

Abstract
Regulation of growth factor signaling involves reversible inactivation of protein tyrosine phosphatases (PTPs) through the oxidation and reduction of their active site cysteine. However, there is limited mechanistic understanding of these redox events and their co-ordination in the presence of cellular antioxidant networks. Here we investigated interactions between PTP1B and the peroxiredoxin 2 (Prx2)/thioredoxin 1 (Trx1)/thioredoxin reductase 1 (TrxR1) network. We found that Prx2 becomes oxidized in PDGF-treated fibroblasts, but only when TrxR1 has first been inhibited. Using purified proteins, we also found that PTP1B is relatively insensitive to inactivation by H2O2 but found no evidence for a relay mechanism in which Prx2 or Trx1 facilitates PTP1B oxidation. Instead, these proteins prevented PTP1B inactivation by H2O2. Intriguingly, we discovered that TrxR1/NADPH directly protects PTP1B from inactivation when present during the H2O2 exposure. This protection was dependent on the concentration of TrxR1 and independent of Trx1 and Prx2. The protection was blocked by auranofin and required an intact selenocysteine residue in TrxR1. This activity likely involves reduction of the sulfenic acid intermediate form of PTP1B by TrxR1 and is therefore distinct from the previously described reactivation of end-point oxidized PTP1B, which requires both Trx1 and TrxR1. The ability of TrxR1 to directly reduce an oxidized phosphatase is a novel activity that can help explain previously observed increases in PTP1B oxidation and PDGF receptor phosphorylation in TrxR1 knockout cells. The activity of TrxR1 is therefore of potential relevance for understanding the mechanisms of redox regulation of growth factor signaling pathways.
Keywords: hydrogen peroxide, peroxiredoxin, redox regulation, thioredoxin reductase, protein tyrosine phosphatase (tyrosine phosphatase), growth factor signaling, thiol oxidation
Introduction
Protein tyrosine phosphatases (PTPs)2 are important regulators of receptor tyrosine kinase (RTK) signaling through dephosphorylation of tyrosine residues. Dysregulated RTK signaling pathways have been implicated in several diseases, including cancer, atherosclerosis, and diabetes (1–4). The activities of classical PTPs depend on a low pKa Cys residue in the active site (1–3, 5) that is susceptible to reversible inhibition by cellular oxidants produced following receptor stimulation (5, 6). Reversible oxidation of PTPs is a well-described contributor to growth factor signaling (5). The mechanism involves activation of membrane-bound NADPH oxidases (NOXs) (7), and possibly mitochondrial sites (8), to increase cytoplasmic superoxide and hydrogen peroxide (H2O2) levels with resultant PTP oxidation. Oxidation of PTPs by H2O2 can generate sulfenic, sulfinic (-SO2) or sulfonic (-SO3) acid forms. In PTP1B, the sulfenic acid is a short-lived intermediate (9, 10) that converts to a sulfenylamide through a covalent bond with the peptide nitrogen of a juxtaposed serine residue (11–13). Rapid conversion from sulfenic acid to sulfenylamide is thought to protect the active-site cysteine from further irreversible oxidation (9, 11, 12, 14–16).
For PTP oxidation to be an effective control mechanism for RTK signaling, both inactivation and reactivation need to be carefully regulated through selective oxidation and efficient reduction. To date, studies have mainly focused on understanding PTP oxidation, although there is still uncertainty about the exact mechanisms. The initial oxidant is widely considered to be H2O2, but it is not clear how PTPs, which, in isolation, react slowly with H2O2 (9), can be oxidized in cells containing peroxidases such as peroxiredoxins (Prxs) and glutathione peroxidases, which are up to a million times more reactive (17). One mechanism could involve a redox relay, with a Prx acting as a mediator of oxidant transfer. Such a mechanism has been described for Prx2-mediated formation of disulfide-linked STAT3 oligomers (18). Our first aim was to establish whether PTP1B oxidation could occur via a relay mechanism involving Prx2. We chose PTP1B because of its importance as a negative regulator of growth factor signaling. Oxidative inhibition of PTP1B is known to positively regulate RTK signaling via PDGF and EGF receptor pathways (8, 19). Although PTP1B resides in the endoplasmic reticulum membrane, its catalytic domain faces the cytosol (20). Therefore, we focused on Prx2 as a major cytoplasmic Prx. Prx2 reacts readily with H2O2 and is recycled primarily by the Trx/TrxR system (21, 22), and evidence for cross-talk between PTPs and Prx2 during PDGF signaling has been described (23).
Our second aim was to characterize reduction pathways that could regulate PTP1B activity. We and others have shown that fully oxidized PTP1B (presumably the sulfenylamide) can be reactivated by the Trx system (Trx1, TrxR1, and NADPH) (19, 24–26) and that PTP1B oxidation and PDGF signaling is enhanced in TrxR1 knock-out cells (24). Here we have used purified proteins to examine how components of the Prx2/Trx1/TrxR1 system affect the oxidation and reduction of PTP1B when present during exposure to H2O2. We found that the Prx2/Trx1/TrxR1 system inhibited inactivation of PTP1B by H2O2, with no evidence of a Prx2 relay system for PTP1B oxidation. Our investigation of the reductive mechanism showed that TrxR1 directly protects PTP1B against inactivation by H2O2. This protection occurred without involvement of Trx or Prx activities, and was fully dependent on NADPH and the active site selenocysteine (Sec) residue of TrxR1. Our findings suggest direct transfer of reducing equivalents from TrxR1 to the sulfenic acid of PTP1B. As far as we are aware, this is the first reported example of TrxR1 reducing a protein sulfenic acid and identifies a potential new mechanism for redox control of growth factor signaling pathways.
Results
Stimulation of mouse embryonic fibroblasts by PDGF-BB induces Prx2 dimer formation in the presence of auranofin
PDGF stimulation has been shown previously to increase cellular oxidant production (27, 28). H2O2 production has been detected with the specific biosensor HyPer, which was shown to undergo a continuous increase in fluorescence for at least 5 min following stimulation (29). As confirmation of this, we expressed HyPer in mouse embryonic fibroblasts (MEFs) expressing PDGFR-β and observed that addition of the PDGF-BB ligand (which activates PDGFR-β) (30) caused a continuous increase in cellular fluorescence for at least 20 min that was significantly greater than in untreated cells (Fig. 1, A and B). To determine whether Prx2 becomes oxidized in this system, we measured its redox state at different times after PDGF-BB stimulation. In unstimulated cells, about half of the Prx2 resolved in the reduced monomeric form and half as disulfide-linked dimers (Fig. 1, C and D). This ratio did not change significantly in the 40 min following stimulation. The disulfide of Prx2 is recycled predominantly by the Trx system. When cells were pretreated with auranofin, a potent TrxR inhibitor (31, 32), the basal level of Prx2 oxidation was unchanged. However, PDGF-BB stimulation caused an increase in Prx2 dimers within 5 min, and these remained almost completely oxidized (Fig. 1, E and F). These findings suggest that PDGF receptor activation can induce oxidation of Prx2, but oxidized Prx2 does not accumulate because it is recycled by the Trx system.
Figure 1.

Prx2 dimer formation is induced by PDGF-BB treatment in MEF cells pretreated with auranofin. A, representative images of MEF cells expressing cytosolic HyPer after 0 and 20 min without treatment (Control) or treatment with 100 ng/ml PDGF-BB. B, change in fluorescence over 20 min in untreated or PDGF-BB–stimulated Hyper-expressing MEF cells. Data from 19 transfected cells were analyzed in five different experiments (mean ± S.E.; *, p < 0.05). C, Prx2 immunoblot of non-reducing SDS-PAGE–resolved lysates from MEF cells treated with 50 ng/ml PDGF-BB for the indicated times. D, quantification of Prx2 dimers (percentage of total Prx2) from densitometry analyses of blots represented in C (n = 7). E, immunoblot as in C of lysates from cells pretreated for 1 h with the TrxR1 inhibitor auranofin (1 μm) before PDGF-BB treatment. F, quantitation of Prx2 dimers in auranofin-treated cells determined as in D (mean ± S.E.; *, p < 0.05).
Recombinant reduced Prx2 protects PTP1B from inactivation by H2O2
In theory, Prx2 could scavenge H2O2 and protect PTPs against oxidation or promote oxidation via a redox relay system. To investigate the interactions of Prx2 with PTP1B, we prepared active recombinant human PTP1B and compared the effect of H2O2 on its activity in the absence and presence of Prx2. PTP1B undergoes different types of cleavage in mammalian tissues and when expressed in Escherichia coli (33–35). Here we confirmed that PTP1B expressed in full-length form in E. coli is cleaved upon lysis (34). We also expressed the isolated catalytic domain (26). The PTP1B cleaved variant was characterized as an ∼50-kDa band (tag and linker included) on SDS-PAGE and was used unless stated otherwise (Fig. 2A). Comparing the two PTP1B forms, we found them to have similar activities. As expected from the measured rate constant (9), initial analyses using a chromogenic substrate, pNPP, showed that direct inactivation of PTP1B was slow and required exposure to 100 μm H2O2 for 30 min or to 1 mm H2O2 for 5 min for maximal effect (Fig. 2B). The effect of reduced Prx2 on PTP1B inactivation was initially examined using equimolar H2O2 and Prx2. Under such conditions, the Prx2 should rapidly react with the H2O2, and if a relay mechanism operated, then it should result in more PTP1B inactivation. However, reduced Prx2 completely inhibited inactivation (Fig. 2C). We also tested whether oxidized Prx2 alone could inactivate PTP1B by disulfide exchange. There was no significant effect on activity (Fig. 2C). Non-reducing SDS-PAGE confirmed that Prx2 remained oxidized upon incubation with reduced PTP1B, and no high-molecular-weight complexes between the two were observed (Fig. 2D). Thus, independent methods for monitoring oxidation of the two proteins showed no evidence of transfer of oxidizing equivalents from dimeric Prx2 to reduced PTP1B. Oxidized Prx1 was also tested for effects on PTP1B activity and showed no significant difference compared with controls (Fig. 2E).
Figure 2.

Reduced Prx2 protects PTP1B from inactivation by H2O2. A, analysis of the PTP1B His-tagged cleaved variant and PTP1B catalytic domain on SDS-PAGE stained with Coomassie Blue. The purity was ∼72% and ∼ 99%, respectively, as determined by densitometry using ImageJ. Side-by-side comparison of activity of the reduced PTP1B cleaved variant and PTP1B catalytic domain showed a mean substrate turnover of 269 and 355 min−1, respectively (n = 3). B, H2O2-dependent inactivation of recombinant PTP1B. Reduced PTP1B (600 nm) was exposed to 100 μm or 1 mm H2O2 for the designated times and subsequently analyzed for PTP activity using a p-nitrophenyl phosphate (pNPP) substrate (n = 3; mean ± S.E.; *, p < 0.05). PTP1B activities are expressed as percentages of untreated controls. C, activity of PTP1B following treatment with H2O2 and reduced or oxidized Prx2. Fully reduced PTP1B (600 nm) was incubated for 30 min with 20 μm reduced Prx2 and 20 μm H2O2 or reduced or oxidized Prx alone and then assayed for PTP activity (n = 3; mean ± S.E.; *, p < 0.05; H2O2-treated compared with controls). D, Prx2 oxidation state after treatment with reduced PTP1B. Equimolar reduced PTP1B (150 nm) and oxidized Prx2 were incubated for 30 min and analyzed by non-reducing SDS-PAGE. A silver-stained gel (representative of three independent experiments) shows the positions of individual proteins as well as reduced Prx2 for comparison. The bottom and top oxidized Prx2 bands correspond to dimers containing one or two disulfides, respectively. E, activity of PTP1B following treatment with H2O2 or oxidized Prx1. Fully reduced PTP1B (600 nm) was incubated for 30 min with 10 μm H2O2 or 10 μm oxidized Prx alone and then assayed for PTP activity (n = 3; mean ± S.E.; *, p < 0.05; H2O2-treated compared with controls).
The Trx system together with Prx2 protects PTP1B from inactivation during exposure to H2O2
To test whether Prx2 could facilitate PTP1B oxidation while undergoing redox cycling, we treated PTP1B with excess H2O2 in the presence of Prx2 plus TrxR1/Trx/NADPH. All concentrations of Prx2 together with TrxR1/Trx1/NADPH showed clear protection, with no evidence of accelerated inactivation (Fig. 3A). Non-reducing SDS-PAGE confirmed that a proportion of Prx2 was initially oxidized to the disulfide at the 2-min time point (black arrow, Fig. 3B) and then subsequently reduced by TrxR1/Trx1/NADPH at 20 and 30 min (gray and white arrows, Fig. 3B). These results collectively suggest that Prx2 protects PTP1B by clearance of H2O2, with no evidence of facilitated oxidation by oxidized forms of Prx2.
Figure 3.

Prx2 redox cycling with the Trx system protects PTP1B against inactivation during exposure to H2O2. A, effects of Prx2 and Trx system components during treatment of PTP1B with H2O2. Reduced PTP1B catalytic domain (600 nm) was preincubated for 30 min with 2 μm Trx1, 50 nm TrxR1 (specific activity, 17 units/mg), 200 μm NADPH, and 0–20 μm Prx2. H2O2 (100 μm) was then added, and samples were taken at 5, 15, and 30 min for measurement of PTP activity (n = 3; mean ± S.E.; *, p < 0.05). B, parallel analysis of Prx2 redox status from the experiment shown in A. Aliquots were taken from the assay shown in A after 2, 20, and 30 min of H2O2 treatment and analyzed by non-reducing SDS-PAGE. A Coomassie-stained gel (representative of three independent experiments) shows the positions of reduced and oxidized Prx2, the latter (arrows) showing two Prx2 bands corresponding to one or two disulfides.
TrxR1 with NADPH alone protects PTP1B from inactivation during exposure to H2O2
PTP activity can be regulated not only by oxidation but also by the rate of reduction of the reversibly oxidized protein. This is apparent in Fig. 3, where the PTP1B was reactivated when the Prx2 became reduced. Furthermore, with a higher TrxR concentration and the full Trx1/TrxR1/NADPH system, transient inactivation was not seen, and the majority (80%) of the PTP1B activity was preserved after 30 min of H2O2 exposure (Fig. 4A). Surprisingly, whether Prx2 was present made no difference regarding the protective effect.
Figure 4.

The Trx system reverses H2O2-inactivated PTP1B. A, reduced PTP1B (600 nm) was preincubated for 30 min either alone or with 2 μm Trx1, 0.5 μm TrxR1 (specific activity, 9.75 units/mg), and 200 μm NADPH, with or without Prx2 (10 μm). H2O2 (100 μm) was then added, and samples were taken at 5, 15, and 30 min for measurement of PTP activity (n = 3; mean ± S.E.; *, p < 0.05). B, H2O2 consumption by PTP1B and components of the Trx/Prx2 system. Combinations of Trx1 (2 μm), TrxR1 (0.5 μm, 18 units/mg), Prx2 (10 μm), and NADPH (200 μm) with and without reduced PTP1B (600 nm) were treated with 100 μm H2O2 at 22 °C, and concentrations of H2O2 at the indicated time points were determined by ferrous oxidation of xylenol orange assay (n = 3; mean ± S.E.; *, p < 0.05). C, reactivation of H2O2-inactivated PTP1B (cleaved form). Reduced PTP1B was treated with 1 mm H2O2 for 5 min and then with catalase to remove residual H2O2 and reactivated with 10 mm DTT or TrxR1 (2.5 μm) (specific activity, 18 units/mg) and NADPH (1 mm) with or without Trx1 (10 μm). After 45 min at 22 °C, samples were analyzed for PTP activity (n = 3; mean ± S.E.; *, p < 0.05). D, reactivation of H2O2-inactivated PTP1B catalytic domain. Reduced PTP1B was treated with 1 mm H2O2 for 5 min and then with catalase and reactivated with 10 mm DTT or TrxR1 (2.5 μm) (specific activity, 22 units/mg), NADPH (300 μm), and Trx1 (10 μm). After 5, 10, and 40 min at 22 °C, samples were analyzed for PTP activity (n = 3; mean ± S.E.; *, p < 0.05).
One explanation for PTP1B not being inactivated could be rapid consumption of the added H2O2. This would be expected with Prx2 present because of its well-characterized peroxidase activity, and indeed, Prx2 with the full Trx system rapidly cleared H2O2 (Fig. 4B). However, the Trx system alone is inefficient at removing H2O2 (36), and no detectable H2O2 consumption was observed without Prx2 with any combinations of PTP1B and the Trx system (Fig. 4B).
Alternatively, the Trx system could reactivate the oxidized PTP1B. We first investigated this with PTP1B that had been preoxidized by H2O2 and confirmed our previous findings (24) that inactivation can be reversed by the complete Trx1/TrxR1/NADPH system (Fig. 4C). This required NADPH and did not occur with TrxR1 in the absence of Trx1. However, with both the cleaved PTP1B protein (Fig. 4C) and the catalytic domain (Fig. 4D), reactivation was slow, with only 30–35% recovery over 30–45 min. Under these conditions, DTT appeared to be more effective than the thioredoxin system. This difference was not observed in previous work (24), perhaps because a different substrate was used or the reaction was performed at a different temperature.
These results suggest that reactivation of the fully oxidized (sulfenylamide) form may be too slow to account for the protection of PTP1B during exposure to H2O2. Rather, they suggest that the Trx system continuously regenerates active PTP1B by reduction of an oxidation intermediate. This is most likely to be the sulfenic acid, which is the initial short-lived intermediate formed in the reaction of H2O2 with PTP1B (10–13).
Further experiments were performed to test how the Trx system protects PTP1B against H2O2-mediated inactivation. Intriguingly, protection by just TrxR1 and NADPH was as efficient as when Trx was present (Fig. 5A). These experiments were performed with the PTP1B cleaved variant, but results were similar with the catalytic domain (Fig. 5B). NADPH was required, and protection increased with increasing TrxR1 concentration (Fig. 5C). Therefore, the observed protection against PTP1B oxidation is likely to involve a direct reaction of TrxR1 with PTP1B.
Figure 5.

TrxR1 activity protects PTP1B inactivation during exposure to H2O2. A, protection of PTP1B from H2O2 requires only TrxR1 and NADPH. PTP1B was treated with H2O2 in the presence of various components of the Trx system and assayed for PTP activity as shown in Fig. 3B. Rates of inactivation with Trx1/TrxR1 or TrxR1 without NADPH were indistinguishable from that with PTP1B alone (Fig. 3B) (n = 3; mean ± S.E.; *, p < 0.05). B, reduced PTP1B cleaved and PTP1B catalytic domain variant (600 nm) were treated with the indicated concentrations of TrxR1 (0.5 μm) and 200 μm NADPH and exposed to 100 μm H2O2 for 5, 15, and 30 min (n = 3; mean ± S.E.; *, p < 0.05). C, concentration-dependent protection of PTP1B activity by TrxR1. Reduced PTP1B (600 nm) was treated with the indicated concentrations of TrxR1 and 200 μm NADPH and exposed to 100 μm H2O2 for 5 min (n = 3; mean ± S.E.; *, p < 0.05). D, reduced DEP-1 (80 nm) was treated with the indicated concentrations of TrxR1 (0.5 μm) and 200 μm NADPH, exposed to 150 μm H2O2 for 5 min, and then assayed for PTP activity as in panel A (n = 3; mean ± S.E.; *, p < 0.05).
To see whether this mechanism applies generally to other PTPs, we examined human receptor–like density-enhanced protein tyrosine phosphatase 1 (DEP-1). The intracellular domain of recombinant GST-tagged human DEP-1 was purified and treated identically to PTP1B. DEP-1 was similar to PTP1B in its sensitivity to H2O2 (54% inactivation after 30-min exposure to 150 μm H2O2), but it was not significantly protected against inactivation by TrxR1 and NADPH (55% inactivation) (Fig. 5D). This implies a degree of specificity of TrxR1 for the reduction of PTP1B.
The active-site selenocysteine residue of TrxR1 is indispensable for protecting PTP1B from H2O2
Further evidence that protection of PTP1B required active TrxR1 was obtained using auranofin, a potent inhibitor of TrxR1 that is presumed to interact with its active-site Sec (37). Auranofin had no effect on PTP1B activity in the absence of H2O2 but abolished the protective effect of TrxR1 (Fig. 6A). Finally, we confirmed the requirement for Sec-dependent activity using TrxR1 mutants with the Sec mutated to either Cys or Ser (38). Measurement of PTP1B activity after exposure to H2O2 showed that the protective effect of wild-type TrxR1 was lost in both mutants (Fig. 6B). Thus, the active-site Sec is indispensable for TrxR1 to protect PTP1B against H2O2-mediated inactivation.
Figure 6.
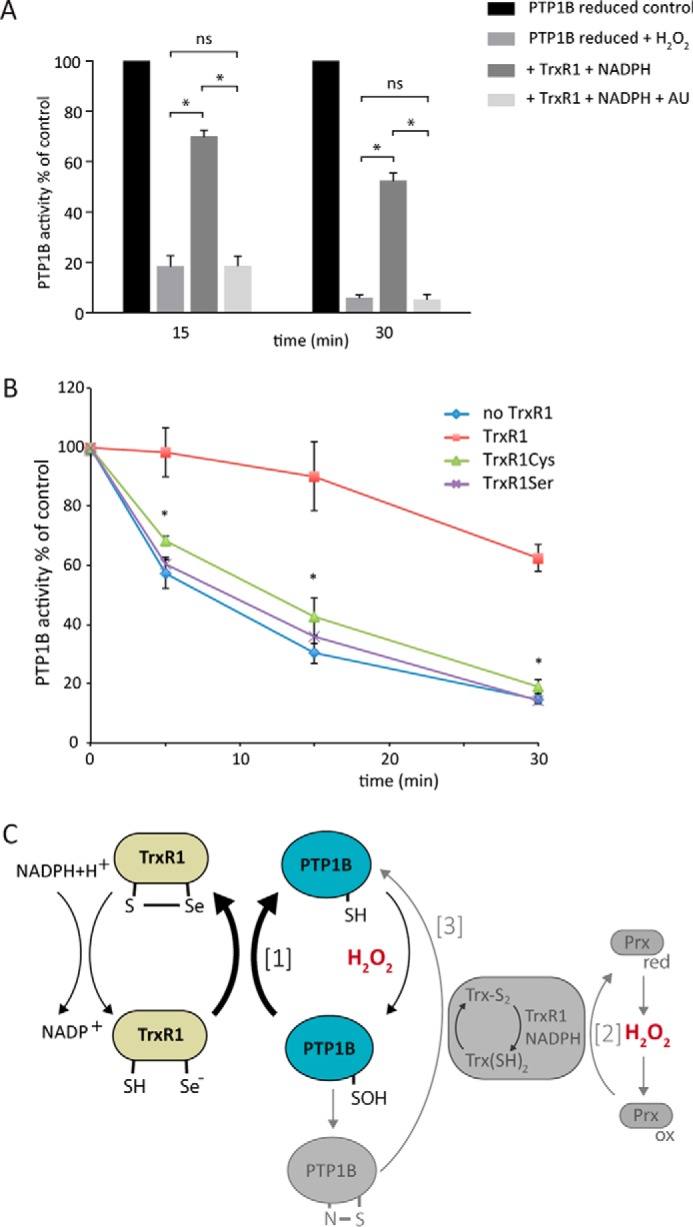
The active-site selenocysteine residue of TrxR1 is indispensable for protection of PTP1B from H2O2-mediated inactivation. A, inhibition of TrxR1 protection by auranofin. Reduced PTP1B (600 nm) was treated with or without TrxR1 (0.5 μm, 9.75 units/mg), NADPH (200 μm), and auranofin (AU, 1 μm). PTP activity was measured at the indicated times after addition of 100 μm H2O2 (n = 3; mean ± S.E.; *, p < 0.05). B, lack of protection by active-site mutants of TrxR1. Reduced PTP1B was exposed to 100 μm H2O2 in the presence of wild-type TrxR1 (18 units/mg) or variants where the active-site SelCys was mutated to Cys (TrxR1Cys) or Ser (TrxR1Ser) and then assayed for PTP activity as in A (n = 3; mean ± S.E.; *, p < 0.05). C, schematic of how the redox activity of the Trx/TrxR/Prx system could regulate PTP1B activity. The bold arrows show the reaction of TrxR1 (green) with the sulfenic acid of PTP1B that, we propose, explains protection of PTP1B activity (blue) by TrxR1 as observed in this study (reaction 1). Additional activities of the Trx system in relation to PTP regulation, as described previously in the literature, are schematically shown in gray. The full scheme is discussed in the text.
Discussion
In this work, we investigated the regulatory mechanisms that influence both oxidation and reduction of PTP1B. Under the conditions used in this study, we found no evidence for Prx or Trx acting as redox relays that transfer oxidizing equivalents from H2O2 to PTP1B. Instead, the Trx system coupled with Prx2 protected PTP1B from oxidation. We also discovered a hitherto unknown direct reaction of TrxR1 with a PTP1B oxidation intermediate that protects against inactivation during exposure to H2O2 (reaction 1, Fig. 6C). These findings have implications for understanding PTP1B regulation.
The prevailing view is that PTP oxidation occurs via H2O2 generated by NOX enzymes (7), although there are relatively few studies where this has been demonstrated unequivocally. Our finding that MEF cells expressing HyPer increased in fluorescence after stimulation with PDGF support other evidence (29) that H2O2 is indeed produced. Despite this, the reversibly oxidized form of the highly H2O2-reactive Prx2 was not significantly altered during stimulation unless TrxR1 was inhibited by auranofin. This implies that Prx2 is oxidized during PDGF stimulation but is efficiently recycled by the Trx system. Prx2 hyperoxidation was not directly analyzed; however, the decrease of the monomer form upon auranofin treatment during stimulation suggests that there is no formation of hyperoxidative forms.
As observed here and by others (9, 26), oxidation of isolated PTP1B requires extended exposure to high concentrations of H2O2. However, PTP1B is readily oxidized in cells (39) where Prx2 and other more reactive targets are present. A mechanism for facilitated oxidation would therefore seem to be essential, and a relay mechanism is an attractive concept (17, 40–42). The observation that Prx2 turnover occurs during signaling raises the question of whether Prx2 acts as a relay protein. To answer this, we set up an in vitro system with protein concentrations estimated to reflect cellular levels (17). We found that reduced Prx2 with or without Trx and redox cycling protected PTP1B against H2O2 rather than enhancing inactivation. This, plus the lack of a detectable disulfide exchange between oxidized Prx2 and reduced PTP1B, is evidence against a redox relay. We cannot, however, exclude a relay mechanism via another sensor or that it requires an additional cellular interaction partner. An alternative mechanism for facilitated PTP oxidation might be that it is localized to where the oxidant source and PTP are in close proximity. In this case, co-localized Prx2 could prevent PTP inactivation by scavenging basal H2O2 (reaction 2, Fig. 6C) and limit global oxidation of cellular proteins. Protection of PTP1B could be overcome when receptor activation resulted in localized inactivation of the Prx, either through oxidation or by peroxidase activity being decreased because of phosphorylation (43, 44). This model has been suggested as an explanation for increased PDGF-dependent phosphorylation in Prx2-deficient cells, with a corresponding decrease in total phosphatase activity of membrane fractions (23). However, it is still unlikely that oxidation could be due to freely diffusible H2O2 (45).
Although oxidative inactivation of PTPs is one mechanism for promulgating RTK pathways, PTPs also need to be recycled as a part of signal regulation. For PTP1B, there is good evidence from our studies of Txnrd1−/− MEF cells that TrxR1 is involved in recycling (22). We found increased basal levels of PTP1B oxidation in these cells compared with wild-type cells, and PDGF-BB caused relatively greater phosphorylation of the PTP1B site of the PDGF receptor and cell proliferation. The phosphorylation changes were mimicked by adding auranofin. Recycling of PTP1B could occur either by reducing the fully oxidized sulfenylamide end product or in a more dynamic process involving reduction of the sulfenic acid intermediate. We and others (19, 24–26) have shown that fully oxidized PTP1B can be at least partially reactivated by the full thioredoxin system (reaction 3, Fig. 6C). Here we confirmed these findings with both the cleaved PTP1B protein used in this study and the isolated catalytic domain used previously.
In cells, during growth factor receptor activation, reductant systems will be present when PTPs are exposed to H2O2 and would have the potential to react with the sulfenic acid. When we simulated this situation by exposing reduced PTP1B to H2O2 together with components of the Trx system, we observed highly efficient protection of PTP1B from H2O2-mediated inactivation. This could not be accounted for by the slower regeneration of the end product of oxidation, and neither was it due to consumption of H2O2 by TrxR1. TrxR1 does react slowly with H2O2, and indeed, calculations from the kinetic data (36) suggest a rate constant of ∼600 M−1 s−1. This is greater than the value of 20 m−1 s−1 for PTP1B (46). However, the TrxR1 rate is not fast enough to consume more than a few micromolar H2O2 during the experiment with PTP1B. Most strikingly, protection was provided by TrxR1 activity with no requirement for Trx. Further characterization revealed that protection requires the active-site Sec, as it was blocked by the TrxR inhibitor auranofin and lost when the Sec was replaced by Cys or Ser. As TrxR1 in the absence of Trx was unable to reactivate PTP1B after its conversion to the sulfenylamide form, a likely explanation of our findings is that the Sec-containing active site of TrxR1 directly reduces the sulfenic acid of PTP1B (reaction 1, Fig. 6C). As seen in Fig. 6, A and B, a gradual loss of PTP1B activity occurred over time in the presence of TrxR1. This is likely due to a minor fraction of PTP1B continuously forming the sulfenylamide, which cannot be reactivated by TrxR1 alone. As far as we are aware, direct reduction of a protein sulfenic acid is a role that has not been described previously for TrxR1. Our results with DEP-1 indicate that this is not a universal mechanism. However, reduction of the sulfenic acid has important implications for TrxR1 as a regulator of PTP1B activity.
TrxR1 could in theory regulate PTP1B activity during cell signaling by several mechanisms (Fig. 6C): by supporting the H2O2-scavenging activity of Prx, by acting with Trx1 to recycle PTP1B when oxidized, or by directly intercepting the sulfenic acid intermediate. These modes of regulation could operate in parallel with increased oxidant production because of NOX activation. Further control could be exerted by down-regulation of the reductive capacity of TrxR; for example, through NADPH depletion or reversible inactivation by nitrosylation (47). TrxR activity with PTP1B would also be decreased if its preferred substrate, Trx, became oxidized because of increased demand for Trx to reduce other substrates, such as oxidized Prx2 (reaction 2, Fig. 6C). This could enable oxidized PTP1B to accumulate. These mechanisms involving TrxR1 are all potential contributors to the enhanced PTP1B inactivation and growth factor signaling that have been observed in Trxrd1−/− cells. We speculated previously that the phenotype of the knock-out cells could be explained by the reduction of fully oxidized PTP1B by the complete Trx system (24). However, the subsequent finding that Trx1 is kept reduced through the GSH system in these cells (48) supports a mechanism such as the one described here for sulfenic acid reduction, where only TrxR1 is required.
Experimental procedures
Preparation of recombinant proteins
cDNA encoding recombinant His6-tagged, full-length human PTP1B (a kind gift from Prof. T. C. Meng, Academia Sinica) was subcloned into the pET28a vector (Novagen) by PCR with the primers 5′-GCGGAATTCATCGAAGGTCGTATGGAGATGGAAAAGGAGTTCGAG-3′ and 5′-GCGTCGACCTATGTGTTGCTGTTGAA-3′ and EcoR1 and Sal1 restriction sites. Although expressed as a full-length protein, we found that human PTP1B was subject to lysis-induced cleavage. This cleavage has been noted before during expression in E. coli (34); therefore, the findings were validated with the catalytic domain PTP1B. The PTP1B catalytic domain (residues 1–322) was subcloned into pD441-H6 by PCR with primers 5′-GTAGGTCTCGGTGGTATGGAGATGGAAAAGGAGTTC-3′ and 5′-GTAGGTCTCTTATTACCCATTGTGTGGCTCCAGG-3′ using Eco31I and Dpn1 restriction sites. Expression, purification, and removal of tags were performed using a protocol described previously (38). His-tagged PTP1B and Prx2 were expressed and purified, and the His tag of Prx2 was cleaved off as described previously (49), except that PTP1B was purified in the presence of 5 mm β-mercaptoethanol to maintain the protein in its reduced form (49). The intracellular domain containing the catalytic center of DEP-1 (a kind gift from Prof. F. Böhmer, Institute for Molecular Cell Biology) was expressed and purified as described previously (50). Recombinant human Trx1 and rat TrxR1 wild-type and mutant proteins were expressed and purified as described previously (38). The His-tagged PTP1B cleaved form, PTP1B catalytic domain, and DEP-1 resolved by SDS-PAGE corresponded to molecular masses of ∼50 kDa, ∼37 kDA and ∼70 kDa, respectively (Fig. 2A and not shown). Prior to all experiments, Prx2 preparations were reduced with 10 mm DTT, and Prx2, PTP1B, and DEP-1 were subjected to buffer exchange to remove the reductant using Bio-Gel P-6DG-Gel (Bio-Rad, catalog no.150-0738) columns (51). Protein concentrations were determined by Direct Detect (Millipore).
PTP activity assay
PTP activity was determined using 15 mm chromogenic substrate p-nitrophenyl phosphate (pNPP) (P4744-1G, Sigma-Aldrich) as described previously (52). The release of pNPP was measured at 410 nm and 22 °C using a Varioskan Flash plate reader (Thermo Scientific). Reduced PTP1B (600 nm, 32.5 μg/ml) or DEP-1 (80 nm, 5.6 μg/ml) was preincubated for 20 min in 20 mm HEPES and 100 mm NaCl buffer (pH 7.4) containing 0.1 mm diethylenetriaminepenta-acetic acid (DTPA), 0.05% BSA, and 1 mm sodium azide with the indicated concentrations of Trx1, TrxR1, NADPH (N7505-100MG, Sigma-Aldrich), and Prx2. Sodium azide was used to inhibit any trace amounts of catalase. Control PTP1B and DEP-1 activities gave a substrate turnover of 2.6–17 (median, 7.1) and 30–33 nmol/min/μg, respectively. Variations in activity were observed between different batches of PTP purifications.
SDS-PAGE analyses
The oxidation state of recombinant Prx2 was determined by separation of monomers and disulfide-bonded dimers by non-reducing SDS-PAGE (12% gel) with subsequent silver or Coomassie staining. The Prx2 oxidation state in MEF cells was determined after PDGF-BB stimulation with and without pretreatment of the cells with auranofin (1 μm for 1 h). Reduced thiols were blocked by adding 10 mm N-ethylmaleimide in buffer (50 mm Tris (pH 7.4) and 150 mm NaCl) and 1 μg/ml catalase to the intact cells, a procedure we have established to provide effective blocking (53). Cells were then trypsinized, washed once with buffer, and lysed with buffer + 1% Nonidet P-40 (Pierce) and Complete protease inhibitors (Roche). Lysates were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore), blocked with 5% milk in Tris-buffered saline, and immunoblotted for Prx2 (Sigma, R8656). Prx2 dimer to total Prx2 ratios were quantified using UV Band analysis software (UVITEC).
Treatment of PTP1B with H2O2
Buffer-exchanged reduced PTP1B was exposed to H2O2 and different components of the Trx system at the indicated time points, followed by addition of substrate and measurement of activity. We initially observed that incubation of PTP1B alone resulted in some time-dependent inactivation that was partially prevented in the presence of BSA, so BSA (0.05%) was added to the buffer. The activity after each H2O2 treatment was related to the activity of untreated PTP1B incubated for the same time. A 0–24% loss of activity was seen under control conditions at 30 min.
Reactivation of H2O2-treated PTP1B
Reactivation was performed using H2O2-inactivated PTP1B. Reduced PTP1B was first buffer-exchanged into reactivation buffer as described previously (26) and inactivated with 100 μm or 1 mm H2O2 for 5 min. Excess H2O2 was removed by adding 20 μg/ml catalase before reactivation was performed by adding components of the Trx system as indicated.
H2O2 measurement
Combinations of PTP1B, Trx1, TrxR1, NADPH, and Prx2 (in 20 mm HEPES, 0.05% BSA, and 1 mm sodium azide (pH 7.4)) were treated with 100 μm H2O2, and the loss of H2O2 was monitored using the ferrous oxidation of xylenol orange assay (54).
Cell culture and transient transfection with HyPer
MEFs (Walter and Elisa Hall Institute) were cultured in DMEM + 10% (v/v) FBS, 2 mm l-glutamine, penicillin, and streptomycin. Cells were stimulated with PDGF-BB (R&D Systems, 220-BB) after overnight serum starvation in DMEM + 1% FBS.
Transient transfection of MEF cells with HyPer
MEF cells were seeded into 6-well plates. Transfections were performed with the pHyPer-Cyto plasmid (6.5 μg, a kind gift from V. Belousov, Russian Academy of Sciences) mixed with 0.5 ml of Opti-MEM reduced-serum medium and 25 μl of 1 mg/ml polyethylenimine (linear, molecular weight 25,000, Polysciences Inc., Warrington, PA). The transfection mixture was incubated for 30 min at room temperature before addition to MEF cells in antibiotic-free medium. The medium was changed the following morning.
Live-cell fluorescence imaging
Live-cell imaging was performed with an Olympus IX81 motorized inverted microscope. pHyper-Cyto–transfected MEF cells were starved overnight and then stimulated with PDGF-BB. Hyper probe fluorescence was monitored at 1-min intervals over 20 min using a FITC channel at 495 nm/519 nm (55). Quantifications were performed using ImageJ as described previously (56).
Statistical analyses
Analyses of data were performed using one-way analysis of variance followed by Tukey post hoc tests for multiple comparisons. HyperCyto-derived fluorescence was analyzed using Student's paired t test.
Author contributions
M. D. performed all experiments and data analyses and contributed to writing the paper. P. E. P. expressed Prx2 and contributed to cloning and expression of PTP1B and to writing the manuscript. Q. C. expressed and purified TrxR1, serine/cysteine variants, and Trx1. J. F. provided intellectual input. A. Ö. provided intellectual input. E. S. J. A. designed experiments, analyzed data, provided essential intellectual input, and contributed to writing the paper. M. B. H. designed experiments, analyzed data, provided intellectual input, and contributed to writing the paper. C. C. W. was responsible for the project overall, contributed to the design of experiments, analyzed data, provided intellectual input, and wrote the paper.
Acknowledgments
We thank Profs. T. C. Meng and F. Böhmer for providing PTP1B and DEP-1 cDNA, respectively, and Drs. Alexander Peskin, Andreas Konigstorfer, and Andrea Betz for advice and fruitful discussions.
This work was supported by Swedish Research Council Grant 537-2014-360, The Swedish Society of Medicine, and the Health Research Council of New Zealand. The authors declare that they have no conflicts of interest with the contents of this article.
- PTP
- protein tyrosine phosphatase
- RTK
- receptor tyrosine kinase
- NOX
- NADPH oxidase
- Prx
- peroxiredoxin
- Sec
- selenocysteine
- MEF
- mouse embryonic fibroblast
- Trx
- thioredoxin
- pNPP
- p-nitrophenyl phosphate.
References
- 1. Tonks N. K. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 2. Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., and Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell 117, 699–711 [DOI] [PubMed] [Google Scholar]
- 3. Andersen J. N., Mortensen O. H., Peters G. H., Drake P. G., Iversen L. F., Olsen O. H., Jansen P. G., Andersen H. S., Tonks N. K., and Møller N. P. (2001) Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 21, 7117–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jayavelu A. K., Müller J. P., Bauer R., Böhmer S. A., Lässig J., Cerny-Reiterer S., Sperr W. R., Valent P., Maurer B., Moriggl R., Schröder K., Shah A. M., Fischer M., Scholl S., Barth J., et al. (2016) NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells. Leukemia 30, 473–483 [DOI] [PubMed] [Google Scholar]
- 5. Frijhoff J., Dagnell M., Godfrey R., and Ostman A. (2014) Regulation of protein tyrosine phosphatase oxidation in cell adhesion and migration. Antioxid. Redox Signal. 20, 1994–2010 [DOI] [PubMed] [Google Scholar]
- 6. Ostman A., Frijhoff J., Sandin A., and Böhmer F. D. (2011) Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 150, 345–356 [DOI] [PubMed] [Google Scholar]
- 7. Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 [DOI] [PubMed] [Google Scholar]
- 8. Frijhoff J., Dagnell M., Augsten M., Beltrami E., Giorgio M., and Östman A. (2014) The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic. Biol. Med. 68, 268–277 [DOI] [PubMed] [Google Scholar]
- 9. Denu J. M., and Tanner K. G. (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642 [DOI] [PubMed] [Google Scholar]
- 10. Garcia F. J., and Carroll K. S. (2014) Redox-based probes as tools to monitor oxidized protein tyrosine phosphatases in living cells. Eur. J. Med. Chem. 88, 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Montfort R. L., Congreve M., Tisi D., Carr R., and Jhoti H. (2003) Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423, 773–777 [DOI] [PubMed] [Google Scholar]
- 12. Salmeen A., Andersen J. N., Myers M. P., Meng T. C., Hinks J. A., Tonks N. K., and Barford D. (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773 [DOI] [PubMed] [Google Scholar]
- 13. Sivaramakrishnan S., Cummings A. H., and Gates K. S. (2010) Protection of a single-cysteine redox switch from oxidative destruction: on the functional role of sulfenyl amide formation in the redox-regulated enzyme PTP1B. Bioorg. Med. Chem. Lett. 20, 444–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huyer G., Liu S., Kelly J., Moffat J., Payette P., Kennedy B., Tsaprailis G., Gresser M. J., and Ramachandran C. (1997) Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 272, 843–851 [DOI] [PubMed] [Google Scholar]
- 15. Krishnan N., Fu C., Pappin D. J., and Tonks N. K. (2011) H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 4, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Y. Y., Chu H. M., Pan K. T., Teng C. H., Wang D. L., Wang A. H., Khoo K. H., and Meng T. C. (2008) Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J. Biol. Chem. 283, 35265–35272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Winterbourn C. C. (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286 [DOI] [PubMed] [Google Scholar]
- 18. Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 [DOI] [PubMed] [Google Scholar]
- 19. Lee S. R., Kwon K. S., Kim S. R., and Rhee S. G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372 [DOI] [PubMed] [Google Scholar]
- 20. Frangioni J. V., Beahm P. H., Shifrin V., Jost C. A., and Neel B. G. (1992) The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell 68, 545–560 [DOI] [PubMed] [Google Scholar]
- 21. Baker L. M., Raudonikiene A., Hoffman P. S., and Poole L. B. (2001) Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: genetic and kinetic characterization. J. Bacteriol. 183, 1961–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perkins A., Nelson K. J., Parsonage D., Poole L. B., and Karplus P. A. (2015) Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 40, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choi M. H., Lee I. K., Kim G. W., Kim B. U., Han Y. H., Yu D. Y., Park H. S., Kim K. Y., Lee J. S., Choi C., Bae Y. S., Lee B. I., Rhee S. G., and Kang S. W. (2005) Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature 435, 347–353 [DOI] [PubMed] [Google Scholar]
- 24. Dagnell M., Frijhoff J., Pader I., Augsten M., Boivin B., Xu J., Mandal P. K., Tonks N. K., Hellberg C., Conrad M., Arnér E. S., and Östman A. (2013) Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-β receptor tyrosine kinase signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 13398–13403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwertassek U., Haque A., Krishnan N., Greiner R., Weingarten L., Dick T. P., and Tonks N. K. (2014) Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J 281, 3545–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parsons Z. D., and Gates K. S. (2013) Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry 52, 6412–6423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sundaresan M., Yu Z. X., Ferrans V. J., Irani K., and Finkel T. (1995) Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270, 296–299 [DOI] [PubMed] [Google Scholar]
- 28. Bae Y. S., Sung J. Y., Kim O. S., Kim Y. J., Hur K. C., Kazlauskas A., and Rhee S. G. (2000) Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J. Biol. Chem. 275, 10527–10531 [DOI] [PubMed] [Google Scholar]
- 29. Malinouski M., Zhou Y., Belousov V. V., Hatfield D. L., and Gladyshev V. N. (2011) Hydrogen peroxide probes directed to different cellular compartments. PLoS ONE 6, e14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heldin C. H., Ostman A., and Rönnstrand L. (1998) Signal transduction via platelet-derived growth factor receptors. Biochim. Biophys. Acta 1378, F79–F113 [DOI] [PubMed] [Google Scholar]
- 31. Angelucci F., Sayed A. A., Williams D. L., Boumis G., Brunori M., Dimastrogiovanni D., Miele A. E., Pauly F., and Bellelli A. (2009) Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: structural and kinetic aspects. J. Biol. Chem. 284, 28977–28985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parsonage D., Sheng F., Hirata K., Debnath A., McKerrow J. H., Reed S. L., Abagyan R., Poole L. B., and Podust L. M. (2016) X-ray structures of thioredoxin and thioredoxin reductase from Entamoeba histolytica and prevailing hypothesis of the mechanism of Auranofin action. J. Struct. Biol. 194, 180–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Charbonneau H., Tonks N. K., Kumar S., Diltz C. D., Harrylock M., Cool D. E., Krebs E. G., Fischer E. H., and Walsh K. A. (1989) Human placenta protein-tyrosine-phosphatase: amino acid sequence and relationship to a family of receptor-like proteins. Proc. Natl. Acad. Sci. U.S.A. 86, 5252–5256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoppe E., Berne P. F., Stock D., Rasmussen J. S., Møller N. P., Ullrich A., and Huber R. (1994) Expression, purification and crystallization of human phosphotyrosine phosphatase 1B. Eur. J. Biochem. 223, 1069–1077 [DOI] [PubMed] [Google Scholar]
- 35. Frangioni J. V., Oda A., Smith M., Salzman E. W., and Neel B. G. (1993) Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. EMBO J. 12, 4843–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhong L., and Holmgren A. (2000) Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 275, 18121–18128 [DOI] [PubMed] [Google Scholar]
- 37. Gromer S., Arscott L. D., Williams C. H. Jr, Schirmer R. H., and Becker K. (1998) Human placenta thioredoxin reductase: isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 273, 20096–20101 [DOI] [PubMed] [Google Scholar]
- 38. Xu J., Eriksson S. E., Cebula M., Sandalova T., Hedström E., Pader I., Cheng Q., Myers C. R., Antholine W. E., Nagy P., Hellman U., Selivanova G., Lindqvist Y., and Arnér E. S. (2015) The conserved Trp114 residue of thioredoxin reductase 1 has a redox sensor-like function triggering oligomerization and crosslinking upon oxidative stress related to cell death. Cell Death Dis. 6, e1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meng T. C., Buckley D. A., Galic S., Tiganis T., and Tonks N. K. (2004) Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 279, 37716–37725 [DOI] [PubMed] [Google Scholar]
- 40. Winterbourn C. C., and Hampton M. B. (2008) Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 45, 549–561 [DOI] [PubMed] [Google Scholar]
- 41. Randall L. M., Ferrer-Sueta G., and Denicola A. (2013) Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods Enzymol. 527, 41–63 [DOI] [PubMed] [Google Scholar]
- 42. Du Y., Zhang H., Zhang X., Lu J., and Holmgren A. (2013) Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J. Biol. Chem. 288, 32241–32247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Woo H. A., Yim S. H., Shin D. H., Kang D., Yu D. Y., and Rhee S. G. (2010) Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 140, 517–528 [DOI] [PubMed] [Google Scholar]
- 44. Rhee S. G., Woo H. A., Kil I. S., and Bae S. H. (2012) Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 287, 4403–4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Travasso R. D. M., Sampaio Dos Aidos F., Bayani A., Abranches P., and Salvador A. (2017) Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 12, 233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., Chen M. W., Ji Y., Hu S. B., and Zhou Y. G. (2016) Kinetic resolution of axially chiral 5- or 8-Substituted quinolines via asymmetric transfer hydrogenation. J. Am. Chem. Soc. 138, 10413–10416 [DOI] [PubMed] [Google Scholar]
- 47. Engelman R., Ziv T., Arnér E. S., and Benhar M. (2016) Inhibitory nitrosylation of mammalian thioredoxin reductase 1: molecular characterization and evidence for its functional role in cellular nitroso-redox imbalance. Free Radic. Biol. Med. 97, 375–385 [DOI] [PubMed] [Google Scholar]
- 48. Peng X., Mandal P. K., Kaminskyy V. O., Lindqvist A., Conrad M., and Arnér E. S. (2014) Sec-containing TrxR1 is essential for self-sufficiency of cells by control of glucose-derived H2O2. Cell Death Dis. 5, e1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nagy P., Karton A., Betz A., Peskin A. V., Pace P., O'Reilly R. J., Hampton M. B., Radom L., and Winterbourn C. C. (2011) Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J. Biol. Chem. 286, 18048–18055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petermann A., Haase D., Wetzel A., Balavenkatraman K. K., Tenev T., Gührs K. H., Friedrich S., Nakamura M., Mawrin C., and Böhmer F. D. (2011) Loss of the protein-tyrosine phosphatase DEP-1/PTPRJ drives meningioma cell motility. Brain Pathol. 21, 405–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pace P. E., Peskin A. V., Han M. H., Hampton M. B., and Winterbourn C. C. (2013) Hyperoxidized peroxiredoxin 2 interacts with the protein disulfide-isomerase ERp46. Biochem. J. 453, 475–485 [DOI] [PubMed] [Google Scholar]
- 52. Montalibet J., Skorey K. I., and Kennedy B. P. (2005) Protein tyrosine phosphatase: enzymatic assays. Methods 35, 2–8 [DOI] [PubMed] [Google Scholar]
- 53. Cox A. G., Winterbourn C. C., and Hampton M. B. (2010) Measuring the redox state of cellular peroxiredoxins by immunoblotting. Methods Enzymol. 474, 51–66 [DOI] [PubMed] [Google Scholar]
- 54. Nourooz-Zadeh J. (1999) Ferrous ion oxidation in presence of xylenol orange for detection of lipid hydroperoxides in plasma. Methods Enzymol. 300, 58–62 [DOI] [PubMed] [Google Scholar]
- 55. Belousov V. V., Fradkov A. F., Lukyanov K. A., Staroverov D. B., Shakhbazov K. S., Terskikh A. V., and Lukyanov S. (2006) Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 3, 281–286 [DOI] [PubMed] [Google Scholar]
- 56. Burgess A., Vigneron S., Brioudes E., Labbé J. C., Lorca T., and Castro A. (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects because of deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. U.S.A. 107, 12564–12569 [DOI] [PMC free article] [PubMed] [Google Scholar]