

Abstract
Mitochondrial calcium plays critical roles in diverse cellular processes ranging from energy metabolism to cell death. Previous studies have demonstrated that mitochondrial calcium uptake is mainly mediated by the mitochondrial calcium uniporter (MCU) complex. However, the roles of the MCU complex in calcium transport, signaling, and dysregulation by oxidative stress still remain unclear. Here, we confirmed that Drosophila MCU contains evolutionarily conserved structures and requires essential MCU regulator (EMRE) for its calcium channel activities. We generated Drosophila MCU loss-of-function mutants, which lacked mitochondrial calcium uptake in response to caffeine stimulation. Basal metabolic activities were not significantly affected in these MCU mutants, as observed in examinations of body weight, food intake, body sugar level, and starvation-induced autophagy. However, oxidative stress-induced increases in mitochondrial calcium, mitochondrial membrane potential depolarization, and cell death were prevented in these mutants. We also found that inositol 1,4,5-trisphosphate receptor genetically interacts with Drosophila MCU and effectively modulates mitochondrial calcium uptake upon oxidative stress. Taken together, these results support the idea that Drosophila MCU is responsible for endoplasmic reticulum-to-mitochondrial calcium transfer and for cell death due to mitochondrial dysfunction under oxidative stress.
Keywords: calcium, calcium channel, calcium transport, cell death, Drosophila, endoplasmic reticulum (ER), inositol trisphosphate receptor (InsP3R), mitochondria, oxidative stress
Introduction
Mitochondrial Ca2+ is a key regulator for cellular metabolic functions by activating Krebs cycle dehydrogenases, metabolite shuttle systems, and ATP synthase. However, inappropriately high Ca2+ levels in the mitochondrial matrix threaten cell survival by increasing reactive oxygen species (ROS)6 production and triggering mitochondrial permeability transition (mPT). The main route for Ca2+ uptake into mitochondria is through mitochondrial calcium uniporter (MCU), a Ca2+-selective ion channel located at the inner mitochondrial membrane (1), which was originally identified as CCDC109A (2, 3). Recent studies have demonstrated that MCU has a homopentameric structure in which the second transmembrane domain forms a hydrophilic pore across the membrane (4) and the N terminus domain modulates MCU activity by protein–protein interaction and binding divalent cations (5, 6). To counteract the Ca2+ influx via MCU, mitochondrial sodium–calcium exchanger (NCLX) provides mitochondrial Ca2+ efflux routes by exchanging Ca2+ for Na+ ions (7). Leucine zipper EF-hand-containing transmembrane protein 1 (LETM1) may also be involved in mitochondrial Ca2+ influx or efflux, which is still in a controversy (8–11).
Previous studies have identified multiple regulators of MCU activity, including mitochondrial calcium uptake 1 and 2 (MICU1/2), mitochondrial calcium uniporter regulator 1 (MCUR1), and essential MCU regulator (EMRE). EMRE is a 10-kilodalton mitochondrial inner membrane protein with a single transmembrane domain, which is an essential component that bridges MICU1/2 with MCU (12). The transmembrane helix of EMRE interacts with MCU, and the C terminus of EMRE binds to MICU1 (13). In addition, silencing of EMRE completely abolishes the channel activity of MCU (14). MICU1/2 are Ca2+-binding EF-hand-containing proteins residing in the mitochondrial intermembrane space. They can modulate MCU activity, depending on cytoplasmic Ca2+ concentration ([Ca2+]i) (15–17). MCUR1 is an inner mitochondrial membrane integral protein binding to MCU and regulates MCU-dependent mitochondrial Ca2+ uptake (18) and Ca2+ threshold for mPT (19).
Over the last few years, studies on the MCU complex have proposed diverse roles of MCU at the cellular and organism levels, using its genetic ablation and/or pharmacologic inhibition models. Although a loss-of-function mutation of MCU in mice exhibited negligible influence on metabolism and cell death (20), further studies revealed that MCU plays an essential role in heart rate acceleration during fight-or-flight response (21), ROS-mediated wound repair (22), and skeletal muscle trophism (23). In pancreatic β-cells, MCU silencing decreases mitochondrial ATP synthesis and impairs the metabolism-secretion coupling (11, 24). In neuronal cells, suppression of MCU relieves ischemia and reperfusion injuries as well as Ca2+ excitotoxicity (25, 26). In cardiac cells, however, a loss-of-function mutation of MCU did not protect from ischemic injury due to [Ca2+]i overload despite preserved mitochondrial membrane potential (ΔΨm) and reduced ROS formation (27).
The main source of mitochondrial Ca2+ is the endoplasmic reticulum (ER), which has a higher Ca2+ level (>400 μm) required for protein folding and Ca2+ signaling. Ca2+ release from the ER is taken up by mitochondria not only to avoid [Ca2+]i accumulation but also to stimulate mitochondrial energy metabolism (28). This ER–mitochondrial interaction is mediated by physical contacts between the two organelles via mitochondria-associated ER membrane (MAM) (29, 30). MAM is composed of several proteins, including inositol 1,4,5-trisphosphate receptor (IP3R), glucose-regulated protein 75 (grp75), porin, and MCU (31). The MAM formation can be expanded in pathologic conditions, such as obesity and diabetes, in which increased ER–mitochondrial Ca2+ connection exacerbates ER Ca2+ depletion and mitochondrial Ca2+ overload (32).
Oxidative stress disrupts cellular Ca2+ homeostasis and consequently induces cytotoxicity. Oxidative stress activates IP3R, stimulates ER Ca2+ release, intensifies ER stress, and leads to apoptosis (33, 34). More importantly, oxidative stress plays significant roles in the pathogenesis of various chronic diseases, including neurodegeneration (35). However, the pathophysiological role of Ca2+ dysregulation by oxidative stress still remains elusive.
In this study, we generated Drosophila MCU loss-of-function mutants for the first time and characterized their phenotypes on metabolism, Ca2+ handling, and cell death. We demonstrated that attenuated Ca2+ transport from the ER to mitochondria in MCU mutants prevents ROS-induced mitochondrial dysfunction and cell death. Because the Drosophila system provides powerful tools for genetic studies, our loss-of-function mutants and transgenic flies for MCU and other components of MCU complex would provide crucial information for understanding the regulatory mechanism of mitochondrial Ca2+ homeostasis.
Results
Drosophila MCU null mutant is viable
According to a recent report, CG18769 is homologous to mammalian MCU (36). Human and mouse MCU protein sequences are similar to the protein sequence encoded by CG18769 (supplemental Fig. S1A). CG18769-encoded protein localizes to mitochondria, and silencing CG18769 decreases mitochondrial Ca2+ entry (36, 37). To assess the in vivo role of MCU, Drosophila MCU loss-of-function mutants were generated by P-element imprecise excision in the 5′-untranslated region of CG18769 using the P(GSV6)GS11565 fly line (Fig. 1A). From the 200 excision alleles obtained, we found MCU52 mutant, which lacked 1,476 bp (3R14578001–14580477) that encoded the transcription start site and the first exon of MCU (Fig. 1, A and B). In the mutant, MCU mRNA and protein were not detected by quantitative RT-PCR (Fig. 1C) and immunoblotting (Fig. 1D). Wild-type MCU protein was expressed weakly in embryo stage but strongly in larva, pupa, and adult stages (Fig. 1E). MCU52 mutant flies were viable and showed a similar survival rate compared with wild-type ones (Fig. 1E).
Figure 1.

MCU mutant shows normal metabolism. A, schematic representation of Drosophila MCU genomic locus and the genomic deletion region of MCU52. Exons are indicated by boxes, and the coding regions are colored black. B, PCR result to detect genomic DNA deletion around the transcription start site of MCU using the primer set in A. C, quantitative RT-PCR analysis of MCU transcript in wild-type control and MCU52 flies (n = 3). D, immunoblotting by anti-MCU and anti-β-tubulin (TUB) antibodies in wild-type control and MCU52 flies. Tubulin was used as a loading control. E, MCU expression and survival rate in various developmental stages. Homogenized samples at the indicated developmental stage were immunoblotted by anti-MCU, anti-porin, and anti-β-tubulin antibodies. Tubulin and porin were used as loading controls. The survival rate of wild-type control and MCU52 mutant during development is shown in the bottom panel. F, body weights of wild-type control and MCU52 (n = 5). G, food intake for 24 h by wild-type control and MCU52 flies (n = 5–6). H, hemolymph concentration of trehalose and glucose in wild-type control and MCU52 flies (n = 3). I, mCherry-ATG8a signal images illustrating starvation-induced autophagy in larval fat body of MCU null flies (Cg>mCherry-ATG8a; MCU52/MCU52) and control flies (Cg>mCherry-ATG8a) in fed and starved conditions. The starvation-induced autophagy assays were conducted using more than 10 flies. Scale bars, 20 μm. ***, p < 0.001. N.S., not significant. Error bars, S.D.
MCU52 mutant does not show significant metabolic phenotypes
To find the exclusive role of MCU in Drosophila physiology, we investigated metabolic phenotypes of MCU52 mutant. First, the body weight of MCU52 mutants was not different from that of wild-type flies in both sexes (Fig. 1F). The amount of food intake of MCU52 mutant was also similar to that of the wild-type fly (Fig. 1G). The concentration of circulating sugars in the hemolymph of MCU52 mutants was not significantly different from that in wild-type flies (Fig. 1H). We also did not observe any detectable changes in starvation-induced autophagy (Fig. 1I). Collectively, these results showed that loss of MCU does not alter basal metabolism in Drosophila.
The Ca2+ channel activity of MCU is conserved in Drosophila
To assess mitochondrial Ca2+ uptake in a physiological context, we measured mitochondrial matrix Ca2+ concentration ([Ca2+]mito) in a larval muscle expressing mitochondria-targeted ratio-pericam (MTRP), a genetically encoded Ca2+ indicator targeted to the mitochondrial matrix (38). MTRP was specifically expressed in muscle tissues by Mef-Gal4 and UAS-MTRP (Fig. 2A). The localization of expressed MTRP to mitochondria was confirmed with two mitochondria markers, streptavidin (Fig. 2B) and ATP5a (supplemental Fig. S2A).
Figure 2.

MCU regulates rapid mitochondrial calcium uptake. A, schematic representation of the muscle region of a dissected Drosophila larva to measure mitochondrial Ca2+ level. MTRP was expressed in larval muscle in Mef>MTRP. Fluorescence intensity was recorded from a well-focused region of body wall muscles 6, 7, 15, 16, and 17 of abdomen segments A2–A6. The central nervous system is marked with solid black. For simplicity, not all muscles and segments are shown. Fluorescence intensity from MTRP expressed in muscle was measured upon stimulation with 10 mm caffeine. B, mitochondrial localization of MTRP. Green, subcellular localization of MTRP in larval muscle. Hoechst (blue) and streptavidin (red) were used to mark DNA and mitochondria, respectively. The genotype is Mef-Gal4/UAS-MTRP. Scale bar, 10 μm. C, immunoblot analyses of endogenous MCU (MCU) and exogenously expressed FLAG-tagged MCU (Flag). D–E, averaged traces (D) and quantitative analysis (E) of [Ca2+]mito signals after treatment of 10 mm caffeine from wild-type, MCU52, and MCU52 mutant with exogenous Drosophila wild-type MCU expression (n = 5–7). Genotypes are as follows: Mef-Gal4/UAS-MTRP (Mef>), Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>), and UAS-MCU-FLAG;Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>MCU). F, immunoblot analyses of endogenous MCU (MCU) and exogenously expressed FLAG-tagged MCU (Flag). G and H, averaged traces (G) and quantitative analysis (H) of [Ca2+]mito signals after treatment with 10 mm caffeine from wild type, MCU52, and MCU52 mutant with human MCU expression (n = 3–6). Genotypes are as follows: Mef-Gal4/UAS-MTRP (Mef>), Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>), and UAS-hMCU-FLAG;Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>hMCU). I–K, a schematic representation of Drosophila wild-type MCU and MCUNIMQ (I). MTS, mitochondrial targeting sequence; CC, coiled-coil domain; TM, transmembrane domain; DIME, DIME motif. Immunoblot analyses of endogenous MCU (MCU) and exogenously expressed FLAG-tagged MCU (Flag) are shown in the bottom panels. J and K, averaged traces (J) and quantitative analysis (K) of [Ca2+]mito signals after 10 mm caffeine treatment from wild type, MCU52, MCU52 mutant with Drosophila wild-type MCU overexpression, and MCU52 mutant with Drosophila MCUNIMQ overexpression (n = 4–10). Genotypes are as follows: Mef-Gal4/UAS-MTRP (Mef>), Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>), UAS-MCU-FLAG;Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>MCUWT), and UAS-MCUNIMQ-FLAG;Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>MCUNIMQ). *, **, and ***, p < 0.05, p < 0.01, and p < 0.001, respectively. N.S., not significant. Error bars, S.D.
To confirm that Drosophila MCU is a functional mitochondrial Ca2+ uptake route, we compared [Ca2+]mito increase in muscle tissues of control, MCU52 mutant, and MCU52 mutant expressing exogenous Drosophila wild-type MCU (Fig. 2C). Caffeine was used to stimulate Ca2+ release from the ER, inducing elevation of cytosolic (supplemental Fig. S3A) and mitochondrial Ca2+ levels (Fig. 2D). In control larvae, [Ca2+]mito was increased sharply after caffeine stimulation and slowly returned to the basal level (Fig. 2D). However, although caffeine-induced [Ca2+]i changes were not significantly different between control and MCU52 mutant larvae (supplemental Fig. S3A), MCU52 mutant failed to elicit any [Ca2+]mito change upon caffeine stimulation (Fig. 2, D and E). Furthermore, when exogenous Drosophila MCU was overexpressed in MCU52 mutant, the absence of caffeine-induced [Ca2+]mito response in the mutant was completely rescued (Fig. 2, D and E). These results consistently demonstrate the indispensable role of Drosophila MCU in mitochondrial Ca2+ uptake.
To test whether Drosophila MCU is functionally equivalent to the mammalian MCU, we expressed human MCU in the muscle of MCU52 mutants (Fig. 2F). Similar to the results with Drosophila MCU, overexpression of human MCU in MCU52 mutant larvae resulted in full recovery of [Ca2+]mito response upon caffeine stimulation (Fig. 2, G and H). These results demonstrate that Drosophila MCU (encoded by CG18769) is a genuine orthologue of human MCU.
MCU has a mitochondrial targeting sequence at its N terminus, two coiled-coil domains, two transmembrane domains, and the DIME motif. Previous studies showed that substitution of two acidic amino acids within the DIME motif resulted in a dominant-negative effect on the uniporter activity of mammalian MCU (39, 40). To test whether the DIME motif in Drosophila MCU is also critical for its Ca2+ channel activity, we generated a mutant of MCU in the DIME motif (MCUNIMQ) (Fig. 2I). Transgenic expression of wild-type MCU in the muscle of MCU52 mutant resulted in full recovery of mitochondrial Ca2+ uptake, as shown (Fig. 2, D and E). In contrast, expression of MCUNIMQ failed to rescue the defective [Ca2+]mito response of MCU52 mutant (Fig. 2, J and K). Therefore, the DIME motif is essential for its Ca2+ transport activity in Drosophila MCU.
EMRE is required for MCU activity in Drosophila
Eye-specific expression of MCU using Gmr-Gal4 led to glazed phenotypes in the eye with partial loss of pigmentation and irregular ommatidial array (Fig. 3A). The activity of MCU appeared critical for these effects because knockdown of MCU completely suppressed the eye phenotype (Fig. 3A). Consistently, a higher MCU expression with two copies of UAS-MCU transgene resulted in more severe phenotypes (Fig. 3A). Overexpression of EMRE, an essential component of the MCU complex, alone did not elicit defective phenotypes (Fig. 3A). Unexpectedly, co-expression of MCU and EMRE led to lethality. However, when those flies were reared at 23 °C instead of 25 °C, few managed to survive with defective eyes marked with black mass tissues, indicating that MCU and EMRE have a strong genetic interaction in vivo (Fig. 3A).
Figure 3.
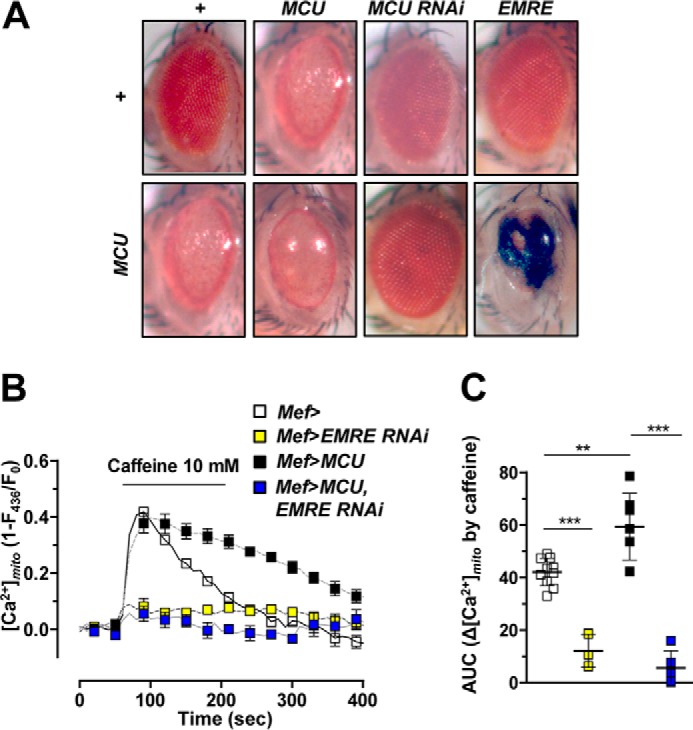
EMRE is required for MCU activity. A, eye-specific expression of transgenes using Gmr-Gal4. Severity of eye defect exacerbated as the level of MCU expression increased. Co-expression of MCU and EMRE in the eye led to lethality when reared in a 25 °C incubator. Several flies escaped the lethality when reared in a 23 °C incubator. The eye of an escaper fly is shown in the right bottom panel. All other flies were reared in a 25 °C incubator. From the left top panel, genotypes are as follows: Gmr-Gal4/+, Gmr-Gal4/+;UAS-MCU-Myc/+, Gmr-Gal4/+;UAS-MCU RNAi/+, Gmr-Gal4/UAS-EMRE-FLAG, Gmr-Gal4/+;UAS-MCU-Myc/+, Gmr-Gal4/+;UAS-MCU-Myc/UAS-MCU-Myc, Gmr-Gal4/+;UAS-MCU RNAi/UAS-MCU-Myc, and Gmr-Gal4/UAS-EMRE-FLAG;UAS-MCU-Myc/+. B and C, averaged traces (B) and quantitative analysis (C) of [Ca2+]mito signals from the flies that express wild-type MCU, EMRE RNAi, or wild-type MCU and EMRE RNAi together (n = 5–10). Genotypes are as follows: Mef-Gal4,UAS-MTRP/+ (Mef>), UAS-EMRE RNAi/+;Mef-Gal4,UAS-MTRP/+ (Mef>EMRE RNAi), UAS-MCU-FLAG/+;Mef-Gal4, UAS-MTRP/+ (Mef>MCU), and UAS-EMRE RNAi/UAS-MCU-FLAG;Mef-Gal4,UAS-MTRP/+ (Mef>MCU, EMRE RNAi). ** and ***, p < 0.01 and p < 0.001, respectively. Error bars, S.D.
To confirm the role of EMRE in Drosophila MCU activity, we used the UAS-EMRE RNAi fly line to knock down EMRE expression. Silencing of EMRE led to impairment of mitochondrial Ca2+ uptake (Fig. 3, B and C), closely resembling the result obtained from MCU52 mutant (Fig. 2, D and E), implying that EMRE is required for mitochondrial Ca2+ uptake. Furthermore, expression of MCU in EMRE knockdown fly failed to increase the caffeine-induced [Ca2+]mito response (Fig. 3, B and C). These results suggest that EMRE interacts with MCU genetically and is an essential and irreplaceable component for Drosophila MCU activity.
Loss of MCU provides resistance to oxidative stress
Oxidative stress is the most common pathogenic mediator for various diseases and is also involved in aging processes. To address whether mitochondrial Ca2+ uptake via MCU has any pathogenic role in inducing cell death under oxidative stress conditions, we first investigated the survival rates of wild-type fly and MCU52 mutant fed on hydrogen peroxide (H2O2)-containing food. Dihydroethidium (DHE) staining was used to detect the level of ROS in the thorax of flies. Elevated level of ROS was detected in the flies fed on 1% H2O2-containing food for the past 72 h, indicating that feeding H2O2 induced oxidative stress in the fly (supplemental Fig. S4A). Interestingly, MCU52 mutant flies survived significantly longer than wild-type flies when fed on 1% H2O2-containing food, whereas normal food caused the mutants to survive slightly less than wild-type flies. These results suggest that MCU52 mutant flies are more resistant to oxidative stress than wild-type flies (Fig. 4A).
Figure 4.

MCU mediates oxidative stress-induced apoptosis. A, survival curves of wild-type and MCU52 male flies on 1% H2O2-containing food (n = 133–149). Log-rank test: p < 0.0001. B–D, detecting apoptotic cells in larval muscle of wild-type and MCU52 flies after treatment of 2% H2O2 for 3 h. The assays were repeated >3 times. B, left panels, TUNEL signals; right panels, merged images of TUNEL (red), Hoechst (blue), and phalloidin (green) staining. C, left panels, GFP reporter expression from hid5′F-WT enhancer; right panels, merged images of GFP reporter (green) and Hoechst (blue) staining. Genotypes are WT (hid5′F-WT-GFP/+) and MCU52 (hid5′F-WT-GFP/+;MCU52/MCU52). D, left panels, cleaved caspase-3; right panels, merged images of cleaved caspase-3 (green) and Hoechst (blue) staining. E, TUNEL assays after treatment of 100 mm H2O2 for 6 h in S2 cells transfected with luciferase dsRNA or Drosophila MCU dsRNA. The TUNEL assay was repeated >3 times. F, the proportion of apoptotic nuclei in S2 cells transfected with luciferase dsRNA or Drosophila MCU dsRNA (n = 12–13). Scale bars, 50 μm (B–D) or 20 μm (E). ***, p < 0.001. Error bars, S.D.
To demonstrate whether ROS-induced apoptosis was attenuated by loss-of-function mutations of MCU, we performed a TUNEL assay under oxidative stress conditions. Wild-type flies showed strong TUNEL signals by 2% H2O2 treatment for 3 h, which was markedly reduced in MCU52 mutants (Fig. 4B). Additionally, we checked hid5′F-WT-GFP reporter expression and cleaved caspase-3 as a marker of early and late phases of apoptosis, respectively (41). Wild-type flies showed up-regulated GFP reporter expression from hid5′F-WT enhancer and increased cleaved caspase-3 staining, but MCU52 mutants displayed markedly reduced signals for both apoptotic markers (Fig. 4 (C and D) and supplemental Fig. S4 (B and C)). Consistently, we also confirmed the reduction of H2O2-induced apoptotic cell death by knockdown of MCU in Drosophila S2 cells (Fig. 4, E and F). Taken together, these results indicate that loss of MCU endows resistance to oxidative stress.
MCU-dependent Ca2+ uptake contributes to mitochondrial dysfunction by oxidative stress
To more clearly demonstrate whether oxidative stress can increase [Ca2+]mito in Drosophila, we applied tert-butyl hydroperoxide (t-BuOOH) to larval muscle and applied H2O2 to S2 cells. First, in control larval muscle in vivo, [Ca2+]mito was increased after treatment of t-BuOOH from 1 to 100 mm in a dose-dependent manner, suggesting that oxidative stress can increase [Ca2+]mito (supplemental Fig. S5A). Compared with in vitro experiments, a higher dose of t-BuOOH is required to induce rapid oxidative stress on inner muscle tissue under the cuticle. Treatment of t-BuOOH (35 mm) on control flies (Mef>) markedly increased [Ca2+]mito (Fig. 5A). As expected, the [Ca2+]mito response induced by t-BuOOH was abolished in MCU52 mutant (MCU52,Mef>), demonstrating the critical role of MCU in oxidative stress-induced [Ca2+]mito increase (Fig. 5, A and B). Transgenic expression of Drosophila MCU (MCU52,Mef>MCU) rescued the abolished [Ca2+]mito response in MCU52 mutant (MCU52,Mef>) (Fig. 5, A and B). By contrast, the [Ca2+]i changes induced by t-BuOOH were not significantly different between control (Mef>) and MCU52 mutant (supplemental Fig. S5B). We also detected [Ca2+]mito rise in S2 cells upon H2O2 stimulation (Fig. 5C). Consistent with the in vivo muscle data, MCU dsRNA-treated S2 cells showed reduced H2O2-induced mitochondrial Ca2+ uptake (−63%) in comparison with control (Luciferase dsRNA-treated) (Fig. 5, C and D). To estimate the functional deterioration of mitochondria as a result of [Ca2+]mito overload, we monitored mitochondrial membrane potential (Ψmito) by using potential-sensitive JC-1 dye. Oxidative stress by H2O2 treatment (3 mm) elicited depolarization of Ψmito in S2 cells (Fig. 5E). Intriguingly, knockdown of MCU strongly prevented H2O2-induced ΔΨmito collapse in S2 cells (Fig. 5, E and F). Based on these results, we suggest that mitochondrial dysfunction and apoptotic cell death induced by oxidative stress are related to MCU-mediated [Ca2+]mito overload.
Figure 5.
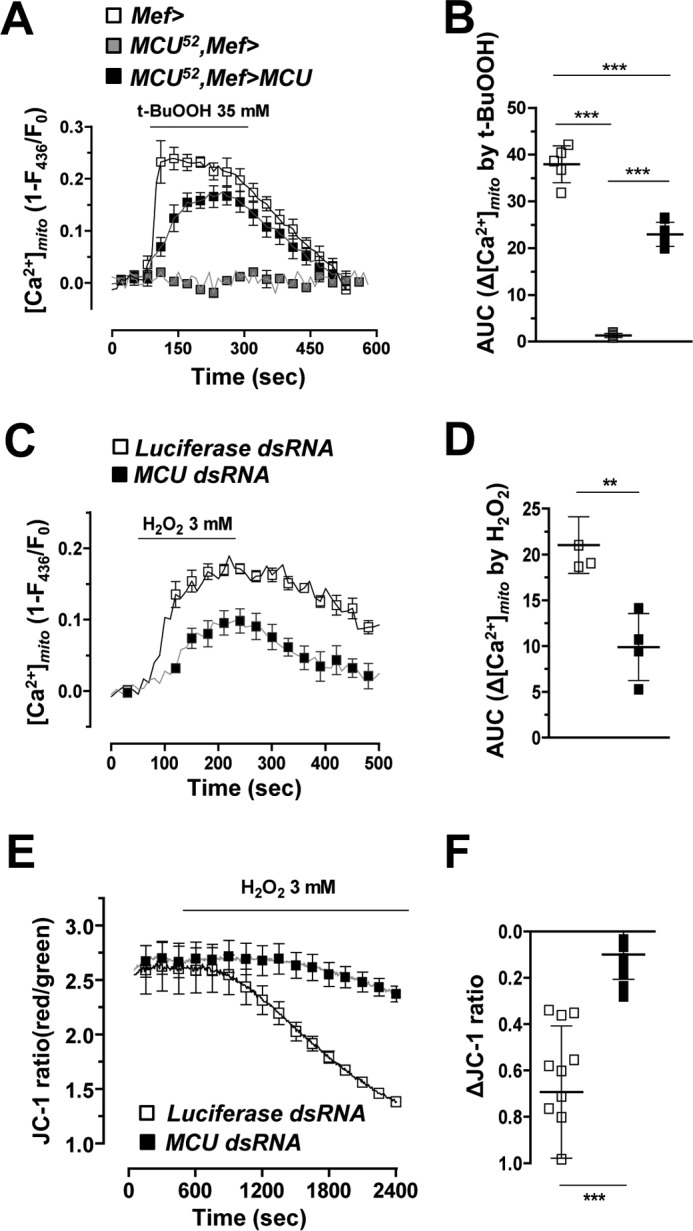
MCU is required for oxidative stress-induced mitochondrial calcium uptake. A and B, averaged traces (A) and quantitative analysis (B) of [Ca2+]mito signals after 35 mm t-BuOOH treatment from wild-type, MCU52, and MCU52 mutant with wild-type Drosophila MCU expression (n = 4–7). Genotypes are as follows: Mef-Gal4/UAS-MTRP (Mef>), Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>), and UAS-MCU-FLAG/+;Mef-Gal4,MCU52/UAS-MTRP,MCU52 (MCU52,Mef>MCU). C and D, averaged traces (C) and quantitative analysis (D) of [Ca2+]mito signals after 3 mm H2O2 treatment from S2 sells transfected with luciferase dsRNA or Drosophila MCU dsRNA (n = 4). E, measurement of Ψmito using JC-1 in S2 cells transfected with luciferase dsRNA or MCU dsRNA upon 3 mm H2O2 treatment. F, quantitative analysis of E (n = 12–16). ** and ***, p < 0.01, and p < 0.001, respectively. Error bars, S.D.
IP3R and MCU participate in oxidative stress-induced ER–mitochondria Ca2+ transfer
Oxidative stress is reported to activate IP3R, a Ca2+ channel in the ER, resulting in ER Ca2+ release (33, 34). Released Ca2+ from the ER provides high Ca2+ level in microdomains of ER-mitochondrial junction called MAM. IP3R is a component of tethering structure between the ER and mitochondria (31). Therefore, we investigated the involvement of IP3R in ER-mitochondria Ca2+ transfer under oxidative stress.
Muscle-specific expression of exogenous MCU using Mef-Gal4 was lethal in pupa stage of the transgenic fly (Fig. 6A). However, this lethality was blocked by knockdown of IP3R, suggesting that MCU and IP3R have a strong genetic interaction in vivo (Fig. 6A). To confirm the role of IP3R in mitochondrial Ca2+ uptake, we compared [Ca2+]mito increase induced by oxidative stress between control (Mef>) and muscle-specific IP3R knockdown flies (Mef>IP3R RNAi). The IP3R knockdown flies showed significantly reduced mitochondrial Ca2+ uptake (−41.2%) upon t-BuOOH stimulation in comparison with control (Mef>) (Fig. 6, B and C). Additionally, when we silenced IP3R in MCU transgenic flies (Mef>MCU, IP3R RNAi), the [Ca2+]mito induced by t-BuOOH was reduced by 34.7% when compared with that of Mef>MCU flies (Fig. 6, B and C). This was consistent with our previous results, where knockdown of IP3R blocked the lethality induced by MCU overexpression (Fig. 6A). In S2 cells, H2O2-induced [Ca2+]mito increase was also strongly inhibited (−64.0%) by transfection of IP3R dsRNA (Fig. 6, D and E). These results strongly indicate that both IP3R and MCU are critical for the Ca2+ transfer from the ER to mitochondria.
Figure 6.

MCU is required for oxidative stress-induced mitochondrial calcium uptake. A, knockdown of IP3R blocked the lethality induced by MCU overexpression in muscle. Genotypes are as follows: Mef-Gal4/+, UAS-MCU-FLAG/+;Mef-Gal4/+, Mef-Gal4/UAS-IP3R RNAi, and UAS-MCU-FLAG/+;Mef-Gal4/UAS-IP3R RNAi. B and C, averaged traces (B) and quantitative analysis (C) of [Ca2+]mito signals from the flies that express IP3R RNAi, Drosophila MCU, or Drosophila MCU and IP3R RNAi upon treatment with 35 mm t-BuOOH (n = 5–9). Genotypes are as follows: Mef-Gal4,UAS-MTRP/+ (Mef>), Mef-Gal4,UAS-MTRP/UAS-IP3R RNAi (Mef> IP3R RNAi), UAS-MCU-FLAG/+;Mef-Gal4,UAS-MTRP/+ (Mef>MCU), and UAS-MCU-FLAG/+;Mef-Gal4, UAS-MTRP/UAS-IP3R RNAi (Mef>MCU, IP3R RNAi). D and E, averaged traces (D) and quantitative analysis (E) of [Ca2+]mito signals from S2 sells transfected with luciferase dsRNA or IP3R dsRNA upon treatment with 3 mm H2O2 (n = 4). F, eye-specific expression of transgenes using Gmr-Gal4. In clockwise order from the top left panel, genotypes are as follows: Gmr-Gal4/+, Gmr-Gal4/UAS-Sod1, Gmr-Gal4/UAS-Sod1 RNAi, Gmr-Gal4/UAS-Sod1 RNAi;UAS-MCU-Myc/+, Gmr-Gal4/UAS-Sod1;UAS-MCU-Myc/+, and Gmr-Gal4/+;UAS-MCU-Myc/+. G, genetic interactions among MCU, IP3R, and Sod1. Genotypes are as follows from left to right: UAS-Sod1 RNAi/+;Mef-Gal4/+, UAS-MCU-FLAG/UAS-Sod1 RNAi;Mef-Gal4/+, UAS-MCU-FLAG/+;Mef-Gal4/UAS-IP3R RNAi, and UAS-MCU-FLAG/UAS-Sod1 RNAi;Mef-Gal4/UAS-IP3R RNAi. ***, p < 0.001. Error bars, S.D.
Finally, we examined the involvement of IP3R and MCU in oxidative stress-induced toxicity using transgenic flies that overexpress or silence superoxide dismutase 1 (Sod1). First, the degenerated eye phenotype in the flies expressing MCU using Gmr-Gal4 driver, as shown in Fig. 3A, was restored by Sod1 expression but was exacerbated by Sod1 knockdown (Fig. 6F). As stated above, the lethality of MCU overexpression using Mef-Gal4 was prevented by the knockdown of IP3R (Fig. 6A). However, interestingly, simultaneous knockdown of both Sod1 and IP3R failed to rescue the lethal phenotype of MCU overexpression using Mef-Gal4 (Fig. 6G). These results suggest that endogenous ROS plays an imperative role in the IP3R- and MCU-mediated mitochondrial Ca2+ overload and toxicity.
Discussion
In this study, we established a Drosophila model system to understand functional roles of MCU in mitochondrial Ca2+ homeostasis in vivo. By generating and characterizing a null mutant of MCU (MCU52), we investigated the physiological roles of MCU in Drosophila. MCU52 mutant did not show significant changes in body weight, metabolism, and autophagic flux compared with wild-type flies. However, caffeine-induced [Ca2+]mito increase was abolished in MCU52 mutant larval muscle, which was completely rescued by transgenic expression of either human or Drosophila MCU. In addition, the DIME amino acid motif, which forms the ion selectivity filter of the MCU channel, was indispensable for the activity of Drosophila MCU, suggesting that MCU is evolutionarily highly conserved. In MCU52 mutant larval muscle and MCU-silenced S2 cells, exogenous ROS-induced [Ca2+]mito increase, ΔΨmito dissipation, and cell death were prevented. Suppression of IP3R, which is a Ca2+ release channel in the ER, also protects from ROS-mediated mitochondrial Ca2+ overload and cytotoxicity. These results demonstrate the critical role of Drosophila MCU in Ca2+ transfer from the ER to mitochondria, contributing to oxidative stress-induced mitochondrial dysfunction and apoptotic cell death.
Mitochondrial Ca2+ is a crucial regulator in energy metabolism, Ca2+ sequestration, and cell death. However, our Drosophila MCU loss-of-function mutant did not exhibit significant metabolic phenotypes. These unexpected results can be explained by undefined compensatory mechanisms for mitochondrial Ca2+ uptake, such as MCU-independent slow Ca2+ channels or exchangers. It is also conceivable that rapid changes in [Ca2+]mito via MCU may not be required for maintaining basal metabolism and daily activities in Drosophila except for exogenous stress or emergency crisis. Mouse MCU knock-out models with outbred CD1 background also did not show any significant phenotypes except for reduced abilities to perform strenuous work (20). However, strangely, MCU deletion within a C57BL/6 background resulted in embryonic lethality (42). This discrepancy suggests that a compensatory mechanism that is absent in C57BL/6 background exist in CD1 mice and allows MCU-independent mitochondrial Ca2+ uptake during animal development. By contrast, up-regulation of MCU augments mitochondrial Ca2+ uptake, leading to aberrantly high [Ca2+]mito level, and this stress accelerates further superoxide production through activation of the electron transport chain or other mechanisms. Together with increased [Ca2+]mito and oxidative stress in mitochondrial matrix, this facilitates opening of mPT and cytochrome c release, leading to apoptotic cell death. In our study, ectopic overexpression of MCU in Drosophila muscle led to pupal lethality (Fig. 6A). Additionally, MCU overexpression in Drosophila eye using Gmr-Gal4 driver resulted in severely destroyed ommatidial array (Fig. 3A). These results imply that overexpression of Drosophila MCU accelerates mitochondrial Ca2+ overload that is detrimental to various tissues and ultimately impairs a viability of organism.
EMRE is an essential auxiliary subunit containing a mitochondrial targeting sequence at its N terminus, a single transmembrane domain located at inner mitochondrial membrane, and an aspartate-rich C terminus (12). The MCU complex requires EMRE for its reconstitution in mammalian cells, but not in Dictyostelium discoideum (43). In mammalian cells, suppression of EMRE abrogates MCU-mediated Ca2+ currents demonstrated by mitoplast patch clamp experiment (12, 14). However, functional consequences of EMRE overexpression have not been studied yet (44). In our study, EMRE showed a strong genetic interaction with MCU in Drosophila. Co-expression of Drosophila EMRE and MCU in fly muscle led to lethality in larva stage. Furthermore, depletion of EMRE in larval muscle abolished caffeine-induced [Ca2+]mito increase regardless of the expression level of MCU, indicating that EMRE is required for MCU activity from human to Drosophila.
Recent studies reported that MCU is involved in Ca2+ excitotoxicity of cortical neurons (26) and oxidative stress-induced cell death of primary cerebellar granule neurons (45). Oxidative stress is proposed as the main causative factor for neurodegenerative, cardiovascular, and other mitochondria-related diseases (46). Previous studies reported that oxidative stress increases [Ca2+]i (47–49), although there are controversies whether the source of Ca2+ is from the extracellular environment or intracellular stores. We observed marked and sustained [Ca2+]mito rises by exogenous oxidative stress inducers, such as H2O2 and t-BuOOH in Drosophila, which have not been investigated previously. Oxidative stress produced by endoplasmic reticulum oxidase 1α (ERO1α) can stimulate Ca2+ release from the ER by activating IP3R (33). Loss of Ca2+ store in the ER by ROS induces ER stress due to impaired Ca2+-sensitive chaperone activities and further ROS production by induction of C/EBP homologous protein (33). In addition, depletion of Ca2+ in the ER stimulates Ca2+ influx from outside of the cell via store-operated Ca2+ entry (50). Both Ca2+ release from the ER and influx from extracellular environment burden cells with the pathology of mitochondrial Ca2+ overload.
Our study clearly showed that loss-of-function mutation of MCU abrogated [Ca2+]mito increases triggered by t-BuOOH in fly muscle. Moreover, knockdown of IP3R also significantly attenuated oxidative stress-induced [Ca2+]mito rises, which explains the protective roles of IP3R RNAi against lethality in MCU-overexpressed flies (Fig. 6A). Oxidative stress can increase ER Ca2+ release by activating not only IP3R but also ryanodine receptor (RyR), both of which are main Ca2+ release channels in the ER (33, 51). Although it has been known that RyR plays more important roles than IP3R in Ca2+ release from the sarcoplasmic reticulum in skeletal muscle, the muscle tissues from larva, pupa, and adult Drosophila express IP3R, which is critical for the muscle development (52). In this study, we could not investigate the interaction between RyR and MCU for oxidative stress-induced mitochondrial Ca2+ overload and lethality because all of the flies with muscle-specific knockdown of RyR died before larval stage. However, we still consider that RyR is another strong candidate involved in mitochondrial Ca2+ overload by oxidative stress in muscle.
In summary, we have established a Drosophila system to study the MCU complex in vivo, demonstrating that oxidative stress induces Ca2+ release from the ER and mitochondrial Ca2+ uptake via MCU, resulting in mitochondrial dysfunction and cell death in vivo. Intriguingly, a recent study showed that ROS modifies MCU directly by S-glutationylation and consequently influences the channel activity of MCU (53). These pieces of evidence indicate both direct and indirect regulation of the Ca2+ channel activity of MCU by ROS. Further genetic studies would enable us to discover novel relationships connecting mitochondrial Ca2+ homeostasis and other cellular activities. In addition, studying the in vivo role of MCU and its related genes in Drosophila will further extend our knowledge of their pathophysiological significance.
Experimental procedures
Fly strains
MCU52 mutant and a revertant were generated using P-element excision of the GS11565 line obtained from the Kyoto Stock Center (Kyoto Institute of Technology, Kyoto, Japan). The revertant with a precise P-element excision was used as a wild-type control. The P-element excisions in MCU52 and the revertant flies were confirmed by PCR using a primer set of the following sequences: 5′-GACGGAATTGCGATGGAAAATC-3′ and 5′-GCCAAAAATCCCATTCTAGTG-3′. The MCU cDNA clone LD26402 and the EMRE cDNA clone RE55001 were purchased from the Drosophila Genomics Resource Center (Indiana University, Bloomington, IN). UAS-MCU-FLAG, UAS-hMCU-FLAG, UAS-MCUNIMQ-FLAG, and UAS-EMRE-FLAG were generated by microinjection of pUAST vector-cloned DNA into w1118 embryos. Cg-Gal4, Mef-Gal4, Gmr-Gal4, and UAS-GCaMP3.T were obtained from the Bloomington Drosophila Stock Center (Indiana University, Bloomington, IN). UAS-MCU RNAi (v9501), UAS-IP3R RNAi (v6484), and UAS-EMRE RNAi (v104493) were provided by the Vienna Drosophila RNAi Center (Vienna, Austria). Other flies were provided with generosity: UAS-MTRP from Dr. G. T. Macleod (University of Texas Health Science Center, San Antonio, TX); UAS-mCherry-ATG8a from Dr. T. P. Neufeld (University of Minnesota, Minneapolis, MN); UAS-MCU-Myc from Dr. S. B. Lee (DGIST, Daegu, Korea); and hid5′F-WT-GFP from Dr. W. Du (University of Chicago).
Quantitative RT-PCR
Wandering larvae were collected, and total RNA was extracted using TRIzol reagent (Invitrogen). Extracted RNAs were reverse transcribed by Moloney murine leukemia virus reverse transcriptase (Promega) and amplified by PCR with primer sets of the following sequences: 5′-GTCTCGCCCTGCGTTTGG-3′ and 5′-CGAAGCTTCTGTGCTGCTG-3′ for MCU and 5′-GCGCTTCTTGGAGGAGACGCCG-3′ and 5′-GCTTCAACATGACCATCCGCCC-3′ for RP49. MCU mRNA levels were normalized by RP49 mRNA levels.
Immunoblotting analysis
For immunoblot samples, tissues were homogenized in ice-cold lysis buffer (20 mm Tris-Cl, pH 7.5, 100 mm NaCl, 1 mm EDTA, 2 mm EGTA, 1 mm Na2VO4, 50 mm β-glycerol phosphate, 50 mm NaF, and 1% Triton X-100). Homogenized samples were incubated in ice for 15 min, centrifuged, and denatured. Rabbit anti-FLAG antibody (Cell Signaling Technology), mouse anti-Drosophila MCU polyclonal antibody (epitope: EDGETDKHKKPTTG) (AbClon), mouse anti-β tubulin antibody (DSHB), and anti-porin antibody (54) were used in immunoblot analyses.
Food intake assay
To measure feeding activity for 24 h, the food intake assay was conducted as described previously (55).
Measurement of trehalose and glucose
Trehalose and glucose in fly body fluid were measured as described previously (56). For each genotype, hemolymph from 5–7 wandering larvae was extracted by tearing up the cuticles. 1 μl of hemolymph was diluted with 99 μl of trehalase buffer (5 mm Tris, pH 6.6, 137 mm NaCl, and 2.7 mm KCl) and incubated at 70 °C for 5 min. Then 40 μl of diluted hemolymph was mixed with either 40 μl of trehalase buffer or 40 μl of trehalase solution. Trehalase solution was prepared by diluting 3 μl of porcine trehalase (Sigma) in 1 ml of trehalase buffer. Samples were incubated at 37 °C overnight, and glucose levels were measured using a glucose assay kit (Sigma-Aldrich).
Starvation-induced autophagy assay
Third instar larvae before wandering stage were rinsed with PBS and either starved in a double-distilled water–containing Petri dish or fed in a food-containing vial for 4 h. Then larvae were dissected and fixed in 4% paraformaldehyde. mCherry-ATG8a signals in larval fat body were observed under a confocal microscope.
Survival curves on H2O2-containing food
120–130 3–5-day-old males were starved 4–6 h in double-distilled water–containing vials and transferred to vials containing either 1% H2O2 or 5% sucrose. Surviving flies were counted every 12 h.
TUNEL assay
To detect H2O2-induced cell death, wandering larvae were dissected and incubated for 3 h in Schneider's medium containing 2% H2O2. Dissected larvae were fixed in 4% paraformaldehyde (PFA) and washed with PBS. Samples were incubated in 0.1 m sodium citrate at 65 °C, and cell death was detected using an in situ cell death detection kit (Roche Applied Science). After TUNEL reaction, the samples were stained by phalloidin and Hoechst to detect filamentous actin and nucleus, respectively. To detect in S2 cells, S2 cells were incubated for 6 h in Schneider's medium-containing 100 mm H2O2. After fixation with 4% PFA for 15 min and washing with PBS, cells were incubated in 0.1 m sodium citrate at room temperature for 10 min, and apoptosis was detected by using an in situ cell death detection kit (Roche Applied Science).
Immunostaining
Third instar larvae were dissected and incubated for 3 h in Schneider's medium containing 2% H2O2. Then samples were fixed in 4% PFA PBST (0.1% Triton X-100 in PBS) for 30 min and washed with PBST. The samples were blocked with 5% FBS, 0.5% BSA PBST (blocking buffer) for 1 h and then incubated with primary antibodies overnight at 4 °C. After three washes for 10 min in blocking buffer, the samples were treated with secondary antibodies and Hoechst 33258 (Invitrogen) in blocking buffer for 45 min. After extensive washes, samples were placed in mounting medium. Primary antibodies used in this study were as follows: anti-GFP rabbit monoclonal antibody (1:500; Thermo Fisher Scientific, A11222) and anti-cleaved caspase-3 polyclonal antibody (1:200; Cell Signaling Technology, 9661). Alexa Fluor 568 streptavidin (Thermo Fisher Scientific) and anti-ATP5a mouse monoclonal antibody (Abcam, ab14748) were used to detect mitochondria. Fluorescein-conjugated F(ab′)2 fragment goat anti-rabbit IgG (Jackson ImmunoResearch, 111-096-144) and rhodamine red-X–conjugated F(ab′)2 fragment goat anti-mouse IgG (Jackson ImmunoResearch, 115-296-146) were used as secondary antibodies.
DHE staining
For ROS detection, adult fly thoraces were dissected in Schneider's medium, incubated for 5 min with 30 μm DHE in the same medium, washed twice, fixed slightly with 4% PFA for 8 min, and rinsed with PBS.
Drosophila S2 cell culture and dsRNA bathing
Drosophila S2-DRSC cells were cultured and dsRNA bathing was conducted as described previously (57). For silencing mRNA expression of MCU and IP3R, dsRNA was synthesized and bathed. The following primers were used for dsRNA synthesis: MCU dsRNA, 5′-TAATACGACTCACTATAGGGTGGAGGATGTGAAGAATCGC-3′ and 5′-TAATACGACTCACTATAGGGTGAATCGTCCAACTGTGGCT-3′; IP3RdsRNA, 5′-TAATACGACTCACTATAGGGCGTTGCTTTATCCTTTGCCA-3′ and 5′-TAATACGACTCACTATAGGGCCGCATAGAGGGACACAATG-3′; luciferase dsRNA, 5′-TAATACGACTCACTATAGGGAGAGGCCCGGCGCCATTCTATC-3′ and 5′-TAATACGACTCACTATAGGGAGAGATTGGGAGCTTTTTTTGCACG-3′. After 4 days of dsRNA bathing, S2 cells were used for a TUNEL assay or fluorescence measurement.
Measurement of mitochondrial matrix or cytosolic calcium
To measure [Ca2+]mito in larval muscle, MTRP was expressed by Mef-Gal4 and UAS-MTRP. Wandering third instar larvae were dissected as described previously (58) with some modifications. Larvae were dissected in perfusion buffer (2 mm CaCl2, 4 mm MgCl2, 2 mm KCl, 2 mm NaCl, 5 mm HEPES, 35.5 mm sucrose, 7 mm l-glutamic acid, pH 7.3, with NaOH) on a stereomicroscope and transferred to a confocal microscope. Transferred larvae were perfused with the same buffer, and fluorescence images were acquired by using an inverted microscope (IX-81, Olympus, Tokyo, Japan) with an array laser confocal spinning disk (CSU10, Yokogawa Electric Corp., Tokyo, Japan) and a cooled charge-coupled device camera (Cascade 512B, Photometrics, Tucson, AZ). Acquired fluorescence images from 435-nm excitation (Ex) and 535-nm emission (Em) were analyzed using Metafluor 6.3 software (Universal Imaging, Molecular Devices) (11). Cytosolic Ca2+ level ([Ca2+]i) was measured by using the confocal system (488-nm Ex/535-nm Em) in GCaMP3-expressed larval muscle (59). To measure [Ca2+]mito in S2 cells, we used a mitochondria-targeted ratio-pericam plasmid (RPmit4.1), generously provided by Dr. Roger Tsien (University of California, San Diego, CA). Cells were transfected with siRNA for 24 h and then transfected with RPmit4.1 using X-tremeGENE (Roche Diagnostics GmbH, Mannheim, Germany). After 48 h of RPmit4.1 transfection, cells were perfused with KRB solution (140 mm NaCl, 3.6 mm KCl, 0.5 mm NaH2PO4, 0.5 mm MgSO4, 1.5 mm CaCl2, 10 mm HEPES, 2 mm NaHCO3, 5.5 mm glucose, pH 7.4, titrated with NaOH), and fluorescence images (435-nm Ex/535-nm Em) were acquired.
Calculation of [Ca2+]mito increases
In every scattered plot, the y axis represents area under the curve of the [Ca2+]mito imaging result under caffeine, t-BuOOH, or H2O2 treatment.
Measurement of mitochondrial membrane potential
To measure the mitochondrial membrane potential (Ψmito), S2 cells seeded onto black-walled 96-well plates (1.2 × 105 cells/well) were loaded with a lipophilic cationic dye, JC-1 (1.5 μm), for 30 min. Cells were washed with KRB solution, and JC-1 fluorescence intensities of red (540-nm Ex/590-nm Em; J-aggregates) and green (490-nm Ex/540-nm Em; monomer) were measured ratiometrically at room temperature using a multiwell fluorescence reader (Flex-Station, Molecular Devices) (60).
Statistics
Values are presented as mean ± S.D., and n is the number of independent experiments. p values were obtained by Student's t test or one-way analysis of variance, and <0.05 was considered to be significant.
Author contributions
S. C., K.-S. P., and J. C. conceived and designed the experiments; S. C., X. Q., S. B., H. Y., and J. K. performed the experiments; K.-S. P. and J. C. analyzed the data; and S. C., J. K., S. B., J. P., K.-S. P., and J. C. wrote the paper.
Supplementary Material
Acknowledgments
We thank Drs. Gregory T. Macleod, Thomas P. Neufeld, Sung-Bae Lee, and Wei Du for providing Drosophila stocks. We are grateful to the Bloomington Stock Center and the Vienna Drosophila RNAi Center.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5.
- ROS
- reactive oxygen species
- MCU
- mitochondrial calcium uniporter
- IP3R
- inositol 1,4,5-trisphosphate receptor
- mPT
- mitochondrial permeability transition
- MAM
- mitochondria-associated ER membrane
- [Ca2+]mito
- mitochondrial matrix Ca2+ concentration
- MTRP
- mitochondria-targeted ratio-pericam
- t-BuOOH
- tert-butyl hydroperoxide
- Ψmito
- mitochondrial membrane potential
- MCU52
- a null mutant of MCU
- RPmit4.1
- mitochondria-targeted ratio-pericam plasmid
- ER
- endoplasmic reticulum
- DHE
- dihydroethidium
- RyR
- ryanodine receptor
- PFA
- paraformaldehyde
- Ex
- excitation
- Em
- emission.
References
- 1. Kirichok Y., Krapivinsky G., and Clapham D. E. (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364 [DOI] [PubMed] [Google Scholar]
- 2. Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., Bao X. R., Strittmatter L., Goldberger O., Bogorad R. L., Koteliansky V., and Mootha V. K. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Stefani D., Raffaello A., Teardo E., Szabò I., and Rizzuto R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oxenoid K., Dong Y., Cao C., Cui T., Sancak Y., Markhard A. L., Grabarek Z., Kong L., Liu Z., Ouyang B., Cong Y., Mootha V. K., and Chou J. J. (2016) Architecture of the mitochondrial calcium uniporter. Nature 533, 269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee S. K., Shanmughapriya S., Mok M. C. Y., Dong Z., Tomar D., Carvalho E., Rajan S., Junop M. S., Madesh M., and Stathopulos P. B. (2016) Structural insights into mitochondrial calcium uniporter regulation by divalent cations. Cell Chem. Biol. 23, 1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee Y., Min C. K., Kim T. G., Song H. K., Lim Y., Kim D., Shin K., Kang M., Kang J. Y., Youn H. S., Lee J. G., An J. Y., Park K. R., Lim J. J., Kim J. H., et al. (2015) Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. 16, 1318–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Stefani D., Rizzuto R., and Pozzan T. (2016) Enjoy the trip: calcium in mitochondria back and forth. Annu. Rev. Biochem. 85, 161–192 [DOI] [PubMed] [Google Scholar]
- 8. De Marchi U., Santo-Domingo J., Castelbou C., Sekler I., Wiederkehr A., and Demaurex N. (2014) NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 289, 20377–20385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nowikovsky K., Pozzan T., Rizzuto R., Scorrano L., and Bernardi P. (2012) Perspectives on: SGP symposium on mitochondrial physiology and medicine: the pathophysiology of LETM1. J. Gen. Physiol. 139, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pendin D., Greotti E., and Pozzan T. (2014) The elusive importance of being a mitochondrial Ca2+ uniporter. Cell Calcium 55, 139–145 [DOI] [PubMed] [Google Scholar]
- 11. Quan X., Nguyen T. T., Choi S. K., Xu S., Das R., Cha S. K., Kim N., Han J., Wiederkehr A., Wollheim C. B., and Park K. S. (2015) Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 290, 4086–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sancak Y., Markhard A. L., Kitami T., Kovács-Bogdan E., Kamer K. J., Udeshi N. D., Carr S. A., Chaudhuri D., Clapham D. E., Li A. A., Calvo S. E., Goldberger O., and Mootha V. K. (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsai M. F., Phillips C. B., Ranaghan M., Tsai C. W., Wu Y., Willliams C., and Miller C. (2016) Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 10.7554/eLife.15545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vais H., Mallilankaraman K., Mak D. O., Hoff H., Payne R., Tanis J. E., and Foskett J. K. (2016) EMRE is a matrix Ca2+ sensor that governs gatekeeping of the mitochondrial Ca2+ uniporter. Cell Rep. 14, 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Csordás G., Golenár T., Seifert E. L., Kamer K. J., Sancak Y., Perocchi F., Moffat C., Weaver D., de la Fuente Perez S., Bogorad R., Koteliansky V., Adijanto J., Mootha V. K., and Hajnóczky G. (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 17, 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Plovanich M., Bogorad R. L., Sancak Y., Kamer K. J., Strittmatter L., Li A. A., Girgis H. S., Kuchimanchi S., De Groot J., Speciner L., Taneja N., Oshea J., Koteliansky V., and Mootha V. K. (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8, e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perocchi F., Gohil V. M., Girgis H. S., Bao X. R., McCombs J. E., Palmer A. E., and Mootha V. K. (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mallilankaraman K., Cárdenas C., Doonan P. J., Chandramoorthy H. C., Irrinki K. M., Golenár T., Csordás G., Madireddi P., Yang J., Müller M., Miller R., Kolesar J. E., Molgó J., Kaufman B., Hajnóczky G., et al. (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chaudhuri D., Artiga D. J., Abiria S. A., and Clapham D. E. (2016) Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. U.S.A. 113, E1872–E1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan X., Liu J., Nguyen T., Liu C., Sun J., Teng Y., Fergusson M. M., Rovira I. I., Allen M., Springer D. A., Aponte A. M., Gucek M., Balaban R. S., Murphy E., and Finkel T. (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu Y., Rasmussen T. P., Koval O. M., Joiner M. L., Hall D. D., Chen B., Luczak E. D., Wang Q., Rokita A. G., Wehrens X. H., Song L. S., and Anderson M. E. (2015) The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 6, 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu S., and Chisholm A. D. (2014) C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev. Cell 31, 48–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mammucari C., Gherardi G., Zamparo I., Raffaello A., Boncompagni S., Chemello F., Cagnin S., Braga A., Zanin S., Pallafacchina G., Zentilin L., Sandri M., De Stefani D., Protasi F., Lanfranchi G., and Rizzuto R. (2015) The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 10, 1269–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tarasov A. I., Semplici F., Ravier M. A., Bellomo E. A., Pullen T. J., Gilon P., Sekler I., Rizzuto R., and Rutter G. A. (2012) The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One 7, e39722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gouriou Y., Bijlenga P., and Demaurex N. (2013) Mitochondrial Ca2+ uptake from plasma membrane Cav3.2 protein channels contributes to ischemic toxicity in PC12 cells. J. Biol. Chem. 288, 12459–12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qiu J., Tan Y. W., Hagenston A. M., Martel M. A., Kneisel N., Skehel P. A., Wyllie D. J., Bading H., and Hardingham G. E. (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 4, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rasmussen T. P., Wu Y., Joiner M. L., Koval O. M., Wilson N. R., Luczak E. D., Wang Q., Chen B., Gao Z., Zhu Z., Wagner B. A., Soto J., McCormick M. L., Kutschke W., Weiss R. M., et al. (2015) Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. U.S.A. 112, 9129–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rizzuto R., De Stefani D., Raffaello A., and Mammucari C. (2012) Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578 [DOI] [PubMed] [Google Scholar]
- 29. Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., and Hajnóczky G. (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tubbs E., Theurey P., Vial G., Bendridi N., Bravard A., Chauvin M. A., Ji-Cao J., Zoulim F., Bartosch B., Ovize M., Vidal H., and Rieusset J. (2014) Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294 [DOI] [PubMed] [Google Scholar]
- 31. Szabadkai G., Bianchi K., Várnai P., De Stefani D., Wieckowski M. R., Cavagna D., Nagy A. I., Balla T., and Rizzuto R. (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arruda A. P., and Hotamisligil G. S. (2015) Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 22, 381–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li G., Mongillo M., Chin K. T., Harding H., Ron D., Marks A. R., and Tabas I. (2009) Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186, 783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bánsághi S., Golenár T., Madesh M., Csordás G., RamachandraRao S., Sharma K., Yule D. I., Joseph S. K., and Hajnóczky G. (2014) Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 289, 8170–8181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin M. T., and Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 [DOI] [PubMed] [Google Scholar]
- 36. Lye C. M., Naylor H. W., and Sanson B. (2014) Subcellular localisations of the CPTI collection of YFP-tagged proteins in Drosophila embryos. Development 141, 4006–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drago I., and Davis R. L. (2016) Inhibiting the mitochondrial calcium uniporter during development impairs memory in adult Drosophila. Cell Rep. 16, 2763–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagai T., Sawano A., Park E. S., and Miyawaki A. (2001) Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. U.S.A. 98, 3197–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patron M., Checchetto V., Raffaello A., Teardo E., Vecellio Reane D., Mantoan M., Granatiero V., Szabò I., De Stefani D., and Rizzuto R. (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raffaello A., De Stefani D., Sabbadin D., Teardo E., Merli G., Picard A., Checchetto V., Moro S., Szabò I., and Rizzuto R. (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tanaka-Matakatsu M., Xu J., Cheng L., and Du W. (2009) Regulation of apoptosis of rbf mutant cells during Drosophila development. Dev. Biol. 326, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murphy E., Pan X., Nguyen T., Liu J., Holmström K. M., and Finkel T. (2014) Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 449, 384–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kovács-Bogdán E., Sancak Y., Kamer K. J., Plovanich M., Jambhekar A., Huber R. J., Myre M. A., Blower M. D., and Mootha V. K. (2014) Reconstitution of the mitochondrial calcium uniporter in yeast. Proc. Natl. Acad. Sci. U.S.A. 111, 8985–8990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jhun B. S., Mishra J., Monaco S., Fu D., Jiang W., Sheu S. S., and O-Uchi J. (2016) The mitochondrial Ca2+ uniporter: regulation by auxiliary subunits and signal transduction pathways. Am. J. Physiol. Cell Physiol. 311, C67–C80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liao Y., Hao Y., Chen H., He Q., Yuan Z., and Cheng J. (2015) Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 6, 434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Giorgi C., Agnoletto C., Bononi A., Bonora M., De Marchi E., Marchi S., Missiroli S., Patergnani S., Poletti F., Rimessi A., Suski J. M., Wieckowski M. R., and Pinton P. (2012) Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 12, 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Doan T. N., Gentry D. L., Taylor A. A., and Elliott S. J. (1994) Hydrogen peroxide activates agonist-sensitive Ca2+-flux pathways in canine venous endothelial cells. Biochem. J. 297, 209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Renard D. C., Seitz M. B., and Thomas A. P. (1992) Oxidized glutathione causes sensitization of calcium release to inositol 1,4,5-trisphosphate in permeabilized hepatocytes. Biochem. J. 284, 507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roveri A., Coassin M., Maiorino M., Zamburlini A., van Amsterdam F. T., Ratti E., and Ursini F. (1992) Effect of hydrogen peroxide on calcium homeostasis in smooth muscle cells. Arch. Biochem. Biophys. 297, 265–270 [DOI] [PubMed] [Google Scholar]
- 50. Xu S., Nam S. M., Kim J. H., Das R., Choi S. K., Nguyen T. T., Quan X., Choi S. J., Chung C. H., Lee E. Y., Lee I. K., Wiederkehr A., Wollheim C. B., Cha S. K., and Park K. S. (2015) Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 6, e1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Terentyev D., Györke I., Belevych A. E., Terentyeva R., Sridhar A., Nishijima Y., de Blanco E. C., Khanna S., Sen C. K., Cardounel A. J., Carnes C. A., and Györke S. (2008) Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103, 1466–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Raghu P., and Hasan G. (1995) The inositol 1,4,5-triphosphate receptor expression in Drosophila suggests a role for IP3 signalling in muscle development and adult chemosensory functions. Dev. Biol. 171, 564–577 [DOI] [PubMed] [Google Scholar]
- 53. Dong Z., Shanmughapriya S., Tomar D., Siddiqui N., Lynch S., Nemani N., Breves S. L., Zhang X., Tripathi A., Palaniappan P., Riitano M. F., Worth A. M., Seelam A., Carvalho E., Subbiah R., et al. (2017) Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol. Cell 65, 1014–1028 e1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park J., Kim Y., Choi S., Koh H., Lee S. H., Kim J. M., and Chung J. (2010) Drosophila Porin/VDAC affects mitochondrial morphology. PLoS One 5, e13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Min S., Chae H. S., Jang Y. H., Choi S., Lee S., Jeong Y. T., Jones W. D., Moon S. J., Kim Y. J., and Chung J. (2016) Identification of a peptidergic pathway critical to satiety responses in Drosophila. Curr. Biol. 26, 814–820 [DOI] [PubMed] [Google Scholar]
- 56. Tennessen J. M., Barry W. E., Cox J., and Thummel C. S. (2014) Methods for studying metabolism in Drosophila. Methods 68, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim W., Kim H. D., Jung Y., Kim J., and Chung J. (2015) Drosophila low temperature viability protein 1 (LTV1) is required for ribosome biogenesis and cell growth downstream of Drosophila Myc (dMyc). J. Biol. Chem. 290, 13591–13604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Budnik V., Gorczyca M., and Prokop A. (2006) Selected methods for the anatomical study of Drosophila embryonic and larval neuromuscular junctions. Int. Rev. Neurobiol. 75, 323–365 [DOI] [PubMed] [Google Scholar]
- 59. Tian L., Hires S. A., Mao T., Huber D., Chiappe M. E., Chalasani S. H., Petreanu L., Akerboom J., McKinney S. A., Schreiter E. R., Bargmann C. I., Jayaraman V., Svoboda K., and Looger L. L. (2009) Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Park K. S., Wiederkehr A., Kirkpatrick C., Mattenberger Y., Martinou J. C., Marchetti P., Demaurex N., and Wollheim C. B. (2008) Selective actions of mitochondrial fission/fusion genes on metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 283, 33347–33356 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.