

Abstract
The World Health Organization recognizes human African trypanosomiasis, Chagas’ disease and the leishmaniases as neglected tropical diseases. These diseases are caused by parasitic trypanosomatids and range in severity from mild and self-curing to near invariably fatal. Public health advances have substantially decreased the impact of these diseases in recent decades, but alone will not eliminate these diseases. Here we discuss why new drugs against trypanosomatids are needed, approaches that are under investigation to develop new drugs and why the drug discovery pipeline remains essentially unfilled. Additionally, we consider the important challenges to drug discovery strategies and the new technologies that can address them. The combination of new drugs, new technologies and public health initiatives are essential for the management and hopefully eventual elimination of trypanosomatid diseases from the human population.
Trypanosomatid parasites cause several neglected diseases of humans and animals, which range in severity from comparatively mild to near invariably fatal [1,2]. The organisms responsible for human diseases are: Trypanosoma brucei spp., which cause human African trypanosomiasis (HAT); T. cruzi, which causes Chagas’ disease; and Leishmania spp., which cause the leishmaniases. Together, these insect-transmitted parasites threaten millions of people. All of these organisms have complex life-cycles, with substantial differences in morphology, cell biology and biochemistry between lifecycle stages and in some cases between species (Box 1).
Box 1. The life cycles of trypanosomatid parasites.
Trypanosomatid parasites have multiple different hosts and are transmitted by insect vectors to humans (see figure, part a). Trypanosoma brucei spp. are transmitted by the tsetse fly (see figure, part b). Following infection at the site of the insect bite, the parasites circulate freely in the bloodstream, and may also accumulate in tissues such as adipose tissue [145] and skin [146]; early stage symptoms of human African trypanosomiasis (HAT) are non-specific, and include fever, headache, fatigue, muscle pain, anaemia and swollen lymph nodes. In second stage disease trypanosomes invade the central nervous system, which gives rise to various neurological symptoms, culminating in coma and death. Diagnosis is frequently only made at this late stage, when treatment options are limited as first stage drugs do not cross the blood brain barrier. Closely related species (in particular T. congolense, T. vivax and T. evansi) also infect domestic and wild animals, causing nagana, a wasting disease, which has a major impact on agricultural animals in Africa, Asia and parts of South America [147–149].
Chagas’ disease is endemic in South and Central America [150], but migration has spread cases to North America, Europe, Japan and Australia [151]. T. cruzi is transmitted by triatomine bugs; following a blood meal, infective parasites in the vector’s faeces can enter at the site of the bite, or through transfer to mucous membranes of the eye, nose or mouth (see figure, part c). Alternative transmission routes include blood transfusion, transplantation, ingestion of contaminated food or drink and maternal vertical transmission. Parasites are predominantly intracellular within mammalian hosts and invade multiple cell types. Chagas’ disease has acute and chronic stages; the acute stage has high fatality in children, but in adults frequently presents with non-specific symptoms, which resolve. Parasites are detectable microscopically in the bloodstream during the acute stage, but are generally absent after progression to the chronic stage, when diagnosis by microscopy is difficult, although xenodiagnostic and serological tests are effective. The infection may remain asymptomatic for life (indeterminant phase), but in a subset of cases the disease progresses to involve the heart or gastrointestinal tract. Patients often only present when they have symptoms, such as cardiac dysfunction, difficulty in swallowing (mega-oesophagus) or in defaecation (mega-colon). Pathology is thought to be either a consequence of the immune response to the ongoing low-grade infection or of an autoimmune response [152]. Differences in disease manifestation are probably due both to genetic variation between T. cruzi strains [153] and host factors [154].
Leishmania spp. cause a set of diseases with varying severity, depending on the species [155]. The parasites are transmitted in the saliva of sandflies; they then invade monocytes and macrophages, where they replicate in parasitophorous vacuoles (see figure, part d). Visceral leishmaniasis is predominantly caused by L. donovani and L. infantum and is a systemic infection that affects the liver, spleen and bone marrow. It is associated with progressive wasting, anaemia and hepatosplenomegaly, and has a high mortality rate unless treated. Mucocutaneous and cutaneous leishmaniasis are characterized by skin and mucosal lesions of varying severity. Co-infection by L. donovani or L. infantum and HIV is a growing concern in Europe.
Parts a and b of the figure were adapted from Ref. 14.
Figure for Box 1.

Control of trypanosomatid diseases has had a mixed history, although public health campaigns are showing success in many instances. For example, the Southern Cone and Andean initiatives are tackling Chagas’ disease with a combination of insecticide spraying of dwellings, improved housing, screening of people in endemic zones and blood bank monitoring [3]. However, South America has considerable numbers of T. cruzi-infected individuals and many infected individuals have migrated to North America and Europe, where the disease is non-endemic. In the case of leishmaniasis, co-infection with Leishmania spp. and HIV can increase the disease burden and severity, and recent refugee movements from the Middle East into Europe are likely to increase the prevalence of leishmaniasis in Europe. In the immediate post-colonial period, HAT resurged, but vector control, active case-finding and treatment have all helped control the disease [4]. However, many trypanosomatid diseases are zoonotic, which will make eradication extremely unlikely. The current target is elimination, which is still an ambitious goal. Despite progress, trypanosomatid diseases remain a substantial public health problem and there is an urgent need for new drugs to tackle them.
None of the available drugs for treatment of trypanosomatid disease (Table 1) are satisfactory and new drugs are needed, especially those suitable for rural health systems with limited resources. The current standard of care is monotherapy, with the exception of nifurtimox-eflornithine combination therapy (NECT) for HAT, although various drug combinations are in clinical trials. Importantly, many of the current treatments require parenteral administration [5], and also suffer from poor efficacy, major side effects and increasing levels of resistance [6–8]. Most of the drugs in use probably have multiple modes of action, due to acting on multiple parasite targets [9]. Goals for drug discovery include the development of completely new classes of therapeutics, reduced host toxicity, improved administration regimens and the development of combination therapies.
Table 1. Current drugs used to treat trypansomatid diseases.
| Drug | Structure | Comments |
|---|---|---|
| Human African trypanosomiasis | ||
| Suramin |
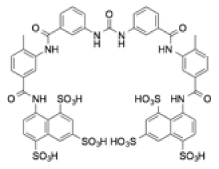
|
|
| Pentamidine |
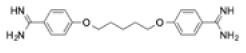
|
|
| Melarsoprol |
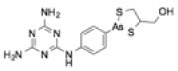
|
|
| Eflornithine |
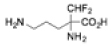
|
|
| NECT (nifurtimox-eflornithine combination therapy) |
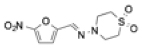
|
|
| Chagas’ disease | ||
| Benznidazole |
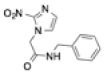
|
|
| Nifurtimox |
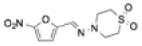
|
|
| Visceral leishmaniasis | ||
| Amphotericin B |
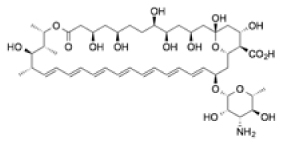
|
|
| Miltefosine |
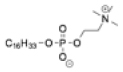
|
|
| Pentavalent antimonials |
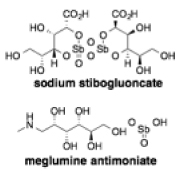
|
|
| Paromomycin |
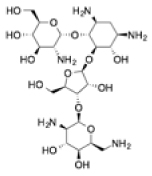
|
|
Vaccine development is a powerful approach to disease management but remains challenging in the trypanosomatid diseases due to efficient immune evasion mechanisms, such as antigenic variation in the African trypanosomes, and the intracellular locations of T. cruzi and Leishmania spp. in the human host. Progress towards human [10] and canine [11] leishmania vaccines and the challenges in developing vaccines for HAT [12] and Chagas’ disease [13] have been reviewed recently and will not be discussed further here.
In this Review, we discuss the potential for the development of new drug therapies against trypanosomatids. We highlight unique biological features of these parasites that suggest potential targets, methods used to identify bioactive compounds and consider some of the outcomes of recent campaigns. We encourage the reader to consider excellent reviews of life cycles, genomes, pathogenesis and more general aspects of the biology of trypanosomatids published elsewhere [14–19].
Drug discovery
A successful drug discovery campaign typically takes 10-15 years (Fig. 1). High attrition rates, together with relatively few organizations working on drug discovery for trypanosomatid parasites mean that the number of new compounds in clinical development is very low (Fig. 2) and unlikely to meet the clinical need. Ideally, the pipeline would contain multiple new agents that are suitable for combination therapy. The advantages of combination therapies are manifold: they can increase the clinical efficacy of treatments; they can reduce side effects by allowing lower dosing of individual agents; and they can reduce the risk of resistance development. Reducing resistance is critical for safeguarding whatever new medicines do emerge from the drug discovery pipeline.
Figure 1. The drug discovery process.

Drug discovery progresses through several stages and each stage involves specific steps and regulations. The failure rate at each stage is high, which underscores the need for an active pipeline of drug discovery projects.
Figure 2. Antitrypanosomatid compounds currently in preclinical and clinical development.

a | Several compounds are currently in preclinical and clinical development for human African trypanosomiasis (HAT), visceral leishmaniasis (VL) and Chagas’ disease (CD). b | Antitrypansomatid compounds identified through phenotypic approaches that have been progressed into clinical trials.
Three broad approaches are used for drug discovery against trypanosomatids: (1) target-based approaches involve screening for inhibitors against a purified protein, for example an enzyme. Compounds identified through the screening (or structure-based) process are subsequently optimized to show efficacy in a cellular model; (2) phenotypic approaches involve screening for growth-inhibitors directly against an intact parasite, usually in an in vitro culture; (3) compound re-positioning is re-deployment of compounds previously developed for an alternative indication as anti-trypanosomatid therapies.
The drug discovery process is ideally driven by target product profiles (TPPs), which define the properties required of a drug for clinical application [20–22]. Such factors include: route of administration (oral, inhaled, intravenous, etc.), acceptable dosing regimen and course of treatment, acceptable safety and tolerability levels, cost and shelf-life. TPPs allow for the development of compound progression criteria, which define parameters for compounds at each stage in the drug discovery process (hit, validated hit, lead, preclinical candidate, etc. –see Fig. 1). Progression criteria include assessment of the physicochemical properties (such as solubility in physiological media, lipophilicity, molecular weight, hydrogen bond donors and acceptors), potency (against the molecular target and intact organism), selectivity, chemical and metabolic stability, pharmacokinetics, efficacy and safety. Additional criteria for parasitic infections can include factors such as cytocidal activity and the rate of parasite killing. The Drugs for Neglected Diseases Initiative (DNDi) is a public private partnership that focuses on drug discovery and clinical development for these organisms. It has developed TPPs and compound progression criteria for trypanosomatid diseases (www.dndi.org) [21].
Target-based approaches
For target-based approaches, the key is careful selection of the most promising molecular targets. A recent review highlights some examples of target-based drug discovery against trypanosomatids [23]. For neglected diseases in general, including the trypanosomatid diseases, there has been very limited success from target-based approaches. This is often due to lack of translation from inhibition of the target (enzyme) in a purified cell-free context to inhibition of proliferation of the parasite and/or subsequent activity in an animal disease model. In part, this reflects the absence of robustly validated targets (for example, enzymes whose activity is essential to the parasite) and highlights the need for fundamental research into trypanosomatid biology and for thorough genetic and chemical validation of potential targets24. However, this is only part of the problem. As will be discussed below, an improved understanding of how to translate compounds that are active in vitro into therapeutics is required, which includes better defining the cellular and animal models (Box 2) that predict clinical efficacy in humans.
Box 2. Animal models.
Currently, human African trypanosomiasis (HAT) is the trypanosomatid disease with the best-evaluated animal models. Peripheral (stage 1) disease is studied in mice that are infected with Trypansosma brucei brucei S427 (infective to animals) or T. b. rhodesiense STIB900 (infective to animals and humans); cure is defined as no parasites in the blood and survival beyond 30 days. Recently, bioluminescence imaging with transgenic parasites that express luciferase has been developed [156,157]; this greatly reduces the number of animals required for monitoring and provides improved longitudinal insight into tissue tropisms and parasite population dynamics within the same mouse; this advance is set to substantially improve in vivo models of HAT. Although most patients with HAT are infected by T. b. gambiense, models for this parasite are more challenging [158].
For central nervous system (CNS) disease (stage 2) , the standard model is infection with T. b. brucei GVR35 [159], which infects the CNS after ˜21 days [160]. As relapse is common, the major issue of this model is the length of time required before cure can be declared (180 days). Bioluminescence imaging may shorten this timeframe [160].
Chagas’ disease has both an acute and chronic stage. There are a number of animal models for the acute stage of infection. Early mouse models of acute Chagas’ disease used reduction in parasitaemia or mean survival time as measures of efficacy [161,162]. More recent models are also using bioluminescence [163–166]. However, treatment does not always cause complete cure and parasite levels rebound after immunosuppression with cyclophosphamide, which indicates that a treatment-refractory reservoir exists [164]. Sterile cure is likely to depend on many factors, including the compound used, treatment regimen and strain of T. cruzi. An animal model that can predict efficacy in humans will be key to avoid failures such as that experienced in the recent clinical trial of posaconazole [166].
Although there are several long-term mouse models for Chagas’ disease, it is unclear if they accurately reflect the human chronic stage and confirmation of complete cure is difficult as parasites can rarely be detected in the blood. Quantitative PCR is problematic as parasites can be found in different tissues, which requires examination of multiple tissues and multiple sampling to minimize false negatives. The new bioluminescent models offer an alternative strategy, which is more direct and only detects live cells. Interestingly, in the bioluminescent models of chronic infection in mice, parasites were mainly detected in the gastrointestinal tract, principally the colon and stomach [163], and essentially no parasites were detected in the heart. Whether these tissue tropisms apply to all strains of T. cruzi is not known.
Mice and hamsters are the most common animal models for visceral leishmaniasis, although other species such as dogs are sometimes used [167]. In the typical mouse model, animals are infected intravenously with amastigotes that are derived from a hamster spleen and treatment is started seven days after infection and usually continued for five days. Animals are euthanized three days after treatment is complete and liver smears are taken.
We have published some criteria to help in selection of molecular targets (Box 3) [9,20,24]. Many target-based drug discovery programmes can be initially viewed as target-validation [25]. It is therefore vital to obtain proof-of-concept (POC) of anti-parasitic activity for new target-derived chemical series at the earliest possible stage, ideally both in cellular and animal models, to minimize waste of resources, should the target fail to progress.
Box 3. Proposed criteria for target selection.
Genetic and chemical validation of the target (essentiality)
Whether the target can be inhibited by drug-like molecules (druggability)
Whether it is possible to establish a high-throughput assay (assayability)
The potential for resistance to emerge against the target
The potential for toxicity by inhibition of human homologues (selectivity)
The availability of structural information of the target
Drug targets with the highest degree of validation
The best validated drug-target in T. brucei is ornithine decarboxylase (ODC), which is the target of eflornithine, a drug used clinically for treatment of HAT. Eflornithine is a suicide inhibitor that was initially developed for the treatment of cancer, but subsequently re-purposed for HAT [26]. Selectivity is thought to arise from the more rapid turnover of human ODC compared to the trypanosome enzyme [27], or due to inhibition of trypanothione biosynthesis [28], which is a metabolite unique to trypanosomatids.
The enzyme N-myristoyltransferase (NMT) has also been well validated as a molecular target for HAT [29–31]. In a programme initiated with a high-throughput screen against NMT, a compound series was identified and subsequently optimized (typified by DDD85646, Fig. 3b) to be active in a mouse model of the first stage of HAT, which does not involve the central nervous system. There was strong evidence that the compounds inhibit NMT in cells and that this inhibition kills parasites, which validates both the target and the mode of action. NMT is also present in humans, but T. brucei is acutely sensitive to NMT inhibition, probably because endocytosis, which occurs at a very high rate in T. brucei, is affected. NMT has also been validated as a target in a second stage mouse model of HAT (K.D.R., personal communication). The challenge with second stage disease is that compounds need to penetrate the blood brain barrier and achieve therapeutic concentrations in the central nervous system without causing host toxicity
Figure 3. Molecular targets in trypanosomatids.

a | trypanosomatids show unique metabolic pathways and cellular functions that are attractive for drug discovery. Many enzymes are divergent from other eukaryotes and they have unique or highly specialized organelles such as the kinetoplast and the glycosome, respectively. b | For some antitrypansomatid compounds the molecular targets are known. DDD85646 targets N-myristoyltransferase (NMT); posaconazole and ravuconazole are CYP51 inhibitors; K777 irreversibly inhibits the cysteine protease cruzipain; and GNF6702 selectively inhibits the trypanosomatid proteasome.
Very recently the proteasome has been shown to have great potential as a target in all three types of trypansomatids [32]. This study used a phenotypic approach to develop a parasite-specific, selective inhibitor (GNF6702) that does not inhibit the human proteasome This is an excellent example of taking a phenotypic hit and subsequently deconvoluting the target. The initial experiments to determine the mode of action involved generating compound-resistant T. cruzi mutants, followed by whole genome sequencing, which revealed mutations in the β4 subunit of the proteasome. Various additional biochemical experiments demonstrated that GNF6703 specifically inhibits the chymotrypsin-like activity of the parasite proteasome.
Biological features of trypanosomatids that might be targeted
Trypanosomatids are one of the most evolutionary divergent eukaryotic lineages from mammals, a feature that is reflected in their distinct biology (Fig. 3a). Conversely, there are many similarities between T. brucei, T. cruzi and Leishmania spp. and many molecular mechanisms are conserved between all three lineages. Trypanosomatid-specific metabolic and cellular pathways (discussed below) should represent excellent drug targets as specificity should be an easier criterion to control, but no candidate drugs have been developed that inhibit such targets. In fact, most potential trypanosome-specific targets remain unexplored for drug discovery and/or are of unknown druggability. Ironically, the best validated targets in trypanosomatids are one repurposed from oncology (ODC) and two pan-eukaryotic essential targets (NMT and the proteasome), discussed above.
Uniquely, trypanosomatids package the first six or seven enzymes of glycolysis into the glycosome, a specialized form of the peroxisome. Glycolysis is especially important for the bloodstream forms of African trypanosomes, which rely exclusively on this pathway for ATP production. The compartmentalization of glycolysis in trypanosomatids is accompanied by fundamental differences in allosteric regulation of the pathway compared to most other eukaryotes. Because of this, phosphofructokinase, for example, is being pursued as a target [33]. However, computational modelling of glycolysis suggests that there is little prospect of killing trypanosomes by suppressing glycolysis unless inhibition is irreversible or uncompetitive, due to the enormous glycolytic flux through the system [34]. Metabolic compartmentalization requires the transport of substrates (glucose), negatively-charged metabolic intermediates (such as 3-phosphoglyceric acid, dihydroxyacetone phosphate and glycerol-3-phosphate) and products (such as pyruvate). The transporters and permeases for these molecules (and other larger charged metabolites and co-factors such as nucleotide di- and tri-phosphates, nucleotide sugars and NAD(H)) remain elusive, but could represent potential drug targets [18]. Similarly, glycosome biogenesis might also have unique and druggable features.
With about 180 members, the kinomes of the trypanosomatids are extensive but lack predicted receptor tyrosine kinases or even general tyrosine kinases and contain disproportionately high numbers of certain enzyme subtypes, for example, STE and NEK kinases [35]. Chemical biology has demonstrated distinct inhibition-profiles for host and parasite kinases [36], which suggests that selective inhibition of parasite kinases is feasible. Furthermore, both genome-wide and kinome-wide RNAi knockdown screens indicate that several of these enzymes are essential [35,37]. However, although potent and selective inhibitors against essential protein kinases in cultured parasites have been developed [38–40], none was sufficiently active in vivo. The repurposing of mammalian kinase inhibitors has shown promise [41], with cure of HAT in an animal model reported for one kinase inhibitor [42]. However, so far it is unknown which (if any) trypanosomatid kinase(s) are being targeted by the repurposed mammalian kinase inhibitor and both chemical and genetic validation of this approach are still required. The recent identification of a highly divergent kinetochore in trypanosomes [43] may provide new kinase targets in this class but their druggability remains to be determined.
Trypanosomatids also have other divergent signaling pathways that offer therapeutic opportunities. For example, whereas trypanosomatids lack identifiable G-protein coupled receptors, they have a large family of membrane-bound adenyl cyclases that modulate the host immune response [44] and are likely involved in parasite differentiation [45] through unconventional downstream cAMP response proteins [46,47]. Similarly, a family of cAMP phosphodiesterases have attracted interest as potential targets [48,49].
Assembly and maintenance of the cell surface is crucial for organisms that interact with, and defend themselves against, their hosts and the immune system. Although fundamentals of protein and membrane synthesis, transport and recycling are well conserved across eukaryotes, there is substantial specialization between species. For example, trypanosomatids have evolved divergent protein N-glycosylation [50,51] and glycosylphosphatidylinositol (GPI) membrane anchor biosynthetic pathways, the latter being a validated target for HAT [52]. Similarly, the machineries for export of glycoproteins and for endocytosis and recycling, are highly divergent in trypanosomes, with several canonical components being replaced by novel factors [53–55]. The major surface glycoproteins are also distinct, and although the functions of many of these glycoproteins remain unknown, they likely are crucial for survival in the host [56]. Furthermore, the endosomal apparatus contains some components that are important for defence against the innate immune response [57]. All of these peculiarities offer the potential for therapeutic exploitation.
Interestingly, endocytosis and transport mediated by transmembrane proteins are important for drug uptake by trypanosomatids. For example, T. brucei aquaglyceroporin-2 is responsible for melarsoprol and pentamidine uptake and the invariant surface glycoprotein-75 is responsible for suramin uptake [58–60].
Divergent gene expression might also be targeted. Transcription in trypanosomatids is almost exclusively polycistronic and several chromatin modifiers are involved in determining the sites of transcription initiation and termination [61]. Bromodomain ‘readers’ in particular, which bind acetylated histones, are potential targets [62] as are the histone acetyltransferases or ‘writers’ [63] and the deacetylases or ‘erasers’ [64]. Novel transcription factors are also potentially druggable, such as class I transcription factor A [65], which is also, unusually, required for the transcription of genes encoding the major surface glycoproteins by RNA polymerase I (Pol1) in the African trypanosome; Pol1 is restricted to ribosomal RNA transcription in most other eukaryotes.
All protein coding mRNAs require trans-splicing in trypanosomatids, which is distinct from the cis-splicing required to remove introns from the vast majority of mammalian mRNAs. Although the splicing mechanism for cis- and trans-splicing is broadly similar, there are substantial differences in the splicing machinery [66]. Polycistronic transcription relies on post-transcriptional control of gene expression and, consistent with this, a large number of trypanosomal RNA binding proteins have key roles in mRNA maturation, stability and translation control [67]. The process of translation itself also presents novel targets at the level of the ribosome [68] and the aminoacyl tRNA synthetases [69].
Examples of target-based drug discovery programmes
There are several examples of trypanosomatid-specific targets that have been investigated. One example involves redox metabolism: trypanosomes have a unique di-thiol, trypanothione. Several enzymes involved in the synthesis and modulation of the trypanothione redox system, including trypanothione reductase (TryR) [70] and synthetase (TryS) [71,72], are essential for parasite survival. A large number of attempts have been made to discover drug-like inhibitors of TryR [73,74]. Multiple series have been identified from several large and medium scale screens of synthetic libraries and natural products, some of which have been used in structure-based drug design. Unfortunately, so far none have delivered compounds suitable for clinical development. A key reason appears to be the large hydrophobic active site of TryR [70], which is difficult to inhibit with a small drug-like molecule. Active compounds have also been designed against the companion biosynthetic enzyme TryS [75].
In Chagas’ disease, sterol biosynthesis has been the focus of multiple drug discovery programmes. Several molecular targets have been investigated, including sterol 14α-demethylase (CYP51) [76] and squalene synthase [77]. Much of this effort involved re-purposing compounds developed as antifungals or as cholesterol-lowering agents. Clinical trials tested two CYP51 inhibitors, posaconazole [78] and fosravuconazole (also known as E1224; a prodrug of ravuconazole) (Fig. 3b). Although there was initial clearance of parasites with posaconazole and fosravuconazole, disease recurred after treatment ceased, indicating that neither agent is suitable for treatment, at least as a monotherapy. The reasons for these failures are not fully understood, but they highlight the need for animal models (Box 2) that can distinguish between compounds that are efficacious in humans and those, such as posaconazole, that are not [79].
Another substantially progressed target in Chagas’ disease is cruzipain, a protease with similarities to cathepsin L. A vinyl sulfone irreversible inhibitor of cruzipain (K777) was advanced to preclinical development [80,81] but abandoned due to poor tolerability even at low dose in primates and dogs.
Folate metabolism has also been the subject of extensive drug discovery programmes, in particular the enzymes dihydrofolate reductase and a trypanosome-specific target, pteridine reductase 1 (PTR1). Both are thought to be essential, at least in T. brucei [82,83]. There are similarities between the substrates for these enzymes and inhibitors have been identified which inhibit both enzymes [84]. Despite extensive work in this area, for reasons that are not fully understood, there is little correlation between activity against the enzyme and activity against the parasite [85]. As far we understand, no inhibitors for these targets have been progressed to preclinical development.
Trypanosomatids lack purine biosynthesis and take up purines from the host. In leishmaniasis, this dependence on external purine has been targeted with allopurinol. Allopurinol is taken up by the parasites and then phosphoribosylated to the corresponding nucleotide, which then acts as a cellular poison [86]. It is used for the treatment of leishmaniasis in dogs and has been in clinical trials in humans, but has not progressed.
Phenotypic approaches
To circumvent the challenges of target-based drug discovery, phenotypic approaches have been widely used for most of the neglected disease agents, including for the trypanosomatids [87]. Here, the key requirements are appropriate chemical libraries for screening [5,88], robust assays and appropriate screening cascades.
Screening cascades
Many different cellular assays are available for analysis of trypanosome responses to compounds (Fig. 4). It is especially important to establish that compounds are effective against the clinically relevant life-cycle stages, which can be problematic for the intracellular stages of Leishmania spp. and T. cruzi. Compounds must cross multiple membranes to reach the parasite in cellular assays; three in the case of Leishmania spp. amastigotes that reside inside an acidic (pH ˜5.5) parasitophorous vacuole within the macrophage. In animal models the situation is more complex still, with additional barriers to cross.
Figure 4. Phenotypic approaches to discover antitrypanosomatid compounds.

Various lifecycle stages can be used for the purpose of hit discovery that range from insect forms to host-stage forms in animal models. The different technologies that can be used for phenotypic assays depend on the parasite form and stage and have specific advantages and disadvantages. Examples of compounds whose antitrypanosomal activity was detected using insect forms, in vitro host-stage forms and animal models are shown.
To identify molecules suitable for drug discovery, it is essential to use an appropriate combination of assays to build confidence in the chemical start points (hits). For example, initial hit finding generally requires a high-throughput assay to access chemical diversity, followed by confirmation by more physiologically relevant, but lower throughput, assays (Fig. 4). Additional cellular assays, which are representative of the in vivo situation, are important to support combined pharmacokinetic and pharmacodynamic analyses in animal models. These provide an indication of the concentration and exposure time of a given compound required to kill the parasites in animals and to predict the likely situation in humans. The best combination and the optimal order of phenotypic assays depends on the parasite in question.
For T. brucei, typical high-throughput screens identify not only favoured cytocidal compounds but also proliferation-slowing and cytostatic compounds. A secondary assay is therefore required to select those hits that are cytocidal, either using washout experiments to demonstrate a lack of reversibility [29,89,90], or direct cell viability assays [91]. A further issue for HAT is that compounds need to penetrate the blood-brain-barrier to be active against second stage disease. Currently there are no reliable in vitro (cell-based) assays for predicting blood brain barrier penetration. However, the physicochemical properties of compounds which are likely to penetrate this barrier have been analyzed [92–94], which can assist in the selection of compounds for screening.
T. cruzi usually replicates well in intracellular amastigote assays [95], which allows the identification of both cidal and static compounds. However, as T. cruzi evades the immune system during chronic infection, cidal compounds are likely essential for cure. Therefore, hits need to be followed up in a cidality assay. There is now also a drive to remove compounds that target CYP51 (see above) and assays directly assessing activity against CYP51 [78,96] need to be added to the cascade.
For Leishmania spp., many of the intracellular assays only report cytocidal compounds as intracellular amastigotes replicate relatively slowly [97,98]. Although this eliminates the need for further cidality assays, the hit-rates are low [21], and throughput can be relatively poor. Furthermore, it is challenging to identify potentially valuable but weak or poorly selective hits. One solution to the low throughput is to use an axenic (free growing) amastigote assay as the primary screen. Axenic amastigotes do not occur naturally, so care must be taken in interpreting the data. Such assays also need to be designed to only identify ytocidal compounds to prevent false positives, as we have recently reported [99]. Hits can then be confirmed in an intracellular assay.
For all trypanosomatids, as in other areas of anti-infective drug discovery, it is also critical to measure activity against a panel of clinical isolates before progressing compound series too far, to be sure that activity is not laboratory-strain specific. For all cell-based assays, replication rate, starting density and rate-of-kill are key factors to correctly interpret compound potency, it is important to define these parameters as clearly as possible before interpreting data on new hits.
To date, phenotypic approaches have been more successful in discovering new developable series compared to target-based screens. In the case of HAT, the two compounds currently in clinical trials, fexinidazole and the oxaborole SCYX-7158, were both derived from phenotypic approaches (Fig. 2b).
Recognizing that nitroheterocycles have anti-trypanosomal activity, DNDi sourced and screened a large number and re-discovered fexinidazole, a compound that had been investigated preclinically by Hoechst, but then abandoned [100]. Nitroheterocycles can be genotoxic, so counter-screening for genotoxicity at an early stage was a key selection criterion [101]. Fexinidazole, like nifurtimox, is a prodrug that requires activation by a nitroreductase [102]. Fexinidazole has also been shown to have potential for the treatment of Chagas’ disease [103] and leishmaniasis. Sulfoxide and the sulfone metabolites of fexinidazole, rather than the parent drug, are the active compounds against the intramacrophage form of Leishmania spp. [104]. Results of a phase II proof-of-concept clinical trial against visceral leishmaniasis are expected soon. For Chagas’ disease, the metabolites are more active than the parent compound [105] and a phase II trial was initiated. Unfortunately, the doses used in this trial caused safety and tolerability issues and the trial was stopped.
Another nitroheterocycle (DNDI-VL-2098) showed activity in animal models of leishmaniasis [106] and was selected for further development from a series of nitroimidazooxazoles being investigated preclinically by DNDi. Unfortunately, toxic effects were noted and the progression of the compound has been stopped. A backup for this compound (DNDi-0690) has now been selected and is in preclinical development (Fig. 2). The antitubercular drug, delamanid, which belongs to the same chemical class, has also been proposed as a possible candidate [107]. A novel nitroreductase (NTR2) has been identified as the activating enzyme for these bicyclic nitroheterocycles in leishmania [108].
From a library of oxaboroles, the benzoxaborole 6-carboxamides were particularly active against T. brucei and, following a lead optimization programme, SCYX-7158 was selected as a clinical candidate for HAT [109]. The mode of action of oxaboroles against HAT is still not understood, but may include polypharmacology [110]. Another oxaborole, DNDi-6148 has recently been moved into preclinical development with DNDi for visceral leishmaniasis.
A series of diamidines showed potent activity against HAT, one of which (pafuramidine) was taken into clinical trials. Pafuramidine is a prodrug that is metabolized by the host into the active compound, diamidine DB [75]. Although the precise mode(s) of action are unknown, like other diamidines, the drug is selectively concentrated within parasites [111]. However, clinical trials were unsuccessful and were stopped due to safety concerns [112].
Trypanosomatids lack purine biosynthesis and take up purines from the host. In leishmaniasis, this dependence on external purine has been targeted with allopurinol. Allopurinol is taken up by the parasites and then phosphoribosylated to the corresponding nucleotide, which then acts as a cellular poison [86]. It is used for the treatment of leishmaniasis in dogs and has been in clinical trials in humans, but has not progressed.
Sitamaquine, an orally bioavailable 8-aminoquinoline, was discovered by the Walter Read Army Institute of Research and has been progressed into clinical trials by GlaxoSmithKline for visceral leishmaniasis [113,114]. The mechanism of its action is not fully understood [115].
Importantly, the mode(s) of action of all of the aforementioned compound series were unknown during the drug discovery process and up to candidate selection, and indeed, remain at best incompletely characterized. Thus, although the absence of a clear understanding of mode of action does not preclude clinical development, it does represent a major gap in knowledge that can hinder the further optimization and the development of back-up series.
Compound repositioning
Recently, there has been considerable interest in repurposing or repositioning drugs and drug-leads for many diseases [116–122]. However, the concept is not new and many drugs currently used for the treatment of neglected tropical diseases were ’repositioned’ from anticancer, antibacterial, antifungal and anti-helminthic indications. These include the antifungal amphotericin B, the anticancer agent miltefosine, and the antibiotic paromomycin, all of which were repurposed for visceral leishmaniasis [123]. Other examples have already been mentioned above. More recently, the nitrofuran, nifurtimox, which was originally developed in the 1960s for the treatment of Chagas’ disease, was repositioned as a combination therapy with eflornithine (NECT) to decrease the cost and duration of treatment of late-stage HAT [124]. Unfortunately, not all repurposing efforts have been successful, for example the CYP51 inhibitors against T. cruzi.
Drug repurposing is not without its drawbacks. For example, the drugs may have been optimized for a different human disease and the initial therapeutic activity may become an undesirable side effect that needs to be reduced or eliminated. A second problem is that repurposed drugs often do not fit the TPP for neglected diseases and many are not fit-for-purpose in resource-poor settings. High cost, marginal safety windows, the need for hospitalization or prolonged treatment, poor stability in conditions of high temperature and high humidity and lack of oral bioavailability are just some of the issues that must be addressed. Nonetheless, Sir James Black’s adage, “the most fruitful basis for the discovery of a new drug is to start with an old drug,” still carries substantial value, as success with NECT, amphotericin B, paromomycin and other drugs attest [125].
Target deconvolution
Phenotypic screening of chemical libraries and existing drugs has produced many chemical start points, particularly for T. brucei and T. cruzi, and to a lesser extent for Leishmania spp. [21]. However, chemical optimization of these phenotypic start points can be challenging due, for example, to pharmacokinetic issues, insufficient potency or off-target toxicity. Without target deconvolution, that is, the identification of the molecular target, target-based screening cannot be used to find alternative chemical scaffolds that might overcome these issues, and structure-based drug design cannot be used for compound optimization [126]. In addition, although not essential, knowledge of the mode-of-action can facilitate the design of combination therapies, surveillance for the emergence and spread of resistance and assessment of the risk of resistance.
Target deconvolution has proved very successful in many therapeutic areas [127], in particular in malaria where a number of new targets have been identified recently from phenotypic hits, including PfATP4 [128], PfPI4K [129], PfeEF2 [130], PfCARL [131] and PfPheRS [132]. Another recent example of validating a trypanosomatid target, the proteasome, through deconvolution of an optimized phenotypic hit was discussed above [32].
Although several approaches to target deconvolution exist [133], further development is needed for the trypanosomatids. Small molecules have many potential cellular targets and unbiased screening approaches can be extremely powerful in identifying genetic, biochemical or metabolic associations with their mode(s) of action. Genetic screens perturb gene-expression by knock-down, knock-out, or over-expression. A particularly powerful approach for T. brucei is RNA interference target sequencing or RIT-seq [37], which has successfully identified genes that contribute to anti-trypanosomal drug action [58]. The CRISPR-Cas9 genome-editing approach is established as a powerful alternative to RNAi for genome-scale loss-of-function screening [134] and is functional in T. cruzi [135,136] and in Leishmania spp [137,138]. Gain-of-function screens have also been used for drug target identification in T. brucei [139] and Leishmania spp. [140], and similar technology is available for T. cruzi [141], but these approaches are yet to be widely applied to target deconvolution.
Chemical proteomics is also useful for target deconvolution. Essentially, proteins from a cell extract are isolated based on affinity for immobilised small-molecule drug leads and then identified by mass spectrometry [142], an approach that has been used to identify potential target kinases in T. brucei [36]. Other approaches, such as the cellular thermal shift assay, also use chemical proteomic profiling but do not require immobilization of the inhibitor on beads [143], which can be problematic for maintaining binding to the target protein. In addition, metabolomics can detect the depletion of a metabolic products and accumulation of substrates, which can point towards the specific target enzymes [144]. Cellular approaches can also contribute in target deconvolution by revealing morphological defects in the cellular compartment(s) that are primarily affected by a drug lead. Similarly, computational approaches may be used for structure-based target prediction.
A combination of largely unbiased orthogonal approaches to target deconvolution (from those outlined above) represents a powerful new strategy to alleviate current bottlenecks in the progression of compounds developed from trypanosomatid phenotypic screens.
Perspectives
In the last decade, drug discovery efforts against neglected tropical diseases have increased. Importantly some pharmaceutical companies have become more engaged in the past decade and several academic centres have established powerful drug discovery capabilities. Public-private partnerships such as DNDi and various charitable and governmental funding agencies have made major financial and other contributions to allow activities to proceed on the scale required for drug discovery. It is exciting that new compounds are undergoing clinical trials for HAT, although attrition in the drug discovery process suggests there is no room for complacency. However, there are currently no new classes of drug in the clinical development pipeline for leishmaniasis or Chagas’ disease and there is still a great need for new (ideally oral) drugs to treat each trypanosomatid disease. Combination therapies to improve efficacy and reduce the risk of resistance, by definition, require two or more drugs, preferably with distinct modes of action, and place even more pressure on the development pipeline. Hence more work is still required.
There are multiple reasons why the drug discovery process has not yet yielded new drugs for trypanosomatid diseases. There is a lack of well-validated molecular targets in the trypanosomatids, which has hampered traditional target-based approaches. Target-based assays have been replaced by more successful phenotypic screens. However, phenotypic screens have their own challenges. For HAT, compounds need to penetrate the blood-brain barrier to treat second stage disease, which limits the compounds which should be screened or progressed. For Chagas’ disease, many of the hits target CYP [51], which is a very promiscuous target and which was unsuccessful in the clinical trials of posaconazole and fosravuconazole. For leishmaniasis, there is a very low hit-rate against the clinically relevant intramacrophage form, for reasons that are not well understood.
One of the key challenges of phenotypic drug discovery is how to address issues, such as potency, toxicity and pharmacokinetic problems, that arise during the hit optimization process. Scaffold hopping and vector optimization become more problematic without knowledge of the molecular target. Identifying the targets of phenotypic hits should facilitate progression of these compounds and also enable more high value target-based drug discovery in the future.
Another major challenge is defining the relevant cellular and animal models (Box 2) that closely mimic human clinical conditions. This is problematic for trypanosomatid diseases as there are very few clinically active compounds which can be used to define these models and many of the clinically active compounds are not conventional; they are reactive (for example, nitro drugs); they are selective due to active transport (for example, melarsoprol and pentamidine); they are active through polypharmacological actions (for example, arsenicals and antimonials); or they are covalent inhibitors (for example, eflornithine). It is possible that in vitro cellular assays which more closely mimic animal and human leishmanial infections could have a higher hit rate in phenotypic screening than the current assays. For Chagas’ disease, we need cellular and animal models that can distinguish between compounds that are active in humans (for example, benznidazole) and those that are not (for example, posaconazole). Each new compound that is taken into the clinic can provide valuable pharmacodynamic insights, which should be fed back into the drug discovery process to refine all these models.
Despite the aforementioned challenges, the development of new in vivo and in vitro technologies, superior methods for genetic manipulation of parasites and increased collaborations between the pharmaceutical industry, academic laboratories, charities and other non-government organizations will start to fill the drug pipeline against these devastating and global diseases.
Key points.
Trypanosomatid parasites cause several neglected diseases of humans and animals, which range in severity from comparatively mild to nearly invariably fatal. The organisms responsible for human diseases are: Trypanosoma brucei spp., which cause human African trypanosomiasis (HAT); T. cruzi, which causes Chagas’ disease; and Leishmania spp., which cause the leishmaniases.
The current drugs for treating trypanosomatid diseases are unsatisfactory due to a number of reasons: poor efficacy, drug resistance, toxic side effects and parenteral administration. Hence new drugs are urgently needed.
The drug discovery process typically takes 10-15 years. This should be guided by target product profiles that define the key features and requirements for a new drug, such as route of administration, length of treatment, cost, and safety margins.
The drug discovery process can start with target-based or phenotypic (whole-cell) approaches. In the former, compounds are screeneds against a molecular target (usually an enzyme); in the latter, compounds are screened directly against the intact parasite growing in culture.
In general, there has been a very poor success rate in target-based approaches against trypanosomatids, despite some unique biochemical and metabolic features in trypanosomatid parasites. There are very few robustly validated drug targets.
Phenotypic approaches have led to some promising compounds, following optimization of the initial hits. Some of these are now in clinical development in the case of HAT.
Another approach for drug discovery is to re-position drugs from other disease areas. In some cases this has been successful.
There is still a need to refine the drug discovery pathway for Chagas’ disease, cellular and animal models in particular, to improve the identification of compounds that can cure patients.
There is still a long way to go, but good progress is being made in drug discovery to find potential new drugs to treat trypansomatid diseases.
Amastigote: The form of Trypanosoma cruzi and Leishmania spp. that resides within cells of the human host. Amastigotes are rounded and lack a free flagellum.
Chemical Series: A series of chemicals that have closely related chemical structure.
CRISPR-Cas9: A prokaryotic immune system that has been repurposed for genome editing in eukaryotic cells; in prokaryotes, the system comprises Clustered Regularly Interspaced Short Palindromic Repeats and the programmable Cas9 nuclease.
Drug-like molecules: Molecules that have the potential to be oral drugs. These will generally follow Lipinski’s rule of 5: molecular weight < 500; clogP (measure of hydrophobicity) < 5; number of hydrogen-bond donors < 5; number of hydrogen bond acceptors < 10.
Druggable: A druggable protein is one that can be inhibited or its function modulated by a drug-like molecule.
Elimination: Zero incidence of infection or disease in a defined geographical area.
Eradication: Permanent reduction to zero of the global incidence of infection or disease.
High-content screening (HCS): Combination of automated microscopy and image analysis that allows multi-parametric extraction and quantification of phenotypes of interest such as intracellular parasite count, cell cycle stage or intracellular protein localisation. HCS allows large-scale compound screening to identify desired cellular phenotypes
Insect vector: Pathogenic trypanosomatids are commonly transmitted by insect species, which are specific for the respective parasite. The geographical distribution of these insects restricts the range of parasite transmission.
Kinetochore: The kinetochore assembles at centromeres of chromosomes and is important for chromosome segregation during cell division.
Kinetoplastids: Kinetoplastids are a class of flagellated protists, which contain a characteristic network of mitochondrial DNA, the kinetoplast. Trypanosomatids are a suborder of the order Kinetoplastida.
Kinome: All protein kinases of a certain organism.
Parenteral administration: Drug application by routes other than through the gastrointestinal tract, generally by injection.
Phenotypic screening: This approach uses a whole cell screen designed to identify effects on a target cell or pathogen without a need for understanding the underlying mode of action. Using high-content screening multiple phenotypes can be detected simultaneously, for example the effects on intracellular parasite viability and host cell viability (that is, toxicity).
Polycistronic: Polycistronic transcription produces mRNA that encodes several polypeptides in one molecule that is then processed into individual polypeptide mRNAs.
Pharmacodynamics: The effects of drugs in the body
Polypharmacology: Drugs that act through inhibiting or modulating more than one molecular target or disease pathway.
Promastigote: The motile, extracellular parasite form. It has a flagellum attached only at the anterior end of the cell body and is associated mainly with infection of insect vectors.
cis-splicing: A step in pre-mRNA maturation during which exons are spliced together and introns are removed.
trans-splicing: Similar to cis-splicing but, in this case, two different mRNA transcripts are spliced together.
Structure-based drug design: The use of three dimensional structures of the inhibitors or modulators bound to their target protein (derived from X-ray crystallography or NMR) and computational chemistry to aid the design and optimization of lead compounds.
Scaffold hopping: modification of the essential core of a molecule to give a new core molecule with broadly similar, but slightly different properties. This is a generally used approach to optimise a hit or lead, improving features such as biological activity, solubility or metabolic stability.
Suicide inhibitor: A compound that is activated by an enzyme to give a reactive intermediate which irreversibly inhibits the enzyme through covalent bond(s).
Trypanosomatid: a member of the order Kinetoplastida, suborderTrypanosomatida , a group of protozoan flagellates that includes many pathogenic species. Frequently used interchangeably with kinetoplastid.
Trypomastigote: A motile, extracellular parasite form with a flagellum attached to the cell body and associated with infection of mammalian hosts.
Vector optimisation: Vectors are the substituents on the core of a molecule. Vector optimisation is modifying these vectors or substituents to improve a property or properties of a lead molecule (for example biological activity, solubility, etc). It may also encompass optimising at which position on the core scaffold a substituent is placed.
Acknowledgements
The authors are grateful to the following for financial support of their work: The Wellcome Trust (Strategic Grants 077705, 083481, 092340, 100476 and 105021), Wellcome Trust Senior Investigator Awards to D.H. and M.A.J.F. (100320/Z/12/Z and 101842/Z/13/Z) and a Wellcome Trust Principal Research Fellowship to A.H.F (079838); Drugs for Neglected Diseases Initiative (DNDi); and the Medical Research Council for grants to MCF (MR/L018853/1), DH (MR/K000500/1) and to M.C.F and D.H. (MR/K008749/1). The authors thank Alessandro Baliani with help in preparing Figure 3.
Biographies
Author biographies
Mark Field
Mark is a cell biologist with specific interests in trypanosomatids and evolution of the eukaryotic cell plan. His focus includes understanding the machinery of protein targeting and gene expression in trypanosomes and how these processes evolved and contribute towards immune evasion, life cycle progression and interactions with drugs. He also works on the origins of eukaryotic cellular compartmentalisation, and especially the manner in which paralogous gene families have influenced the evolution of the endomembrane system and the nuclear envelope.
David Horn
David is a parasitologist specialising in studies initiated using high-throughput genetic screens. He and his colleagues develop genetic tools and use those tools to decode the modes-of-action and resistance mechanisms for drugs that target trypanosomatid parasites. In other work, they have identified and characterised factors and mechanisms of allelic exclusion and DNA recombination, which underpin antigenic variation and immune evasion in African trypanosomes.
Alan Fairlamb
Alan is a Wellcome Principal Research Fellow at the University of Dundee. As a clinician scientist, he has published widely on the mode of drug action, mechanisms of drug resistance, and chemical and genetic validation of novel drug targets in parasitic protozoa. His research career has been devoted to discovery of better treatments for neglected diseases of poverty, serving as an expert scientific advisor to organizations such as WHO/TDR, DNDi and the Tres Cantos Open Lab Foundation. Together with colleagues at Dundee, he played a pivotal role in establishing the Drug Discovery Unit and was awarded a CBE in 2005, for services to medical science.
Michael Ferguson
Mike is a parasitologist specialising in glycobiology. He and his colleagues determine the chemical structures of cell-surface glycoconjugates. These include the first glycosylphosphatidylinositol (GPI) anchor, leishmania lipophosphoglycans (LPG) and the N-, O- and phosphate-linked glycans of trypanosomatid glycoproteins. Using the structures as roadmaps, his group delineates their biosynthetic pathways, using synthetic chemistry to generate necessary substrates, and enter essential enzymes into drug discovery programs. Mike is a founder-member of the Dundee Drug Discovery Unit and serves on the Boards of The Wellcome Trust and Medicines for Malaria Venture.
David Gray
David is Head of Biology within the Drug Discovery Unit. His role is to develop strategies for people, facilities, equipment and IT, including compound and data management to allow the effective support of hit discovery, hits to leads and lead optimisation programs. Prior to joining the DDU, David held roles of increasing responsibility within GlaxoWellcome and GlaxoSmithKline becoming Director, Screening & Compound Profiling in 1996. During his time with GlaxoSmithKline, David was responsible for the development of enabling approaches to finding drugs for 7 transmembrane, integrin and nuclear hormone receptors. David has a BSc in pharmacology for the University of Glasgow and a PhD from University College London. David has published over 30 peer-reviewed papers.
Kevin Read
Kevin’s background is in drug metabolism and pharmacokinetics, with extensive experience of early phase drug discovery, lead optimisation, project leadership and preclinical development gained from over 25 years in the pharmaceutical industry and in the Drug Discovery Unit at the University of Dundee (DDU). During his 17 years at GlaxoSmithKline, Kevin achieved a considerable track record of success in drug discovery, playing a significant part in 7 compounds entering pre-clinical development, 4 of which entered clinical trials. Kevin then moved to Dundee in 2008, joining the management team of the DDU, to fully integrate the DDU as an early drug discovery engine for neglected tropical diseases. He co-led the malaria project team that delivered the first pre-clinical development candidate for the DDU.
Manu De Rycker
Manu is Team Leader for Parasite Screening in the Drug Discovery Unit at the University of Dundee. Manu started his career with a degree in biotechnological engineering from the University of Ghent, Belgium. He moved to the University of Cincinnati where he did a PhD in Molecular Genetics. Manu then carried out post-doctoral work in the Cancer Research UK laboratories in Lincoln’s Inn Field in London. He then moved to the DDU, where he was initially involved in the development of high-content screening assays for intracellular parasites. Manu now oversees parasite screening at the Drug Discovery Unit.
Leah Torrie
Leah has been a lead biologist on trypanosomatid target-based projects in the Drug Discovery Unit (DDU) at the University of Dundee for 9 years. Leah and her team support early stage drug discovery projects by developing high-throughput-compatible screening assays for enzyme targets, carrying out hit discovery campaigns and performing detailed mode of inhibition studies on key compounds. Prior to joining the DDU in 2007, Leah graduated from the University of Dundee with a B.Sc. (Hons.) in Pharmacology before completing an M.Res. and PhD at the University of Glasgow. She then moved to industry and spent 3 years working for Upstate Ltd before joining the DDU.
Paul Wyatt
Paul is Head of the Drug Discovery Unit (DDU) at the University of Dundee, where he is responsible for the overall direction and management of the unit. The DDU was founded to bring together drug discovery expertise with excellent academic research in biology with the ultimate aim of developing new treatments for a variety of diseases. The DDU has two main focuses: translating novel biology into drug discovery programmes and tackling neglected diseases such the trypanosomatid diseases, TB and malaria. Prior to joining the University of Dundee in 2006, Paul worked in the BioPharma industry for 23 years; playing a significant part in seven compounds entering pre-clinical development. Paul started his career at the University of Birmingham where he obtained a BSc and PhD in Chemistry.
Susan Wyllie
Susan has studied the biology of trypanosomatid parasites for the last 15 years. Specifically, her research has focused on drug mechanisms of action and mechanism of drug resistance. She has co-authored more than 50 peer-reviewed articles in this research area. Currently, she leads a research group devoted to understanding the mechanism of action of phenotypically-active anti-trypanosomal compounds.
Ian Gilbert
Ian is Head of Medicinal Chemistry in the Drug Discovery Unit. Ian moved to the University of Dundee in 2005, when he helped to set up the Drug Discovery Unit. In the DDU Ian has worked in multiple drug discovery projects, including against the trypanosomatid parasites. He led the project which developed a preclinical candidate for malaria. Ian started his scientific career at the University fo Cambridge, where he obtained a PhD in synthetic chemistry. Following a post-doctoral fellowship with Parke-Davis Research, Ian spent a year teaching chemistry at the University of Zambia. After further post-doctoral experience at the University of Cambridge, Ian moved to t the Welsh School of Pharmacy in Cardiff University, where he set up his own medicinal chemistry research group focusing on anti-infectives
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Subject categories
Biological sciences / Microbiology / Parasitology / Parasite biology
[URI /631/326/417/1716]
Biological sciences / Microbiology / Antimicrobials / Antiparasitic agents
[URI /631/326/22/1294]
Health sciences / Diseases / Infectious diseases / Parasitic infection
[URI /692/699/255/1715]
Health sciences / Medical research / Drug development
[URI /692/308/153]
ToC blurb
Trypanosomatid parasites can cause life-threatening diseases such as human African trypanosomiasis, leishmaniasis and Chagas’ disease. In this Review, Gilbert and colleagues discuss the drug discovery landscape and describe some of the challenges in developing new drugs to treat these diseases.
References
- 1.McCall LI, McKerrow JH. Determinants of disease phenotype in trypanosomatid parasites. Trends Parasitol. 2014;30:342–349. doi: 10.1016/j.pt.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 2.WHO. Investing to overcome the global impact of neglected tropical diseases: Third WHO report on neglected diseases. World Health Organisation; 2015. [Google Scholar]
- 3.Dias JC. Southern Cone Initiative for the elimination of domestic populations of Triatoma infestans and the interruption of transfusional Chagas’ disease. Historical aspects, present situation, and perspectives. Mem Inst Oswaldo Cruz. 2007;102(Suppl 1):11–18. doi: 10.1590/s0074-02762007005000092. [DOI] [PubMed] [Google Scholar]
- 4.Khyatti M, et al. Infectious diseases in North Africa and North African immigrants to Europe. Eur J Public Health. 2014;24(Suppl 1):47–56. doi: 10.1093/eurpub/cku109. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert IH. Target-based drug discovery for human African trypanosomiasis: selection of molecular target and chemical matter. Parasitology. 2014;141:28–36. doi: 10.1017/S0031182013001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh N, Kumar M, Singh RK. Leishmaniasis: current status of available drugs and new potential drug targets. Asian Pac J Trop Med. 2012;5:485–497. doi: 10.1016/S1995-7645(12)60084-4. [DOI] [PubMed] [Google Scholar]
- 7.Clayton J. Chagas’ disease: pushing through the pipeline. Nature. 2010;465:S12–S15. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- 8.Fairlamb AH, Gow NAR, Matthews KR, Waters AP. Drug resistance in eukaryotic microorganisms. Nature Microbiol. 2016;1:e16092. doi: 10.1038/nmicrobiol.2016.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert IH. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J Med Chem. 2013;56:7719–7726. doi: 10.1021/jm400362b. [This paper compares molecular target-based and phenotypic approaches for drug discovery and highlights the strengths and weaknesses of each approach in the context of neglected diseases.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava S, Shankar P, Mishra J, Singh S. Possibilities and challenges for developing a successful vaccine for leishmaniasis. Parasit Vectors. 2016;9:277. doi: 10.1186/s13071-016-1553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gharbi M, et al. Leishmaniosis (Leishmania infantum infection) in dogs. Rev Sci Tech. 2015;34:613–626. doi: 10.20506/rst.34.2.2384. [DOI] [PubMed] [Google Scholar]
- 12.La Greca F, Magez S. Vaccination against trypanosomiasis: can it be done or is the trypanosome truly the ultimate immune destroyer and escape artist? Hum Vaccin. 2011;7:1225–1233. doi: 10.4161/hv.7.11.18203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Morales O, et al. Experimental Vaccines against Chagas’ Disease: A Journey through History. J Immunol Res. 2015;2015:489758. doi: 10.1155/2015/489758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langousis G, Hill KL. Motility and more: the flagellum of Trypanosoma brucei. Nat Rev Microbiol. 2014;12:505–518. doi: 10.1038/nrmicro3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by Trypanosoma cruzi during acute infection. Front Immunol. 2015;6:659. doi: 10.3389/fimmu.2015.00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welburn SC, Molyneux DH, Maudlin I. Beyond Tsetse - Implications for Research and Control of Human African Trypanosomiasis Epidemics. Trends Parasitol. 2015 doi: 10.1016/j.pt.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Imhof S, Roditi I. The Social Life of African Trypanosomes. Trends Parasitol. 2015;31:490–498. doi: 10.1016/j.pt.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Haanstra JR, Gonzalez-Marcano EB, Gualdron-Lopez M, Michels PA. Biogenesis, maintenance and dynamics of glycosomes in trypanosomatid parasites. Biochim Biophys Acta. 2016;1863:1038–1048. doi: 10.1016/j.bbamcr.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 19.McConville MJ, Naderer T. Metabolic pathways required for the intracellular survival of Leishmania. Annu Rev Microbiol. 2011;65:543–561. doi: 10.1146/annurev-micro-090110-102913. [DOI] [PubMed] [Google Scholar]
- 20.Wyatt PG, Gilbert IH, Read KD, Fairlamb AH. Target Validation: Linking Target and Chemical Properties to Desired Product Profile. Curr Topics Med Chem. 2011;11:1275–1283. doi: 10.2174/156802611795429185. [A key paper discussing the selection of molecular targets for neglected disease drug discovery.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Don R, Ioset J-R. Screening strategies to identify new chemical diversity for drug development to treat kinetoplastid infections. Parasitology. 2014;141:140–146. doi: 10.1017/S003118201300142X. [A summary of screening strategies for drug discovery in kinetoplastids.] [DOI] [PubMed] [Google Scholar]
- 22.Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. 2006;5:941–955. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- 23.Nagle AS, et al. Recent Developments in Drug Discovery for Leishmaniasis and Human African Trypanosomiasis. Chem Rev. 2014;114:11305–11347. doi: 10.1021/cr500365f. [A comprehensive survey of some target-based drug discovery programmes against trypanosomatids.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frearson JA, Wyatt PG, Gilbert IH, Fairlamb AH. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007;23:589–595. doi: 10.1016/j.pt.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Workman P, Collins I. Probing the Probes: Fitness Factors For Small Molecule Tools. Chem Biol. 2010;17:561–577. doi: 10.1016/j.chembiol.2010.05.013. [A detailed discussion of what is required for a chemical probe.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Priotto G, et al. Safety and effectiveness of first line eflornithine for Trypanosoma brucei gambiense sleeping sickness in Sudan: cohort study. BMJ (Clinical research ed) 2008;336:705–708. doi: 10.1136/bmj.39485.592674.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brun R, Don R, Jacobs RT, Wang MZ, Barrett MP. Development of novel drugs for human African trypanosomiasis. Future Microbiol. 2011;6:677–691. doi: 10.2217/fmb.11.44. [DOI] [PubMed] [Google Scholar]
- 28.Fairlamb AH, Henderson GB, Bacchi CJ, Cerami A. In vivo effects of difluoromethylornithine on trypanothione and polyamine levels in blood-stream forms of Trypanosoma brucei. Mol Biochem Parasitol. 1987;24:185–191. doi: 10.1016/0166-6851(87)90105-8. [DOI] [PubMed] [Google Scholar]
- 29.Frearson JA, et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature. 2010;464:728–732. doi: 10.1038/nature08893. [Validation of N-myristoyltransferase as a drug target for Trypanosoma brucei.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brand S, et al. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J Med Chem. 2012;55:140–152. doi: 10.1021/jm201091t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand S, et al. Lead optimization of a pyrazole sulfonamide series of Trypanosoma brucei N-myristoyltransferase inhibitors: identification and evaluation of CNS penetrant compounds as potential treatments for stage 2 human African trypanosomiasis. J Med Chem. 2014;57:9855–9869. doi: 10.1021/jm500809c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khare S, et al. Proteasome inhibition for treatment of leishmaniasis, Chagas’ disease and sleeping sickness. Nature. 2016;537:229–233. doi: 10.1038/nature19339. [Validation of the proteasome as a drug target for the trypanosomatids.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brimacombe KR, et al. Identification of ML251, a potent inhibitor of T. brucei and T. cruzi phosphofructokinase. ACS Med Chem Lett. 2014;5:12–17. doi: 10.1021/ml400259d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenthal R, Cornish-Bowden A. Prospects for antiparasitic drugs. The case of Trypanosoma brucei, the causative agent of African sleeping sickness. J Biol Chem. 1998;273:5500–5505. doi: 10.1074/jbc.273.10.5500. [DOI] [PubMed] [Google Scholar]
- 35.Jones NG, et al. Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a kinome-wide RNAi screen. PLoS Pathog. 2014;10:e1003886. doi: 10.1371/journal.ppat.1003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urbaniak MD, et al. Chemical proteomic analysis reveals the drugability of the kinome of Trypanosoma brucei. ACS Chem Biol. 2012;7:1858–1865. doi: 10.1021/cb300326z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alsford S, et al. High Throughput Phenotyping using Parallel Sequenceing of RNA Interference Targets in the African Trypanosome. Genome Res. 2011;21:915–924. doi: 10.1101/gr.115089.110. [Describes a genome-scale loss-of-function RNAi approach that facilitates drug-target prioritisation for a trypanosomatid] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodland A, et al. From on-target to off-target activity: identification and optimisation of Trypanosoma brucei GSK3 inhibitors and their characterisation as anti-Trypanosoma brucei drug discovery lead molecules. ChemMedChem. 2013;8:1127–1137. doi: 10.1002/cmdc.201300072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urich R, et al. The design and synthesis of potent and selective Inhibitors of Trypanosoma brucei Glycogen Synthase Kinase 3 for the treatment of human African trypanosomiasis. J Med Chem. 2014;57:7536–7549. doi: 10.1021/jm500239b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma J, et al. Nuclear DBF-2-related Kinases Are Essential Regulators of Cytokinesis in Bloodstream Stage Trypanosoma brucei. J Biol Chem. 2010;285:15356–15368. doi: 10.1074/jbc.M109.074591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amata E, et al. Identification of “Preferred” Human Kinase Inhibitors for Sleeping Sickness Lead Discovery. Are Some Kinases Better than Others for Inhibitor Repurposing? ACS Infect Dis. 2016;2:180–186. doi: 10.1021/acsinfecdis.5b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diaz R, et al. Identification and characterization of hundreds of potent and selective inhibitors of Trypanosoma brucei growth from a kinase-targeted library screening campaign. PLoS Negl Trop Dis. 2014;8:e3253. doi: 10.1371/journal.pntd.0003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akiyoshi B, Gull K. Discovery of unconventional kinetochores in kinetoplastids. Cell. 2014;156:1247–1258. doi: 10.1016/j.cell.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salmon D, et al. Adenylate cyclases of Trypanosoma brucei inhibit the innate immune response of the host. Science. 2012;337:463–466. doi: 10.1126/science.1222753. [DOI] [PubMed] [Google Scholar]
- 45.Mony BM, et al. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature. 2014;505:681–685. doi: 10.1038/nature12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gould MK, et al. Cyclic AMP effectors in African trypanosomes revealed by genome-scale RNA interference library screening for resistance to the phosphodiesterase inhibitor CpdA. Antimicrob Agents Chemother. 2013;57:4882–4893. doi: 10.1128/AAC.00508-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tagoe DN, Kalejaiye TD, de Koning HP. The ever unfolding story of cAMP signaling in trypanosomatids: vive la difference! Front Pharmacol. 2015;6:185. doi: 10.3389/fphar.2015.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Koning HP, et al. Pharmacological validation of Trypanosoma brucei phosphodiesterases as novel drug targets. J Infect Dis. 2012;206:229–237. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veerman J, et al. Synthesis and evaluation of analogs of the phenylpyridazinone NPD-001 as potent trypanosomal TbrPDEB1 phosphodiesterase inhibitors and in vitro trypanocidals. Bioorg Med Chem. 2016;24:1573–1581. doi: 10.1016/j.bmc.2016.02.032. [DOI] [PubMed] [Google Scholar]
- 50.Izquierdo L, et al. Distinct donor and acceptor specificities of Trypanosoma brucei oligosaccharyltransferases. Embo J. 2009;28:2650–2661. doi: 10.1038/emboj.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Damerow M, et al. Identification and functional characterization of a highly divergent N-acetylglucosaminyltransferase I (TbGnTI) in Trypanosoma brucei. J Biol Chem. 2014;289:9328–9339. doi: 10.1074/jbc.M114.555029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith TK, Crossman A, Brimacombe JS, Ferguson MAJ. Chemical validation of GPI biosynthesis as a drug target against African sleeping sickness. Embo J. 2004;23:4701–4708. doi: 10.1038/sj.emboj.7600456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adung'a VO, Gadelha C, Field MC. Proteomic analysis of clathrin interactions in trypanosomes reveals dynamic evolution of endocytosis. Traffic (Copenhagen, Denmark) 2013;14:440–457. doi: 10.1111/tra.12040. [DOI] [PubMed] [Google Scholar]
- 54.Manna PT, Gadelha C, Puttick AE, Field MC. ENTH and ANTH domain proteins participate in AP2-independent clathrin-mediated endocytosis. J Cell Sci. 2015;128:2130–2142. doi: 10.1242/jcs.167726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manna PT, Kelly S, Field MC. Adaptin evolution in kinetoplastids and emergence of the variant surface glycoprotein coat in African trypanosomatids. Mol Phylogenet Evol. 2013;67:123–128. doi: 10.1016/j.ympev.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gadelha C, et al. Architecture of a Host-Parasite Interface: Complex Targeting Mechanisms Revealed Through Proteomics. Mol Cell Proteomics. 2015;14:1911–1926. doi: 10.1074/mcp.M114.047647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bart JM, et al. Localization of serum resistance-associated protein in Trypanosoma brucei rhodesiense and transgenic Trypanosoma brucei brucei. Cell Microbiol. 2015;17:1523–1535. doi: 10.1111/cmi.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alsford S, et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. 2012;482:232–236. doi: 10.1038/nature10771. [Genome-scale loss-of-function RNAi approach to identify mechanisms of drug-resistance and potential target pathways in Trypanosoma brucei.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zoltner M, Leung KF, Alsford S, Horn D, Field MC. Modulation of the Surface Proteome through Multiple Ubiquitylation Pathways in African Trypanosomes. PLoS Pathog. 2015;11:e1005236. doi: 10.1371/journal.ppat.1005236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song J, et al. Pentamidine is not a permeant but a nanomolar inhibitor of the Trypanosoma brucei aquaglyceroporin-2. PLoS Pathog. 2016;12:e1005436. doi: 10.1371/journal.ppat.1005436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siegel TN, et al. Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev. 2009;23:1063–1076. doi: 10.1101/gad.1790409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schulz D, et al. Bromodomain Proteins Contribute to Maintenance of Bloodstream Form Stage Identity in the African Trypanosome. PLoS Biol. 2015;13:e1002316. doi: 10.1371/journal.pbio.1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawahara T, et al. Two essential MYST-family proteins display distinct roles in histone H4K10 acetylation and telomeric silencing in trypanosomes. Mol Microbiol. 2008;69:1054–1068. doi: 10.1111/j.1365-2958.2008.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q, et al. Targeting Lysine Deacetylases (KDACs) in Parasites. PLoS Negl Trop Dis. 2015;9:e0004026. doi: 10.1371/journal.pntd.0004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brandenburg J, et al. Multifunctional class I transcription in Trypanosoma brucei depends on a novel protein complex. Embo J. 2007;26:4856–4866. doi: 10.1038/sj.emboj.7601905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gunzl A. The pre-mRNA splicing machinery of trypanosomes: complex or simplified? Eukaryot Cell. 2010;9:1159–1170. doi: 10.1128/EC.00113-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clayton CE. Networks of gene expression regulation in Trypanosoma brucei. Mol Biochem Parasitol. 2014;195:96–106. doi: 10.1016/j.molbiopara.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 68.Hashem Y, et al. High-resolution cryo-electron microscopy structure of the Trypanosoma brucei ribosome. Nature. 2013;494:385–389. doi: 10.1038/nature11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalidas S, et al. Genetic validation of aminoacyl-tRNA synthetases as drug targets in Trypanosoma brucei. Eukaryot Cell. 2014;13:504–516. doi: 10.1128/EC.00017-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patterson S, et al. Dihydroquinazolines as a novel class of Trypanosoma brucei trypanothione reductase inhibitors: discovery, synthesis, and characterization of their binding mode by protein crystallography. J Med Chem. 2011;54:6514–6530. doi: 10.1021/jm200312v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wyllie S, et al. Dissecting the essentiality of the bifunctional trypanothione synthetase-amidase in Trypanosoma brucei using chemical and genetic methods. Mol Microbiol. 2009;74:529–540. doi: 10.1111/j.1365-2958.2009.06761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Torrie LS, et al. Chemical Validation of Trypanothione Synthetase. J Biol Chem. 2009;284:36137–36145. doi: 10.1074/jbc.M109.045336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bernardes LS, Zani CL, Carvalho I. Trypanosomatidae diseases: from the current therapy to the efficacious role of trypanothione reductase in drug discovery. Curr Med Chem. 2013;20:2673–2696. doi: 10.2174/0929867311320210005. [DOI] [PubMed] [Google Scholar]
- 74.Beig M, Oellien F, Krauth Siegel RL, Selzer PM. In: Trypanosomatid Diseases: Molecular Routes to Drug Discovery. Jager T, Koch O, Flohe L, editors. Wiley-VCH Verlag GmbH & Co. KGaA; 2013. pp. 385–404. [Google Scholar]
- 75.Spinks D, et al. Design, synthesis and biological evaluation of Trypanosoma brucei trypanothione synthetase inhibitors. ChemMedChem. 2012;7:95–106. doi: 10.1002/cmdc.201100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Urbina JA. Chemotherapy of Chagas’ disease: the how and the why. J Mol Med. 1999;77:332–338. doi: 10.1007/s001090050359. [DOI] [PubMed] [Google Scholar]
- 77.Urbina JA, Concepcion JL, Rangel S, Visbal G, Lira R. Squalene synthase as a chemotherapeutic target in Trypanosoma cruzi and Leishmania mexicana. Mol Biochem Parasitol. 2002;125:35–45. doi: 10.1016/s0166-6851(02)00206-2. [DOI] [PubMed] [Google Scholar]
- 78.Molina I, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. 2014;370:1899–1908. doi: 10.1056/NEJMoa1313122. [A clinical trial of posaconazole for Chagas’ disease.] [DOI] [PubMed] [Google Scholar]
- 79.Francisco AF, et al. Limited Ability of Posaconazole To Cure both Acute and Chronic Trypanosoma cruzi Infections Revealed by Highly Sensitive In Vivo Imaging. Antimicrob Agents Chemother. 2015;59:4653–4661. doi: 10.1128/AAC.00520-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choy JW, et al. Chemical-biological characterization of a cruzain inhibitor reveals a second target and a mammalian off-target. Beilstein J Org Chem. 2013;9:15–25. doi: 10.3762/bjoc.9.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barr SC, et al. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob Agents Chemother. 2005;49:5160–5161. doi: 10.1128/AAC.49.12.5160-5161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sienkiewicz N, Jaroslawski S, Wyllie S, Fairlamb AH. Chemical and genetic validation of dihydrofolate reductase-thymidylate synthase as a drug target in African trypanosomes. Mol Microbiol. 2008;69:520–533. doi: 10.1111/j.1365-2958.2008.06305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sienkiewicz N, Ong HB, Fairlamb AH. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice. Mol Microbiol. 2010;77:658–671. doi: 10.1111/j.1365-2958.2010.07236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hardy LW, Matthews W, Nare B, Beverley SM. Biochemical and genetic tests for inhibitors of Leishmania pteridine pathways. Exp Parasitol. 1997;87:157–169. [PubMed] [Google Scholar]
- 85.Gibson MW, Dewar S, Ong HB, Sienkiewicz N, Fairlamb AH. Trypanosoma brucei DHFR-TS revisited: Characterisation of a bifunctional and highly unstable recombinant dihydrofolate reductase-thymidylate synthase. PLoS Negl Trop Dis. 2016;10:e0004714. doi: 10.1371/journal.pntd.0004714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Looker DL, Marr JJ, Berens RL. Mechanisms of action of pyrazolopyrimidines in Leishmania donovani. J Biol Chem. 1986;261:9412–9415. [PubMed] [Google Scholar]
- 87.Gilbert IH, Leroy D, Frearson JA. Finding new hits in neglected disease projects: Target or phenotypic based screening? Curr Topics Med Chem. 2011;11:1284–1291. doi: 10.2174/156802611795429176. [DOI] [PubMed] [Google Scholar]
- 88.Brenk R, et al. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem. 2008;3:435–444. doi: 10.1002/cmdc.200700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mercer L, et al. 2,4-Diaminopyrimidines as Potent Inhibitors of Trypanosoma brucei and Identification of Molecular Targets by a Chemical Proteomics Approach. PLoS Negl Trop Dis. 2011;5:e956. doi: 10.1371/journal.pntd.0000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shibata S, et al. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei Infection in Mice. Antimicrob Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Rycker M, et al. A static-cidal assay for Trypanosoma brucei to aid hit prioritisation for progression into drug discovery programmes. PLoS Negl Trop Dis. 2012;6:e1932. doi: 10.1371/journal.pntd.0001932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wager TT, et al. Defining desirable Central Nervous System drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem Neurosci. 2010;1:420–434. doi: 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: The development of a Central Nervous System Multiparameter Optimization (CNS MPO) approach to enable aignment of drug-like properties. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wager TT, Villalobos A, Verhoest PR, Hou XJ, Shaffer CL. Strategies to optimize the brain availability of central nervous system drug candidates. Expert Opin Drug Discov. 2011;6:371–381. doi: 10.1517/17460441.2011.564158. [DOI] [PubMed] [Google Scholar]
- 95.Ley V, Andrews NW, Robbins ES, Nussenzweig V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. J Exp Med. 1988;168:649–659. doi: 10.1084/jem.168.2.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Riley J, et al. Development of a Fluorescence-based Trypanosoma cruzi CYP51 Inhibition Assay for Effective Compound Triaging in Drug Discovery Programmes for Chagas’ Disease. PLoS Negl Trop Dis. 2015;9:e0004014. doi: 10.1371/journal.pntd.0004014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.De Rycker M, et al. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrob Agents Chemother. 2013;57:2913–2922. doi: 10.1128/AAC.02398-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berman JD, Dwyer DM, Wyler DJ. Multiplication of Leishmania in human macrophages in vitro. Infect Immun. 1979;26:375–379. doi: 10.1128/iai.26.1.375-379.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nuhs A, et al. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Negl Trop Dis. 2015;9:e0004094. doi: 10.1371/journal.pntd.0004094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Torreele E, et al. Fexinidazole--a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl Trop Dis. 2010;4:e923. doi: 10.1371/journal.pntd.0000923. [The development of fexinidazole for the treatment of HAT.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tweats D, Bourdin Trunz B, Torreele E. Genotoxicity profile of fexinidazole--a drug candidate in clinical development for human African trypanomiasis (sleeping sickness) Mutagenesis. 2012;27:523–532. doi: 10.1093/mutage/ges015. [DOI] [PubMed] [Google Scholar]
- 102.Wyllie S, et al. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J Antimicrob Chemother. 2016;71:625–634. doi: 10.1093/jac/dkv376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bahia MT, et al. Fexinidazole: a potential new drug candidate for Chagas’ disease. PLoS Negl Trop Dis. 2012;6:e1870. doi: 10.1371/journal.pntd.0001870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wyllie S, et al. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci Transl Med. 2012;4:119re111. doi: 10.1126/scitranslmed.3003326. [Repositioning fexinidazole for leishmaniasis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bahia MT, et al. Antitrypanosomal activity of fexinidazole metabolites, potential new drug candidates for Chagas’ disease. Antimicrob Agents Chemother. 2014;58:4362–4370. doi: 10.1128/AAC.02754-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gupta S, et al. Nitroimidazo-oxazole compound DNDI-VL-2098: an orally effective preclinical drug candidate for the treatment of visceral leishmaniasis. J Antimicrob Chemother. 2015;70:518–527. doi: 10.1093/jac/dku422. [A preclinical candidate for visceral leishmaniasis.] [DOI] [PubMed] [Google Scholar]
- 107.Patterson S, et al. The anti-tubercular drug delamanid as a potential oral treatment for visceral leishmaniasis. eLife. 2016;5:e09744. doi: 10.7554/eLife.09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wyllie S, et al. Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in Leishmania. PLoS Pathog. 2016;12:e1005971. doi: 10.1371/journal.ppat.1005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jacobs RT, et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [A clinical candidate for HAT.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jones DC, et al. Genomic and Proteomic Studies on the Mode of Action of Oxaboroles against the African Trypanosome. PLoS Negl Trop Dis. 2015;9:e0004299. doi: 10.1371/journal.pntd.0004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mathis AM, et al. Accumulation and Intracellular Distribution of Antitrypanosomal Diamidine Compounds DB75 nad DB820 in African Trypanosomes. Antimicrob Agents Chemother. 2006;50:2185–2191. doi: 10.1128/AAC.00192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barrett MP, Croft SL. Management of trypanosomiasis and leishmaniasis. Br Med Bull. 2012;104:175–196. doi: 10.1093/bmb/lds031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Loiseau PM, Cojean S, Schrevel J. Sitamaquine as a putative antileishmanial drug candidate: from the mechanism of action to the risk of drug resistance. Parasite (Paris, France) 2011;18:115–119. doi: 10.1051/parasite/2011182115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sundar S, et al. Pharmacokinetics of oral sitamaquine taken with or without food and safety and efficacy for treatment of visceral leishmaniais: A randomized study in Bihar, India. Am J Trop Med Hyg. 2011;84:892–900. doi: 10.4269/ajtmh.2011.10-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Coimbra ES, et al. Mechanism of interaction of sitamaquine with Leishmania donovani. J Antimicrob Chemother. 2010;65:2548–2555. doi: 10.1093/jac/dkq371. [DOI] [PubMed] [Google Scholar]
- 116.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 117.Jin G, Wong ST. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today. 2014;19:637–644. doi: 10.1016/j.drudis.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nzila A, Ma Z, Chibale K. Drug repositioning in the treatment of malaria and TB. Future Med Chem. 2011;3:1413–1426. doi: 10.4155/fmc.11.95. [DOI] [PubMed] [Google Scholar]
- 119.Cavalla D. Predictive methods in drug repurposing: gold mine or just a bigger haystack? Drug Discov Today. 2013;18:523–532. doi: 10.1016/j.drudis.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 120.Cragg GM, Grothaus PG, Newman DJ. New horizons for old drugs and drug leads. J Nat Prod. 2014;77:703–723. doi: 10.1021/np5000796. [DOI] [PubMed] [Google Scholar]
- 121.Cavalla D. Therapeutic switching: a new strategic approach to enhance R&D productivity. IDrugs. 2005;8:914–918. [PubMed] [Google Scholar]
- 122.O'Connor OA. Developing new drugs for the treatment of lymphoma. Eur J Haematol Suppl. 2005:150–158. doi: 10.1111/j.1600-0609.2005.00470.x. [DOI] [PubMed] [Google Scholar]
- 123.Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Priotto G, et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet. 2009;374:56–64. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- 125.Raju TN. The Nobel chronicles. 1988: James Whyte Black, (b 1924), Gertrude Elion (1918-99), and George H Hitchings (1905-98) Lancet. 2000;355:1022. doi: 10.1016/s0140-6736(05)74775-9. [DOI] [PubMed] [Google Scholar]
- 126.Skinner-Adams TS, et al. Defining the targets of antiparasitic compounds. Drug Discov Today. 2016;21:725–739. doi: 10.1016/j.drudis.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 127.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 128.Rottman M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McNamara CW, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504:248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baragana B, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meister S, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science. 2011;334:1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kato N, et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature. 2016;538:344–349. doi: 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Horn D, Duraisingh MT. Antiparasitic chemotherapy: from genomes to mechanisms. Annu Rev Pharmacol Toxicol. 2014;54:71–94. doi: 10.1146/annurev-pharmtox-011613-135915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Peng D, Kurup SP, Yao PY, Minning TA, Tarleton RL. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 2015;6:e02097–02014. doi: 10.1128/mBio.02097-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lander N, Li ZH, Niyogi S, Docampo R. CRISPR/Cas-9-Induced Disruption of Paraflagellar Rod Protein 1 and 2 Genes in Trypanosoma cruzi Reveals Their Role in Flagellar Attachment. mBio. 2015;6:e01012. doi: 10.1128/mBio.01012-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sollelis L, et al. First efficient CRISPR-Cas9-mediated genome editing in Leishmania parasites. Cell Microbiol. 2015;17:1405–1412. doi: 10.1111/cmi.12456. [DOI] [PubMed] [Google Scholar]
- 138.Zhang WW, Matlashewski G. CRISPR-Cas9-Mediated Genome Editing in Leishmania Parasites. mBio. 2015;21:e00861. doi: 10.1128/mBio.00861-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Begolo D, Erben E, Clayton C. Drug target identification using a trypanosome overexpression library. Antimicrob Agents Chemother. 2014;58:6260–6264. doi: 10.1128/AAC.03338-14. [Validation of a high-throughput gain-of-function genetic approach for drug-target identification in T. brucei.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gazanion E, Fernandez-Prada C, Papadopoulou B, Leprohon P, Ouellette M. Cos-Seq for high-throughput identification of drug target and resistant mechanisms in the protozoan parasite Leishmania. Proc Natl Acad Sci USA. 2016;113:E3012–3021. doi: 10.1073/pnas.1520693113. [Validation of a high-throughput gain-of-function genetic approach for drug-target identification in Leishmania spp..] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kelly JM, Das P, Tomas AM. An approach to functional complementation by introduction of large DNA fragments into Trypanosoma cruzi and Leishmania donovani using a cosmid shuttle vector. Mol Biochem Parasitol. 1994;65:51–62. doi: 10.1016/0166-6851(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 142.Wang K, et al. Chemistry-based functional proteomics for drug target deconvolution. Expert Rev Proteomics. 2012;9:293–310. doi: 10.1586/epr.12.19. [DOI] [PubMed] [Google Scholar]
- 143.Martinez Molina D, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 144.Creek DJ, Anderson J, McConville MJ, Barrett MP. Metabolomic analysis of trypanosomatid protozoa. Mol Biochem Parasitol. 2012;181:73–84. doi: 10.1016/j.molbiopara.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 145.Trindade S, et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe. 2016 doi: 10.1016/j.chom.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Capewell P, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. eLife. 2016;5:e17716. doi: 10.7554/eLife.17716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Desquesnes M, et al. Trypanosoma evansi and surra: a review and perspectives on origin, history, distribution, taxonomy, morphology, hosts, and pathogenic effects. Biomed Res Int. 2013:194176. doi: 10.1155/2013/194176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carnes J, et al. Genome and phylogenetic analyses of Trypanosoma evansi reveal extensive similarity to T. brucei and multiple independent origins for dyskinetoplasty. PLoS Negl Trop Dis. 2015;9:e3404. doi: 10.1371/journal.pntd.0003404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Auty H, Torr SJ, Michoel T, Jayaraman S, Morrison LJ. Cattle trypanosomosis: the diversity of trypanosomes and implications for disease epidemiology and control. Rev Sci Tech. 2015;34:587–598. doi: 10.20506/rst.34.2.2382. [DOI] [PubMed] [Google Scholar]
- 150.Strosberg AM, et al. CHAGAS’ DISEASE: A Latin American Nemesis. Institute for OneWorld Health; 2007. [Google Scholar]
- 151.Gascon J, Bern C, Pinazo MJ. Chagas’ disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010;115:22–27. doi: 10.1016/j.actatropica.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 152.Bonney KM, Engman DM. Autoimmune pathogenesis of Chagas’ heart disease: looking back, looking ahead. Am J Pathol. 2015;185:1537–1547. doi: 10.1016/j.ajpath.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zingales B, et al. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance and research applications. Infect Genet Evol. 2012;12:240–253. doi: 10.1016/j.meegid.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 154.Cunha-Neto E, Chevillard C. Chagas’ disease cardiomyopathy: immunopathology and genetics. Mediators Inflamm. 2014;2014:683230. doi: 10.1155/2014/683230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hussain H, Al-Harrasi A, Al-Rawahi A, Green IR, Gibbons S. Fruitful Decade for Antileishmanial Compounds from 2002 to Late 2011. Chem Rev. 2014;114:10369–10428. doi: 10.1021/cr400552x. [DOI] [PubMed] [Google Scholar]
- 156.McLatchie AP, et al. Highly sensitive in vivo imaging of Trypanosoma brucei expressing “red-shifted” luciferase. PLoS Negl Trop Dis. 2013;7:e2571. doi: 10.1371/journal.pntd.0002571. [A key mouse model for HAT.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Burrell-Saward H, Rodgers J, Bradley B, Croft SL, Ward TH. A sensitive and reproducible in vivo imaging mouse model for evaluation of drugs against late-stage human African trypanosomiasis. J Antimicrob Chemother. 2015;70:510–517. doi: 10.1093/jac/dku393. [DOI] [PubMed] [Google Scholar]
- 158.Giroud C, et al. Murine Models for Trypanosoma brucei gambiense disease progression--from silent to chronic infections and early brain tropism. PLoS Negl Trop Dis. 2009;3:e509. doi: 10.1371/journal.pntd.0000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jennings FW, Urquhart GM, Murray PK, Miller BM. Treatment with suranim and 2-substituted 5-nitroimidazoles of chronic murine Trypanosoma brucei infections with central nervous system involvement. Trans R Soc Trop Med Hyg. 1983;77:693–698. doi: 10.1016/0035-9203(83)90207-9. [DOI] [PubMed] [Google Scholar]
- 160.Myburgh E, et al. In vivo imaging of trypanosome-brain interactions and development of a rapid screening test for drugs against CNS stage trypanosomiasis. PLoS Negl Trop Dis. 2013;7:e2384. doi: 10.1371/journal.pntd.0002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Romanha AJ, et al. In vitro and in vivo experimental models for drug screening and development for Chagas’ disease. Mem Inst Oswaldo Cruz. 2010;105:233–238. doi: 10.1590/s0074-02762010000200022. [DOI] [PubMed] [Google Scholar]
- 162.Bustamante JM, Tarleton RL. Methodological advances in drug discovery for Chagas’ disease. Expert Opin Drug Discov. 2011;6:653–661. doi: 10.1517/17460441.2011.573782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lewis MD, et al. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell Microbiol. 2014;16:1285–1300. doi: 10.1111/cmi.12297. [A key mouse model for Chagas’ disease.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bustamante JM, Craft JM, Crowe BD, Ketchie SA, Tarleton RL. New, combined, and reduced dosing treatment protocols cure Trypanosoma cruzi infection in mice. J Infect Dis. 2014;209:150–162. doi: 10.1093/infdis/jit420. [A key mouse model for Chagas’ disease.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Canavaci AM, et al. In vitro and in vivo high-throughput assays for the testing of anti-Trypanosoma cruzi compounds. PLoS Negl Trop Dis. 2010;4:e740. doi: 10.1371/journal.pntd.0000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lewis MD, Francisco AF, Taylor MC, Kelly JM. A new experimental model for assessing drug efficacy against Trypanosoma cruzi infectin based on highly sensitive in vivo imaging. J Biomol Screen. 2015;20:36–43. doi: 10.1177/1087057114552623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gupta S. Visceral leishmaniaisis: experimental models for drug discovery. Indian J Med Res. 2011;133:27–39. [PMC free article] [PubMed] [Google Scholar]