-Ku70 antibodies. (B) Recombinant BLM protein or HeLa nuclear extract was incubated for the indicated times at 30°C with 30 ng caspase-3. After electrophoresis and transfer to nitrocellulose, the resulting blot was probed with an anti-N-terminal BLM antibody. (C) Control HL-60 cells or cells grown in the presence of 68 µM etoposide for 4 h were analysed in a western blot together with control HeLa nuclear extract (50 µg) and extract that had been incubated in the presence of 30 ng caspase-3 at 30°C for 30 min. The blot was probed with an anti-N-terminal BLM antibody. Arrowheads indicate the main degradation products. (D) Location of the main caspase-3 cleavage site in BLM. The two hatched boxes at the N-terminal end of the protein indicate acidic regions, the central black box indicates the helicase domain and the grey C-terminal box indicates the nuclear localisation sequence. The numbers indicate the amino acid position around the cleavage site. The arrow shows the actual cleavage site.
Next, we treated recombinant BLM or HeLa cell nuclear extracts with recombinant caspase-3 and analysed BLM cleavage by western blot analysis with antibodies directed against the N-terminal region of the protein. This revealed progressive degradation over time (Fig. 4B). At very short treatment times (2 and 5 min), bands of ∼70 and ∼40 kDa were seen. By 10 min incubation, the amount of the 70 kDa band had decreased, with a concomitant increase in the 40 kDa band. Finally, when the caspase-3 treatment was extended to 30 min, the main products were ∼40 and ∼25 kDa (indicated by arrowheads in Fig. 4C), the same size bands as those observed in vivo (Fig. 4A and the first two panels of Fig. 4C). These results suggest that BLM is preferentially cleaved at ∼70 kDa from the N-terminal end of the protein and that subsequent cleavage of this fragment produces smaller fragments of ∼25 and ∼40 kDa. Our failure to detect the 70 kDa band in vivo may indicate that it is unstable and is rapidly degraded to the 40 kDa fragment.
To characterise the principal BLM cleavage site at the amino acid level, we incubated 5 µg recombinant BLM with an excess of caspase-3 and carried out N-terminal sequencing of the 120 kDa C-terminal fragment (see Materials and Methods for preparation details). The sequenced amino acids correspond to the cleavage site indicated in Figure 4D by the vertical arrow. Specifically, proteolytic cleavage takes place between amino acid residues 436 and 437, a cleavage which would produce an N-terminal fragment of ∼50 kDa. This probably corresponds to the ∼70 kDa band seen by western blot analysis, since this fragment includes an acidic region that might cause anomalous migration during electrophoresis on an SDS–polyacrylamide gel (acidic regions are known to cause anomalous migration on such gels; 39). As shown in Figure 4C, cleavage is immediately C-terminal to an aspartate residue in the sequence DSLD, which conforms reasonably well to the canonical site for caspase-3 (DEXD) and to known target sites for caspase-3 in other DNA repair proteins such as DNA-PKcs and ATM (13–15). According to our experimental data, there should be at least two additional cleavage sites for caspase-3 within the 50 kDa N-terminal region. Proteolysis at the first site should produce a fragment with SDS–PAGE mobility consistent with a mass of ∼40 kDa. A further cleavage within this fragment should then produce the fragment with an apparent molecular weight of 25 kDa. We were unable to map these sites by N-terminal sequencing because the ∼25 kDa band co-migrates on gels with other contaminating bands and, under the digestion conditions used, the 40 kDa band had been further cut to yield the 25 kDa band. However, inspection of the published amino acid sequence of BLM revealed several consensus caspase-3 sites in this region, including a DERD/N site at positions 288–292, which could be responsible for the 40 kDa fragment observed both in vitro and in vivo. The sequence at position 163–171 is DWDDMDDFD, including several caspase-3 putative cleavage sites, and could be responsible for the 25 kDa band observed.
BLM helicase activity is not affected by cleavage
Since the activities of several other DNA repair-associated proteins have been reported to be inhibited as a consequence of caspase-3 cleavage, we were prompted to test whether this was also the case for BLM. One of the salient features of BLM is a RecQ-like helicase domain in the centre of the protein (Fig. 4D). The main cleavage site in BLM according to our analysis is located between the N-terminal region and this helicase central domain, raising the possibility that this proteolysis event could modulate its helicase function. To see whether this was the case, we compared the helicase activity of purified recombinant BLM that had been incubated with caspase-3 with that of a mock-treated control sample. Figure 5 shows that the full-length protein and the cleaved protein had similar helicase activities. Therefore, in contrast with other DNA repair proteins that are cleaved during apoptosis, the intrinsic biochemical enzymatic activity of BLM is not markedly impaired following proteolysis by caspase-3.
Figure 5.
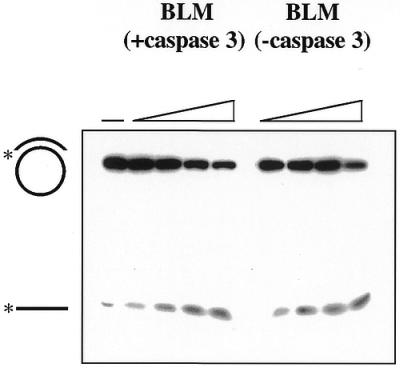
Caspase-3-treated BLM retains helicase activity. Helicase assays were performed with either caspase 3-treated or mock-treated recombinant BLM as described in Materials and Methods. The amount of BLM included in each reaction ranged from 7.5 to 50 ng. Positions of the starting substrate and the unwound oligonucleotide product are indicated on the left.
The BLM–topoisomerase IIIα interaction is affected by apoptosis
To see whether caspase-3-mediated cleavage of BLM during apoptosis alters the interaction between BLM and topoisomerase IIIα, we immunoprecipitated BLM with the anti-BLM C-terminal antibody from untreated HL-60 cells and from cells that had been treated with etoposide. The immunoprecipitates were then subjected to SDS–PAGE followed by sequential western immunoblot analysis with anti-BLM C-terminal and anti-topoisomerase IIIα antibodies (30). Figure 6 shows that the amount of topoisomerase IIIα associated with BLM was greater for the control extracts than for extracts of the cells that had been treated with etoposide, whereas the amount of BLM immunoprecipitated did not change significantly. This result, therefore, suggests that the interaction of BLM with topoisomerase IIIα is compromised by removal of the N-terminus of BLM.
Figure 6.
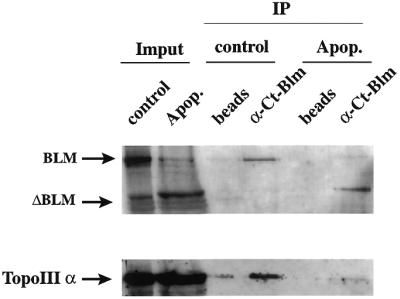
The main apoptotic degradation product of BLM shows impaired topoisomerase IIIα interaction. Nuclear extracts were made from control HL-60 cells or from cells which had been treated with 68 µM etoposide for 5 h. Nuclear extracts (150 µg) were incubated in the presence of 50 µg anti-C-terminal BLM antibody for 1 h at 4°C and immunoprecipitated using Dynabeads coupled to anti-rabbit antibodies. Control experiments were carried out using uncoupled beads. The western blot was probed with anti-C-terminal-BLM and anti-topoisomerase IIIα antibodies as indicated in the figure.
To further demonstrate that the reduction in BLM–topoisomerase IIIα interaction was due to lack of the BLM N-terminus, we carried out an in vitro binding assay using a BLM recombinant fragment lacking the first 212 amino acids (BLMΔN) and full-length BLM as a control. Purified BLMΔN and BLM were blotted into a membrane and incubated with the indicated radiolabelled proteins (Fig 7A). When topoisomerase IIIα was used as a probe, a strong reduction in its binding capacity to BLMΔN was observed in comparison to the full-length BLM protein (Fig. 7A, left). As positive and negative controls, a protein that interacts with the C-terminal region of BLM (MLH1; Fig 7A, centre) and a C-terminal version of it (MLH1ΔC; Fig 7A, right) were also incubated with the blotted proteins, respectively (G.Pedrazzi and I.Stagljar, manuscript in preparation). hMLH1 binds with the same affinity to BLMΔN and BLM, and deletion of its C-terminus abolishes binding in both cases, indicating that the effect observed with topoisomerase IIIα is specific. Figure 7B shows the purity of all the radiolabelled proteins that were used in the in vitro binding assay in Figure 7A.
Figure 7.

The BLM N-terminus is important for topoisomerase IIIα interaction. (A) Aliquots of 0, 100, 200 and 400 ng full-length BLM, an N-terminal truncation (BLMΔN) and BSA were blotted on a nitrocellulose membrane and probed against the indicated radiolabelled proteins obtained in an in vitro coupled transcription/translation system as indicated in Materials and Methods. (B) SDS–PAGE electrophoresis analysis of the radiolabelled proteins used for the assay.
DISCUSSION
Programmed cell death is a complex process in which the final steps involve activation of specific proteases (caspases) that cleave a select range of proteins, resulting in dysregulation of their function. The identification of caspase substrates is therefore of fundamental importance if we are to fully understand the mechanism and consequences of apoptosis. To date, only a relatively small number of substrates (∼20 for caspase-3 and caspase-6) has been identified. In this study we have identified a new target for caspase-3, the BLM protein, which is mutated in individuals with BS. Specifically, by use of antibodies against the C- and N-terminal ends of BLM, we have shown that BLM is specifically cleaved during apoptosis near its N-terminus. Notably, the time course of BLM cleavage is at least as rapid as that of the well-studied caspase substrate PARP, meaning that BLM proteolysis is an early marker for apoptotic progression. Furthermore, we have shown that BLM cleavage occurs in response to different apoptotic signals and in both HL-60 cells and HeLa cells, suggesting that it is likely to be a general feature of the apoptotic response.
By the use of recombinant caspase-3, we have shown that this enzyme generates BLM products that are electrophoretically indistinguishable from the products arising within apoptotic cells. Furthermore, we then showed that the main caspase-3 cleavage site in BLM is between amino acid residues 436 and 437. The sequence of the cleavage site thus identified is in agreement with the consensus cleavage site for caspase-3 (40,41) and is similar to those found in the DNA-PKcs and ATM proteins (13–15). Cleavage at this site uncouples the helicase and C-terminal domains of BLM from the N-terminus of the protein. From our western blot analysis, we also know that two other caspase-3 sites must exist in the N-terminal region of BLM in order to produce the observed fragments of ∼25 and ∼40 kDa. Our in vivo and in vitro data suggest that cleavage at the main site between residues 436 and 437 is the first event after induction of apoptosis, and that degradation of the 70 kDa N-terminal fragment occurs later to yield the other two products. We suggest that the additional N-terminal cleavage sites could provide a mechanism to ensure complete BLM cleavage in vivo even if the main site is inaccessible, for example through it being involved in interactions with another protein or with another part of the BLM polypeptide. Alternatively, or in addition, cleavage at these other sites could be required for the disruption of folding within the N-terminal domain of BLM and to prevent the N-terminal domain reassociating with the C-terminal domain to inappropriately reconstitute BLM activity. Finally in this regard, it is interesting to note that the secondary cleavage events only occur at late time points after the apoptotic cascade is triggered, raising the possibility that BLM function is modulated in two distinct, sequential ways during apoptotic progression.
Recently, another group reported that BLM is cleaved during apoptosis (42). Our data both confirm and extend their data. In our in vivo studies, we used a model in which apoptosis was triggered by DNA damage (etoposide), whereas Bischoff et al. (42) looked at Fas-induced apoptosis in Jurkat cells. Taken together, the available data give strong support to the proposition that cleavage of BLM is a common event in apoptosis independent of cell type or apoptotic stimulus. In agreement with the work of Bischoff et al. (42), our in vitro data indicate that caspase-3 is the protease responsible for BLM cleavage. Interestingly, however, we mapped the main cleavage site to amino acid position 436–437, whereas Bischoff et al. (42) report that cleavage occurs at position 415–416. These two sites are very close to one another and might constitute complementary sites for BLM degradation in the N-terminal portion of the protein. If cleavage does indeed occur at both of these sites, the main C-terminal product resulting from BLM cleavage would still start at position 437. Unlike Bischoff et al. (42), we observed two additional caspase-3-mediated cleavage sites for BLM, both in vivo and in vitro. Since we found that cleavage at these sites is only evident at late times after etoposide treatment or when caspase-3 digestion in vitro is allowed to go to completion, we suspect that the conditions used by Bischoff et al. (42) may not have been sufficient to lead to production of these small fragments.
Many of the other proteins involved in the repair or signalling of DNA damage that are cleaved during apoptosis have been shown to lose activity after caspase-3 cleavage. For example, DNA-PKcs and ATM lose their protein kinase activity (13–15). Similarly, PARP loses its poly(ADP-ribose) polymerasation activity (10,11) and PARG loses its poly(ADP-ribose) glycohydrolase activity (12). Such inactivations could ensure that repair activities are not inappropriately triggered in response to CAD-mediated degradation of genomic DNA. Both ourselves and Bischoff et al. have observed no change in the in vitro helicase activity of BLM after caspase-3 treatment, however, which is consistent with the sites of BLM cleavage not being within the defined helicase domain of the protein. Instead, and as discussed further below, our data suggest that the aspect of BLM function that is modulated by caspase cleavage is its interaction with DNA topoisomerase IIIα.
We have found that the ability of BLM to interact with topoisomerase IIIα, as assessed by co-immunoprecipitation analysis, is abrogated when BLM has been cleaved during apoptosis. Moreover, we have demonstrated that the first 212 amino acids of BLM are important for topoisomerase IIIα interaction using an in vitro binding assay. Taken together with the proteolytic site mapping data, this suggests that the N-terminal region of BLM plays a key role in mediating the topoisomerase IIIα interaction. Furthermore, in vitro data using the far western approach have suggested that both the N- and C-terminal portions of BLM can interact with human topoisomerase IIIα (30), raising the possibility that caspase cleavage in the N-terminal region of BLM also causes structural alterations in the C-terminal part of the protein. It is interesting to note that the N-terminal domain of Sgs1p, the yeast homologue of BLM, also interacts with Top3p and the yeast two-hybrid approach has indicated that the first 500 amino acid residues of Sgs1p are required for this interaction (20), while biochemical and genetic approaches have further narrowed the interaction site to the N-terminal 107 residues (28). Since the biological functions of BLM and Sgs1p have been strongly associated with an ability to interact with topoisomerase III, it seems likely that ablation of the BLM–topoisomerase IIIα interaction during apoptosis will impair many or all of the biological functions of BLM, including its roles in replication and recombination. In this regard, it may be relevant to note that BLM knockout mice show increased apoptosis during development (43). It will clearly be of great interest to further define the BLM–topoisomerase interaction and determine how this and possibly other functions of BLM are altered during apoptosis. It is tempting to speculate that such investigations will not only inform us about the mechanism of apoptotic progression but may also provide insights into the diverse physiological functions of BLM and how loss of these can lead to the pathological states associated with BS.
Acknowledgments
ACKNOWLEDGEMENTS
We thank members of the S.P.J. laboratory for their advice and support and also thank J. M. Bradbury for valuable scientific and editorial input. Thanks are also due to E. Salido and J. R. Murguia for critical reading of the manuscript. The caspase-3 expression construct was a kind gift from G. Cohen at the Universtity of Leicester. We thank Mike Waldon at the Protein Nucleic Acid Chemistry Facility, Biochemisty Department, University of Cambridge, for technical assistance with N-terminal sequence analysis. This work was funded by grants from the Cancer Research Campaign and the Association for International Cancer Research. R.F. is currently supported by a FIS (Fondo Investigaciones Sanitarias) contract.
References
- 1.Green D. (1998) Apoptotic pathways: the roads to ruin. Cell, 94, 695–698. [DOI] [PubMed] [Google Scholar]
- 2.Strasser A., O’Connor,L. and Dixit,V. (2000) Apoptosis signaling. Annu. Rev. Biochem., 69, 217–245. [DOI] [PubMed] [Google Scholar]
- 3.Cryns V. and Yuan,J. (1998) Proteases to die for. Genes Dev., 12, 1551–1570. [DOI] [PubMed] [Google Scholar]
- 4.Hengartner M. (2000) The biochemistry of apoptosis. Nature, 407, 770–776. [DOI] [PubMed] [Google Scholar]
- 5.Salvesen G. and Dixit,V. (1997) Caspases: intracellular signaling by proteolysis. Cell, 14, 443–446. [DOI] [PubMed] [Google Scholar]
- 6.Enari M., Sakahira,H., Yokoyama,H., Okawa,K., Iwamatsu,A. and Nagata,S. (1998) A caspase-activated DNase that degrades DNA during apoptosis and its inhibitor ICAD. Nature, 391, 43–50. [DOI] [PubMed] [Google Scholar]
- 7.Sakahira H., Enari,M. and Nagata,S. (1998) Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature, 391, 96–99. [DOI] [PubMed] [Google Scholar]
- 8.Cardone M., Salvesen,G., Widmann,C., Johnson,G. and Frisch,S. (1997) The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell, 90, 315–23. [DOI] [PubMed] [Google Scholar]
- 9.Kothakota S., Azuma,T., Reinhard,C., Klippel,A., Tang,J., Chu,K., McGarry,T., Kirschner,M., Koths,K., Kwiatkowski,D. and Williams,L. (1997) Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science, 278, 294–298. [DOI] [PubMed] [Google Scholar]
- 10.Kaufmann S., Desnoyers,S., Ottaviano,Y., Davidson,N. and Poirier,G. (1993) Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res., 53, 3976–3985. [PubMed] [Google Scholar]
- 11.Lazebnik Y., Kaufmann,S., Desnoyers,S., Poirier,G. and Earnshaw,W. (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature, 371, 346–347. [DOI] [PubMed] [Google Scholar]
- 12.Affar E., Germain,M., Winstall,E., Vodenicharov,M., Shah,R., Salvesen,G. and Poirier,G. (2001) Caspase-3-mediated processing of poly(ADP-ribose) glycohydrolase during apoptosis. J. Biol. Chem., 276, 2935–2942. [DOI] [PubMed] [Google Scholar]
- 13.Casciola-Rosen L., Anhalt,G. and Rosen,A. (1995) DNA-dependent protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J. Exp. Med., 182, 1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song Q., Lees-Mille,R.S., Kumar,S., Zhang,Z., Chan,D., Smith,G., Jackson,S., Alnemri,E., Litwack,G., Khanna,K. et al. (1996) DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO J., 15, 3238–3246. [PMC free article] [PubMed] [Google Scholar]
- 15.Smith G., di Fagagna,F., Lakin,N. and Jackson,S. (1999) Cleavage and inactivation of ATM during apoptosis. Mol. Cell. Biol., 19, 6076–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.German J. (1993) Bloom Syndrome: a mendelian prototype of somatic mutational disease. Medicine, 71, 393–406. [PubMed] [Google Scholar]
- 17.Ellis N., Groden,J., Ye,T.-Z., Straughen,J., Lennon,D., Ciocci,S., Proytcheva,M. and German,J. (1995) The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 18.Yu C., Oshima,J., Fu,Y., Wijsman,E., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S. et al. (1996) Positional cloning of the Werner’s syndrome gene. Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 19.Kitao S., Shimamoto,A., Goto,M., Miller,R., Smithson,W., Lindor,N. and Furuichi,Y. (1999) Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 20.Gangloff S., McDonald,J., Bendixen,C., Arthur,L. and Rothstein,R. (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol., 14, 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watt P., Louis,E., Borts,R. and Hickson,I. (1995) Sgs1: a eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell, 81, 253–260. [DOI] [PubMed] [Google Scholar]
- 22.Stewart E., Chapman,C., Al-Khodairy,F., Carr,A. and Enoch,T. (1997) rqh1+, a fission yeast gene related to the Bloom’s and Werner’s syndrome genes, is required for reversible S phase arrest. EMBO J., 16, 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray J., Lindsay,H., Munday,C. and Carr,A. (1997) Role of Schizosaccharomyces pombe RecQ homolog, recombination and checkpoint genes in UV damage tolerance. Mol. Cell. Biol., 17, 6868–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karow J., Wu,L. and Hickson,I. (2000) RecQ family helicases: roles in cancer and aging. Curr. Opin. Genet. Dev., 10, 32–38. [DOI] [PubMed] [Google Scholar]
- 25.Frei C. and Gasser,S. (2000) RecQ-like helicases: the DNA replication checkpoint connection. J. Cell Sci., 113, 2641–2646. [DOI] [PubMed] [Google Scholar]
- 26.Wu L., Karow,J. and Hickson,I. (1999) Genetic recombination: helicases and topoisomerases link up. Curr. Biol., 15, R518–R520. [DOI] [PubMed] [Google Scholar]
- 27.Watt P., Hickson,I., Borts,R. and Louis,E. (1996) SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics, 144, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett R., Noirot-Gros,M. and Wang,J. (2000) Interaction between yeast sgs1 helicase and DNA topoisomerase III. J. Biol. Chem., 275, 26898–26905. [DOI] [PubMed] [Google Scholar]
- 29.Yamagata K., Kato,J.-I., Shimamoto,A., Goto,M., Furuichi,Y. and Ikeda,H. (1998) Bloom’s and Werner’s syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu L., Davies,S., North,P., Goulaouic,H., Riou,J., Turley,H., Gatter,K. and Hickson,I. (2000) The Bloom’s syndrome gene product interacts with topoisomerase III. J. Biol. Chem., 275, 9636–9344. [DOI] [PubMed] [Google Scholar]
- 31.Johnson F., Lombard,D., Neff,N., Mastrangelo,M., Dewolf,W., Ellis,N., Marciniak,R., Yin,Y., Jaenisch,R. and Guarente,L. (2000) Association of the Bloom syndrome protein with topoisomerase IIIalpha in somatic and meiotic cells. Cancer Res., 1162–1167. [PubMed] [Google Scholar]
- 32.Moens P., Freire,R., Tarsounas,M., Spyropoulos,B. and Jackson,S. (2000) Expression and nuclear localization of BLM, a chromosome stability protein mutated in Bloom’s syndrome, suggest a role in recombination during meiotic prophase. J. Cell Sci., 113, 663–672. [DOI] [PubMed] [Google Scholar]
- 33.Jackson S. (1993) Identification and characterization of eukaryotic transcription factors. In Hames,B.D. and Higgins,S.J. (eds), Gene Transription: A Practical Approach. Oxford University Press, Oxford, UK, pp. 189–242.
- 34.Karow J., Chakraverty,R. and Hickson,I.D. (1997) The Bloom’s syndrome product is a 3′→5′ DNA helicase J. Biol. Chem., 272, 30611–30614. [DOI] [PubMed] [Google Scholar]
- 35.Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 36.Raschle M., Marra,G., Nystrom-Lahti,M., Schar,P. and Jiricny,J. (1999) Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1 apoptosis. J. Biol. Chem., 274, 32368–32375. [DOI] [PubMed] [Google Scholar]
- 37.Han Z., Chatterjee,D., He,D., Early,J., Pantazis,P., Wyche,J. and Hendrickson,E. (1995) Evidence for a G2 checkpoint in p53-independent apoptosis induction by X-irradiation. Mol. Cell. Biol., 15, 5849–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han Z., Malik,N., Carter,T., Reeves,W., Wyche,J. and Hendrickson,E. (1996) DNA-dependent protein kinase is a target for a CPP32-like apoptotic protease. J. Biol. Chem., 271, 25035–25040. [DOI] [PubMed] [Google Scholar]
- 39.Critchlow S., Bowater,R. and Jackson,S. (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol., 8, 588–598. [DOI] [PubMed] [Google Scholar]
- 40.Talanian R., Quinlan,C., Trautz,S., Hackett,M., Mankovich,J., Banach,D., Ghayur,T., Brady,K. and Wong,W. (1997) Substrate specificities of caspase family proteases. J. Biol. Chem., 272, 9677–9682. [DOI] [PubMed] [Google Scholar]
- 41.Thornberry N., Rano,T., Peterson,E., Rasper,D., Timkey,T., Garcia-Calvo,M., Houtzager,V., Nordstrom,P., Roy,S., Vaillancourt,J. et al. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem., 272, 17907–17911. [DOI] [PubMed] [Google Scholar]
- 42.Bischof O., Galande,S., Farzane,F., Kohwi-Shigematsu,T. and Campisi,J. (2001) Selective cleavage of BLM, the Bloom syndrome protein, during apoptotic cell death. J. Biol. Chem., 276, 12068–12075. [DOI] [PubMed] [Google Scholar]
- 43.Chester N., Kuo,F., Kozak,C., O’Hara,C. and Leder,P. (1998) Stage-specific apoptosis, developmental delay and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev., 12, 3382–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]