

Abstract
Reduced autophagy may be associated with normal and pathological aging. Here we report a link between autophagy and Werner protein (WRNp), mutated in Werner syndrome, the human premature aging Werner syndrome (WS). WRN mutant fibroblast AG11395 and AG05229 respond weakly to starvation induced autophagy compared to normal cells. While the fusion of phagosomes with lysosome is normal, WS cells contain fewer autophagy vacuoles. Cellular starvation autophagy in WS cells is restored after transfection with full length WRN. Further, siRNA mediated silencing of WRN in the normal fibroblast cell line WI-38 results in decreased autophagy and altered expression of autophagy related proteins. Thus, our observations suggest that WRN may have a role in controlling autophagy and hereby cellular maintenance.
Keywords: Autophagy, Aging, Beclin-1, RecQ helicase, Werner protein, Werner syndrome
1. Introduction
RecQ helicases are ubiquitous in life and are found in a broad range of prokaryotes, as well as in yeast and human cells [1,2]. Among the five RecQ helicases found in humans, defects in three give rise to clinical disorders associated with cancer predisposition and/or variable symptoms of premature aging. Of these Werner syndrome (WS) and Rothmund Thompson syndrome (RTS) are associated with premature aging and often develop age associated diseases including cancer [3]. Along with the conserved 3′–5′ helicase domain, the WRN gene has other conserved catalytic domains such as 3′–5′ exonuclease, 27 aa direct repeats, a helicase-and-ribonuclease D/C-terminal (HRDC) and a C-terminal NLS [4]. WRN protein (WRNp, 1432 aa) participates in replication [5], transcription [6] and base excision repair, homologous recombination and non-homologous end joining [7–9]. It has been proposed that accumulation of DNA damage in WS cells may be responsible for the accelerated aging and age associated diseases seen among the patients. However, the exact role of WRNp in vivo, in preventing accelerated aging is not yet understood.
Autophagy, specifically macroautophagy is an evolutionarily conserved catabolic process which degrades cellular proteins and damaged or excess organelles [10]. This process is a survival strategy for starving cells involving recycling amino acids [10–12]. Degradation and recycling of building blocks of the organelles or protein are also important for maintenance of cellular homeostasis [13]. Damaged macromolecules or organelles block various cellular functions and their chewed up components are recycled to keep cellular homeostasis. Formation of double membrane autophagic vacuoles, also known as autophagosomes, and transportation of damaged protein to the lysosome for degradation require additional proteins [14,15]. Some of these are highly conserved from flies to mammals. Failure of autophagy accumulates damaged proteins inside the cells, which are responsible for the development of different neurodegenerative disorders [16], autoimmunity [17] and cancer [18]. Similarly, accumulating evidence suggests that autophagy may play an important role in cellular aging [19]. Autophagy decreases with age and this reduced function may underlie the accumulation of damaged non-functional proteins and cause oxidative stress [20]. This perturbs many cellular functions and contributes to the development of many age associated diseases including cancer [13].
Besides the already known impact of WRN on DNA metabolism and cell cycle regulation, defects in transcription have also been observed in WS cells implicating that WRNp may have a role in transcriptional control [21,22]. WRNp participates in transcription of genes induced after stress [23]. Moreover, WRNp involvement in RNA pol I and RNA pol II mediated transcription has been reported [21,24]. WRNp also affects the expression of genes involved in adipogenesis and inflammation [22]. Using array analysis approach, it was shown that both normal old and WS cells lack expression of Beclin-1 by 1.5 fold [25]. Beclin-1 plays a crucial role at the initial stage of autophagic vesicle formation [26–28]. Additionally, proteasomal degradation is also defective in WS cells resulting in the accumulation of damaged proteins [25]. Though autophagy and WS both are hallmarks of aging, these two have not been clearly connected. Thus, we were prompted to investigate autophagy in WS cells. We found that WS cells respond very weakly to starvation induced autophagy and that this was complemented by transfection with full length WRN which restored the expression of genes responsible for the induction of autophagy. Similarly, depletion of WRN from normal cells results in diminished autophagy and down regulation of autophagy related genes. Thus our results suggest a role of WRN in the induction of autophagy.
2. Materials and methods
2.1. Materials
Earle’s balanced salt solution (EBSS) and L-glutamine were purchased from Himedia. Fetal bovine serum (FBS), penicillin_streptomycin, MEM NEAA, MEM amino acids, MEM vitamin solution, and Dulbecco’s modified eagle medium (DMEM) were obtained from Life Technologies, USA. Bovine serum albumin (BSA) was purchased from SRL (India). 3-MA, monodansylcadaverine (MDC), and osmiumtetraoxide (OsO4) were obtained from Sigma-Aldrich (USA). Anti-LC3B and anti-Atg5 were purchased from Abcam (Cambridge, England). Anti-beclin-1, anti-β-actin, anti-mTOR, and anti-p-mTOR (Ser2448) antibodies were purchased from Cell Signaling Technologies. Anti-Werner antibody and horse radish peroxidase-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology, USA. The EGFP-LC3B plasmid, which encodes a fusion protein of EGFP and LC3B, was a kind gift from Prof. Tamatsu Yoshimori (Japan). pBABE-puro mCherry-EGFP-LC3B which encodes a fusion protein EGFP, mCherry and LC3B, was a generous gift from Dr. Jayanta Debnath (Department of Pathology, University of California, San Francisco). siRNA against human WRN was purchased from Invitrogen, USA.
2.2. Cell lines and culture conditions
WS cells AG11395 (SV40 transformed fibroblast; age: 60 year, biopsy source: skin; tissue source: skin; gender: male; ethnicity: Caucasia) and WS primary fibroblasts AG05229 (untransformed fibroblast; age: 25 year, biopsy source: thigh; tissue source: skin; gender: male; ethnicity: Caucasian) cells were cultured in minimal essential medium (MEM) supplemented with 10% FBS, 1% penicillin_streptomycin, 1% L-glutamine, 1% MEM NEAA, 1% MEM amino acids, and 1% MEM vitamin solution. WI-38 (SV40 transformed fibroblast; age: 3 months gestation fetus; biopsy source: lung; tissue source: lung; gender: female; ethnicity: Caucasian) cells were cultured in Dulbecco’s modified eagle medium (DMEM) with 10% FBS. All the cells were maintained at 37 °C, 5% CO2 and 95% relative humidity (RH). Starvation was performed by incubating exponential growing cells in Earle’s balanced salt solution (EBSS).
2.3. MDC staining
AG11395, AG05229 or WI-38 cells (2 × 104) were seeded in 35 mm plate for overnight. After 16 h cells were starved with EBSS for different time points (2 to 24 h). The cells were then washed with 1× PBS thrice and incubated with 50 mmole/L MDC for 10 min at 37 °C [29,30] After washing with 1× PBS the cells were mounted on glass slides and viewed under a fluorescence microscope (Leica DM 2500).
2.4. Green fluorescent protein-light chain 3 plasmid transfection
AG11395 or WI-38 cells (2 × 104) were seeded in 35 mm plate for overnight. After 16 h cells were transfected 1 μg of EGFP-LC3B plasmid [31] using FuGENE6 as per the manufacturer’s instructions (Roche). Next day cells were starved with EBSS for 24 h. The cells were then washed with 1×PBS thrice and mounted on glass slides. Finally cells were observed with a fluorescence microscope (Leica DM 2500).
2.5. Autophagic flux measurement
After 16 h of growth cells were transfected with pBABE-puro mCherry-EGFP-LC3B using Fugene 6 as per manufacturer’s instructions (Roche). Next day cells were starved with EBSS for 8 h. Then cells were washed with 1×PBS thrice and mounted on glass slides followed by fluorescence microscopic observation (Leica DM 2500).
2.6. Western bloting
Whole cell lysates were prepared from the cells with lysis buffer containing 1% Triton X-100, 50 mM NaCl, 50 mM NaF, 20 mM Tris (pH 7.4), 1 mM EGTA, 1 mM sodium vanadate, 0.2 mM PMSF, 0.5% NP-40 and protease inhibitors (Bio vision). The supernatant was collected and protein concentration was estimated using Bradford’s reagent. Cell lysates containing equal amount of protein (80 μg) were solubilized in Lamellae buffer, boiled for 5 min, and electrophoresed on a 12% SDS-polyacrylamide gel in Tris–glycine buffer (pH 8.8). Proteins were then transferred to polyvinylidine difluoride (PVDF) membrane (Bio-Rad). Nonspecific binding was blocked with 5% non-fat dry milk and 0.05% Tween-20 in 20 mM Tris–Cl, pH 7.6 (TBS-T). After incubation with the appropriate primary antibody, membrane was washed with TBS-T and blot was reincubated with secondary antibodies conjugated with horse radish peroxidase (HRP). Bound antibodies were detected by the ECL detection reagent (Santa Crutz).
2.7. Small interfering RNA (siRNA) mediated silencing of WRN
WI-38 cells were transfected with WRN siRNA (ID: s14907) and scramble (negative control #2 siRNA, Invitrogen, USA) using invitrogen transfection reagent according to the manufacturer’s instructions. After 24 h, knockdown efficacy was determined by Western blotting with anti-WRN antibody.
2.8. Transmission electron microscopy (TEM)
AG11395 or WI-38 cells were starved with Earle’s balanced salt solution (EBSS) for 24 h. Cells were then collected and prefixed with 2.5% glutaraldehyde. These cells were then post fixed with 1% osmium tetraoxide for 1 h in dark. Cells were then dehydrated by increasing concentrations of acetone. These cells were then embedded with epoxy resin. Polymerization of these cells was done by placing it gradually in oven for 42 °C for 2 h, 52 °C for overnight and then 62 °C for another overnight. Ultrathin sections (50–70 nm) of these blocks were cut using a Leica Ultramicrotome EM UC6. These ultrathin sections were collected form 10% ethanol turf. The sections were contrasted using 1% aqueous uranyl acetate for 5 min and lead citrate in a CO2-depleted atmosphere for approximate 2 to 4 min. A FEI TECNAI G2 Spirit BioTWIN (120 kV) electron microscope (Netherland) was used to study the sections in 100 kV.
2.9. Statistical analysis
Non parametric Mann Whitney’s U was used to calculate the statistical differences between the groups. P < 0.05 was considered as statistically significant. Error bars represent the means ± SD for all plots. Data analysis was performed using the Origin pro v. 8 software (Origin Lab).
3. Results
3.1. WS cells show poor starvation induced autophagy
Starvation induced autophagy was measured in WS cell lines (AG11395 and AG05229) and in normal skin fibroblasts WI-38 by MDC staining. After starvation by incubating cells in Earle’s balanced salt solution (EBSS) cells were collected at different time points and stained for autophagic vacuoles with MDC followed by analysis under a fluorescence microscope. As seen in (Fig. 1A), following starvation there were fewer autophagic cells amongst AG11395 and AG05229 cells than WI-38 cells at all time points tested. Since LC3B is an essential component of the autophagosome we also analyzed LC3B. Cells were allowed to express EGFP-LC3B for 24 h and then starved for another 24 h. As seen in (Fig. 1B and C) AG11395 cells showed fewer EGFP-LC3B foci than WI-38 cells. We also confirmed our observation by immunoblotting total cellular extract for autophagy marker proteins from WS, and normal cell lines. As seen in (Fig. 1D and E), the band corresponding to LC3BII is more intense in the normal cell line after starvation compared to WS cells. Similarly, autophagy marker proteins Beclin-1 and Atg5 increased more after starvation in WI-38 than in WS cells. Finally, we also used electron microscopy to directly visualize autophagic vacuoles. As seen in (Fig. 1F) we observed large numbers of autophagic vacuole in WI-38 cells (Fig. 1F. b) whereas AG11395 cells showed few autophagic vacuoles (Fig. 1F. d) after 24 h starvation.
Fig. 1.
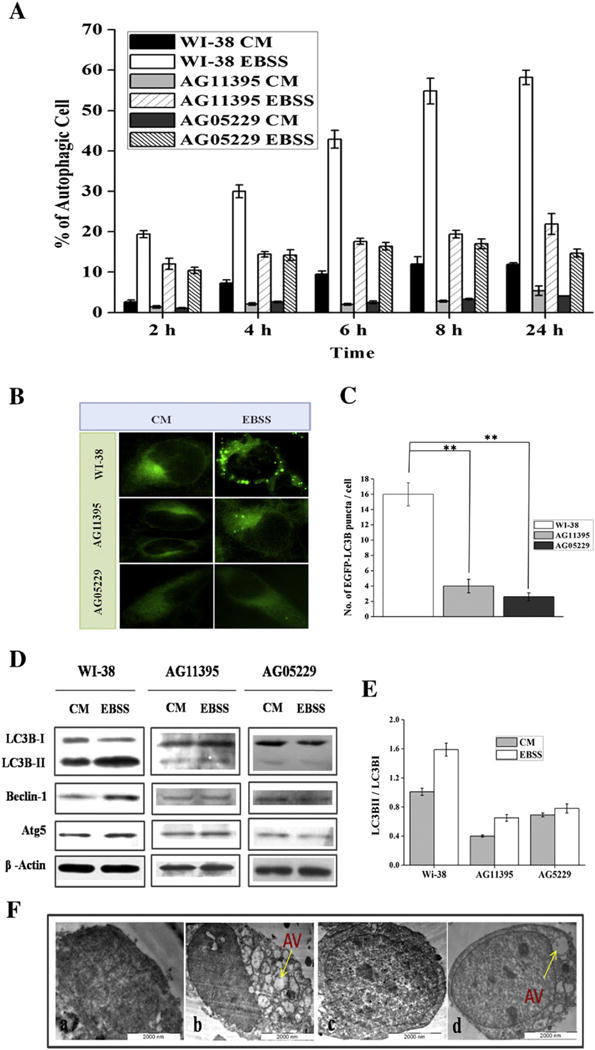
Normal fibroblast cell (WI-38) and Werner syndrome fibroblast cells (AG11395, AG05229) were starved with Earle’s balanced salt solution (EBSS) along with complete medium (CM). (A) Graphical representation of MDC staining at different time points 2 to 24 h of all the cells. More than 600 cells were analyzed for each condition. (B) WI-38 cell and WS cells (AG11395, AG05229) cells are transfected with EGFP-LC3B and then starved for 24 h. Images were taken under fluorescence microscopy (100× magnification). (C) Graphical representation of number of EGFP-LC3 dots per cell during 24 h starvation. ** P < 0.005, mean ± SD, n = 3. More than 600 cells were analyzed for each condition. (D) Immunoblotting of total cellular extract for autophagy marker proteins of normal fibroblast WI-38 and both WS cells AG11395, AG05229. (E) Graphical representation of ratio LC3BII and LC3BI of all the cell lines. 3 Western blots were examined for each condition. (F) Electron microscopic images of autophagic vacuoles during 24 h starvation of WI-38 and AG11395 cell lines. a. WI-38 control, b. WI-38 starved, c. AG11395 control, and d. AG11395 starved. Arrow indicates autophagic vacuole (AV).
3.2. WS cells show normal autophagic flux
In the autophagy process the autophagosome is fused with lysosomes and different cellular proteins regulate this process. To check whether there was any defect in WS cells in this fusion process, we transfected these cells with mCherry-EGFP-LC3 plasmid. As the green color is acid sensitive, fusion with acidic lysosomes results in only red color and un-fused vacuoles with lysosome gives yellow color (Fig. 2A). As seen in (Fig. 2B) the ratio of red to yellow dots for WI-38 (0.46) was almost similar with AG11395 (0.25) or AG05229 (0.3) cells indicating that the rate of subsequent fusion with acidic lysosomes was almost similar for both the cell lines but the total amount of autophagic vacuoles was fewer in AG11395 or AG05229 cells.
Fig. 2.
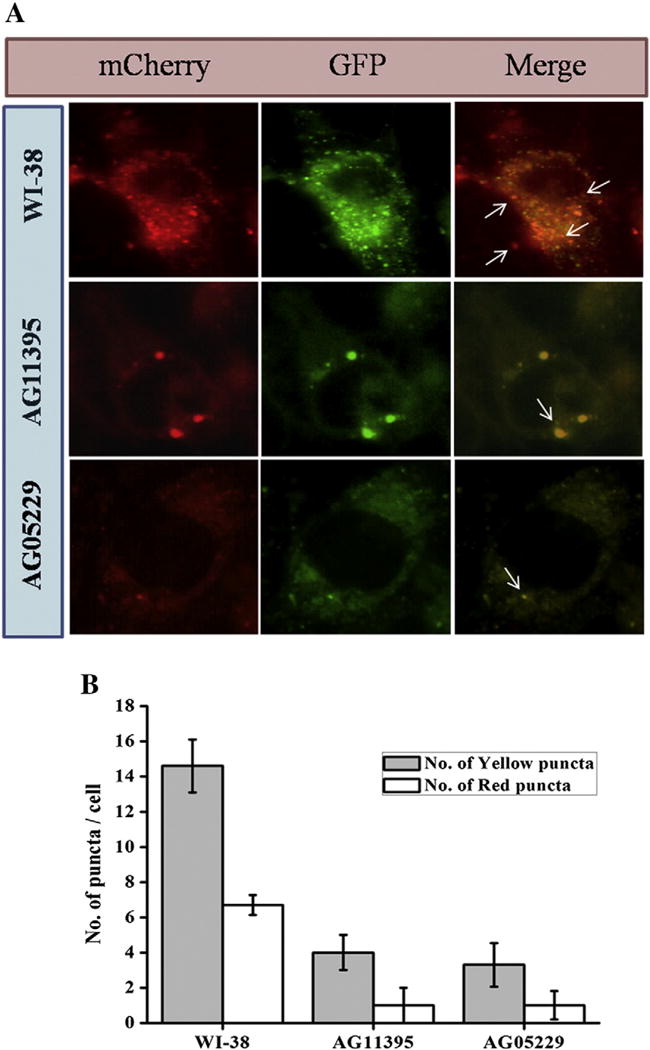
WI-38 and WS cells (AG11395, AG05229) were transfected with pBABE-puro mCherry-EGFP-LC3B and starved with EBSS for 8 h. (A) mCherry and EGFP signal were observed under fluorescence microscopy. Arrow heads indicate fusion of autophagosome and lysosome. At least 450 cells were examined for different conditions. (B) Graphical representation of autophagic flux of all cell lines. The ratio of red puncta over yellow puncta for AG11395 is 0.25, for AG05229 0.3 and for WI38 is 0.46.
3.3. Transient overexpression of WRNp enhances starvation induced autophagy
Given that WRN cell lines are defective in starvation induced autophagy we next transfected AG11395 cells with full length WRN to investigate whether starvation induced autophagy could be rescued. We observed enhanced numbers of autophagic foci during starvation (Fig. 3A) compared to the cells transfected with empty vector (EGFPC1). Moreover, the number of autophagic vacuoles after starvation gradually increased with time. After 24 h, WRN over-expression in AG11395 cells resulted in 55% autophagic cells (Fig. 3B). When normal WI-38 cells transfected with WRN (Fig. 3C) it was observed that the level of autophagic gene expression increased slightly, compared to the untransfected WI-38 cells (Fig. 1D) [21,25]. During starvation the ratio of LC3BII/LC3BI for untransfected cells was 1.59 (Fig. 1D) whereas for WRN transfected cells it was 1.6 and for Atg5 cells it was 1.68 and 1.7; for Beclin-1 the corresponding values were 1.42 and 1.7. Expression of autophagy related genes in WI38 cells transfected with WRN and followed by incubation in complete medium did not increase significantly compared to untransfected cells. Transfection and subsequent expression of WRN during 24 h in starved AG11395 cells were confirmed by immunobloting the total cellular extract with WRN antibody (Fig. 3D). Total cellular extract was also used for immunobloting of autophagic proteins at different time points after initiation of starvation. As seen in (Fig. 3E), full length WRN transfected cells showed a higher amount of LC3BII, Beclin-1 and Atg5 compared to empty vector transfected cells. The ratio of LC3BII and I is plotted in (Fig. 3F) and the level of Beclin-1, and Atg5 compared to β-actin is plotted in (Fig. 3G and H respectively).
Fig. 3.
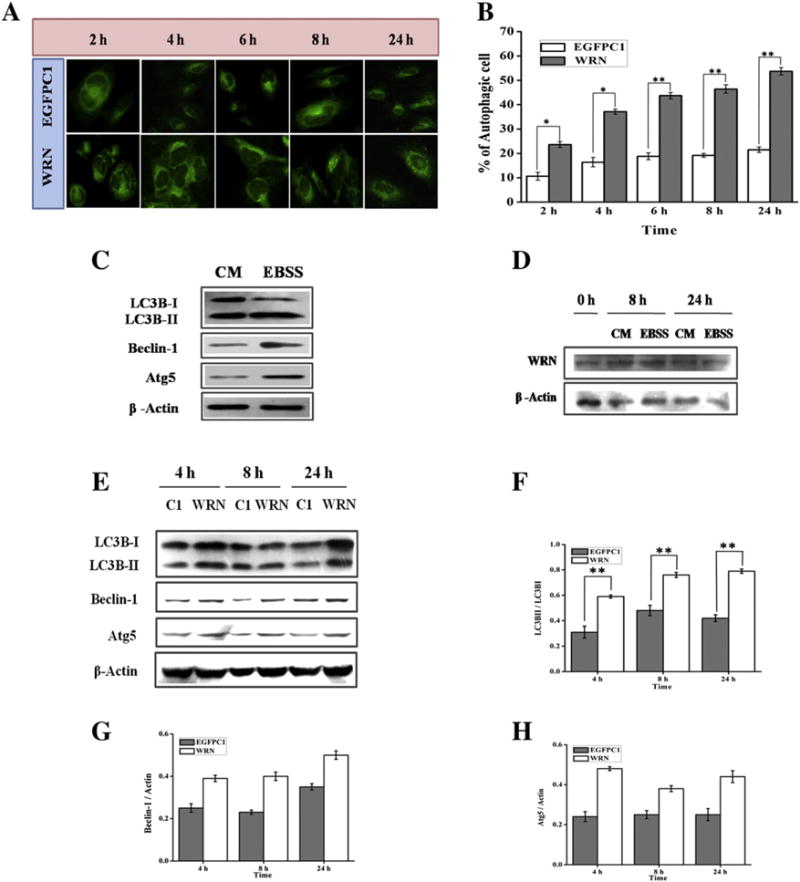
AG11395 cells were transfected with empty vector (EGFPC1) and vector containing full length WRN and starved with EBSS for 2 to 24 h. (A) Images of MDC staining taken with a fluorescence microscope under 40× magnification. (B) Graphical representation of % of autophagic cell during the time period (2 to 24 h). More than 500 cells per transfection were examined. * p < 0.05, ** p < 0.005, mean ± SD, n = 3. (C) WI-38 cells were transfected with full length WRN and allowed to express in complete medium (CM). After 24 h one set remains in complete medium (CM) and the other set was allowed for starvation for 24 h. (D) AG11395 cells were transfected with plasmid containing full length WRN and allowed to express WRN in complete medium (CM) for 24 h followed by starvation for 0, 8 and 24 h. Whole cell lysate was examined by immunoblotting with anti-WRN antibody. Here β-actin was used as loading control. (E) AG11395 cell was transfected with empty vector (EGFPC1) and full length WRN plasmid and starved after 24 h for 4, 8 and 24 h. Total cell lysate was immunoblotted with anti-LC3B, anti-beclin-1, and anti-Atg5 antibodies. Here β-actin was used as a loading control. (F–H) Band intensity was measured and graphically represents the ratio of LC3B-II and LC3B-I, beclin-1 and Atg5 at different time points respectively. ** p < 0.005, mean ± SD, n = 3.
3.4. WRNp slows down mTOR status in AG11395 cell
Cellular autophagy is regulated by mTOR and phosphorylation of mTOR at Ser2448 is an activator of mTORC1[32] resulting inhibition of autophagy [33]. We analyzed mTOR and its phosphorylation at Ser2448 in normal cells, WS cells and WS cells transfected with full length WRN. As seen in Fig. 4 transfection of full length WRN in WS cells resulted in a reduced amount of mTOR and p-mTOR during starvation (Fig. 4 compare lanes 5 and 6).
Fig. 4.

Normal cells, WS cells and WS cells transfected with full length WRN were starved and lysate was immunoblotted with anti-mTOR and anti p-mTOR. Here β-actin was used as loading control.
3.5. Inhibition of WRNp using siRNA decreases autophagic genes
Finding that full length WRNp rescued starvation induced autophagy in AG11395 cells, we became curious to determine the cellular fate during starvation by silencing WRN from WI-38 cells. Using the RNA interference we depleted WRN in WI-38 cells. More than 60% knock down of WRNp was achieved (Fig. 5A and B) in WI-38 cells. Starvation induced autophagy was diminished in cells treated with WRN siRNA compared to scrambled siRNA in WI-38 cells, as determined by MDC staining (Fig. 5C) and the percentage of autophagic cells are plotted in (Fig. 5D). Also, the level of Beclin-1 and Atg5 decreased in siRNA treated WI-38 cells immunoblotting (Fig. 5E). The corresponding ratio of different proteins in siRNA treated WI-38 cells was compared with scramble RNA treated cells and unmodified WI-38 cells were plotted in Fig. 5F (LC3BII to LC3BI), Fig. 5G (Beclin-1: Actin), and Fig. 5H (Atg5: Actin).
Fig. 5.
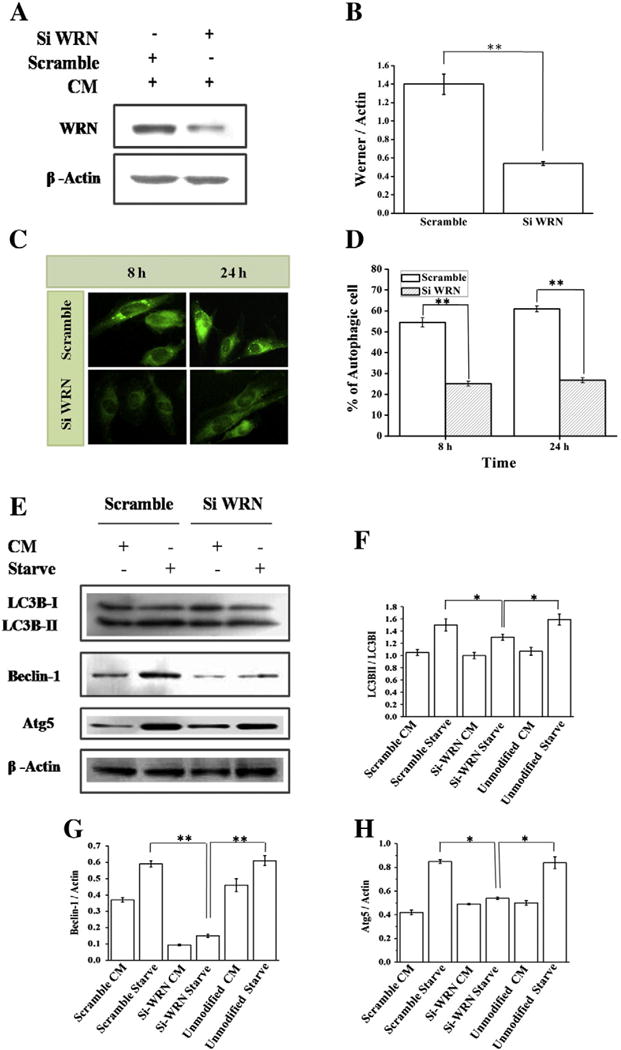
Silencing of WRN protein using WRN siRNA in WI-38 cell. (A) Western blot analysis of expression of WRN protein in siWRN and scrambled siRNA transfected cell. (B) Graphical representation of inhibition of WRN protein. ** p < 0.005, mean ± SD, n = 3. (C) WI-38 cell transfected with siWRN or scrambled siRNA and starved for 8 and 24 h. Images of MDC staining were taken under a fluorescence microscope (40×). (D) % of autophagic cells are represented in graph. ** p < 0.005, mean ± SD, n = 3. (E) After transfection with siWRN or scrambled siRNA WI-38 cells were treated with complete medium (CM) for 24 h followed by starvation using EBSS. Western blot of total cell lysate was performed using anti-LC3B, anti-beclin-1, and anti-Atg5 antibodies. Here β-actin was used as a loading control. (F–H) Quantification of band intensity was performed using Image J and the ratio of LC3BII and LC3BI, beclin-1 and Atg5 represents respectively. Graphical representation includes unmodified WI-38 cell. * p < 0.05, ** p < 0.005, mean ± SD, n = 3.
4. Discussion
Here we observed that WS cells showed a reduced amount of starvation induced autophagy. Transfection with full length WRN rescued the starvation induced autophagy in WS cells. Furthermore, overexpression of WRN resulted in increased expression of proteins which are known as positive regulators of autophagy.
WS cells may have a general deficiency in the autophagic response as in complete medium we detect less autophagy in WS cells. Thus, deficiency of autophagy in WS cells indicates that damaged proteins and organelles may accumulate in these cells and this may lead to cellular senescence and premature aging. Our results clearly demonstrated that in WS cells, fewer autophagic vacuoles formed but fusion with lysosomes seemed to be normal. This is primarily due to the lack of response of the autophagic initiation process which depends on several proteins including Beclin-1, Atg5 [34] etc. We have also demonstrated that the expression of Beclin-1 and Atg5 is reduced in WS cells and can be complemented by transfection of full length WRN. Our results are consistent with a previous study, which showed that WS cells possess reduced expression of specific genes including Beclin-1 [25]. Additionally, transcriptional activation of several genes by the 27 aa repeat residues of WRN has been documented [21]. Thus, taken together, it is possible that expression of WRN is necessary for the subsequent expression of autophagy proteins like Beclin-1 and Atg5. In normal fibroblasts, expression of these proteins is constitutive but when WRN is depleted in normal cells the reduced expression of these proteins may be responsible for the lack of autophagy. Furthermore, involvement of other proteins in this process is likely. Additionally, WRN mediated transcriptional activation of other genes related to cellular energy production is another possible explanation for the reduced autophagy in WS cells. In WRN helicase mutant mouse embryonic fibroblasts reduced production of ATP has been documented [34]. Reduced ATP production in mitochondria in mutant WRN cells was proposed to be associated with disturbance in mitochondrial inner membrane potential [35]. Energy production decline with aging is one of the reasons for the lack of autophagy in aged cells [36]. Additionally, autophagy mediated by mitochondrial dysregulation may also be related to aging and a role of WRNp in regulation of mitochondrial ROS has been suggested [37]. Much attention has been given to the role of WRN in the DNA damage response pathway and lack of successful DNA repair renders accumulation of damaged DNA which in turn is the cause for the accelerated aging. It is likely that WRN also has a role in a DNA damage induced autophagic pathway [38,39]. Our results demonstrate that not only in starved condition but also in the presence of complete medium, the percentage of autophagy in WS cells is less compared to WI-38 cell. This indicates a linkage between WRN and autophagy.
Apart from transcriptional regulation of autophagy related genes direct involvement of WRNp in autophagy is possible. WRNp regulates hypoxia-inducible factor-1 (HIF-1) by affecting mitochondrial ROS production [40] and mitochondrial generated ROS can induce autophagy via the AMPK pathway [41]. Thus, accumulation of damaged proteins and lack of autophagy to remove them may be responsible for the accelerated aging for WS cells.
Introduction of WRN in WS cells resulted in down regulation of mTOR which is consistent with the observation that PI3 kinase is upregulated in normal aging and in WS cells [25]. Suppression of mTOR activates association of Atg13 and Atg1 which is important for autophagy [42,43]. mTOR activity is also associated with protein aggregation [39] which is reversed when treating with the mTOR inhibitor, rapamycin. Thus mTORC1 pathway was suggested as a potential therapeutic target in WS [38]. Similar reports are also available for Hutchinson–Gilford progeria syndrome (HGPS), where activation of mTOR is associated with reduced autophagy and increased protein aggregation [44]. Accumulation of insoluble protein aggregates and increased oxidative damage in WS cells have been reported [39]. This is in contradiction with observed increased autophagy in WS cells or in WRN knock down cells [38] and also upregulation of mTOR in WS cells was noted [39]. There is only one report demonstrating simultaneous activation of mTOR and autophagy in 6-thoiguanine treated cells [45]. Currently, this apparent discrepancy is not clear. But, since mTOR is regulated by different pathways it is likely that under different conditions cellular mTOR is differentially activated. Most of the literature shows an inverse relation between mTOR and autophagy. We depleted WRN from normal fibroblast and found a reduction of starvation induced autophagy. Thus our findings explain that the upregulation of mTOR inhibits autophagy under starvation. DNA damage induced autophagy may differ and WS cells are sensitive to DNA damaging agents. Thus, WRN enhances autophagy perhaps by two different pathways. WRNp suppresses mTOR, a negative regulator of autophagy. On the other hand and probably more significantly WRN interacts with RNA pol II and subsequently trans-activates autophagic genes including LC3B, Beclin-1, and Atg5 under starvation. While the exact mechanisms by which WRN induces autophagy are speculative. Our results indicate that lack of autophagy is one of the possible reasons for the premature aging in WS cells.
5. Conclusion
Our report finds a relationship between WRNp and starvation induced autophagy. In WS cells or WRN knock down normal fibroblast cells autophagy is reduced concomitant with downregulation of Beclin-1, Atg5 and LC3B. Increased level of mTOR and p-mTOR is also observed in these cells. Taken together we suggest that WRNp positively regulates autophagy related genes and thus promotes autophagy to prevent cellular aging.
Supplementary Material
Acknowledgments
The authors would like to acknowledge for financial support for this research work the Council for Scientific and Industrial Research (CSIR), project no: [37/(1442)/10/EMR-II] Government of India. We sincerely acknowledge Dr. Jayanta Debnath (Department of Pathology, University California, San Francisco) for pBABE-puro mCherry-EGFP-LC3B and Prof. Tamatsu Yoshimori (Osaka, Japan) for EGFP-LC3 plasmid. We also express sincere thanks to Prof. S. Roy, Director, Indian Institute of Chemical Biology, Kolkata, India, for his permission to use the transmission electron microscope. This project was partially supported by funds from the Intramural program of the National Institute on Aging, NIH.
Abbreviations
- ATG
Autophagy-related gene
- EBSS
Earle’s balanced salt solution
- EGFP-LC3
Enhanced green fluorescent protein-microtubule-associated protein-1 light chain 3
- MDC
Monodansylcadaverine
- mTOR
Mammalian target of rapamycin
- WS
Werner syndrome
- WRNp
Werner protein
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbadis.2014.09.007.
References
- 1.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 2.Croteau DL, Popuri V, Opresko PL, Bohr VA. RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutat Res. 2005;577:237–251. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 4.Kamath-Loeb AS, Shen JC, Loeb LA, Fry M. Werner syndrome protein characterization of the integral 3′-5′ DNA exonuclease. J Biol Chem. 1998;273:34145–34150. doi: 10.1074/jbc.273.51.34145. [DOI] [PubMed] [Google Scholar]
- 5.Sidorova JM, Li N, Folch A, Monnat RJ., Jr The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle. 2008;7:796–807. doi: 10.4161/cc.7.6.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn B, Harrigan JA, Indig FE, Wilson DM, III, Bohr VA. Regulation of WRN helicase activity in human base excision repair. J Biol Chem. 2004;279:53465–53474. doi: 10.1074/jbc.M409624200. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Huang S, Lee L, Davalos A, Schiestl RH, Campisi J, Oshima J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;4:191–199. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 9.Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous recombination resolution defect in Werner syndrome. Mol Cell Biol. 2002;20:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N. Autophagy: process and function. Gene Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 15.Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;12:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 17.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. 2013. [DOI] [PubMed] [Google Scholar]
- 18.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4:422–424. doi: 10.1016/s1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 19.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 21.Balajee AS, Machwe A, May A, Gray MD, Oshima J, Martin GM, Nehlin JO, Brosh R, Orren DK, Bohr VA. The Werner syndrome protein is involved in RNA polymerase II transcription. Mol Biol Cell. 1999;10:2655–2668. doi: 10.1091/mbc.10.8.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turaga RV, Paquet ER, Sild M, Vignard J, Garand C, Johnson FB, Masson JY, Lebel M. The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell Cycle. 2009;8:2080–2092. doi: 10.4161/cc.8.13.8925. [DOI] [PubMed] [Google Scholar]
- 23.Lachaud AA, Auclair-Vincent S, Massip L, Audet-Walsh E, Lebel M, Anderson A. Werner’s syndrome helicase participates in transcription of phenobarbital-inducible CYP2B genes in rat andmouse liver. Biochem Pharmacol. 2010;79:463–470. doi: 10.1016/j.bcp.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Shiratori M, Suzuki T, Itoh C, Goto M, Furuichi Y, Matsumoto T. WRN helicase accelerates the transcription of ribosomal RNA as a component of an RNA polymerase I-associated complex. Oncogene. 2002;21:2447–2454. doi: 10.1038/sj.onc.1205334. [DOI] [PubMed] [Google Scholar]
- 25.Kyng KJ, May A, Kølvraa S, Bohr VA. Gene expression profiling in Werner syndrome closely resembles that of normal aging. Proc Natl Acad Sci U S A. 2003;100:12259–12264. doi: 10.1073/pnas.2130723100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 27.He C, Levine B. The beclin 1 interactome. Curr Opin Cell Biol. 2014;2:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;122:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biederbick A, Kern HF, Elsässer HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- 30.Laha D, Pramanik A, Maity J, Mukherjee A, Pramanik P, Laskar A, Karmakar P. Interplay between autophagy and apoptosis mediated by copper oxide nanoparticles in human breast cancer cells MCF7. Biochim Biophys Acta. 2014;1840:1–9. doi: 10.1016/j.bbagen.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Chiang Gary G, Abraham Robert T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- 33.Parkhitko AA, Favorova OO, Khabibullin DI, Anisimov VN, Henske EP. Kinase mTOR: Regulation and Role in Maintenance of Cellular Homeostasis. Vol. 79. Tumor Development, and Aging Pleiades Publishing, Ltd.; 2014. pp. 88–101. [DOI] [PubMed] [Google Scholar]
- 34.Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- 35.Labbé A, Turaga RV, Paquet ER, Garand C, Lebel M. Expression profiling of mouse embryonic fibroblasts with a deletion in the helicase domain of the Werner Syndrome gene homologue treated with hydrogen peroxide. BMC Genomics. 2010;11:127. doi: 10.1186/1471-2164-11-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, Michaud M, Madeo F, Tavernarakis N, Kroemer G. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labbé A, Lafleur VN, Patten DA, Robitaille GA, Garand C, Lamalice L, Lebel M, Richard DE. The Werner syndrome gene product (WRN): a repressor of hypoxia-inducible factor-1 activity. Exp Cell Res. 2010;318:1620–1632. doi: 10.1016/j.yexcr.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Saha B, Cypro A, Martin GM, Oshima J. Rapamycin decreases DNA damage accumulation and enhances cell growth of WRN deficient human fibroblasts. Aging Cell. 2013;13:573–575. doi: 10.1111/acel.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talaei F, van Praag VM, Henning RH. Hydrogen sulfide restores a normal morphological phenotype in Werner syndrome fibroblasts, attenuates oxidative damage and modulates mTOR pathway. Pharmacol Res. 2013;74:34–44. doi: 10.1016/j.phrs.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013;1:50–65. doi: 10.1016/j.cellsig.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 41.Voit R, Grummt I. Phosphorylation of UBF at serine 388 is required for interaction with RNA polymerase I and activation of rDNA transcription. Proc Natl Acad Sci U S A. 2001;98:13631–13636. doi: 10.1073/pnas.231071698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 43.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graziotto John J, Cao Kan, Collins Francis S, Krainc Dimitri. Rapamycin activates autophagy in Hutchinson–Gilford progeria syndrome. Autophagy. 2012;8:147–151. doi: 10.4161/auto.8.1.18331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng X, Kinsella TJ. Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 2008;68:2384–2390. doi: 10.1158/0008-5472.CAN-07-6163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.