

Abstract
Macrophages use various cell-surface receptors to sense their environment and undergo polarized responses. The cytokines, interleukin (IL)-4 and IL-13, released from T-helper type 2 (Th2) cells, drive macrophage polarization toward an alternatively activated phenotype (M2). This phenotype is associated with the expression of potent pro-resolving mediators, such as the prostaglandin (PG) D2-derived cyclopentenone metabolite, 15d-PGJ2, produced by the cyclooxygenase (Ptgs; Cox) pathway. Interestingly, IL-4 treatment of bone marrow-derived macrophages (BMDMs) significantly down-regulates Cox-2 protein expression, whereas Cox-1 levels are significantly increased. This phenomenon not only challenges the dogma that Cox-1 is only developmentally regulated, but also demonstrates a novel mechanism in which IL-4-dependent regulation of Cox-1 involves the activation of the mechanistic target of rapamycin complex (mTORC). Using specific chemical inhibitors, we demonstrate here that IL-4-dependent Cox-1 up-regulation occurs at the post-transcriptional level via the Fes-Akt-mTORC axis. Activation of AMP-activated protein kinase (AMPK) by metformin, inhibition of mTORC by torin 1, or CRISPR/Cas9-mediated genetic knock-out of tuberous sclerosis complex-2 (Tsc2) blocked the IL-4-dependent expression of Cox-1 and the ability of macrophages to polarize to M2. However, use of 15d-PGJ2 partially rescued the effects of AMPK activation, suggesting the importance of Cox-1 in macrophage polarization as also observed in a model of gastrointestinal helminth clearance. In summary, these findings suggest a new paradigm where IL-4-dependent up-regulation of Cox-1 expression may play a key role in tissue homeostasis and wound healing during Th2-mediated immune responses, such as parasitic infections.
Keywords: metformin, mTOR complex (mTORC), parasite, post-transcriptional regulation, prostaglandin
Introduction
Macrophages utilize various cell-surface receptors to sense their environment and elicit a polarized response. Based on the stimuli, macrophages can be polarized to either a classically activated (M1) pro-inflammatory phenotype or an alternatively activated (M2) anti-inflammatory phenotype that corresponds to two ends of a spectrum with many intermediate phenotypes (1, 2). IL-4 or IL-13 released by T-helper type 2 (Th2)3 cell responses during allergies, asthma, or parasitic infection (as seen during helminth Nippostrongylus brasiliensis infection), activate the IL-4 receptor on macrophages to drive M2 polarization (3–6). Binding of IL-4 to its receptor leads to the recruitment of various tyrosine kinases, such as Janus kinases (Jak1–3) and Feline sarcoma oncogene kinase (Fes). Jak1 and Jak3 activation of signal transducer and activator of transcription-6 (Stat6) leads to nuclear translocation and transcriptional activation of M2 macrophage genes including peroxisome proliferator-activated receptor γ (PPAR-γ), arginase-1 (Arg-1), mannose receptor-1 (Mrc-1; CD206), and IL-10 (7).
In addition to signaling through the Jak/Stat pathway, IL-4 receptor engagement also up-regulates protein translation through the recruitment of Fes (8). Fes activates phosphatidylinositide 3-kinase (PI3K) leading to the generation of phosphatidylinostol (3,4,5)-triphosphate. 3-Phosphoinositide-dependent protein kinase-1 (Pdk-1) is activated by membrane phospholipids and is a major regulator of protein kinase B/Akt signaling (9, 10). Additionally, mTORC activity increases the phosphorylation of Akt. Akt inhibits tuberous sclerosis complex (TSC), activating mTORC. mTORC increases translation by activating ribosomal protein S6 kinase β-1 (p70S6K) and releasing elongation factor eIF4E from its inhibitor 4EBP. S6 kinase negatively feeds back to inhibit PI3K activity (11). Therefore Akt is both regulated and regulates mTORC signaling pathways. Finally, mTORC has been shown to play a critical role in M2 macrophage polarization in both peritoneal macrophages and BMDMs with IL-4 stimulation increasing the expression of Tsc1 and Tsc2 protein in stimulated macrophages (12).
Metformin, a commonly used anti-diabetic drug, increases AMP-activated protein kinase (AMPK) activity, which up-regulates the Tsc1/2 complex (13, 14). Treatment with metformin has been shown to significantly reduce the IL-13-induced expression of M2 macrophage markers, such as CD206 (14). However, little is known about how metformin treatment affects the production of endogenous ligands, Δ12-prostaglandin (PG) J2 and 15-deoxy-Δ-12,14-PGJ2 (15d-PGJ2) that are known to activate PPAR-γ and affect polarization of macrophages (15–18). This is particularly interesting because IL-4/IL-13 significantly down-regulate the expression of Cox-2 (19, 20). IL-4 receptor signaling converges with arachidonic acid metabolism during an inflammatory stimulus at PPAR-γ signaling to drive M2 macrophage polarization (21). Upon stimulation, arachidonic acid is metabolized by Cox-1 and Cox-2 in a two-step conversion to PGH2. PGH2 is converted to PGE2, PGI2, PGF2α, PGD2, and thromboxane (TxA2) by specific synthases that play various roles in pathophysiology (22–24). Although PGE2 and TxA2 exacerbate inflammation, PGD2, a product of hematopoietic PGD2 synthase (H-PGDS) and lipocalin-type PGD2 synthase (L-PGDS), mediates resolution of inflammation, primarily through two metabolites, Δ12-PGJ2 and 15d-PGJ2. More importantly, Cox-1 functionally couples with H-PGDS to form PGD2 and its downstream products (25, 26). Treatment with indomethacin, a nonselective Cox inhibitor, led to decreased M2 macrophage markers in vivo as well as increased N. brasiliensis burden, which was reversed by exogenous treatment with 15d-PGJ2 (17).
Even though Cox-1 (Ptgs-1) and Cox-2 (Ptgs-2) have a high degree of sequence homology (∼63%), they differ in many respects from gene structure to enzymatic activity. Cox-1 has been generally considered as a constitutive enzyme, with some exceptions such as up-regulation of transcription in fibroblasts by TGFβ and IL-1β and during cellular differentiation of monocytes with phorbol ester in THP-1 cells (27). Cox-2 is subjected to transcriptional and post-translational regulation by diverse stimuli (28–31). Although Ptgs-2 promoter contains many regulatory elements present in “early immediate genes,” the Ptgs-1 promoter is more reminiscent of “housekeeping” genes lacking TATA and CAAT boxes, high GC content, and multiple transcriptional start sites (TSS) (28–31). Additionally, Ptgs-2 mRNA, as well as protein, has a short half-life of 2–7 h, whereas Ptgs-1 mRNA and protein is relatively stable (29). The instability of Cox-2 protein is due to a 27-amino acid cassette at the C terminus, not present in Cox-1, which targets it for degradation through the endoplasmic reticulum-associated protein degradation system followed by ubiquitination and proteolysis by 26S proteasome (29, 32).
Here we describe for the first time the post-transcriptional regulation of Cox-1 expression by IL-4 in primary macrophages (both human and mouse) to be mediated through the Fes-Akt-mTORC axis rather than the Jak/Stat pathway. Activation of AMPK or direct inhibition of mTORC by torin 1 blocked the IL-4-dependent expression of Cox-1 and affected macrophage polarization toward the M2 phenotype. Use of 15d-PGJ2 partially rescued the effects of AMPK activation in mice infected with N. brasiliensis, suggesting the importance of Cox-1 in the polarization of macrophages. In summary, these studies suggest a novel paradigm where IL-4-dependent post-transcriptional control of Cox-1 expression may play a key role in tissue homeostasis and wound healing during Th2-mediated immune responses, such as parasitic infections.
Results
IL-4 stimulation increases the expression of Cox-1
To determine the effects of IL-4 stimulation on Cox expression, murine BMDMs were treated with or without recombinant IL-4 for 20 h prior to collection. Cells were collected and analyzed by real-time PCR and Western immunoblot for Ptgs-1 (Cox-1) and Ptgs-2 (Cox-2) expression, and by flow cytometry to quantify M2 macrophages (CD206+Arg-1+F480+CD11b+). In addition, lipids were extracted from cell culture media supernatants to quantify the production of 15d-PGJ2. Ptgs-1 transcript levels were unaffected by IL-4 treatment (Fig. 1A). Cox-1 protein levels were increased 2.5-fold with IL-4 stimulation compared with untreated control BMDMs (Fig. 1B). Interestingly, the Ptgs-2 transcript was unaffected (data not shown) and Cox-2 protein was not detected in either untreated control or IL-4-treated BMDMs by Western immunoblot (Fig. 1C). Additionally, there was no increase in mPges-1 (microsomal PGE2 synthase) or Txas-1 (thromboxane synthase) expression with IL-4 stimulation compared with untreated control BMDMs (supplemental Fig. S1, A and B). LC-MS/MS analysis of BMDM culture media supernatants showed a statistically significant increase in the production of 15d-PGJ2 20 h post-IL-4 treatment as compared with untreated control BMDMs (Fig. 1D). IL-4 stimulation did not lead to an increase in the production of PGE2 or TxB2 compared with untreated control BMDMs (supplemental Fig. S1, C and D). BMDMs treated with the TxAS inhibitor, ozagrel, where able to polarize toward an M2 phenotype in response to IL-4 at similar levels to DMSO-treated BMDMs (supplemental Fig. S1E). Treatment with ozagrel lead to a decrease in the production of TxB2 (supplemental Fig. 1F). Finally, IL-4 stimulation led to approximately a 10-fold increase in the expression of M2 macrophages that was previously shown to be sensitive to indomethacin (Fig. 1E) (17). Macrophages were first gated by their forward and side scatter characteristics followed by CD11b+F480+ cells, which consisted of ∼98% of the macrophage population, and were further gated on CD206+ and Arg-1+ (supplemental Fig. 2A). Furthermore, human macrophages differentiated from frozen peripheral blood mononuclear cells (PBMCs) (33) also showed an increase in the expression of COX-1 protein upon treatment with IL-4 as with murine BMDMs. Differentiated human macrophages were confirmed by flow cytometry to be ∼95% CD68+ (supplemental Fig. 2B). PBMCs from subject 1 and subject 2 had a 2.5- and 2.1-fold increase in COX-1 after IL-4 stimulation, respectively (Fig. 1F). This data confirmed a previously reported increase in COX-1 expression after IL-4 stimulation (34).
Figure 1.

IL-4 stimulation increased Cox-1 protein expression. A, real-time PCR of Ptgs-1 expression in murine BMDMs stimulated with or without IL-4 and 0.1% DMSO for 20 h. B, representative Western immunoblot of Cox-1 and Gapdh expression in murine BMDMs stimulated with or without IL-4 for 20 h. C, representative Western immunoblot of Cox-2 and Gapdh expression in murine BMDMs stimulated with or without IL-4 or LPS for 20 h (n = 1–2 biological replicates per group). D, 15d-PGJ2 production 20 h post IL-4 stimulation. E, percentage of CD206+Arg-1+F480+CD11b+ murine BMDMs stimulated with or without IL-4 relative to untreated control. F, Western immunoblots of COX-1 and GAPDH from human PBMC-derived macrophages stimulated with or without human IL-4. Unpaired two-tailed t test. **, p < 0.01; ****, p < 0.0001. n = 4–7 biological replicates per group. All experimental data are expressed as mean ± S.E.
Cox-1 is post-transcriptionally up-regulated by IL-4 stimulation
Based on the results above, to further examine whether regulation of Cox-1 expression was at the transcriptional or translational level, BMDMs were pre-treated with either actinomycin D or cycloheximide for 30 min or 4 h, respectively, prior to IL-4 stimulation. BMDMs treated with actinomycin D were collected 2 h post-IL-4 stimulation due to impact on cell viability. BMDMs treated with cycloheximide were collected 20 h post-IL-4 stimulation. Arg-1 expression, which is known to be transcriptionally regulated by IL-4 stimulation, was evaluated to determine the efficacy of the actinomycin D treatment. BMDMs treated with actinomycin D had a 4-fold decrease in Arg-1 expression compared with DMSO untreated control (Fig. 2A). Cox-1 protein expression was not down-regulated after actinomycin D treatment (Fig. 2, B and C). Strikingly, Cox-1 protein was completely absent in both untreated control and IL-4-treated cells compared with DMSO controls after cycloheximide treatment (Fig. 2D). Because Cox-1 appears to be translationally regulated, we reasoned that Ptgs-1 likely has a 5′ terminal oligopyrimidine (TOP)-like motif, which enables eIF4E to enhance translation. To this end, sequence analysis of PTGS-1 mRNA in both murine and human cells revealed the presence of TOP sequences (Fig. 2E). Interestingly, polysome profiling of murine BMDMs suggested that Ptgs-1 translation is modulated by mTORC signaling, based on a loss of Ptgs-1 mRNA from the heaviest polysome fractions (26% with IL-4 treatment) after torin 1 treatment (19% with IL-4 + torin 1 treatment), but is not mTORC exclusive (Fig. 2F). As a comparison, Rpl-13a, a bona fide TOP mRNA (35), revealed a classical polysome profile for a typical mTORC-dependent mRNA, where all polysome fractions were sensitive to torin 1 treatment (30% with IL-4 treatment to 14% with IL-4 + torin 1 treatment) (Fig. 2F).
Figure 2.

Cox-1 is post-transcriptionally up-regulated by IL-4. A, real-time PCR on Arg-1 expression in murine BMDMs stimulated with or without IL-4 and DMSO or actinomycin D. B, representative Western immunoblot for Cox-1 and Gapdh expression in murine BMDMs stimulated with or without IL-4 and DMSO or actinomycin D. C, protein density of Cox-1 relative to DMSO untreated control. D, representative western immunoblot for Cox-1 and Gapdh expression in murine BMDMs stimulated with or without IL-4 and DMSO or cycloheximide. E, bioinformatic analysis of TOP sequences in murine and human PTGS-1 mRNA. F, polysome profile on BMDMs stimulated with or without IL-4 and torin 1 for Ptgs-1 and Rpl-13a mRNA expression, n = 1 per group. Two-way ANOVA with Fisher's LSD test. *, p < 0.05; **, p < 0.01; ****, p < 0.0001. n = 2–4 biological replicates per group. All experimental data are expressed as mean ± S.E.
The role of Fes/Akt/mTORC signaling pathway in IL-4-dependent up-regulation of Cox-1 expression
The above data suggested that IL-4 treatment led to increased levels of Cox-1 protein. Therefore, we examined the underlying mechanisms downstream of the IL-4 receptor. BMDMs were treated with or without Jak inhibitor I 4 h prior to IL-4 stimulation. Inhibitor efficacy was confirmed by the significant decrease in the expression of the Jak-dependent genes Arg-1 and Cd36 (supplemental Fig. S3, A and B). Surprisingly, Cox-1 was up-regulated upon IL-4 treatment regardless of the presence or absence of Jak inhibitor I (supplemental Fig. S3, C and D). However, M2 macrophages were decreased by ∼25-fold compared with untreated control BMDMs (supplemental Fig. S3E).
Because IL-4-mediated expression of Cox-1 was not dependent on the Jak/Stat signaling pathway, we explored if the proto-oncogene protein tyrosine kinase Fes was key in transducing the signal downstream of the IL-4R to effect increased Cox-1 translation. Western immunoblot analysis of BMDMs confirmed the expression of Fes (supplemental Fig. 4A). Incubation of BMDMs with the Fes inhibitor, herbimycin A, followed by treatment with IL-4 significantly decreased expression of the Cox-1 protein in BMDMs (Fig. 3A).
Figure 3.

Cox-1 expression is controlled by the Fes/Akt/mTORC signaling pathway. A, representative Western immunoblot for Cox-1 and Gapdh expression in murine BMDMs stimulated with or without IL-4 and DMSO or herbimycin A. B, representative Western immunoblot for Cox-1 and Gapdh expression in murine BMDMs stimulated with or without IL-4 and DMSO or Akt inhibitor VIII. C, representative Western immunoblot for Cox-1 and Gapdh expression in murine BMDMs stimulated with or without IL-4 and DMSO or torin 1. D, production of 15d-PGJ2 from murine BMDMs stimulated with or without IL-4 (20 h) and DMSO or torin 1. E, percentage CD206+Arg-1+F480+CD11b+ murine BMDMs stimulated with or without IL-4 or torin 1 relative to DMSO untreated control. F, Western immunoblots of COX-1 and GAPDH from human PBMC-derived macrophages stimulated with or without human IL-4 and DMSO or torin 1. G, representative Western immunoblot for Cox-1 and Gapdh expression in Cas9 murine BMDMs transduced with either Ntc or Tsc2-specific CRISPR gRNA and stimulated with or without IL-4 (n = 1 per group). Two-way ANOVA with Fisher's LSD test. *, p < 0.05; **, p < 0.01; ****, p < 0.0001. n = 3–6 biological replicates per group. All experimental data are expressed as mean ± S.E.
Fes has been shown to signal through the Akt/mTORC pathway (8–10). To examine activation of the Akt/mTORC pathway, we first examined if PI3K was involved upstream of Akt. BMDMs were pretreated with the PI3K inhibitor, Ly294002, 4 h prior to IL-4 stimulation. Treatment of BMDMs with Ly294002 and IL-4 did not decrease Cox-1 protein (supplemental Fig. S4, B and C). BMDMs were next pre-treated with the Akt inhibitor VIII 4 h prior to IL-4 stimulation. Western immunoblot analysis clearly showed that Akt inhibitor VIII significantly decreased the IL-4-dependent expression of Cox-1 protein (Fig. 3B). Because polysome profiling suggested a loss of Ptgs-1 mRNA from the heaviest polysome fractions following torin 1 treatment of IL-4-stimulated BMDMs, we examined if torin 1 treatment also impacted Cox-1 expression at the protein level. BMDMs were pretreated with torin 1 4 h prior to IL-4 stimulation. Torin 1, completely inhibited Cox-1 protein expression in both untreated control and IL-4-treated BMDMs (Fig. 3C). Treatment with torin 1 also significantly inhibited the phosphorylation of ribosomal protein S6 kinase, which was slightly more phosphorylated in IL-4-treated BMDMs (supplemental Fig. 4D). Torin 1 significantly decreased the production of 15d-PGJ2 compared with DMSO + IL-4-treated cells (Fig. 3D). Torin 1 treatment also led to a significant decrease in IL-4-dependent skewing of BMDMs toward an M2 phenotype (Fig. 3E). Human PBMC-derived macrophages pre-treated with DMSO followed by IL-4 stimulation had a significant increase in COX-1, which was significantly decreased upon torin 1 pre-treatment (Fig. 3F). IL-4 stimulation of Ntc BMDMs led to a 2.8-fold increase in Cox-1 expression as seen throughout this study (Fig. 3G). Knock-out of Tsc2 in Cas9 BMDMs led to a 2.4-fold decrease in Cox-1 expression compared with Ntc Cas9 BMDMs (Fig. 3G). Tsc2 genomic mutation was confirmed by PCR followed by a surveyor assay (data not shown). This further confirmed the role of mTORC signaling in Cox-1 protein expression through use of the CRISPR/Cas9 gene editing system. Fluorescent microscopy confirmed >70% population purity of the Ntc CRISPR gRNA and Tsc2 CRISPR gRNA (mCherry+) into Cas9 GFP+ bone marrow (supplemental Fig. S5, A and B).
Activation of AMPK suppresses mTORC signaling and Cox-1 expression
We examined if metformin, a widely prescribed anti-diabetic drug that also activates AMPK, which inhibits mTORC, affected Cox-1 expression in BMDMs. We reasoned that if metformin affected Cox-1 expression, it would translate to adversely affecting the production of 15d-PGJ2 and consequent polarization of macrophages. BMDMs were treated with or without metformin 4 h prior to IL-4 stimulation (36). BMDMs treated with IL-4 following metformin pre-treatment had a decrease in Cox-1 expression (Fig. 4A). LC-MS/MS analysis of the culture media supernatants showed a decrease in 15d-PGJ2 production upon metformin pre-treatment and IL-4 stimulation (Fig. 4B). In addition, BMDMs treated with metformin and IL-4 had a slight but significantly decreased M2 macrophage phenotype compared with IL-4-stimulated BMDMs that were not treated with metformin (Fig. 4C).
Figure 4.
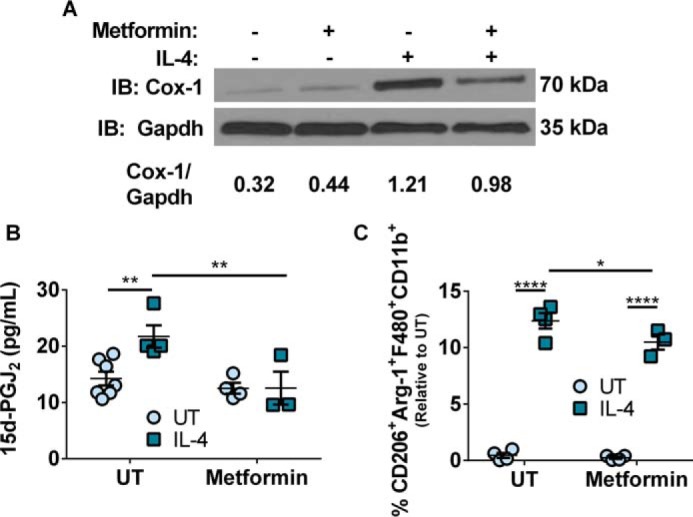
AMPK activation in vitro reduced Cox-1 expression and M2 macrophages. A, representative Western immunoblot for Cox-1 and Gapdh expression in murine BMDMs stimulated with or without metformin or IL-4. B, production of 15d-PGJ2 from murine BMDMs stimulated with or without metformin or IL-4 (20 h). C, percentage of CD206+Arg-1+F480+CD11b+ murine BMDMs stimulated with or without metformin or IL-4 relative to untreated control. Two-way ANOVA with Fisher's LSD test. *, p < 0.05; **, p < 0.01; ****, p < 0.0001. n = 3–7 biological replicates per group. All experimental data are expressed as mean ± S.E.
In vivo AMPK activation exacerbates parasitic infection
To examine if inhibition of mTORC through activation of AMPK by metformin treatment of mice impacted their ability to clear N. brasiliensis and if Cox-1-derived 15d-PGJ2 played any role, mice were administered 300 mg/kg of metformin water with or without 0.05 mg/kg/day 15d-PGJ2 treatment 1 day prior to inoculation with infective N. brasiliensis third-stage larvae (L3) and continued on treatment through day 8 of the infection. Control mice either received Milli-Q water with sterile PBS intraperitoneal (i.p.) injections or metformin water with sterile PBS i.p. injections. Immunoblot analysis of the jejunal extracts indicated a significant decrease in Cox-1 expression compared with the control jejunum (Fig. 5A). Interestingly, jejunal extracts of mice revealed no Cox-2 expression in any of the treatment groups suggesting that Cox-1 was the predominant enzyme during a Th2 cyokine (IL-4/IL-13) response to infection with N. brasiliensis (supplemental Fig. S6A) (37). Mice receiving Milli-Q water with sterile PBS i.p. injections had the least parasitic burden as determined by number of adult N. brasiliensis worms in the small intestines (Fig. 5B). However, metformin administration and sterile PBS i.p. injections led to a 3-fold higher adult worm burden compared with mice on Milli-Q water and sterile PBS i.p. injections (Fig. 5B). Furthermore, mice on metformin water that were treated with exogenous 15d-PGJ2 had a significant decrease in adult worm burden (Fig. 5B). Interestingly, there was a strong negative correlation between the amount of Cox-1 protein expression and number of adult worms present in mice (Fig. 5C). The expression of Arg-1, a M2 macrophage marker, was slightly decreased in the jejunum of mice treated with metformin; however, treatment of mice with metformin and 15d-PGJ2 restored the jejunal expression of Arg-1 (Fig. 5D).
Figure 5.

AMPK activation in vivo delayed parasitic clearance. A, representative Western immunoblot for Cox-1 and Gapdh expression in the jejunum of mice treated with either water + PBS or metformin + PBS day 8 post-inoculation with infective larvae. B, number of adult worms per mouse based on treatment day 8 post-inoculation. C, correlation between Cox-1 protein expression and number of adult worms per mouse based on treatment day eight post-inoculation. D, real-time PCR on Arg-1 expression in the jejunum of infected mice based on treatment day 8 post-inoculation. ROUT method (Q = 5.0%). One-way ANOVA with Fisher's LSD test. Linear regression. *, p < 0.05; **, p < 0.01. n = 5–6 biological replicates per group. All experimental data are expressed as mean ± S.E.
Discussion
Cyclooxygenase-1 has been generally regarded as a constitutive enzyme regulated developmentally and key in the production of prostanoids, which effect homeostasis in the kidney, stomach, and intestine. Previous studies have shown that various stimuli including TNFα, IL-1β, and LPS leads to the production of TxA2/TxB2 in monocytes/macrophages as well as human umbilical vein cells (38, 39). In unstimulated human umbilical vein cells, COX-1 is the predominant isoform leading to the production of TxA2, however, upon IL-1β stimulation, COX-2 is significantly increased with a corresponding increase in PGE2 and PGI2 (38). IL-4 treatment has been shown in other studies to decrease pro-inflammatory mediators such as TxB2 and PGE2, which is also decreased in our study compared with LPS treatment. Additionally, we have shown that resting and IL-4-stimulated BMDMs produce baseline levels of TxB2 and PGE2, which appear to not play a significant role in M2 macrophage polarization. Our study describes a novel mechanism of up-regulation of Cox-1 expression by a Th2 cytokine, IL-4, in macrophages. Interestingly, Cox-2, which is a highly inducible gene regulated by diverse stimuli via mechanisms involving transcriptional and translational control of expression, was significantly down-regulated by IL-4, highlighting the importance of Cox-1 in a Th2 environment, such as during helminth infection.
Furthermore, the Akt-mTORC axis was involved in increased translation of Ptgs-1 mRNA upon IL-4 treatment. The presence of TOP sequences in the vicinity of the TSS in the 5′ UTR of both murine and human PTGS-1 mRNA further supported the involvement of mTORC. Surprisingly, Ptgs-1 appears to not be exclusively regulated by mTORC in the same manner as traditional TOP mRNA sequences. The use of CRISPR/Cas9 to knock-out Tsc2 led to a decrease in Cox-1 expression. It is known that mTORC1 is negatively regulated by Tsc, whereas mTORC2 is positively regulated by the Tsc complex. Additionally, mTORC2 is not sensitive to rapamycin treatment (data not shown) but is torin 1 sensitive. Finally, because Cox-1 does not behave as a bona fide TOP sequence in regards to the polysome data, it is likely that mTORC2 may affect Cox-1 protein expression. However, the downstream targets of mTORC2 are unknown and further work is needed to delineate the functional differences between mTORC1 and mTORC2 in regulating Cox-1 protein expression. That said, mTORC signaling appears to play a role in the accumulation of Cox-1 protein over the course of IL-4 stimulation suggesting other mechanisms potentially involving protein stability.
Interestingly, when monocytes differentiate into macrophages various heat shock protein molecular chaperones are differentially expressed and vary depending on the macrophage polarization state (40, 41). IL-4-treated (M2) macrophages differentially express five HSPs (DNAJB5, HSPA13, HSPBAP1, HSPH1, and HSPB1) compared with unpolarized macrophages (41). Additionally, HSP27 (HSPB1) has been shown to have anti-inflammatory and “anti-danger signal” activities (40). There are a few studies that have examined a relationship between Cox-2 and HSP27 during myofibroblast migration (42, 43). Additionally, a recent study reported that HSP90 inhibition decreased Cox-2 mRNA expression and protein level in a colorectal cancer cell line (44). In light of the fact that IL-4 treatment of macrophages increases the expression of these chaperones, whereas significantly down-regulating Cox-2 protein and increasing Cox-1 protein, it is plausible that Cox-1 interacts with HSPs, due to high structural similarities between Cox-1 and Cox-2, leading to the stabilization of Cox-1 protein. Further work is warranted to investigate this phenomenon in macrophages and its overall impact on the pathophysiology of IL-4/IL-13-dependent intestinal clearance of N. brasiliensis.
The canonical pathway of signaling downstream of IL-4Rα typically involves Jak/Stat signaling and the PI3K/Akt/mTORC pathway (7, 11). Although treatment of BMDMs with herbimycin A, Akt inhibitor VIII, or torin 1 blocked Cox-1 protein expression; treatment with Ly294002 (PI3K inhibitor), had no effect on Cox-1 expression implicating an alternate pathway of activation of Akt that involves Fes, as in T-cells and early myeloid progenitor cells (Fig. 6) (8–10). Further work is warranted to characterize the under-appreciated interaction between IL-4Rα, Fes, and Akt in the context of mTORC activation. To extend the analysis of mTORC signaling and Cox-1 in macrophages, we showed that the production of 15d-PGJ2 was significantly increased with IL-4 stimulation, which was inhibited upon loss of Cox-1 expression by torin 1 as well as metformin. Our data suggested that Cox-1 functionally couples with H-PGDS to likely provide the ligand (15d-PGJ2) to PPARγ, which acts in concert with Stat6 to activate transcription of PPARγ target genes to aid in macrophage (M2) polarization (15–17, 45). More importantly, inhibition of mTORC by metformin, which negatively impacted M2 macrophage polarization through decrease in Cox-1 expression, was rescued by exogenous treatment with 15d-PGJ2 suggesting the importance of Cox-1 activity in macrophage function.
Figure 6.

Schematic illustration of IL-4-mediated post-transcriptional control of Cox-1. In addition to the previously identified Jak/Stat6 pathway that is downstream of IL-4R, our studies suggested translational control of Cox-1 expression through the IL-4R, Fes, Akt, and mTORC axis to effect M2 polarization and resolution of helminth infection. Metformin-dependent activation of AMPK inhibited mTORC to decrease M2 macrophages in addition to Cox-1 translation which reduced 15d-PGJ2 and subsequent PPARγ activation. Dotted lines represent post-transcriptional mechanisms of Cox-1 regulation downstream of mTORC that is yet to be identified.
We utilized a helminth model to induce Th2-type inflammation to test the role of Cox-1 and metformin in helminth clearance. Mice treated with metformin had a significantly higher adult parasite burden compared with control mice, which was partially rescued with exogenous administration of 15d-PGJ2. As in the helminth model, Cox-1 could also play a critical role in asthma and airway function. Cox-1−/− mice are hyper-responsive to bronchial constrictors and non-selective Cox inhibitors increased bronchial constriction in wild type mice; whereas rofecoxib (a Cox-2 selective inhibitor) had no effect on airway hyper-responsiveness despite the expression of Cox-2 in epithelial tissue (46, 47). This clearly suggested Cox-1, and not Cox-2, activity regulated airway function. However, metformin administration in asthmatics has proven to be beneficial (13) perhaps given its ability to impact a large set of mTORC targets, in addition to COX-1. In light of our studies demonstrating the regulation of Cox-1 by IL-4 in macrophage polarization, it is necessary to revisit the role of Cox-1 in the context of Th2 diseases in an effort to reconcile such apparent differences. Additionally, Langenbach et al. (27) have shown that Cox-1−/− mice exhibit slightly reduced resolution and macrophage recruitment in addition to delayed parturition, which all have varying extents of involvement of M2 macrophages. Thus, we can only speculate that there could be an impact on M2 polarization in Cox-1−/− mice, which will need to be further tested.
In conclusion, our studies provide evidence for a novel mechanism involving post-transcriptional regulation of Cox-1 expression upon IL-4 stimulation in human and murine macrophages (Fig. 6). This is relevant because Cox activity is required to generate bioactive prostanoids, particularly PGJ2 metabolites, that serve as endogenous ligands for PPARγ and drive macrophage polarization (15–17, 48). As shown here, only Cox-1, and not Cox-2, was expressed both in vitro and in vivo during a Th2-mediated response emphasizing the need to better understand the role of Cox-1 to enable development of new and more specific therapeutic regimens in addition to understanding how current drugs, such as metformin, may have unintentional effects on Cox regulation. Specifically, because inhibition of Cox-1 with indomethacin (17) or metformin was rescued by exogenous treatment with 15d-PGJ2 to significantly alleviate nematode burden, it is clear that further studies are necessary to unravel the role of Cox-1 in promoting resolution in Th2-mediated inflammation.
Materials and methods
Mice
C57BL/6 male age-matched mice were purchased from Taconic Biosciences, Inc. (Hudson, NY) and maintained on an AIN-76-based semi-purified diet from Harlan-Teklad (Madison, WI). All studies were pre-approved by the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee at Penn State University.
Bone marrow-derived macrophage culture
Femurs were harvested and bone marrow was collected from C57BL/6 male age-matched mice. Bone marrow was separated into a single cell suspension. Cells were plated in DMEM (Life Technologies) with 5% (v/v) fetal bovine serum (Atlanta Biologicals), 2 mm l-glutamine (Corning), 100 IU/ml of penicillin (Corning), 100 μg/ml of streptomycin (Corning), and 10% (v/v) L929 fibroblast-conditioned DMEM. L929 fibroblasts were purchased from the American Type Culture Collection (Manassas, VA).
Treatments
Various compounds were used at the following concentrations: 7.5 μm herbimycin A (Santa Cruz), 10 μm Ly294002 (Cayman), 10 μm Akt inhibitor VIII (Cayman), 250 nm torin 1 (Selleck Chemicals), 80 nm actinomycin D (Sigma), 10 μm cycloheximide (gift from Dr. Gary Perdew, Penn State University), 50 nm Jak inhibitor I (Santa Cruz), 20 μm metformin (Sigma), and 30 nm ozagrel (Cayman). Metformin was reconstituted in cell culture grade water, whereas all other compounds were reconstituted in cell culture grade DMSO (Sigma) and added to cells at concentrations not exceeding 0.1% (v/v). The above mentioned compounds were added to BMDM cultures 4 h prior to IL-4 stimulation, except for actinomycin D, which was added 30 min prior to IL-4 stimulation due to impact on cell viability. IL-4 (10 ng/ml; R&D Systems) was added for 20 h, unless described otherwise in text, and cells were collected for analysis as described below. Cell viability was confirmed by trypan blue staining.
RNA extraction and real-time PCR
Cell pellets were stored in 1 ml of Tri-reagent (Thermo) at −80 °C until RNA extraction. RNA extraction was performed according to the manufacturer's protocol as described previously (21). RNA was reverse transcribed to cDNA using a high capacity cDNA reverse transcription kit (Life Technologies). cDNA was used in quantitative PCR with specific TaqMan probes for Ptgs-1, Ptgs-2, Arg-1, Cd36, mPges-1, Txas-1, or Gapdh (Life Technologies) in a StepOnePlus-Real Time PCR System (Thermo Applied Biosystems). Gene abundance was analyzed and 2−ΔΔCt was calculated to represent changes in gene expression relative to controls (49).
Western immunoblot
Mammalian protein extraction reagent (Thermo) or tissue protein extraction reagent (Thermo) containing protease inhibitor mixture (Roche Applied Science) and 5 mm sodium orthovanadate (Sigma) was added to cell pellets or tissue samples, respectively, and incubated on ice for 20 min and vortexed every 5 min. Cell debris was pelleted by centrifugation at 20,817 × g for 10 min. Protein concentration in the supernatant was measured using a BCA protein assay kit (Thermo). Protein was loaded onto a discontinuous SDS-PAGE gel (t = 12.5%). Nitrocellulose blotting membranes were blocked with a 5% (w/v) skim milk solution made with Tris-buffered saline containing 0.1% Tween 20 (Sigma) for 1 h prior to the addition of primary antibodies. Primary antibodies were added to the blots for 12 h at 4 °C at the following dilutions: 1:2,000 Cox-1 (Cayman), 1:2,000 Cox-2 (Cayman), 1:2,000 Fes (Abcam), 1:1,000 phospho-S6K (R&D Systems), 1:1,000 total S6K (R&D Systems), 1:20,000 β-actin (Fitzgerald), and 1:600,000 Gapdh (Fitzgerald). Appropriate secondary antibodies conjugated to horseradish peroxidase were used at dilutions: 1:2,000 goat anti-rabbit (Thermo), or 1:5,000 goat anti-mouse (Thermo) and developed using West Pico reagent (Thermo). Autoradiograms densities were evaluated using Image-J (National Institutes of Health).
Flow cytometry
Cells were enumerated by trypan blue staining using a hemocytometer. 100,000–200,000 cells were resuspended in 100 μl of flow buffer (phosphate-buffered saline (Life Technologies) containing 100 IU/ml of penicillin (Corning), 100 μg/ml of streptomycin (Corning), and 2% (v/v) fetal bovine serum (Atlanta Biologicals)). Extracellular staining was performed by incubating cells with 0.25 μg of Fc block (BD Biosciences) for 10 min followed 30 min at 4 °C with the following antibodies: 0.25 μg of FITC-conjugated CD206 (Biolegend), 0.10 μg of PE-Cy7-conjugated CD11b (BD Biosciences), and 0.15 μg of APC-conjugated F4/80 (Miltenyi). Cells were then fixed by adding 2% formaldehyde solution (Sigma), and incubated for 20 min at room temperature. To perform intracellular staining, cells were resuspended in a permeability buffer (48.5 ml of phosphate-buffered saline containing 1.5 ml of fetal bovine serum (Atlanta Biologicals), 50 mg of saponin (Sigma), and 45 mg of sodium azide (J.T. Baker)) and incubated at room temperature for 15 min. Cells were resuspended in 100 μl of permeability buffer and 0.125 μg of PE-conjugated arginase-1 (R&D Systems) antibody or 2.5 μl of Alexa Fluor 647-conjugated CD68 (BD Biosciences) and incubated for 30 min at room temperature. Cells were collected on the C6 Accuri Flow Cytometer (BD Biosciences) in addition to the following controls: unstained, OneComp eBeads (eBioscience) single stained, and fluorescence minus one (FMO). Data were analyzed using FlowJo. Gating was based on fluorescence minus one staining controls.
Lipid extraction and liquid chromatography-mass spectrometry
Endogenous lipids were extracted from cell culture media samples using C-18 Sep-Pak cartridges (Waters, Milford, MA). Briefly, cell-free culture supernatants were acidified with 6 n HCl and loaded on to the Sep-Pak cartridge, bound lipids were eluted with methanol, evaporated, and stored in ethyl acetate, until analyzed at −80 °C. Lipids were suspended in 70% methanol and analyzed by LC-MS/MS on an API 2000 LC-MS/MS system with Turbo V source and an electrospray ionization probe in negative ion mode (15). Quantitative analysis of 15d-PGJ2 was performed using a calibration curve generated by respective standards in multiple reaction monitoring mode using three transitions for 15d-PGJ2 (315.1/271.1, 315.1/203.0, and 315.1/158.0 m/z). Quantitative analysis of PGE2 was performed as described above using transitions for PGE2 (351.2/315.0, 351.2/271.0, and 351.2/189.0 m/z). Quantitative analysis of TxB2, was performed as described above using transitions for TxB2 (369.0/195.1 and 369.0/168.9 m/z). Data acquisition and analysis was performed using Analyst software (AB Sciex; version 1.5).
Polysome profile assay
Polysome profiling and mRNA distribution analysis was performed as described previously (50). Briefly, BMDMs were seeded in 150-mm Petri plates and grown up to 70% confluence (∼1.5 × 107 cells). Following DMSO or IL-4 treatment for 20 h, cycloheximide (CHX) was added to cells in 100 μg/ml for 10 min at 37 °C. Cells were collected at 1,500 × g for 10 min and washed twice with cold PBS containing 100 μg/ml of CHX. The cell pellets were suspended in 300 μl of lysis buffer (10 mm HEPES-KOH (pH 7.5), 2.5 mm MgCl2, 100 mm KCl, 0.25% Nonidet P-40, 100 μg/ml of CHX, 1 mm DTT, 200 units/ml of RNase inhibitor (RNaseOUT, Invitrogen) and EDTA-free protease inhibitor (Roche)), kept on ice for 20 min, and then passed 15 times through a 23-gauge needle. Lysates were cleared by centrifugation at 16,000 × g for 20 min and supernatants (cytosolic cell extracts) were collected and absorbance was measured at 260 nm. Approximately 7 absorbance units (at 260 nm) of lysates were layered over 15–54% cold sucrose gradients in buffer (10 mm HEPES-KOH (pH 7.5), 2.5 mm MgCl2, and 100 mm KCl). Gradients were centrifuged at 121,000 × g in a Beckman SW28 rotor for 4 h at 4 °C. After centrifugation, 12 equal size fractions (1.2 ml/fraction) were collected. RNA from each fraction was isolated using TRIzol LS reagent (Invitrogen) and an equal volume of RNA from each fraction was used for cDNA synthesis with SuperScript III First-Strand Synthesis Supermix (Invitrogen). The relative quantities of specific mRNAs were measured by quantitative PCR using the VeriQuest SYBR Green qPCR master Mix (Affymetrix) on a StepOnePlus Real-Time PCR system (Applied Biosystem).
Infection with N. brasiliensis
Mice were pretreated with sterile PBS or 0.05 mg/kg/day of 15d-PGJ2 (formulated in sterile PBS) intraperitoneally and administered metformin (300 mg/kg in Milli-Q water) or Milli-Q water alone starting 1 day prior to subcutaneous inoculation with 500 third stage N. brasiliensis larvae (L3) (17, 51). Metformin water was made fresh and changed every other day throughout infection. Mice received i.p. injections daily. Treatment was continued throughout the infection until sacrifice on day 8 post-inoculation when adult worms in the jejunum were enumerated (52).
Human macrophage culture
Frozen human PBMC were purchased from AllCells (Alameda, CA) from donors. Upon arrival, cells were thawed in a 37 °C water and enumerated with trypan blue staining using a hemocytometer to determine cell viability. Cells were transferred to a 50-ml tube and 20 ml of RPMI 1640 (Corning) containing 2 mm l-glutamine (Corning), 10% (v/v) fetal bovine serum (Atlanta Biologicals), 100 IU of penicillin (Corning), and 100 μg/ml of streptomycin (Corning) was added dropwise. Cells were centrifuged at 200 × g for 15 min at room temperature. This was repeated and PBMCs were finally resuspended in 2 ml of media and enumerated by trypan blue using a hemocytometer. PBMCs were brought to a concentration of 3.0 × 106 cells/ml and plated in a 6-well culture plate. Cells were cultured with 10 ng/ml of recombinant human macrophage colony-stimulating factor (GoldBio) for 24 h, then at 25 ng/ml for remaining culturing period. Media was changed every 3 days for 8 days adding fresh macrophage colony-stimulating factor with each change (33). Adherent cells (comprising of 95% macrophages) were pre-treated with either DMSO or 250 nm torin 1 followed by 20 h with or without recombinant human IL-4 (R&D Systems) stimulation. Cells were harvested and analyzed by flow cytometry and Western immunoblot analysis as described above.
Transfection and transduction
HEK293TN cells (System Biosciences; 350,000 cells/2 ml) were plated in DMEM (Life Technologies) with 1 mm sodium pyruvate (Life Technologies), 2 mm l-glutamine (Corning), 100 IU/ml of penicillin (Corning), 100 μg/ml of streptomycin (Corning), and 10% (v/v) fetal bovine serum (Atlanta Biologicals) in a 6-well plate and allowed to adhere overnight. Cells were then transfected three times, once every 24 h with 2.45 μg of pMD2.G (Addgene), 4.9 μg of psPAX-2 (Addgene), 4.9 μg of lentiviral CRISPR gRNA plasmid (Ampr/Purr) expressing mCherry (Vector Builder), 26.6 μl of TransIT 293 transfection reagent (Mirus), and 700 μl of serum-free DMEM (Life Technologies). The gRNA sequences for non-targeting control and Tsc2 were GCACTACCAGAGCTAACTCA and CAGGAGGACCTGCGCGCGAA, respectively. Viruses were harvested and passed through a 0.45-μm filter. Bone marrow was harvested from Cas9 GFP+ mice and incubated with 1 part viral supernatant to 1 part BMDM. After 48 h of viral transduction, bone marrow cells were centrifuged at 100 × g for 5 min at 4 °C and resuspended in DMEM containing 2 μg/ml of puromycin (GoldBio) and continued for 4 days with puromycin, changing media every 2 days. Cells were imaged by fluorescent microscopy to detect GFP+ and mCherry+. After 4 days of puromycin selection, cells were cultured in media without puromycin for an additional 2 days. BMDMs were stimulated with or without 10 ng/ml of recombinant murine IL-4 (R&D Systems) for 20 h and analyzed for Tsc2 knock-out and Cox-1 expression by surveyor assay and Western immunoblot analysis, respectively.
Bioinformatics and statistics
The TSS of murine and human PTGS-1 were analyzed with Promoter 2.0 (cbs.dtu.dk) and DBTSS (dbtss.hgc.jp) that predict TSS of vertebrate PolII promoters in DNA sequences (35, 53). The most common TSS in PTGS-1 that was predicted by Promoter 2.0 corroborated with consensus sequences defined by earlier studies (GenBankTM AF440204.1 and AH015269.2). All experimental data are expressed as mean ± S.E. Outliers were removed using ROUT method (Q = 5.0%), where appropriate (Fig. 5). Either an unpaired one-tailed t test, an unpaired two-tailed t test, a one-way ANOVA with Fisher's LSD test, or a two-way ANOVA with Fisher's LSD test was used, where appropriate, to compare the mean of each treatment group with the mean of each other treatment group using GraphPad Prism (GraphPad Software). A linear regression analysis was also performed, where appropriate. p values ≤ 0.05 were considered statistically significant.
Author contributions
A. E. S. and K. S. P. conceived and coordinated the study as well as wrote the manuscript and created the schematic. A. E. S. designed, performed, and analyzed the data shown in Figs. 1–6 and supplemental Figs. S1–S6. B. T. D. performed and analyzed the data shown in Figs. 1C, 3D, 4B, supplemental Figs. S1, D, E, and G, and 4D and assisted with manuscript preparation. B. J. G. performed and analyzed the data shown in Fig. 2F and assisted with manuscript preparation. V. N. cultured, stimulated, and harvested the BMDMs required for the polysome assay in Fig. 2F and assisted with manuscript preparation. J. F. U. provided L3 stage N. brasiliensis larvae and assisted with manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Wanying Dai and Svenjita Berry for help with lipid extractions, Michael Quickel and Laura Bennett for help with the CRISPR/Cas9 model, Dr. Ramesh Ramachandran for fluorescence microscopy, and Dr. Robert F. Paulson for helpful discussions.
This work was funded in part by the United States Public Health Service National Institutes of Health and Office of Dietary Supplements Grant R01DK077152, and by USDA Hatch Funds Grant 4605 (to K. S. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S6.
- Th2
- T-helper type 2
- Fes
- Feline sarcoma oncogene kinase
- TSC
- tuberous sclerosis complex
- mTORC
- mechanistic target of rapamycin complex
- BMDM
- bone marrow-derived macrophage
- AMPK
- AMP-activated protein kinase
- PG
- prostaglandin
- PPAR
- peroxisome proliferator-activated receptor
- Tx
- thromboxane
- PBMC
- peripheral blood mononuclear cells
- TOP
- terminal oligopyrimidine
- TSS
- transcription start site
- CHX
- cycloheximide
- ANOVA
- analysis of variance
- Cox
- cyclooxygenase
- PE
- phycoerythrin.
References
- 1. Martinez F. O., and Gordon S. (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Das A., Sinha M., Datta S., Abas M., Chaffee S., Sen C. K., and Roy S. (2015) Monocyte and macrophage plasticity in tissue repair and regeneration. Am. J. Pathol. 185, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gordon S. (2003) Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 [DOI] [PubMed] [Google Scholar]
- 4. Martinez F. O., Helming L., and Gordon S. (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483 [DOI] [PubMed] [Google Scholar]
- 5. Kreider T., Anthony R. M., Urban J. F. Jr., and Gause W. C. (2007) Alternatively activated macrophages in helminth infections. Curr. Opin. Immunol. 19, 448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reece J. J., Siracusa M. C., and Scott A. L. (2006) Innate immune responses to lung-stage helminth infection induce alternatively activated alveolar macrophages. Infect. Immun. 74, 4970–4981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang H., Harris M. B., and Rothman P. (2000) IL-4/IL-13 signaling beyond JAK/STAT. J. Allergy Clin. Immunol. 105, 1063–1070 [DOI] [PubMed] [Google Scholar]
- 8. Izuhara K., Feldman R. A., Greer P., and Harada N. (1994) Interaction of the c-fes proto-oncogene product with the interleukin-4 receptor. J. Biol. Chem. 269, 18623–18629 [PubMed] [Google Scholar]
- 9. Izuhara K., Feldman R. A., Greer P., and Harada N. (1996) Interleukin-4 induces association of the c-fes proto-oncogene product with phosphatidylinositol-3 kinase. Blood 88, 3910–3918 [PubMed] [Google Scholar]
- 10. Jiang H., Foltenyi K., Kashiwada M., Donahue L., Vuong B., Hehn B., and Rothman P. (2001) Fes mediates the IL-4 activation of insulin receptor substrate-2 and cellular proliferation. J. Immunol. 166, 2627–2634 [DOI] [PubMed] [Google Scholar]
- 11. Byles V., Covarrubias A. J., Ben-Sahra I., Lamming D. W., Sabatini D. M., Manning B. D., and Horng T. (2013) The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 4, 2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu L., Yang T., Li L., Sun L., Hou Y., Hu X., Zhang L., Tian H., Zhao Q., Peng J., Zhang H., Wang R., Yang Z., Zhang L., and Zhao Y. (2014) TSC1 controls macrophage polarization to prevent inflammatory disease. Nat. Commun. 5, 4696. [DOI] [PubMed] [Google Scholar]
- 13. Park C. S., Bang B. R., Kwon H. S., Moon K. A., Kim T. B., Lee K. Y., Moon H. B., and Cho Y. S. (2012) Metformin reduces airway inflammation and remodeling via activation of AMP-activated protein kinase. Biochem. Pharmacol. 84, 1660–1670 [DOI] [PubMed] [Google Scholar]
- 14. Ding L., Liang G., Yao Z., Zhang J., Liu R., Chen H., Zhou Y., Wu H., Yang B., and He Q. (2015) Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 6, 36441–36455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gandhi U. H., Kaushal N., Ravindra K. C., Hegde S., Nelson S. M., Narayan V., Vunta H., Paulson R. F., and Prabhu K. S. (2011) Selenoprotein-dependent up-regulation of hematopoietic prostaglandin D2 synthase in macrophages is mediated through the activation of peroxisome proliferator-activated receptor (PPAR) γ. J. Biol. Chem. 286, 27471–27482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vunta H., Davis F., Palempalli U. D., Bhat D., Arner R. J., Thompson J. T., Peterson D. G., Reddy C. C., and Prabhu K. S. (2007) The anti-inflammatory effects of selenium are mediated through 15-deoxy-Δ12,14-prostaglandin J2 in macrophages. J. Biol. Chem. 282, 17964–17973 [DOI] [PubMed] [Google Scholar]
- 17. Nelson S. M., Shay A. E., James J. L., Carlson B. A., Urban J. F. Jr., and Prabhu K. S. (2016) Selenoprotein expression in macrophages is critical for optimal clearance of parasitic helminth Nippostrongylus brasiliensis. J. Biol. Chem. 291, 2787–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang J. T., Welch J. S., Ricote M., Binder C. J., Willson T. M., Kelly C., Witztum J. L., Funk C. D., Conrad D., and Glass C. K. (1999) Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382 [DOI] [PubMed] [Google Scholar]
- 19. Cho W., Kim Y., Jeoung D. I., Kim Y. M., and Choe J. (2011) IL-4 and IL-13 suppress prostaglandins production in human follicular dendritic cells by repressing COX-2 and mPGES-1 expression through JAK1 and STAT6. Mol. Immunol. 48, 966–972 [DOI] [PubMed] [Google Scholar]
- 20. Vane J. R., Bakhle Y. S., and Botting R. M. (1998) Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 38, 97–120 [DOI] [PubMed] [Google Scholar]
- 21. Nelson S. M., Lei X., and Prabhu K. S. (2011) Selenium levels affect the IL-4-induced expression of alternative activation markers in murine macrophages. J. Nutr. 141, 1754–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lawrence T., Willoughby D. A., and Gilroy D. W. (2002) Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2, 787–795 [DOI] [PubMed] [Google Scholar]
- 23. Serhan C. N. (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Serhan C. N., Chiang N., and Van Dyke T. E. (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ueno N., Murakami M., Tanioka T., Fujimori K., Tanabe T., Urade Y., and Kudo I. (2001) Coupling between cyclooxygenase, terminal prostanoid synthase, and phospholipase A2. J. Biol. Chem. 276, 34918–34927 [DOI] [PubMed] [Google Scholar]
- 26. Kihara Y., Gupta S., Maurya M. R., Armando A., Shah I., Quehenberger O., Glass C. K., Dennis E. A., and Subramaniam S. (2014) Modeling of eicosanoid fluxes reveals functional coupling between cyclooxygenases and terminal synthases. Biophys. J. 106, 966–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langenbach R., Loftin C., Lee C., and Tiano H. (1999) Cyclooxygenase knockout mice: models for elucidating isoform-specific functions. Biochem. Pharmacol. 58, 1237–1246 [DOI] [PubMed] [Google Scholar]
- 28. Fitzpatrick F. A. (2004) Cyclooxygenase enzymes: regulation and function. Curr. Pharm. Des. 10, 577–588 [DOI] [PubMed] [Google Scholar]
- 29. Kang Y. J., Mbonye U. R., DeLong C. J., Wada M., and Smith W. L. (2007) Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 46, 108–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Williams C. S., Mann M., and DuBois R. N. (1999) The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 18, 7908–7916 [DOI] [PubMed] [Google Scholar]
- 31. Morita I. (2002) Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 68–69, 165–175 [DOI] [PubMed] [Google Scholar]
- 32. Mbonye U. R., Wada M., Rieke C. J., Tang H. Y., Dewitt D. L., and Smith W. L. (2006) The 19-amino acid cassette of cyclooxygenase-2 mediates entry of the protein into the endoplasmic reticulum-associated degradation system. J. Biol. Chem. 281, 35770–35778 [DOI] [PubMed] [Google Scholar]
- 33. Ramos P., Casu C., Gardenghi S., Breda L., Crielaard B. J., Guy E., Marongiu M. F., Gupta R., Levine R. L., Abdel-Wahab O., Ebert B. L., Van Rooijen N., Ghaffari S., Grady R. W., Giardina P. J., and Rivella S. (2013) Macrophages support pathological erythropoiesis in polycythemia vera and β-thalassemia. Nat. Med. 19, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martinez F. O., Gordon S., Locati M., and Mantovani A. (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 [DOI] [PubMed] [Google Scholar]
- 35. Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., and Sabatini D. M. (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Erices R., Bravo M. L., Gonzalez P., Oliva B., Racordon D., Garrido M., Ibañez C., Kato S., Brañes J., Pizarro J., Barriga M. I., Barra A., Bravo E., Alonso C., Bustamente E., Cuello M. A., and Owen G. I. (2013) Metformin, at concentrations corresponding to the treatment of diabetes, potentiates the cytotoxic effects of carboplatin in cultures of ovarian cancer cells. Reprod. Sci. 20, 1433–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oeser K., Schwartz C., and Voehringer D. (2015) Conditional IL-4/IL-13-deficient mice reveal a critical role of innate immune cells for protective immunity against gastrointestinal helminths. Mucosal Immunol. 8, 672–682 [DOI] [PubMed] [Google Scholar]
- 38. Caughey G. E., Cleland L. G., Penglis P. S., Gamble J. R., and James M. J. (2001) Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J. Immunol. 167, 2831–2838 [DOI] [PubMed] [Google Scholar]
- 39. Kabashima K., Murata T., Tanaka H., Matsuoka T., Sakata D., Yoshida N., Katagiri K., Kinashi T., Tanaka T., Miyasaka M., Nagai H., Ushikubi F., and Narumiya S. (2003) Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat. Immunol. 4, 694–701 [DOI] [PubMed] [Google Scholar]
- 40. Miller-Graziano C. L., De A., Laudanski K., Herrmann T., and Bandyopadhyay S. (2008) HSP27: an anti-inflammatory and immunomodulatory stress protein acting to dampen immune function. Novartis Found. Symp. 291, 196–208; discussion 208–111, 221–224 [DOI] [PubMed] [Google Scholar]
- 41. Fagone P., Di Rosa M., Palumbo M., De Gregorio C., Nicoletti F., and Malaguarnera L. (2012) Modulation of heat shock proteins during macrophage differentiation. Inflamm. Res. 61, 1131–1139 [DOI] [PubMed] [Google Scholar]
- 42. Saini S., Liu T., and Yoo J. (2016) TNF-α stimulates colonic myofibroblast migration via COX-2 and Hsp27. J. Surg. Res. 204, 145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chu E., Saini S., Liu T., and Yoo J. (2017) Bradykinin stimulates protein kinase D-mediated colonic myofibroblast migration via cyclooxygenase-2 and heat shock protein 27. J. Surg. Res. 209, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohammadi A., Yaghoobi M. M., Gholamhoseinian Najar A., Kalantari-Khandani B., Sharifi H., and Saravani M. (2016) HSP90 inhibition suppresses PGE2 production via modulating COX-2 and 15-PGDH expression in HT-29 colorectal cancer cells. Inflammation 39, 1116–1123 [DOI] [PubMed] [Google Scholar]
- 45. Szanto A., Balint B. L., Nagy Z. S., Barta E., Dezso B., Pap A., Szeles L., Poliska S., Oros M., Evans R. M., Barak Y., Schwabe J., and Nagy L. (2010) STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 33, 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harrington L. S., Lucas R., McMaster S. K., Moreno L., Scadding G., Warner T. D., and Mitchell J. A. (2008) COX-1, and not COX-2 activity, regulates airway function: relevance to aspirin-sensitive asthma. FASEB J. 22, 4005–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peebles R. S. Jr., Hashimoto K., Morrow J. D., Dworski R., Collins R. D., Hashimoto Y., Christman J. W., Kang K. H., Jarzecka K., Furlong J., Mitchell D. B., Talati M., Graham B. S., and Sheller J. R. (2002) Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 165, 1154–1160 [DOI] [PubMed] [Google Scholar]
- 48. Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A. W., and Chawla A. (2007) Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−δΔ CT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 50. Guan B. J., Krokowski D., Majumder M., Schmotzer C. L., Kimball S. R., Merrick W. C., Koromilas A. E., and Hatzoglou M. (2014) Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2α. J. Biol. Chem. 289, 12593–12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Incio J., Suboj P., Chin S. M., Vardam-Kaur T., Liu H., Hato T., Babykutty S., Chen I., Deshpande V., Jain R. K., and Fukumura D. (2015) Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS ONE 10, e0141392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Camberis M., Le Gros G., and Urban J. Jr. (2003) Animal model of Nippostrongylus brasiliensis and Heligmosomoides polygyrus. Curr. Protoc. Immunol. Chapter 19, Unit 19 12 [DOI] [PubMed] [Google Scholar]
- 53. Knudsen S. (1999) Promoter2.0: for the recognition of PolII promoter sequences. Bioinformatics 15, 356–361 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.