

Abstract
Free Fatty Acid receptor 4 (FFA4), also known as GPR120, is a G-protein-coupled receptor (GPCR) responsive to long-chain fatty acids that is attracting considerable attention as a potential novel therapeutic target for the treatment of type 2 diabetes mellitus (T2DM). Although no clinical studies have yet been initiated to assess efficacy in this indication, a significant number of primary publications and patents have highlighted the ability of agonists with potency at FFA4 to improve glucose disposition and enhance insulin sensitivity in animal models. However, the distribution pattern of the receptor suggests that targeting FFA4 may also be useful in other conditions, ranging from cancer to lung function. Here, we discuss and contextualise the basis for these ideas and the results to support these conclusions.
Keywords: G protein-coupled receptor, free fatty acid, diabetes, cancer, lung function inflammation
Trends
Substantial focus on the therapeutic potential of FFA4/GPR120 is currently directed towards type 2 diabetes.
Progress in the identification and characterisation of FFA4/GPR120 agonist ligands is apparent in both the primary scientific and patent literatures.
In models of glucose handling, FFA4/GPR120 agonists appear highly effective.
Recent indications provide support for consideration of FFA4/GPR120 ligands in areas of cancer treatment.
High levels of expression of FFA4/GPR120 in the lung suggest utility in analysis of the potential therapeutic roles of FFA4/GPR120 ligands in both acute and chronic airway inflammatory conditions.
Receptors for Long-Chain Fatty Acids
In recent years, it has become clear that many components of foodstuffs, or metabolites derived thereof, act as homeostatic monitors of nutrient availability [1]. In many cases, such metabolites do so by binding to, and activating, members of the rhodopsin-like or ‘class A’ family of GPCRs [1]. Among such metabolites is the group of long-chain, nonesterified or ‘free’ fatty acids. A broad range of long-chain fatty acids of varying chain length and position and extent of unsaturation (Box 1) are able to activate a pair of these GPCRs 2, 3. Initially designated GPR40 [4] and GPR120 [5], upon acceptance that long-chain FFAs are indeed the key endogenous activators of these receptors, they were systematically renamed FFA1 (GPR40) [6] and FFA4 (GPR120) [7], although the initial, colloquial terminologies remain in widespread use. FFA1 has been validated clinically as a therapeutic target able to control blood glucose levels. Although the potential for FFA4 to also be a therapeutic target for regulating blood glucose levels and improving tissue insulin sensitivity appears clear from rodent model studies, this remains to be addressed in a clinical setting. However, recent studies exploring further roles of FFA4 in lung function and in the development of resistance to platinum-containing chemotherapeutics suggest that interest in FFA4 should be expanded beyond metabolic diseases and should also consider potential therapeutic applications of FFA4 antagonists as well as agonists.
Box 1. Fatty Acids.
A fatty acid is a carboxylic acid with a linked aliphatic chain and may be either saturated or unsaturated. Most naturally occurring fatty acids contain a linear, unbranched aliphatic chain that lacks further modifications. However, in recent times, several modified forms, including hydroxy fatty acids [77] and branched fatty acid esters of hydroxy fatty acids 78, 79, have been suggested to have important biological roles via the activation of fatty acid-responsive GPCRs. Although both short-chain (C2–C5) and medium-chain (C9–C11)-length fatty acids are also known to activate specific GPCRs (short-chain fatty acids, FFA2, FFA3; medium chain fatty acids, GPR84), the longer-chain fatty acids activate both FFA1 and FFA4. While most long-chain fatty acids can be synthesised by the body, a pair of unsaturated long-chain fatty acids, α-linolenic acid [18:3(n-3)] and linoleic acid [18:2(n-6)] cannot, due to the lack of an appropriate desaturase enzyme. Therefore, these are defined as being ‘essential’ fatty acids. While both α-linolenic acid and linoleic acid contain 18 carbon atoms, they differ in the number of unsaturated bonds within the aliphatic chain (α-linolenic acid has three, whereas linoleic acid has two). Given that different fatty acids vary in chain length, to allow consistency of nomenclature the terminal carbon atom is designated ‘omega’ (n), after the last letter in the Greek alphabet. An ‘omega-3’ fatty acid has the first position of unsaturation three carbon atoms from the tail, while,for an ‘omega-6’ fatty acid, this is located six carbon atoms from the tail. As such, α-linolenic acid is coded as ‘18:3(n-3)’, while linoleic acid is coded as ‘18:2(n-6)’. Omega-3 fatty acids have attracted considerable attention as being beneficial for health, not least due to the high levels of such fatty acids in ‘oily’ fish, including mackerel and salmon. Before systematic redesignation as FFA4, in some publications this receptor (FFA4/GPR120) was highlighted as a (the) receptor for omega-3 fatty acids [80] and, although such fatty acids are among the most potent at this receptor, other long-chain saturated and unsaturated fatty acids are also able to activate this receptor [3].
FFA1
FFA1 is highly expressed by pancreatic β cells and, because fatty acids are known, at least in acute settings, to promote insulin release from pancreatic islets, synthetic small-molecule activators of FFA1 were initially assessed for their ability to mimic this effect. Rapid translation to show that such ligands were also efficacious in various glucose tolerance tests, resulted in the optimisation of potentially drug-like FFA1 agonist ligands and the introduction into first-in-human clinical trials of fasiglifam {TAK-875, 2-[(3S)-6-({3-[2,6-dimethyl-4-(3-methylsulfonylpropoxy)phenyl]phenyl}methoxy)-2,3-dihydro-1-benzofuran-3-yl]acetic acid} 8, 9, 10 as a potential antidiabetic medication. Although substantial efficacy was noted in both Phase II and initial Phase III studies, development of fasiglifam was discontinued in late 2013 based on concerns of potential liver toxicity [11]. Subsequent reports have indicated that these adverse effects are likely related to the build-up of high concentrations of fasiglifam and an acyl glucuronide derivative of the ligand in the bile, reflecting blockage of various bile acid transporters including, but not limited to, the bile salt export pump (BSEP) 12, 13. Given the clinical validation of targeting FFA1, there remains significant interest in the potential of novel ligands at this receptor for the treatment of T2DM 14, 15, 16. Recent publications from various pharmaceutical companies several, of what appear, at least in animal models, to be highly effective and potent FFA1 ligands 17, 18, 19, support this. Clearly, issues akin to those that resulted in the removal of fasiglifam from clinical development, including inhibition of BSEP, would need to be addressed directly before further clinical studies commence.
FFA4
GPR120 was deorphanised as the second receptor for long-chain fatty acids in 2005 [5]. Initial focus highlighted expression in the lower gut, the capacity of unsaturated fatty acids to promote release of the incretin glucagon-like peptide-1 (GLP-1) from the enteroendocrine cell line STC-1, and that both fatty acid-mediated elevation of phosphorylated extracellular regulated kinase (ERK) 1/2 MAP kinases and internalisation of the receptor from the surface of transfected cells could be used effectively as means to screen for, and identify, synthetic agonists at the receptor [5]. These initial studies also highlighted the importance of the carboxylate of the fatty acids to their function because equivalent methyl esters were inactive. Although the authors also reported [5] the ability of α-linolenic acid [18:3(n-3); Box 1] to increase levels of both GLP-1 and insulin in both the portal vein and inferior vena cava of mice, there remains uncertainty over the contribution of such released GLP-1 to the observed increase in circulating insulin levels and whether this is also the case in humans. Of particular interest, although not explored further in the initial studies, was the observation of particularly high levels of receptor mRNA in the lung of both humans and mice [5]. The potential relevance of this is discussed below. Although only distantly related in terms of sequence to FFA1, acceptance of GPR120 as a bona fide GPCR responsive to long-chain fatty acids resulted in its systematic re-classification as FFA4 [7].
Although detailed and comprehensive analyses have shown that a broad swathe of saturated as well as unsaturated fatty acids are able to activate FFA4 [20] (reviewed in [3]), initial description of the capacity of α-linolenic acid to activate this receptor paved the way for a particular focus on the effects of this and other health-beneficial omega-3 fatty acids acting either predominantly, or even exclusively, via FFA4 21, 22, 23, 24, 25, 26, 27, 28. This is undoubtedly an oversimplification. For example, there have been suggestions that several beneficial effects of omega-3 fatty acids do not require FFA4 29, 30, 31, 32, 33. However, it is important to note equivalent studies where using combinations of FFA4 expression knockout and animals that synthesise high levels of polyunsaturated omega-3 fatty acids, resulted in the suggestion that such fatty acids regulate beneficially vascular inflammation and neointimal hyperplasia via FFA4 [34]. A challenge for the translation of these ideas to humans is whether, even with dietary supplementation, levels of such fatty acids are likely sufficient to engage the receptor to a substantial level [35]. This question is further complicated by the fact that quantitatively more prevalent fatty acids are also able to activate FFA4 [20], while specific, but relatively uncommon, fatty acids or their derivatives, which do not display substantially greater potency at FFA4 in vitro, are capable of generating biological functions with apparent high potency and that are lacking in FFA4-knockout animals [36] or are reduced with knock down of this receptor in model cell systems [37].
Signalling Mechanisms of FFA4
The ability of FFA4 to elevate intracellular levels of Ca2+ 5, 38, 39, 40 provided early predictions of a key role of the phosphoinositidase C-linked G proteins Gq and/or G11 in transduction of signals from this receptor, while later studies that examined production of inositol phosphates [41] provided further support. The importance of this group of G proteins to key aspects of FFA4 function has been confirmed by the ability of selective Gq/G11 inhibitors to block such signals [41]. When expressed in HEK293 cells that had been genome-edited to lack expression of both Gq and G11, FFA4 was unable to induce elevation of either inositol phosphates or intracellular Ca2+ levels [41]. Although this appears to be the dominant mode of G protein-mediated signalling for FFA4 (Figure 1), several reports suggest that treatment with pertussis toxin eliminates the ability of this receptor to regulate the release of the satiety hormone ghrelin [42] (Figure 1) and also the release of somatostatin from delta cells of the pancreas [43]. This indicates a key role for Gi-family G proteins in these processes. Segerstolpe et al. [44] noted expression of mRNA encoding FFA4 in delta cells isolated from both healthy individuals and those with T2DM that was higher than expression of this receptor in pancreatic β cells derived from the same individuals. Interestingly, and by contrast, FFA4 mRNA expression was not detected in either α or γ cells [44]. This expression profile in cell subtypes may have substantial significance and could be exploited if FFA4 agonists can be identified that show ‘bias’ between promoting signalling via Gq/11 and Gi-family G proteins. In initial deorphanisation studies, Hirasawa et al. [5] used measures of the internalisation of FFA4 from the surface of transfected cells. Such agonist-induced internalisation of FFA4 is both robust and extensive 39, 40 and, at least in the context of HEK293 cells, is almost entirely dependent on interactions with an arrestin adapter protein [41] (Figure 1). Non-canonical, non-G-protein-mediated, signalling role(s) of such a FFA4/arrestin complex remain to be fully defined. For example, although arrestins are often linked to aspects of the temporal profile of GPCR-mediated regulation of the ERK1/2 MAP kinases, Alvarez-Curto et al. [41] did not identify a substantial role for arrestins in FFA4-mediated phosphorylation of ERK1/2 when using HEK293 cells genome-edited to lack expression of either Gαq plus Gα11, or of β-arrestin 1 + β-arrestin 2. Moreover, the key role of arrestins in FFA4 signalling in these cell backgrounds was their more traditional role in acting to desensitise G protein-mediated signalling because their elimination resulted in Ca2+ ‘spikes’ being generated repetitively with maintained exposure to an agonist [41]. Despite this, the now well-established anti-inflammatory roles of FFA4 expressed within immune cell populations where, in mice, particularly high levels are reported in thymus CD8+ dendritic cells and in lung-resident macrophages (Immunological Genome Projecti) has focused attention on potential contributions of interactions of FFA4-associated arrestins with the transforming growth factor beta (TGF-β)-activated kinase 1 binding protein 1 (TAB-1). This is believed to limit TAB-1 interacting with TGF-β-activated kinase 1 (TAK1), a complex that is important for transmitting signals from activated cell surface Toll-like receptors (TLRs), and the receptor for tumour necrosis factor alpha (TNFα), for the production and release of proinflammatory mediators [45]. The sustained interaction between agonist-occupied FFA4 and an arrestin is based on agonist-promoted phosphorylation of several serine and threonine residues located in the intracellular, C terminal of the receptor 46, 47, 48 (Figure 2). Conversion of these residues to non-hydroxyl amino acids greatly reduces such interactions, while production of antisera that recognise amino acids within the C-terminal region specifically only when they are phosphorylated 41, 47, 48 has provided reagents able to assess the state of receptor activation. In humans, splice variation within the third intracellular loop of FFA4 produces a ‘long’ isoform containing 16 additional amino acids [49]. Although expression of the long variant seems to be restricted, it appears to act as a ‘biased’ receptor, unable to engage with G protein-mediated signalling systems, but is able to interact with β-arrestins as the short isoform and to undergo internalisation in an agonist-dependent manner [39]. The broader implications of this remain unclear because there is no obvious tissue situation in which the long isoform is uniquely expressed and in which only β-arrestin- or non-G protein-mediated signalling might therefore be anticipated.
Figure 1.

Free Fatty Acid Receptor 4 (FFA4) Can Engage Multiple Signalling Pathways to Regulate Distinct Physiological Outcomes. Agonist-induced interactions with members of the Gq/G11 G protein family, resulting in elevated intracellular [Ca2+], are observed following expression of FFA4 in heterologous cell systems. This pathway is also central to many effects of FFA4 in a physiological context [2]. However, release of the satiety hormone ghrelin is sensitive to treatment with pertussis toxin [42], defining a role for Gi-family G proteins in this endpoint. Many efforts to identify synthetic agonists at FFA4 have used receptor-β-arrestin interaction assays, and key physiological roles for such β-arrestin-mediated interactions include regulation of production of anti-inflammatory mediators from macrophages [21]. Although often interlinked, effects defined by agonist-induced phosphorylation of FFA4 can be resolved from effects generated following FFA4 with arrestin [41]. Further work, potentially involving the use of novel transgenic mouse lines expressing phosphorylation-resistant (see Figure 2 in the main text) variants of FFA4 are likely to help unravel distinct roles of these regulatory interactions.
Figure 2.

Regulation of the Phosphorylation State of Free Fatty Acid Receptor 4 (FFA4): Potential Physiological Roles. Both mouse (m) [48], as illustrated here (red circles in the cartoon of the seven transmembrane domain organisation of the receptor structure, and red font in the sequence of specific amino acids within the indicated region of its primary amino acid sequence) and human [47] FFA4 become rapidly phosphorylated on two groups of Ser/Thr residues within the intracellular C-terminal tail of the receptor upon addition of an agonist ligand. Alteration to generate the phosphorylation-defective form (ph-def) results in extended maintenance of elevated Ca2+ levels when either wild-type mouse FFA4 (wt; black) or ph-def mouse FFA4 (grey) are expressed in HEK293 cells and exposed to an agonist ligand (red arrow). Generation of transgenic mice in which wild-type FFA4 is replaced with either a C-terminally HA epitope-tagged form of FFA4 (left-hand side) or a similarly epitope-tagged form of ph-def FFA4 (right-hand side) is allowing analysis of receptor expression patterns (see Figure 4 in the main text) and specific roles of agonist-mediated phosphorylation of FFA4. Studies using these animals are still to be reported in the primary literature.
Synthetic Agonists for FFA4
Long-chain fatty acids can be rapidly converted to different biologically active species that function at GPCRs other than FFA1 and/or FFA4. Given that such interconversion is challenging to limit in vivo, direct studies with fatty acids can be difficult to interpret unequivocally. As such, the identification and characterisation of synthetic ligands with affinity for FFA4 have been integral to better understanding the biological functions of this receptor (Table 1). However, owing to the strongly overlapping profile of fatty acids as activators of FFA4 and FFA1, many of the initially described synthetic ligands with activity at FFA4 are also able to activate FFA1 (Table 1). This reflects their structural similarity to fatty acids. More recently described ligands display improved selectivity for FFA4 over FFA1 (Table 1).
Table 1.
Structures of Key Ligands with Activity at FFA4
| Ligand | Structure | Mode of action | Selectivity | Other comments | Refs |
|---|---|---|---|---|---|
| TAK-875 | 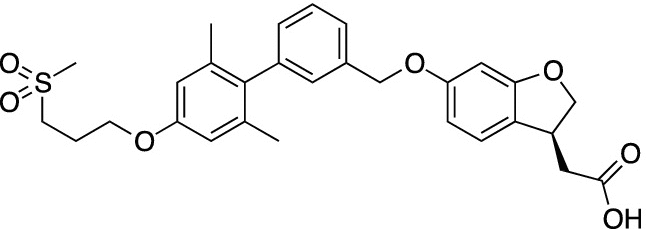 |
Agonist | FFA1 | [11] | |
| GW9508 | 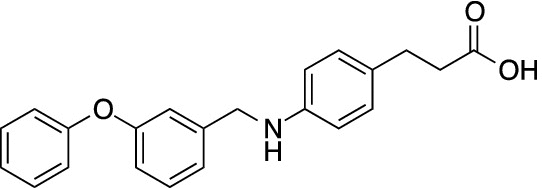 |
Agonist | FFA1 >>FFA4 | 38, 21 | |
| NCG21 | 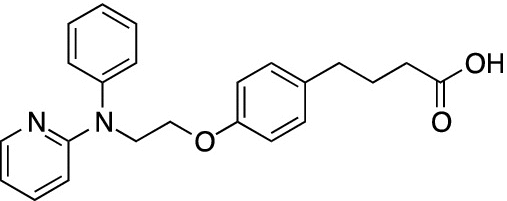 |
Agonist | FFA1 = FFA4 | 50, 51 | |
| TUG-891 | 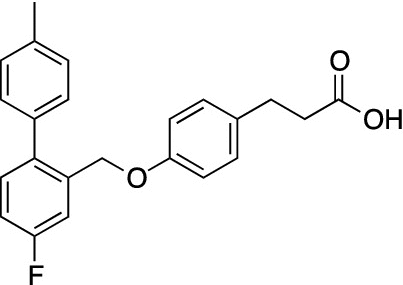 |
Agonist | FFA4 > FFA1 | Degree of selectivity is species dependent | 52, 40 |
| GSK137647A | 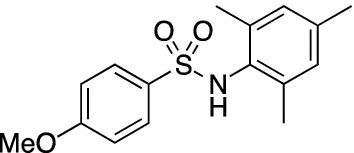 |
Agonist | FFA4 | [56] | |
| TUG-1197 | 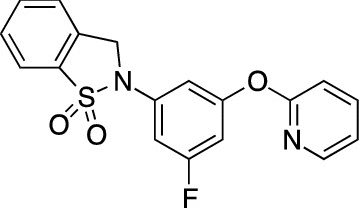 |
Agonist | FFA4 | [57] | |
| AH-7614 | 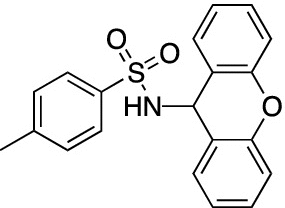 |
Antagonist | FFA4 | 56, 60 | |
| TUG-1387 | 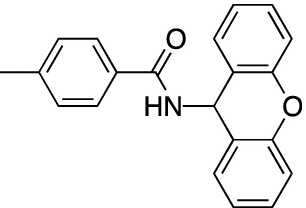 |
Inactive | – | [60] |
The first described synthetic FFA1 active agonist, GW9508 (4-{[(3-phenoxyphenyl)methyl]amino}benzenepropanoic acid), was immediately shown to also activate FFA4, although with some 100-fold lower potency [38]. Therefore, in the initial absence of FFA4-selective synthetic agonist ligands, GW9508 was used as a FFA4 agonist in several studies using cells and tissues that lacked detectable levels of co-expressed FFA1 (e.g., [21]). Probably because this ligand is available commercially, this approach has continued (reviewed in [2]). Early efforts to produce FFA4-selective ligands reported only modest success. For example, Suzuki et al. [50] modified PPARγ-active molecules to generate a ligand, 4-{4-[2-(phenyl-pyridin-2-yl-amino)-ethoxy]-phenyl}-butyric acid, later named NCG21 (Table 1), which displayed modest selectivity for FFA4 over FFA1, but also only modest potency [51]. With appropriate recognition that it probably acts as a combined FFA4 and FFA1 activator, this compound has been used recently alongside other more-selective ligands to help unravel the contribution of each long-chain fatty acid receptor to the ability of the omega-3 fatty acid, hexadeca-4,7,10,13-tetraenoic acid [16:4(n-3)], to generate systemic resistance to the DNA-damaging chemotherapeutic cisplatin [36] (discussed below). In the first significant advance in developing FFA4-selective agonist ligands, Shimpukade et al. [52] reported the ortho-biphenyl ligand 4-{[4-fluoro-4′-methyl(1,1′-biphenyl)-2-yl]methoxy}-benzenepropanoic acid (TUG-891) (Table 1). This molecule showed good potency at both human and mouse FFA4 and 1000-fold selectivity over human FFA1 in assays based on the induced interactions between the receptor and β-arrestin 2. These studies also reported loss of agonist function of TUG-891 at an Arg99 to Gln mutant of human FFA4 and, alongside the parallel mutagenesis studies of Watson et al. [39], provided the first direct evidence of the key role of this arginine residue in coordinating the carboxylate of fatty acids and fatty acid-like synthetic ligands.
GPCRs responsive to fatty acids have been shown to display substantial variation in pharmacology between species orthologues [53]. More extensive studies with TUG-891 illustrated that selectivity reported between human FFA4 and FFA1 was significantly less pronounced at mouse orthologues [40] and also varied when measuring G protein-mediated Ca2+ elevation versus receptor interactions with an arrestin [40]. Although there is no comprehensive analysis of these features for many ligands, which are rarely reported beyond humans versus mice, the major reason for the lower selectivity between the mouse orthologues is their higher potency at mouse FFA1 rather than reduced potency at mouse FFA4. In practise, although a potent and selective FFA4 agonist in human cells and tissues, TUG-891 is best described as a dual FFA4 and FFA1 agonist in equivalent tissues from rodents [36]. More recently reported compounds have provided greater levels of selectivity. For example, Adams et al. [54] reported a chromane propionic acid-based agonist series where specific members are at least 300-fold more selective for both human and mouse FFA4 compared with FFA1. Similarly, Sparks et al. [55] described a phenylpropanoic acid series with an exemplar compound showing between 40- and 130-fold selectivity over FFA1 across human, mouse, and rat orthologues.
Similar to free fatty acids, all of the compounds described above contain a carboxylate that has been shown directly (or at least modelled) to interact with Arg99 of FFA4. However, a pair of recent reports has also described sulfonamide-containing FFA4 agonists 56, 57. GSK137647A [4-methoxy-N-(2,4,6-trimethylphenyl)benzenesulfonamide] (Table 1) is reported to have greater than 50-fold selectivity for FFA4 over FFA1 and that this is preserved across species [56]. Similarly, a potent nonacidic sulfonamide FFA4 agonist, TUG-1197 {2-[3-(pyridin-2-yloxy)phenyl]-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide} (Table 1) is described as having no detectable activity at FFA1 [57]. Despite the nonacidic nature of the compound, both mutational and modelling studies indicated that it likely binds within the same orthosteric binding pocket as the carboxylate-containing agonists that resemble synthetic fatty acids [57]. Clearly, the more selective nature of several recently disclosed ligands offers potential for more defined analysis of FFA4-mediated functions, and several such ligands (e.g., GSK137647A) are now available from commercial sources. However, not all of the more recently described ligands are suitable for in vivo studies due to poor pharmacokinetic and pharmacodynamic properties [56]. By contrast, although not currently available from commercial suppliers, phenylpropanoic acid ‘compound 29’ [55], nonacidic sulfonamide ‘compound 34’ [57], and chromane propionic acid ‘compound 18’ [54] have each been used for rodent in vivo studies to explore glucose handing and aspects of regulation of insulin sensitivity. In each case, these have provided clear support for an important role of FFA4 in the regulation of glucose homeostasis. The emergence of chemically distinct series of FFA4 agonists allows the possibility of using pairs of compounds from different series to provide greater support for specific roles of FFA4 [58]. Even if the full off-target profile of each ligand is not currently available, it is reasonable to assume that compounds derived from different chemotypes will produce varying non-FFA4-mediated effects. Although no FFA4 agonist has yet entered clinical studies, there is considerable expectation that such ligands may offer novel combinations of benefits in T2DM 53, 59.
Synthetic Antagonists for FFA4
To date, only compounds from a single chemical series have been reported as FFA4 ‘antagonists’ (Table 1). ‘Compound 39’ (4-methyl-N-9H-xanthen-9-yl-benzenesulfonamide), now available from commercial vendors as AH-7614, was initially reported as an antagonist at this receptor [56]. This compound, and a closely related molecule, 4-methyl-N-(9H-thioxanthen-9-yl)benzenesulfonamide (TUG-1506) [60] (Table 1), act as noncompetitive, negative allosteric modulators of the action of a range of FFA4 agonist chemotypes [60]. Although neither competitive nor displaying more than moderate affinity (approximately 10 nM) [60] for the receptor, AH-7614 has recently been used in a range of studies; for example, to confirm a specific role for FFA4 as the long-chain fatty acid receptor on splenic macrophages responsible for release of a lysophosphatidic acid species that is able to produce systemic resistance to platinum-containing chemotherapeutics [36]; to identify a role of the receptor in activation of brown fat [61]; to explore whether various effects of arachidonic acid are produced via FFA4 [62]; and the contribution of FFA4 to docosahexaenoic acid [20:6(n-3)]-mediated effects in GnRH-producing neurones [63]. However, as noted by Watterson et al. [60], AH-7614 is a simple xanthene-containing chemical and, although this molecule does not block agonist effects at FFA1 [60], other potential sites of action have not been explored. Therefore, it is noteworthy that, in studies in rat round spermatids, AH-7614 itself induced an increase in [Ca2+]i in the absence of FFA4 receptor-activating ligands [64], akin to those produced by the omega-6 fatty acid arachidonic acid [20:4(n–6)]. Given such concerns, Watterson et al. [60] suggested as a control the parallel use of 4-methyl-N-(9H-xanthen-9-yl)benzamide (TUG-1387) (Table 1), an analogue of both AH-7614 and TUG-1506 that has no activity at FFA4. Whereas AH-7614 blocked autocrine differentiation of mouse preadipocytes towards an adipocyte phenotype, TUG-1387 did not [60], providing stronger evidence for a direct role of FFA4 in this process.
Therapeutic Opportunities for FFA4 in Cancer
Beyond T2DM, the therapeutic area in which FFA4 has perhaps attracted greatest attention to date is cancer. In part, this reflects appreciated roles for fatty acids and fat-rich diets in either promoting cancer cell growth and motility or the capacity of health-beneficial fatty acids, including omega-3 fatty acids, to reduce the growth of several types of tumour. The significance of a number of the reported studies is hard to define, in that many have focused largely on the effects of fatty acids alone and have frequently suggested potential contributions of both FFA4 and FFA1. Several of these studies have recently been reviewed elsewhere [65]. However, growing appreciation of the developing pharmacology at FFA4 and at FFA1 has provided new insights into the potential for FFA4 ligands. A key example is the contribution of FFA4 to the development of systemic resistance to cisplatin-based chemotherapy. Mesenchymal stem cells produce a pair of polyunsaturated fatty acids [66], 12-S-hydroxy-5,8,10-heptadecatrienoic acid (12-S-HHT), an activator of the leukotriene B4 receptor 2 [67], and hexadeca-4,7,10,13-tetraenoic acid [16:4(n-3)], which, similar to many other fatty acids, can activate both FFA4 and FFA1 with similar potency [36], at least in vitro. Recent studies used a combination of the genetic elimination of FFA4 in mice and a range of both markedly selective and dual-acting FFA4 and FFA1 agonists, in concert with selective antagonists of each receptor, to show that, although both FFA4 and FFA1 are expressed by a key population of splenic macrophages, activation by 16:4(n-3) of FFA4 on these cells specifically induced a signalling cascade that, via activation of cytosolic phospholipase A2, resulted in the release of several species of lysophosphatidic acid into the medium (Figure 3). Such ‘conditioned medium’ was able to induce resistance to cisplatin-induced DNA damage in tumour cells and, subsequently, to limit the effectiveness of cisplatin to inhibit tumour growth when injected into tumour-bearing mice [36]. Moreover, a single lysophosphatidic acid species, C24:1, was able to replicate the effect of 16:4(n-3)-conditioned medium, suggesting C24:1 as a likely end-effector, although the molecular basis for the effect of lysophosphatidic acid C24:1 remains undefined [36]. Addition of either dual-acting FFA4/FFA1 agonists, including TUG-891 [52] and NCG21 [51], but, more importantly, also the highly selective FFA4 agonist TUG-1197 [57], to splenic macrophages isolated from wild-type mice generated conditioned medium that was as effective in producing chemoresistance as treatment with 16:4(n-3) [36]. By contrast, addition of 16:4(n-3) to splenic macrophages isolated from mice lacking expression of FFA4 did not generate conditioned medium that was able to replicate this effect. Moreover, co-addition of the FFA4 antagonist AH-7614 blocked the ‘conditioning’ effect of 16:4(n-3) in cells isolated from wild-type animals [36]. Together, these results suggest the potential of FFA4 antagonists to either limit the development of resistance to platinum-containing chemotherapeutic agents or to spare the dose of such agents required for efficacy. However, the effect of the second platinum-induced fatty acid, 12-S-HHT, was not lost in cells isolated from FFA4 knockout mice [36]. As such, blockade of FFA4 with a synthetic antagonist is unlikely to be fully effective in vivo if used in isolation.
Figure 3.

A Role for Free Fatty Acid Receptor 4 (FFA4) in Mediating Resistance to Platinum-Containing Chemotherapeutics. Cisplatin and other platinum-based therapeutics are integral to chemotherapy. However, the development of resistance to such agents limits their effectiveness. In a mouse model, the fatty acid 16:4(n-3) was identified to contribute to the development of resistance in a manner linked to a key subpopulation of splenocytes. 16:4(n-3) is able to activate both FFA4 and FFA1, and these two G-protein-coupled receptors are co-expressed by this splenocyte population. Medium ‘conditioned’ by exposure of splenocytes to 16:4(n-3) is able to induce resistance to cisplatin when injected into tumour-bearing mice and both pharmacological and receptor ‘knockout’ studies demonstrated the effect of 16:4(n-3) to be mediated specifically by FFA4 [36]. The mechanism was shown to involve FFA4-mediated activation of splenocyte cytosolic (c)PLA2, resulting in the release of a specific lysophosphatidic acid species C24:1, with this isolated lipid species able to mimic the effect of conditioned medium [36].
As well as the above studies, Meier and coworkers used TUG-891, alongside omega-3 fatty acids, to show a potential role for FFA4 in inhibiting proliferation of DU145 prostate cancer cells [68]. Given that these cells express both FFA4 and FFA1 and the current view that TUG-891 may not be sufficiently selective to fully differentiate between the two fatty acid receptors, the fact that FFA4 knockdown prevented TUG-891-induced inhibition of growth and migration provided extra support for a key role of FFA4 [68]. However, the obvious conclusion from these studies is that FFA4 agonism, rather than antagonism, as suggested in limiting the development of induced chemoresistance to cisplatin treatment, might be effective in this context. In a subsequent study, the same group used a pair of FFA1/FFA4-active agonists to examine possible roles of these GPCRs in the proliferation of a pair of breast cancer cell lines. However, although the pharmacological studies were unable to clearly discriminate between the effects of the ligands as reflecting activation of FFA1 or FFA4, both the MCF-7 and MDA-MD-231 cell lines appeared to express significantly higher levels of mRNA encoding FFA1 than FFA4. Moreover, although immunoblotting studies potentially detected two variants of FFA4 in MCF-7cells, equivalent forms were lacking in MDA-MD-231 cells. Based on these observations, the authors concluded that the major contributions of GW9508 and TUG-891 were likely to be mediated via FFA1 [69].
Other Potential Therapeutic Opportunities in Targeting FFA4
Both mRNA expression patterns and availability of selective FFA4 antibodies have revealed that this receptor is expressed in a variety of tissues, pointing to a range of functions yet to be fully defined [5]. For example, FFA4 was found to be highly expressed in murine lung [70] (Figure 4), where clues to its function are only just beginning to emerge. Expression in this organ appears restricted to the airway epithelium [70], which primarily comprises mucous-secreting goblet cells and ciliate columnar epithelial cells. The role of FFA4 in these various cell types is unknown, but it is of interest that the dietary-derived omega-3 fatty acids, docosahexaenoic acid [22:6(n-3)] and eicosapentaenoic acid [20:5(n-3)], have been reported to be enriched in airway mucosa [71], suggesting that there is a ‘store’ of endogenous ligands for the FFA4 receptor located at the lung epithelium. Furthermore, a recent study suggested that FFA4, acting on epithelial club cells, promotes bronchial epithelial repair following naphthalene-induced epithelial injury [72]. This study may provide a potential explanation for the observed benefits of clinical administration of omega-3 fatty acid-rich fish oils in human lung injury [73]. Further studies on the role of FFA4 in lung function are clearly warranted.
Figure 4.

Immunohistochemical Detection of Free Fatty Acid receptor 4 (FFA4) in Mouse Lung. Antigen-retrieved, formalin-fixed paraffin-embedded lung tissue slices obtained from wild-type mice were incubated with anti-FFA4 antibodies [48]. They were incubated subsequently with Alexa 488-conjugated secondary antibody (green) and cy3-conjugated anti α-actin (to detect smooth muscle) (red). Following further washes, samples were placed on coverslips with a mounting medium containing the nuclear stain DAPI (blue). Images were taken using a confocal microscope. FFA4 appears to be expressed predominantly within the epithelial layer.
Studies on the possibility that FFA4 regulates central functions are similarly in their infancy. However, FFA4 immunoreactive neurones have been identified in the hypothalamus, where they are thought to protect against hypothalamic dysfunction in obesity [74]. The mechanism appears to have two distinct components. The first is a reduction in hypothalamic inflammation mediated by downregulation of TLR4 and TNF-induced inflammatory pathways [74]. The second is via neuropeptide Y-expressing hypothalamic neurones that co-express FFA4, through which omega-3 fatty acids are seen to reverse obesity-induced resistance to leptin [74]. These central FFA4-mediated mechanisms to regulate food intake and body mass likely work in concert with peripheral pathways, as exemplified by studies on the release of ghrelin, a key hormone that mediates food-seeking behaviour and food intake as well as adiposity. Dietary-derived long-chain fatty acids targeting FFA4 on ghrelin-expressing cells act to inhibit the secretion of ghrelin [75], thereby providing a negative feedback loop to reduce food intake.
Concluding Remarks
Although potential therapeutic opportunities from targeting FFA4 are currently focussed firmly on T2DM and other metabolic indications, including nonalcoholic steatohepatitis, the broader expression pattern of the receptor suggests wider roles in (patho)physiology. Full understanding of the repertoire of physiological roles of FFA4 has been restricted by the focus on T2DM and the beguiling prospect that it may result in an entirely novel therapeutic entity. However, the continuing development of tool compounds, vital for detailed pharmacological analysis, in concert with the use of novel genetically engineered mice where in vivo FFA4 signalling is modulated (Figure 2), as used for other GPCRs (e.g., the muscarinic acetylcholine M3 receptor [76]), is likely to highlight further opportunities (see Outstanding Questions). Two opportunities that are beginning to emerge are the role of FFA4 in cancer and the possibility that the high level of expression of FFA4 in lung will be linked to key physiological endpoints associated with airway function and dysfunction.
Conflict of Interest
Both GM and BDH are shareholders in Caldan Therapeutics, a company exploring potential novel treatments for type 2 diabetes.
Outstanding Questions.
Will FFA4 become a validated therapeutic target in T2DM? Although FFA4 is considered a candidate therapeutic target for T2DM, to date no clinical studies have been initiated. Key outstanding questions include whether agonist ligands with appropriate drug-like characteristics can be identified, whether these will provide features and outcomes above and beyond glucose-lowering in rodent models and whether potential safety issues associated with clinically used FFA1/GPR40 agonists can be mitigated against or overcome.
How important will the anti-inflammatory effects of FFA4 be to clinical delivery? Although activation of FFA4 expressed by macrophages results in the regulation of proinflammatory mediator release via a mechanism reported to involve interaction of the receptor with an arrestin adaptor protein, the broader roles of both non-G protein-mediated signaling and of activation of G protein-dependent pathways other than those involving the phosphoinositidase C-linked G proteins Gq/G11 remains to be fully elucidated and understood. Clear analysis of these aspects of FFA4 signaling will define whether a search for agonist ligands that display ‘bias’ might be useful.
Might FFA4 become a therapeutic target for treatment of nonalcoholic steatohepatitis? Although little direct information is yet available on this topic, considerable interest in this possibility has been voiced
Will FFA4 antagonists provide a mean to enhance the chemotherapeutic index of platinum-based chemotherapeutics? Recent studies suggest that ‘platinum’ chemotherapy induces the generation of the fatty acid hexadeca-4,7,10,13-tetraenoic acid and that, by activating FFA4/GPR120 expressed by a splenocyte subpopulation, this fatty acid promotes release of a isoform of lysophosphatidic acid that appears to act as the final mediator of the development of resistance to platinum-based chemotherapeutics. Defining the molecular target for this lysophosphatidic acid may be key in providing novel approaches to either limit the development of such chemoresistance or to allow the effective use of lower doses.
What therapeutic opportunities may be identified for regulation of FFA4 in airway function? Early studies of the distribution of FFA4/GPR120 highlighted that mRNA encoding the receptor was expressed abundantly in lung. Assessment of the role of FFA4/GPR120 in lung is currently at an early stage, but is likely to provide a rich source of novel insights.
Acknowledgments
Work described herein was supported by Biotechnology and Biological Sciences Research Council [grant BB/K019864/1)] (to GM) and [grant BB/K019856/1] (to ABT).
Contributor Information
Graeme Milligan, Email: Graeme.Milligan@glasgow.ac.uk.
Andrew B. Tobin, Email: Andrew.Tobin@glasgow.ac.uk.
Resources
References
- 1.Blad C.C. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat. Rev. Drug Discov. 2012;11:603–619. doi: 10.1038/nrd3777. [DOI] [PubMed] [Google Scholar]
- 2.Milligan G. Complex pharmacology of free fatty acid receptors. Chem. Rev. 2017;117:67–110. doi: 10.1021/acs.chemrev.6b00056. [DOI] [PubMed] [Google Scholar]
- 3.Ulven T., Christiansen E. Dietary fatty acids and their potential for controlling metabolic diseases through activation of FFA4/GPR120. Annu. Rev. Nutr. 2015;35:239–263. doi: 10.1146/annurev-nutr-071714-034410. [DOI] [PubMed] [Google Scholar]
- 4.Briscoe C.P. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 5.Hirasawa A. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 6.Stoddart L.A. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, −2, and −3: pharmacology and pathophysiological functions. Pharmacol. Rev. 2008;60:405–417. doi: 10.1124/pr.108.00802. [DOI] [PubMed] [Google Scholar]
- 7.Davenport A.P. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol. Rev. 2013;65:967–986. doi: 10.1124/pr.112.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leifke E. A multiple-ascending-dose study to evaluate safety, pharmacokinetics, and pharmacodynamics of a novel GPR40 agonist, TAK-875, in subjects with type 2 diabetes. Clin. Pharmacol. Ther. 2012;92:29–39. doi: 10.1038/clpt.2012.43. [DOI] [PubMed] [Google Scholar]
- 9.Kaku K. Randomized, double-blind, dose-ranging study of TAK-875, a novel GPR40 agonist, in Japanese patients with inadequately controlled type 2 diabetes. Diabetes Care. 2013;36:245–250. doi: 10.2337/dc12-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayer M. Evaluation of the pharmacokinetics and safety of a single oral dose of fasiglifam in subjects with normal or varying degrees of impaired renal function. Drugs R D. 2014;14:273–282. doi: 10.1007/s40268-014-0066-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaku K. Long-term safety and efficacy of fasiglifam (TAK-875), a G-protein-coupled receptor 40 agonist, as monotherapy and combination therapy in Japanese patients with type 2 diabetes: a 52-week open-label phase III study. Diabetes Obes. Metab. 2016;18:925–929. doi: 10.1111/dom.12693. [DOI] [PubMed] [Google Scholar]
- 12.Wolenski F.S. Fasiglifam (TAK-875) alters bile acid homeostasis in rats and dogs: a potential cause of drug induced liver injury. Toxicol. Sci. 2017;157:50–61. doi: 10.1093/toxsci/kfx018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otieno M.A. Fasiglifam (TAK-875): mechanistic investigation and retrospective identification of hazards for drug induced liver injury (DILI) Toxicol. Sci. 2017 doi: 10.1093/toxsci/kfx040. Published online February 16, 2017. [DOI] [PubMed] [Google Scholar]
- 14.Chen C. GPR40 agonists for the treatment of type 2 diabetes mellitus: the biological characteristics and the chemical space. Bioorg. Med. Chem. Lett. 2016;26:5603–5612. doi: 10.1016/j.bmcl.2016.10.074. [DOI] [PubMed] [Google Scholar]
- 15.Ghislain J., Poitout V. The role and future of FFA1 as a therapeutic target. Handb. Exp. Pharmacol. 2017;236:159–180. doi: 10.1007/164_2016_51. [DOI] [PubMed] [Google Scholar]
- 16.Sharma N. Recent advances in development of GPR40 modulators (FFA1/FFAR1): an emerging target for type 2 diabetes. Mini Rev. Med. Chem. 2017;17:947–958. doi: 10.2174/1389557517666170120152917. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y. A selective GPR40 (FFAR1) agonist LY2881835 provides immediate and durable glucose control in rodent models of type 2 diabetes. Pharmacol. Res. Perspect. 2016;4:e00278. doi: 10.1002/prp2.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurica E.A. Discovery of pyrrolidine-containing GPR40 agonists: stereochemistry effects a change in binding mode. J. Med. Chem. 2017;60:1417–1431. doi: 10.1021/acs.jmedchem.6b01559. [DOI] [PubMed] [Google Scholar]
- 19.Agarwal S. Identification of an orally efficacious GPR40/FFAR1 receptor agonist. ACS Med. Chem. Lett. 2016;7:1134–1138. doi: 10.1021/acsmedchemlett.6b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christiansen E. Activity of dietary fatty acids on FFA1 and FFA4 and characterisation of pinolenic acid as a dual FFA1/FFA4 agonist with potential effect against metabolic diseases. Br. J. Nutr. 2015;113:1677–1688. doi: 10.1017/S000711451500118X. [DOI] [PubMed] [Google Scholar]
- 21.Oh D.Y. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X. Cyclooxygenase-2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4) FASEB J. 2013;27:4987–4997. doi: 10.1096/fj.13-235333. [DOI] [PubMed] [Google Scholar]
- 23.Wellhauser L., Belsham D.D. Activation of the omega-3 fatty acid receptor GPR120 mediates anti-inflammatory actions in immortalized hypothalamic neurons. J. Neuroinflamm. 2014;11:60. doi: 10.1186/1742-2094-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lager S. Expression and localization of the omega-3 fatty acid receptor GPR120 in human term placenta. Placenta. 2014;35:523–525. doi: 10.1016/j.placenta.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim N. Endogenous ligand for GPR120, docosahexaenoic acid, exerts benign metabolic effects on the skeletal muscles via AMP-activated protein kinase pathway. J. Biol. Chem. 2015;290:20438–20447. doi: 10.1074/jbc.M115.657379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furutani A. Fish oil accelerates diet-induced entrainment of the mouse peripheral clock via GPR120. PLoS One. 2015;10:e0132472. doi: 10.1371/journal.pone.0132472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arantes E.L. Topical docosahexaenoic acid (DHA) accelerates skin wound healing in rats and activates GPR120. Biol. Res. Nurs. 2016;18:411–419. doi: 10.1177/1099800415621617. [DOI] [PubMed] [Google Scholar]
- 28.Della Corte C. Docosahexaenoic acid and Its role in G-protein-coupled receptor 120 activation in children affected by nonalcoholic fatty liver disease. Endocr. Dev. 2016;30:29–36. doi: 10.1159/000439324. [DOI] [PubMed] [Google Scholar]
- 29.Bjursell M. The beneficial effects of n-3 polyunsaturated fatty acids on diet induced obesity and impaired glucose control do not require Gpr120. PLoS One. 2014;9:e114942. doi: 10.1371/journal.pone.0114942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung H. Omega-3 fatty acids reduce obesity-induced tumor progression independent of GPR120 in a mouse model of postmenopausal breast cancer. Oncogene. 2015;34:3504–3513. doi: 10.1038/onc.2014.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belchior T. Omega-3 fatty acids protect from diet-induced obesity, glucose intolerance, and adipose tissue inflammation through PPARγ-dependent and PPARγ-independent actions. Mol. Nutr. Food Res. 2015;59:957–967. doi: 10.1002/mnfr.201400914. [DOI] [PubMed] [Google Scholar]
- 32.Pærregaard S.I. FFAR4 (GPR120) Signaling is not required for anti-inflammatory and insulin-sensitizing effects of omega-3 fatty acids. Mediators Inflamm. 2016;2016:1536047. doi: 10.1155/2016/1536047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shewale S.V. In vivo activation of leukocyte GPR120/FFAR4 by PUFAs has minimal impact on atherosclerosis in LDL receptor knockout mice. J. Lipid Res. 2017;58:236–246. doi: 10.1194/jlr.M072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X. Endogenously generated omega-3 fatty acids attenuate vascular inflammation and neointimal hyperplasia by interaction with free fatty acid receptor 4 in mice. J. Am. Heart Assoc. 2015;4:e001856. doi: 10.1161/JAHA.115.001856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connell T.D. ω3-Polyunsaturated fatty acids for heart failure: effects of dose on efficacy and novel signaling through free fatty acid receptor 4. J. Mol. Cell Cardiol. 2016;103:74–92. doi: 10.1016/j.yjmcc.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Houthuijzen J.M. Fatty acid 16:4(n-3) stimulates a GPR120-induced signaling cascade in splenic macrophages to promote chemotherapy resistance. FASEB J. 2017;31:2195–2209. doi: 10.1096/fj.201601248R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yore M.M. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014;159:318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Briscoe C.P. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br. J. Pharmacol. 2006;148:619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson S.J. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol. Pharmacol. 2012;81:631–642. doi: 10.1124/mol.111.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudson B.D. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol. Pharmacol. 2013;84:710–725. doi: 10.1124/mol.113.087783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Curto E. Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signaling. J. Biol. Chem. 2016;291:27147–27159. doi: 10.1074/jbc.M116.754887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engelstoft M.S. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Mol. Metab. 2013;2:376–392. doi: 10.1016/j.molmet.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stone V.M. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia. 2014;57:1182–1191. doi: 10.1007/s00125-014-3213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segerstolpe Å. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell. Metab. 2016;24:593–607. doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talukdar S. Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol. Sci. 2011;32:543–550. doi: 10.1016/j.tips.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burns R.N. Mechanisms of homologous and heterologous phosphorylation of FFA receptor 4 (GPR120): GRK6 and PKC mediate phosphorylation of Thr34 7, Ser35 0, and Ser35 7 in the C-terminal tail. Biochem. Pharmacol. 2014;87:650–659. doi: 10.1016/j.bcp.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butcher A.J. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J. Biol. Chem. 2014;289:18451–18465. doi: 10.1074/jbc.M114.568816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prihandoko R. Distinct phosphorylation clusters determine the signaling outcome of free fatty acid receptor 4/G protein-coupled receptor 120. Mol. Pharmacol. 2016;89:505–520. doi: 10.1124/mol.115.101949. [DOI] [PubMed] [Google Scholar]
- 49.Moore K. Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: comparison with human GPR120 splice variants. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009;154:419–426. doi: 10.1016/j.cbpb.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki T. Identification of G protein-coupled receptor 120-selective agonists derived from PPARgamma agonists. J. Med. Chem. 2008;51:7640–7644. doi: 10.1021/jm800970b. [DOI] [PubMed] [Google Scholar]
- 51.Sun Q. Structure-activity relationships of GPR120 agonists based on a docking simulation. Mol. Pharmacol. 2010;78:804–810. doi: 10.1124/mol.110.066324. [DOI] [PubMed] [Google Scholar]
- 52.Shimpukade B. Discovery of a potent and selective GPR120 agonist. J. Med. Chem. 2012;55:4511–4515. doi: 10.1021/jm300215x. [DOI] [PubMed] [Google Scholar]
- 53.Suckow A.T., Briscoe C.P. Key questions for translation of FFA receptors: from pharmacology to medicines. Handb. Exp. Pharmacol. 2017;236:101–131. doi: 10.1007/164_2016_45. [DOI] [PubMed] [Google Scholar]
- 54.Adams G.L. Discovery of chromane propionic acid analogues as selective agonists of GPR120 with in vivo activity in rodents. ACS Med. Chem. Lett. 2016;8:96–101. doi: 10.1021/acsmedchemlett.6b00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sparks S.M. Exploration of phenylpropanoic acids as agonists of the free fatty acid receptor 4 (FFA4): Identification of an orally efficacious FFA4 agonist. Bioorg. Med. Chem. Lett. 2017;27:1278–1283. doi: 10.1016/j.bmcl.2017.01.034. [DOI] [PubMed] [Google Scholar]
- 56.Sparks S.M. Identification of diarylsulfonamides as agonists of the free fatty acid receptor 4 (FFA4/GPR120) Bioorg. Med. Chem. Lett. 2014;24:3100–3103. doi: 10.1016/j.bmcl.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 57.Azevedo C.M. Non-acidic free fatty acid receptor 4 agonists with antidiabetic activity. J. Med. Chem. 2016;59:8868–8878. doi: 10.1021/acs.jmedchem.6b00685. [DOI] [PubMed] [Google Scholar]
- 58.Hansen S.V., Ulven T. Pharmacological tool compounds for the free fatty acid receptor 4 (FFA4/GPR120) Handb. Exp. Pharmacol. 2017;236:33–56. doi: 10.1007/164_2016_60. [DOI] [PubMed] [Google Scholar]
- 59.Scheen A.J. Investigational insulin secretagogues for type 2 diabetes. Expert Opin. Investig. Drugs. 2016;25:405–422. doi: 10.1517/13543784.2016.1152260. [DOI] [PubMed] [Google Scholar]
- 60.Watterson K.R. Probe-dependent negative allosteric modulators of the long-chain free fatty acid receptor FFA4. Mol. Pharmacol. 2017;91:630–641. doi: 10.1124/mol.116.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quesada-López T. The lipid sensor GPR120 promotes brown fat activation and FGF21 release from adipocytes. Nat. Commun. 2016;7:13479. doi: 10.1038/ncomms13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villegas-Comonfort S. Effects of arachidonic acid on FFA4 receptor: signaling, phosphorylation and internalization. Prostaglandins Leukot. Essent. Fatty Acids. 2017;117:1–10. doi: 10.1016/j.plefa.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 63.Tran D.Q. Induction of Gnrh mRNA expression by the ω-3 polyunsaturated fatty acid docosahexaenoic acid and the saturated fatty acid palmitate in a GnRH-synthesizing neuronal cell model, mHypoA-GnRH/GFP. Mol. Cell Endocrinol. 2016;426:125–135. doi: 10.1016/j.mce.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 64.Paillamanque J. Arachidonic acid triggers [Ca2+]i increases in rat round spermatids by a likely GPR activation, ERK signalling and ER/acidic compartments Ca2+ release. PLoS One. 2017;12:e0172128. doi: 10.1371/journal.pone.0172128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Houthuijzen J.M. For better or worse: FFAR1 and FFAR4 signaling in cancer and diabetes. Mol. Pharmacol. 2017;90:738–743. doi: 10.1124/mol.116.105932. [DOI] [PubMed] [Google Scholar]
- 66.Roodhart J.M. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. 2011;20:370–383. doi: 10.1016/j.ccr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 67.Houthuijzen J.M. Lysophospholipids secreted by splenic macrophages induce chemotherapy resistance via interference with the DNA damage response. Nat. Commun. 2014;5:5275. doi: 10.1038/ncomms6275. [DOI] [PubMed] [Google Scholar]
- 68.Liu Z. Omega-3 fatty acids and other FFA4 agonists inhibit growth factor signaling in human prostate cancer cells. J. Pharmacol. Exp. Ther. 2015;352:380–394. doi: 10.1124/jpet.114.218974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hopkins M.M. Eicosopentaneoic acid and other free fatty acid receptor agonists inhibit lysophosphatidic acid- and epidermal growth factor-induced proliferation of human breast cancer cells. J. Clin. Med. 2016;5:E16. doi: 10.3390/jcm5020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miyauchi S. Distribution and regulation of protein expression of the free fatty acid receptor GPR120. Naunyn Schmiedebergs Arch. Pharmacol. 2009;379:427–434. doi: 10.1007/s00210-008-0390-8. [DOI] [PubMed] [Google Scholar]
- 71.Freedman S.D. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N. Eng. J. Med. 2004;350:560–569. doi: 10.1056/NEJMoa021218. [DOI] [PubMed] [Google Scholar]
- 72.Lee K.P. ω-3 polyunsaturated fatty acids accelerate airway repair by activating FFA4 in club cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017;312:L835–L844. doi: 10.1152/ajplung.00350.2016. [DOI] [PubMed] [Google Scholar]
- 73.Wiedegmann H.P. Fish oil is not the fix for acute lung injury. Crit. Care Med. 2011;39:1829–1830. doi: 10.1097/CCM.0b013e31821b82bb. [DOI] [PubMed] [Google Scholar]
- 74.Cintra D.E. Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS One. 2012;7:e30571. doi: 10.1371/journal.pone.0030571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu X. Postprandial inhibition of gastric ghrelin secretion by long-chain fatty acid through GPR120 in isolated gastric ghrelin cells and mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G367–G376. doi: 10.1152/ajpgi.00541.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bradley S.J. Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction. Proc. Natl. Acad. Sci. U. S. A. 2016;113:4524–4529. doi: 10.1073/pnas.1521706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Susuki M. Medium-chain fatty acid-sensing receptor, GPR84, is a proinflammatory receptor. J. Biol. Chem. 2013;288:10684–10689. doi: 10.1074/jbc.M112.420042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee J. Branched fatty acid esters of hydroxy fatty acids (FAHFAs) protect against colitis by regulating gut innate and adaptive immune responses. J. Biol. Chem. 2016;291:22207–22217. doi: 10.1074/jbc.M115.703835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yore M.M. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014;159:318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh D.Y., Olefsky J.M. Omega 3 fatty acids and GPR120. Cell Metab. 2012;15:564–565. doi: 10.1016/j.cmet.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]