

Abstract
Lipid oxidation due to oxidative stress plays an important role in the pathogenesis of inflammatory and thrombotic cardiovascular diseases. Several findings suggest that lipid peroxidation can alter the function of coagulation proteins and contribute to a hypercoagulable state, but the molecular mechanisms are unclear. Here, we report that oxidized phospholipids suppress the anticoagulant function of the serpin, protein Z-dependent protease inhibitor (ZPI), a specific inhibitor of membrane-associated factor Xa (FXa) that requires protein Z (PZ), phospholipid, and calcium as cofactors. We found that this suppression arises from a diminished ability of the oxidized membrane to function as a cofactor to promote ZPI inhibition of membrane-bound FXa, due fully or in part to the susceptibility of the bound ZPI-PZ complex to oxidative inactivation. Surprisingly, free ZPI was also susceptible to inactivation by oxidized membrane vesicles in the absence of calcium. Oxidized vesicles containing both phosphatidylserine and polyunsaturated fatty acids were required to promote inactivation of the ZPI-PZ complex or free ZPI, indicating that binding of the PZ-complexed or free ZPI to peroxide-modified phospholipid vesicles mediates the inactivation. Heparin protected the ZPI-PZ complex and free ZPI from inactivation, suggesting that blocking the heparin-binding site on ZPI interferes with ZPI binding to lipid or to PZ. This was confirmed by direct lipid-binding experiments. Native PAGE indicated that oxidization induced dissociation of the ZPI-PZ complex and increased the negative charge of ZPI. We conclude that compromised ZPI anticoagulant function could contribute to thrombus initiation and growth in oxidative stress-induced cardiovascular diseases.
Keywords: coagulation factor, lipid oxidation, protease inhibitor, serpin, thrombosis
Introduction
Lipid peroxidation arising from oxidative stress is a known contributor to the pathogenesis of cardiovascular diseases such as atherosclerosis and thrombosis (1–3). It is well established that lipid peroxidation and lipid peroxidation products cause oxidative damage to blood vessels as well as circulating blood cells, lipoproteins, and coagulation proteins that can perturb the hemostatic balance and produce a hypercoagulable state (4, 5).
Lipid peroxidation may perturb hemostasis by modifying cell membranes upon which blood coagulation proteins assemble to perform their function. Oxidative modifications of phospholipid membrane vesicles have been shown to enhance prothrombinase procoagulant function and activated protein C (aPC)4 anticoagulant function as well as promote the down-regulation of aPC function by the serpin, protein C inhibitor (6–8). Lipid peroxidation products can additionally perturb hemostasis by activating platelets or endothelial cells or by modifying critical residues of coagulation proteins that impair their function. Lipid peroxidation products of low density lipoprotein (LDL) have been shown to chemically modify and impair the anticoagulant function of bound tissue factor pathway inhibitor (TFPI) (9, 10). Moreover, lipid peroxidation products contribute to oxidative modifications of fibrinogen that result in both a reduced ability of fibrinogen to clot and a decreased susceptibility of fibrin clots to plasmin lysis (11). Together, such findings suggest that lipid peroxidation can alter the function of multiple coagulation proteins and contribute to the hypercoagulable state associated with oxidative stress.
Protein Z-dependent protease inhibitor (ZPI) is a serpin superfamily protein circulating in plasma as a complex with the vitamin K-dependent protein cofactor, protein Z (PZ) (12–14). ZPI functions as a key anticoagulant by rapidly inhibiting factor Xa (FXa) in the presence of PZ, calcium, and procoagulant membranes (15, 16). PZ acts as a cofactor of ZPI by binding ZPI through its pseudoprotease and EGF1 domains and by binding membrane lipids through its N-terminal Gla domain so as to present ZPI to FXa on the membrane surface and promote rapid FXa inhibition (17, 18). The importance of ZPI and PZ as anticoagulant regulators of FXa is supported by the thrombosis phenotype of mouse knockouts lacking either ZPI or PZ, especially when combined with the Factor V Leiden mutation (19, 20).
We have noted in past studies that ZPI anticoagulant function was reduced in the presence of aged procoagulant lipid vesicles. We now report a systematic investigation of this phenomenon that reveals a novel mechanism of ZPI inactivation by oxidized procoagulant lipids. The inactivation is rapid in the presence of PZ and calcium cofactors, but surprisingly is also efficient in the absence of these cofactors. The importance of ZPI as a critical anticoagulant regulator of hemostasis suggests that under pathologic conditions where oxidative stress and lipid peroxidation are common, the loss of ZPI function could contribute to altering the hemostatic balance to generate a prothrombotic state.
Results
Oxidation of phospholipid vesicles
ZPI and protein Z function to regulate the activity of factor Xa on procoagulant cell membrane surfaces. To investigate whether membrane oxidation affects ZPI anticoagulant function, phospholipid vesicles composed of 30% PS, 70% PC or 100% PC and containing linolenic (18:3), linoleic (18:2), or oleic (18:1) fatty acids in the PC component but only oleic acid in the PS component were oxidized under either strong conditions with 200 μm Cu2+, 2 mm H2O2 or mild conditions with 10 μm Cu2+ in pH 7.4 Tris buffer at 37 °C as in past studies (7, 21, 22). Oxidation was monitored by ultraviolet difference spectroscopy employing tandem cuvettes at 234 nm, reflecting the formation of the conjugated diene precursors of lipid peroxidation (21). The 234-nm absorbance increased sigmoidally to a plateau for vesicles with 18:2 and 18:3 fatty acids, whereas vesicles with 18:1 fatty acids showed only a minor increase in absorbance over the same time (Fig. 1A). Addition of the antioxidant, butylated hydroxytoluene (BHT), to 18:3 vesicles at 100 μm abolished the absorbance increase. Maximal absorbance values corresponding to 0.3, 12, and 8% conversion of unsaturated fatty acids in 18:1, 18:2, and 18:3 PS/PC, respectively, to conjugated dienes (23) were reached after ∼45 min for strong conditions and ∼300 min for mild conditions (Fig. 1B), consistent with previous reports (21). Oxidation was continued at 4 °C for strong conditions or at 37 °C for mild conditions for up to 50 h to correlate the extent of oxidation with effects on ZPI activity. Based on these results, most studies were done with 18:3 PS/PC oxidized under strong conditions for 45 min at 37 °C and then at 4 °C for ∼20 h (OxPS/PC). Analysis of the light scattering properties of these vesicles revealed a scattering intensity ∼25% that of non-oxidized vesicles, suggesting that oxidation had reduced the size of the vesicles (Fig. 1C).
Figure 1.

Time course of PS/PC oxidation. A and B, oxidation of 500 μm 18:1, 18:2, or 18:3 PS/PC vesicles as indicated with either 200 μm CuSO4, 2 mm H2O2 (panel A) or 10 μm CuSO4 (panel B) in pH 7.4 Tris buffer at 37 °C monitored by conjugated diene formation at 234 nm. The effect of including 100 μm BHT on the oxidation of 18:3 vesicles is shown by open squares in panel A. C, light scattering at 320 nm produced by increasing concentrations of 18:3 OxPS/PC (●) or 18:3 PS/PC (○) in Tris buffer, pH 7.4. Lines drawn through the data are least squares fits.
Oxidation impairs PS/PC promotion of ZPI-PZ anti-FXa activity
18:3 OxPS/PC vesicles showed a reduced ability to function as a membrane cofactor in promoting ZPI-PZ complex inhibition of FXa in the presence of calcium relative to non-oxidized 18:3 PS/PC. This was evident from the reduced enhancement of the rate of ZPI-PZ complex inhibition of factor Xa by oxidized vesicles relative to non-oxidized vesicles in calcium buffer (Fig. 2, A and B). The reduced membrane cofactor function of OxPS/PC relative to PS/PC was more pronounced at 50 μm than 25 μm lipid. Notably, OxPS/PC-promoted reactions of the ZPI-PZ complex with factor Xa showed an increasing residual factor Xa activity end point as the lipid concentration was increased, whereas PS/PC-promoted reactions resulted in essentially complete inhibition of factor Xa at these lipid concentrations. This suggested a time-dependent loss of ZPI-PZ complex inhibitory function during the OxPS/PC-promoted reaction. Factor Xa activity was not significantly inhibited by the ZPI-PZ complex in the absence of lipid, confirming that inactivation of factor Xa by the ZPI-PZ complex was dependent on lipid and calcium cofactors. Moreover, factor Xa activity was unaffected by OxPS/PC or PS/PC in calcium buffer in the absence of ZPI-PZ complex, indicating that neither oxidized or unoxidized lipid directly affected factor Xa activity in the presence of calcium.
Figure 2.

Oxidation impairs PS/PC cofactor promotion of ZPI-PZ anti-FXa activity. Progress curves of the inhibition of 0.6 nm FXa by: A, 20 nm ZPI and equimolar PZ plus 25 μm 18:3 PS/PC (●) or 18:3 OxPS/PC (▴) and B, 12 nm ZPI and equimolar PZ plus 50 μm 18:3 PS/PC (●) or 18:3 OxPS/PC (▴), both in buffer containing 5 mm Ca2+. Control reactions of 0.6 nm FXa without ZPI and PZ but with added 18:3 PS/PC (○) or 18:3 OxPS/PC (▵) or with added ZPI and equimolar PZ but without lipid (♦). Solid lines are exponential fits for reactions with PS/PC and empirical lines with OxPS/PC. 18:1 and 18:3 PS/PC behaved indistinguishably as lipid cofactors. C, clotting times measured in human plasma diluted 10-fold in buffer containing 5 mm calcium after activation with either 50 μm 18:3 PS/PC and 0.7 pm TF (●, ○) or 50 μm 18:3 OxPS/PC and 2 pm TF (▴, ▵) as a function of increasing concentrations of added ZPI and equimolar PZ without (closed symbols) or with (open symbols) preincubation of ZPI/PZ with a neutralizing PZ antibody as described under “Experimental procedures.” Values represent the mean ± S.E. of 3 independent measurements. PS/PC vesicles (500 μm) were oxidized with Cu2+/H2O2 for 45 min at 37 °C and then for an additional ∼20 h at 4 °C before diluting 10–20-fold into in vitro or clotting assays.
OxPS/PC promotion of ZPI-PZ complex anti-FXa activity in the presence of calcium was similarly impaired in plasma clotting assays initiated by tissue factor (Fig. 2C). OxPS/PC vesicles required a higher tissue factor concentration than PS/PC vesicles to produce an equivalent clotting time in these assays. Addition of increasing concentrations of ZPI-PZ complex to plasma resulted in a prolongation of the clotting time in assays with both PS/PC and OxPS/PC vesicles, but the prolongation was considerably less for OxPS/PC than for PS/PC. Preincubating ZPI-PZ complex with a neutralizing PZ antibody abolished the prolongation for assays with both oxidized and non-oxidized lipids, consistent with it resulting from the anticoagulant effect of the added ZPI-PZ.
OxPS/PC inactivates ZPI anticoagulant function
To determine whether the reduced membrane cofactor function of OxPS/PC resulted from the oxidized lipid altering ZPI-PZ complex activity, the ZPI-PZ complex was preincubated with 18:3 OxPS/PC vesicles in calcium buffer and then aliquots were removed for assay of ZPI-PZ complex inhibition of FXa in the presence of unoxidized PS/PC and calcium. ZPI-PZ complex anti-FXa activity was rapidly lost by preincubating the complex with OxPS/PC (Fig. 3). Surprisingly, preincubation of free ZPI with OxPS/PC in the absence of calcium caused an even faster inactivation of the PZ-dependent anti-FXa activity of ZPI. Free ZPI in the presence of calcium or ZPI-PZ complex in the absence of calcium were inactivated much slower. Preincubating ZPI-PZ complex or free ZPI with non-oxidized PS/PC in buffer with or without calcium had no effect on ZPI-PZ complex anti-FXa activity. Incubation of PZ alone with OxPS/PC in the absence or presence of calcium resulted in minimal losses in PZ cofactor activity over the time frame of these experiments (not shown).
Figure 3.
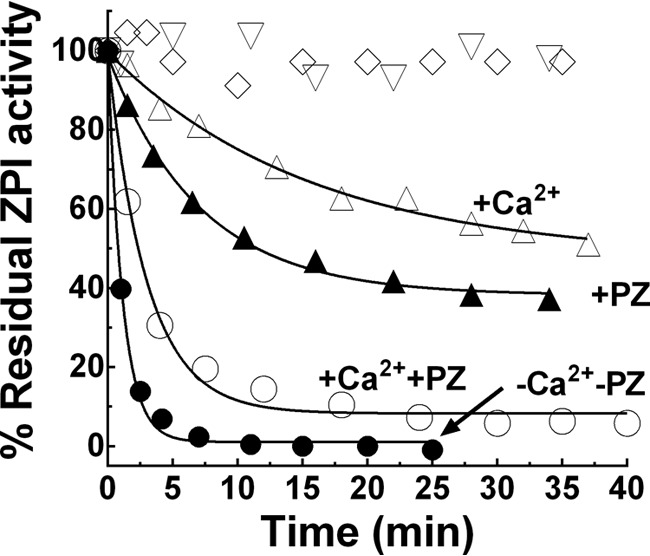
OxPS/PC inactivates ZPI and ZPI-PZ complex anticoagulant activity. Progress curves of inactivation of 150 nm ZPI by 50 μm 18:3 OxPS/PC (●) in calcium-free buffer, (▵) plus 5 mm Ca2+, (▴) plus 150 nm PZ, or (○) plus both 5 mm Ca2+ and 150 nm PZ. Vesicles (500 μm) were oxidized with Cu2+/H2O2 and diluted 10-fold into ZPI. Residual ZPI activity was measured as described under “Experimental procedures.” Effect of nonoxidized 18:3 PS/PC on the activity of ZPI in the presence of PZ/Ca2+ cofactors (▿) or in the absence of cofactors (♢). Lines are empirically drawn through the data points.
The effect of fatty acid composition of PS/PC vesicles and the oxidizing conditions on the potency of OxPS/PC inactivation of the ZPI-PZ complex or free ZPI was assessed by comparing 18:1, 18:2, and 18:3 PS/PC vesicles oxidized under either strong (200 μm Cu2+, 2 mm H2O2) or mild (10 μm Cu2+) conditions. 18:2 or 18:3 OxPS/PC achieved a similar maximal inactivating potency when oxidized under strong or mild conditions, but this potency was reached only after oxidation was allowed to proceed for 20–50 h beyond the time at which the conjugated diene markers of fatty acid hydroperoxides had reached their maximum (Figs. 1 and 4, A–E). Oxidation of vesicles prepared from a natural mixture of brain phospholipids or a defined mixture of heart phospholipids similarly rendered them rapid inactivators of free ZPI in the absence of calcium (Fig. 4, F and G) or of the ZPI-PZ complex in the presence of calcium (not shown) after 70 h oxidation. 18:1 PS/PC vesicles oxidized similarly showed a weak potency in the inactivating ZPI-PZ complex or free ZPI, consistent with the resistance of 18:1 fatty acids to oxidation (Fig. 5). Notably, 18:3 PC vesicles oxidized identically to 18:3 PS/PC showed only a weak ability to inactivate the ZPI-PZ complex or free ZPI despite a similar extent of conjugated diene formation, indicating a requirement for PS in the oxidized vesicles for rapid inactivation. The antioxidant, BHT, was ineffective in blocking OxPS/PC inactivation of the ZPI-PZ complex or free ZPI, whereas the peroxide reactive compound, KI (24, 25), was highly effective in this regard (Fig. 5).
Figure 4.
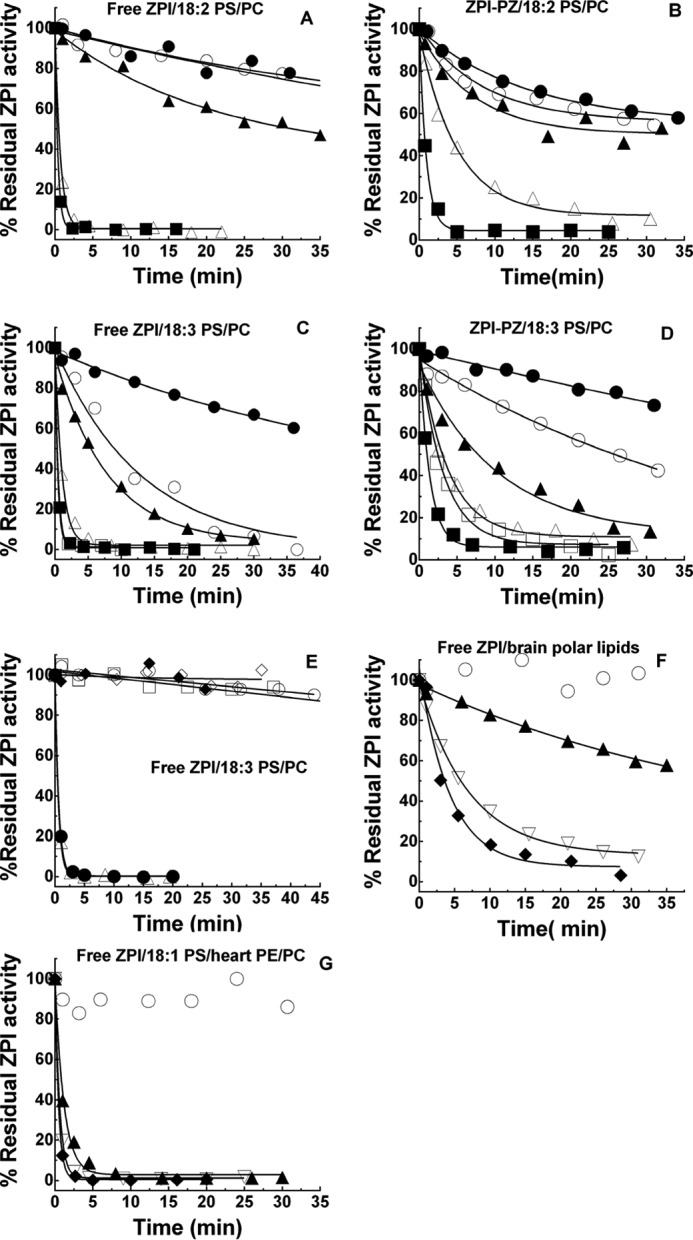
Correlation of PS/PC oxidation with potency in ZPI inactivation. Progress curves of the inactivation of 150 nm free ZPI in calcium-free buffer or 150 nm ZPI-PZ complex in buffer plus 5 mm Ca2+ by 50 μm lipid vesicles after varying times of oxidation are shown. Vesicles (500 μm) were oxidized with 200 μm CuSO4, 2 mm H2O2 for 45 min at 37 °C and then at 4 °C (panels A–D, F, and G) or with 10 μm CuSO4 at 37 °C (panel E) for the times indicated before diluting 10-fold into ZPI or ZPI-PZ complex. A, free ZPI reactions with 18:2 PS/PC oxidized for 0.5 (●), 3.7 (○), 6.1 (▴), 24 (▵), and 49 h (■); B, ZPI-PZ complex reactions with 18:2 PS/PC oxidized for 0.14 (●), 1.8 (○), 5.4 (▴), 24 (▵), and 51 h (■); C, free ZPI reactions with 18:3 PS/PC oxidized for 0.6 (●), 2.9 (○), 5.7 (▴), 24 (▵), 30 (□), and 48 h (■); D, ZPI-PZ complex reactions with 18:3 PS/PC oxidized for 0.2 (●), 1.8 (○), 4.5 (▴), 23 (▵), 28 (□), and 48 h (■); E, free ZPI reactions with 18:3 PS/PC vesicles oxidized for 2.7 (○), 5.2 (□), 18 (▵), and 26 h (●) or incubated without oxidant for 3 (♦) or 23 h (♢); F, free ZPI reactions with natural porcine brain polar lipid vesicles oxidized for 2 (○), 22 (▴), 45 (▿), and 70 h (♦); G, free ZPI reactions with vesicles comprised of synthetic 18:1 PS and natural bovine heart PE and PC after oxidation for 3 (○), 28 (▴), 51 (▿), and 74 h (♦). Residual ZPI activity was measured as described under “Experimental procedures.” Lines are empirically drawn through the data points.
Figure 5.
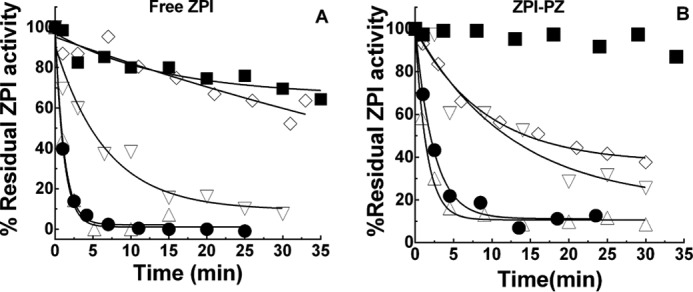
Requirements for OxPS/PC inactivation of ZPI and ZPI-PZ complex. A and B, progress curves of the inactivation of 150 nm free ZPI in calcium-free buffer (panel A) or 150 nm ZPI-PZ complex in buffer plus 5 mm Ca2+ (panel B) by 50 μm phospholipid vesicles of the following compositions: 18:3 OxPS/PC (●); 18:3 OxPC (■); 18:3 OxPS/PC (▿) in the presence of 10 mm KI; 18:3 OxPS/PC in the presence of 100 μm BHT (▵) and 18:1 OxPS/PC (♢). Vesicles (500 μm) were oxidized with Cu2+/H2O2 and diluted 10-fold into ZPI. Lines are empirically drawn through the data points.
Several control experiments demonstrated that Cu2+ or H2O2 carried over in the preincubation of ZPI with OxPS/PC made no contribution to the observed inactivation of ZPI. Thus, (i) inclusion of 1 mm EDTA in preincubations to chelate carried over Cu2+ or extensive dialysis of OxPS/PC to remove Cu2+ had no effect on the kinetics of OxPS/PC inactivation of ZPI; and (ii) preincubation of ZPI with 40 μm Cu2+ and 2 mm H2O2 produced no inactivation of ZPI anti-FXa activity (Fig. 6, A and B). Finally, enzymatic oxidation of 18:3 PS/PC vesicles with soybean lipoxygenase in the presence of octyl glucoside (7) resulted in a maximal 234 nm absorbance indistinguishable from that using other oxidation methods and after exhaustive dialysis for ∼40 h to remove detergent and reform vesicles produced oxidized vesicles that were potent inactivators of ZPI (Fig. 7).
Figure 6.
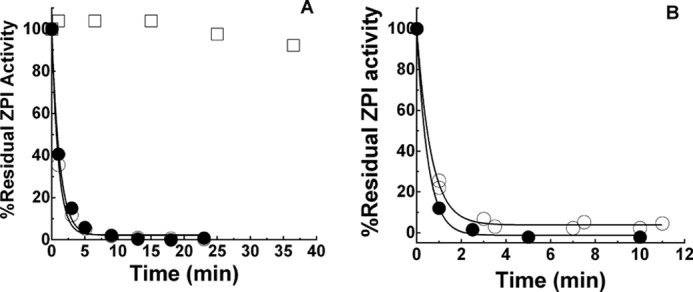
Effect of EDTA or lipid dialysis on ZPI inactivation by OxPS/PC. A, progress curves of the inactivation of 150 nm ZPI by 40 μm 18:3 OxPS/PC in calcium-free buffer in the absence (○) or presence of 1 mm EDTA (●). Control incubation of ZPI in the absence of lipid, but in the presence of 40 μm Cu2+ and 2 mm H2O2 (□). Vesicles (500 μm) were oxidized with 200 μm Cu2+, 2 mm H2O2 for 45 min at 37 °C and then incubated for ∼20 h at 4 °C before dilution into ZPI. B, progress curves of the inactivation of 150 nm ZPI by 50 μm OxPS/PC in calcium-free buffer after dialysis of OxPS/PC to remove Cu2+ (○) or without dialysis (●). Vesicles were oxidized with 10 μm Cu2+ for ∼20 h at 37 °C followed by dialysis for ∼40 h with several buffer changes at 4 °C or incubated at 4 °C without dialysis for the same time and then diluted 10-fold into buffer containing ZPI.
Figure 7.
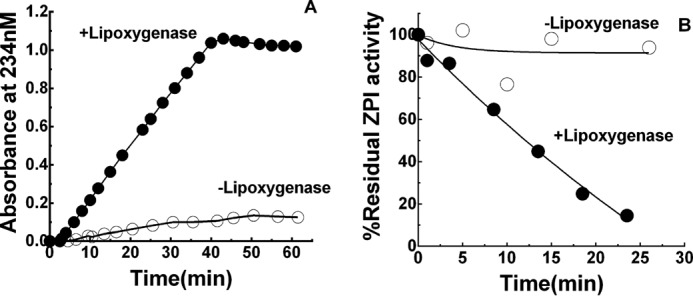
ZPI inactivation by PS/PC vesicles oxidized with lipoxygenase. A, ●, oxidation of 500 μm 18:3 PS/PC with 40 μg/ml soybean lipoxygenase in 100 mm borate, 20 mm octyl glucoside, pH 9.0, monitored by conjugated diene formation at 234 nm. ○, spontaneous oxidation in the absence of lipoxygenase. B, progress curves of the inactivation of 150 nm ZPI by ∼200 μm 18:3 OxPS/PC (●) or unoxidized 18:3 PS/PC vesicles (○). Vesicles were oxidized with or without lipoxygenase as shown in panel A. After 1 h, oxidized and non-oxidized vesicles were dialyzed against pH 7.4 Tris buffer containing Chelex resin at 4 °C for ∼40 h with several buffer changes and then extruded several times with a 100-nm filter.
We further investigated whether oxidized low density lipoprotein, an established inactivator of the anticoagulant protein, TFPI (10), was capable of inactivating ZPI anticoagulant function. Oxidation of LDL with 10 μm Cu2+ as in previous reports resulted in no significant inactivation of free ZPI in the absence of calcium or the ZPI-PZ complex in the presence of calcium (not shown). Because PC is the predominant phospholipid in LDL, such findings were consistent with the requirement for PS in OxPS/PC to mediate ZPI inactivation.
The rate of ZPI-PZ complex or free ZPI inactivation by OxPS/PC as well as the reaction extent increased by different degrees as the OxPS/PC concentration was increased. However, the extent and rate of ZPI inactivation decreased when the OxPS/PC concentration was fixed and the ZPI-PZ complex or free ZPI concentration was increased. This suggested a limited capacity of OxPS/PC to inactivate the ZPI-PZ complex or free ZPI (Fig. 8, A–E). Plasma ZPI-PZ complex was inactivated by OxPS/PC with kinetics indistinguishable from the recombinant ZPI-PZ complex, whereas free plasma ZPI (pZPI) was inactivated slower than free recombinant ZPI (rZPI). Notably, rapid and complete inactivation of the ZPI-PZ complex or free ZPI was observed at plasma concentrations of the serpin (∼50 nm) and OxPS/PC concentrations as low as 25 μm, consistent with the inactivation occurring under physiologically relevant conditions.
Figure 8.

Lipid and ZPI concentration dependence of the kinetics of OxPS/PC inactivation of ZPI and ZPI-PZ complex. A–E, progress curves of inactivation of free ZPI in calcium-free buffer (panels A, C, and E) or ZPI-PZ complex in buffer plus 5 mm Ca2+ (panels B and D) by 18:3 OxPS/PC. A and B, fixed concentrations (100 nm) of rZPI (A), rZPI and PZ (B, closed symbols), or pZPI and PZ (B, open symbols) and varied concentrations of 18:3 OxPS/PC as indicated; C and D, fixed concentration (50 μm) of 18:3 OxPS/PC and varied concentrations of rZPI (C) or rZPI plus PZ (D) as indicated; E, fixed concentrations (40 nm) of rZPI (○) or pZPI (●) and 15 μm 18:3 OxPS/PC. Vesicles (500 μm) were oxidized with Cu2+/H2O2 and diluted 10–33-fold into ZPI or ZPI-PZ complex. Lines are empirically drawn through the data points.
ZPI binding to lipid
The PS requirement for OxPS/PC inactivation of ZPI-PZ complex or free ZPI implied that both PZ-complexed and free ZPI must bind to oxidized lipid vesicles to promote the inactivation. Because free ZPI has not been thought to bind PS/PC vesicles, we sought evidence for such binding. Gel filtration chromatography of ZPI alone or after mixing with non-oxidized vesicles in calcium-free buffer showed that PS/PC but not PC vesicles dose-dependently shifted the ZPI elution peak to a higher molecular weight peak corresponding to the vesicle elution peak, as revealed by chromatography of NBD-labeled vesicles (Fig. 9A and Table 1). Calcium did not affect the PS/PC-induced shift in ZPI elution. ZPI complexation with PZ abrogated the ability of PS/PC vesicles to shift the complexed ZPI elution peak in calcium-free buffer, but promoted a complete shift of the PZ-complexed ZPI peak in buffer containing calcium (Table 1). These findings suggested a calcium-independent weak binding of free ZPI to PS/PC vesicles that is blocked by PZ binding to ZPI in the absence but not in the presence of calcium due to the calcium-dependent binding of ZPI-PZ complex to PS/PC through PZ. Notably, neither the heparin-binding serpin, antithrombin, nor the A- and B-clade serpins, antitrypsin and ovalbumin, showed shifts in their elution when mixed with the same concentrations of PS/PC vesicles (not shown), suggesting that ZPI binding to lipid was specific.
Figure 9.

ZPI binds PS/PC vesicles through the heparin-binding site. A, gel filtration chromatography profiles of 1.5 μm ZPI alone (cyan) or after mixing with 250 μm PS/PC (red) or 1000 μm PS/PC (black) in calcium-free buffer on a Superdex 200 column with detection of ZPI elution by protein fluorescence as described under “Experimental procedures.” B and D, progress curves of inactivation of free ZPI in calcium-free buffer or ZPI-PZ complex in buffer plus 5 mm Ca by 50 μm 18:3 OxPS/PC in the absence and presence of the indicated concentrations of 50-saccharide heparin. Lines are empirically drawn through the data points. C, gel filtration profiles of 1.5 μm ZPI alone (black) and after mixing with 28 μm 50-saccharide heparin (blue), 1000 μm 18:3 PS/PC (red), or both 28 μm heparin and 1000 μm PS/PC (green) in calcium-free buffer on a Superdex 200 column with detection of ZPI elution by protein fluorescence. E, gel filtration chromatography profiles of 1.7 μm PZ alone (green) after adding 1.5 μm ZPI (cyan) and after adding 1.5 μm ZPI and 50 (blue) or 170 μm (black) 50-saccharide heparin (H50) in calcium-free buffer on a Superdex 200 column with detection of proteins by protein fluorescence. The reduction in the ZPI-PZ complex peak upon H50 addition results in an increase in the free PZ peak and presumably also a ZPI-H50 complex peak, which migrates close to the position of PZ.
Table 1.
Binding of ZPI and ZPI-PZ complex to phospholipid vesicles
15 μg of rZPI with or without 20 μg of PZ was mixed with buffer or with 18:1 PS/PC or 18:1 PC vesicles at the indicated concentrations in calcium-free Tris buffer, pH 7.1, when PZ was absent or buffer plus 5 mm Ca2+ when PZ was present. 200 μl was then applied to a Superdex 200 column and the proteins were eluted at 0.4 ml/min with buffer and detected by protein fluorescence as described under “Experimental procedures.” Areas under peaks were quantitated with Waters HPLC software. Data represent averages ± S.E. of 2–3 independent measurements.
| Proteins | Ca2+ | Lipid | Concentration | Residual peak area |
|---|---|---|---|---|
| μm | % | |||
| ZPI | − | − | − | 100 |
| ZPI | − | PC | 250 | 107 ± 4 |
| ZPI | − | PC | 1000 | 88 ± 7 |
| ZPI | − | PS/PC | 250 | 73 ± 4 |
| ZPI | − | PS/PC | 1000 | 28 ± 3 |
| ZPI | + | − | − | 100 |
| ZPI | + | PS/PC | 250 | 77 ± 1 |
| ZPI/PZ | − | − | − | 100 |
| ZPI/PZ | − | PS/PC | 250 | 94 ± 2 |
| ZPI/PZ | − | PS/PC | 1000 | 91 ± 1 |
| ZPI/PZ | + | PS/PC | 250 | 3 ± 3 |
| PZ | + | − | − | 100 |
| PZ | + | PS/PC | 250 | 5 ± 0a |
a Residual peak likely reflects inactive protein.
Heparin protects ZPI from OxPS/PC inactivation
The negatively charged glycosaminoglycan, heparin, binds ZPI through a basic site and promotes ZPI inhibition of FXa in the absence of PZ and lipid (26, 27). To determine whether ZPI bound negatively charged PS/PC vesicles through the heparin-binding site, we examined the effect of heparin on the inactivation of free ZPI by OxPS/PC and on the binding of ZPI to PS/PC. Heparin dose-dependently protected ZPI from inactivation by OxPS/PC (Fig. 9B). Gel filtration experiments further showed that heparin reduced the amount of ZPI eluting with PS/PC vesicles and caused the free ZPI peak to shift to a higher molecular weight peak corresponding to that observed when ZPI was mixed with heparin alone (Fig. 9C). Such results were consistent with calcium-independent binding of ZPI to lipid through the heparin-binding site.
Surprisingly, heparin also protected the ZPI-PZ complex from OxPS/PC inactivation in the presence of calcium, although not as effectively as free ZPI (Fig. 9D). Gel filtration experiments showed that addition of heparin to the ZPI-PZ complex dose-dependently reduced the ZPI-PZ complex peak and increased the peak corresponding to elution of free PZ, consistent with heparin protecting the ZPI-PZ complex from OxPS/PC inactivation as a result of competitive binding of heparin and PZ to ZPI (Fig. 9E).
Products of ZPI inactivation
The products of ZPI and ZPI-PZ complex inactivation by OxPS/PC were analyzed by native PAGE. Inactivation of free ZPI by OxPS/PC resulted in an increased mobility of ZPI and a multiple banding pattern indicative of ZPI polymerization (Fig. 10A). Immunoblotting confirmed that the multiple bands arose from OxPS/PC modifications of ZPI (Fig. 10B). Nonreducing SDS-PAGE showed that inactivated ZPI migrated as a single band with the same mobility as native ZPI, indicating that the ZPI polymers on native PAGE were noncovalent (Fig. 10C). Inactivation of the ZPI-PZ complex by OxPS/PC resulted in loss of the higher mobility ZPI-PZ complex band on native PAGE (16), appearance of a free PZ band and multiple faster migrating ZPI bands similar to those seen in the inactivation of free ZPI (Fig. 10A). The mobilities of free ZPI or the ZPI-PZ complex were unaffected by non-oxidized PC vesicles, but were shifted by non-oxidized PS/PC vesicles in a manner consistent with binding to both gel-impermeant and permeant vesicles.
Figure 10.
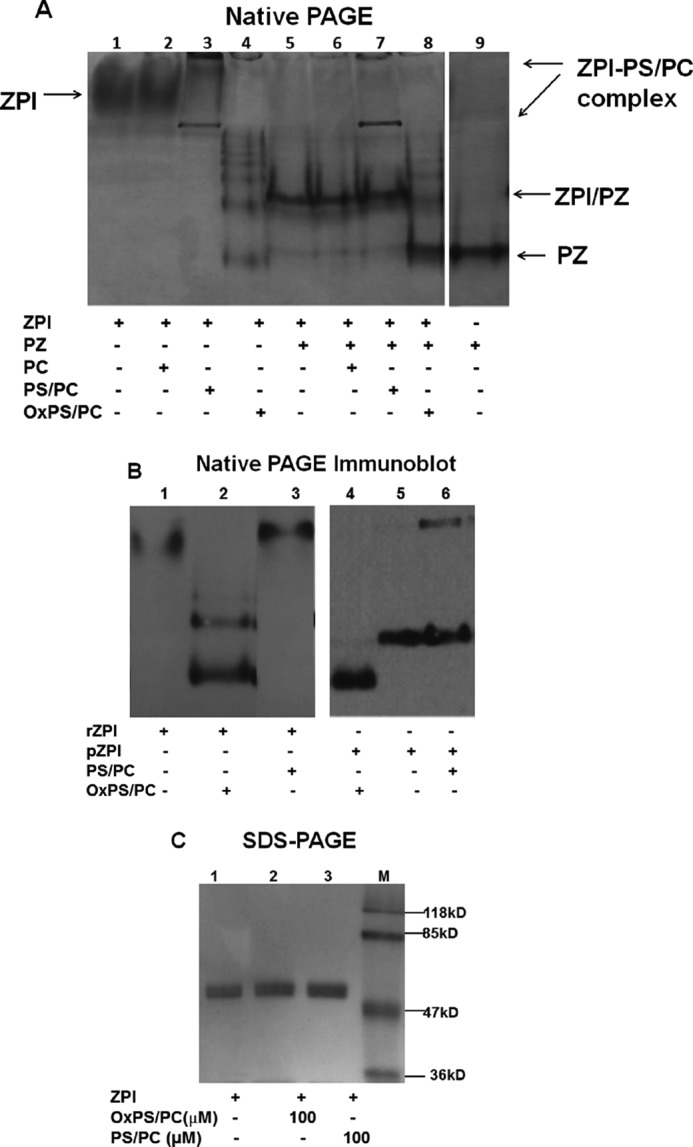
PAGE analysis of the products of OxPS/PC inactivation of ZPI. A, 5.5% native PAGE analysis of 2 μm ZPI (4 μg) alone (lanes 1–4) or with added PZ (1.7 μm, 4.3 μg) (lanes 5–8) after incubation without lipid or with 200 μm PC, PS/PC, or OxPS/PC vesicles in buffer without calcium when PZ was absent and with 5 mm Ca2+ when PZ was present for 25 min. Lane 9, PZ alone. Protein bands were detected by Coomassie Blue staining. B, 5.5% native PAGE analysis of 60 nm rZPI (lanes 1–3) or pZPI (lanes 4–6) after incubation in the absence or presence of 50 μm 18:3 OxPS/PC or PS/PC in calcium-free buffer at 25 °C for 25 min. ZPI bands were detected by immunoblotting with an anti-ZPI antibody as described under “Experimental procedures.” C, SDS-PAGE of 1.5 μm ZPI (3 μg) after incubation without lipid or with 100 μm OxPS/PC or PS/PC vesicles for 25 min in calcium-free buffer. Protein bands were stained with Coomassie Blue. Molecular weight markers are in lane M.
Immunoblotting analyses comparing the products of recombinant and plasma forms of free ZPI inactivated by OxPS/PC on native PAGE showed that inactivation increased the mobilities of both ZPI forms relative to native ZPI mobilities, the latter differing due to differential glycosylation (Fig. 10B). Interestingly, inactivation of only recombinant ZPI and not plasma ZPI resulted in the formation of polymers. Control PS/PC vesicles shifted the mobility of recombinant and plasma forms of ZPI in accord with ZPI binding to the vesicles.
Discussion
We have demonstrated a marked susceptibility of the anticoagulant serpin, ZPI, either free or in its physiologically relevant complex with the cofactor, PZ, to oxidative inactivation by lipid peroxidation products known to be generated by oxidative stress and to contribute to the pathogenesis of atherosclerosis and thrombosis. The protein Z-dependent anti-FXa activity of ZPI was rapidly inactivated by low concentrations of oxidized PS/PC vesicles containing 18:2 or 18:3 polyunsaturated fatty acid chains but minimally by non-oxidized vesicles or oxidized PS/PC vesicles with 18:1 fatty acid chains that are resistant to oxidation (21, 28). The comparable effects of oxidized 18:2 and 18:3 fatty acids on ZPI inactivation implies that oxidation of the Δ9Δ12 double bonds common to these fatty acids accounts for their similar abilities to inactivate ZPI and that such oxidation is relevant to the 18:2 fatty acids commonly found in human membrane lipids (29). The ability of oxidized brain or heart phospholipids with natural fatty acid compositions to rapidly inactivate free ZPI or ZPI-PZ complex reinforces the physiologic significance of ZPI inactivation by oxidized natural lipids. Oxidized phospholipids containing PS have been documented in vivo in apoptotic cells of atherosclerotic lesions or in erythrocytes under conditions of oxidative stress at levels expected to cause ZPI inactivation (30, 31). Both recombinant and plasma ZPI-PZ complexes were similarly susceptible to oxidative inactivation at plasma concentrations (∼50 nm). Because PZ is limiting and acts catalytically (15, 16), free ZPI also circulates in blood under physiologic conditions and its susceptibility to oxidative inactivation is thus likely to also be physiologically significant.
The observation that oxidized PC vesicles were much less effective in inactivating the ZPI-PZ complex or free ZPI despite similar extents of lipid peroxidation indicated that PS was required for rapid ZPI inactivation. This was confirmed from the finding that both PZ-complexed and free ZPI bound to non-oxidized PS/PC vesicles but not PC vesicles based on the vesicle-induced shifts in elution of the ZPI-PZ complex or free ZPI from a size exclusion column or in mobility on native PAGE. Free ZPI binding to PS/PC vesicles was independent of calcium and weaker than that of the ZPI-PZ complex, the latter which is calcium-dependent and mediated by binding of the complex through the Gla domain of PZ (32). Evidence that free ZPI is inactivated by OxPS/PC by binding vesicles through the heparin-binding site of the serpin was obtained by the findings that heparin dose-dependently protects ZPI from inactivation by OxPS/PC and heparin displaces ZPI from its complex with PS/PC vesicles. That the ZPI-PZ complex is inactivated by OxPS/PC through the calcium-dependent binding of the complex to the vesicles via PZ was supported by the finding that in the absence of calcium, ZPI complexation with PZ blocks the ability of the complex to bind PS/PC vesicles and to be inactivated by OxPS/PC. Heparin also protected the ZPI-PZ complex from inactivation and this was correlated with the ability of heparin to compete with PZ for binding to ZPI. Such findings suggest that the PZ and heparin-binding sites on ZPI may be contiguous.
Interestingly, calcium alone protected free ZPI from inactivation by OxPS/PC even though calcium had only a modest effect on the direct interaction of ZPI with lipid. Calcium similarly antagonizes the ability of heparin to promote ZPI inhibition of factor Xa despite having little effect on ZPI binding to heparin (26). This observation has suggested that calcium interactions with heparin alter the mode of ZPI binding to heparin and thereby the ability of heparin to effectively bridge the ZPI-factor Xa interaction. Calcium interactions with lipid may thus likewise alter the mode of free ZPI binding to lipid and affect its reactivity with oxidants on the lipid surface.
Recombinant and plasma ZPIs were inactivated by OxPS/PC with similar kinetics when complexed with PZ in the presence of calcium but different kinetics when free in the absence of calcium, implying that glycosylation differences affect the susceptibility of ZPI to OxPS/PC inactivation when ZPI is directly bound to the lipid surface but not when it is indirectly bound through PZ. Other quantitative differences in the kinetics of the reactions of the ZPI-PZ complex and free ZPI likely arise from the different affinities and modes of interaction of free ZPI and the ZPI-PZ complex with the lipid surface. The faster extent and rate of inactivation of the ZPI-PZ complex than free ZPI at low OxPS/PC concentrations thus correlates well with the greater affinity of complexed than free ZPI for PS/PC vesicles.
Native PAGE analyses revealed that oxidative inactivation of free or PZ-complexed recombinant or plasma forms of ZPI modifies residues that result in an increased negative charge of the serpin and in the case of recombinant ZPI also non-covalent polymerization. Because of their metastability, all serpins are prone to noncovalent polymerization (33). The polymerization of inactivated recombinant ZPI but not plasma ZPI suggests that glycosylation differences confer differential stability and susceptibility to polymerization (34). Moreover, it implies that polymerization is a secondary consequence of recombinant ZPI inactivation and that oxidative modification of the ZPI monomer accounts for the inactivation of anticoagulant function. Inactivation of the ZPI-PZ complex resulted in displacement of PZ and modifications of ZPI similar to those observed with free ZPI, suggesting that common oxidative modifications of free ZPI and the ZPI-PZ complex that impair PZ binding are responsible for inactivation of ZPI anticoagulant activity (Fig. 11). Such modifications could involve the oxidation of tyrosine and methionine residues known to be critical for PZ binding and the inhibition of FXa (17, 18, 35).
Figure 11.
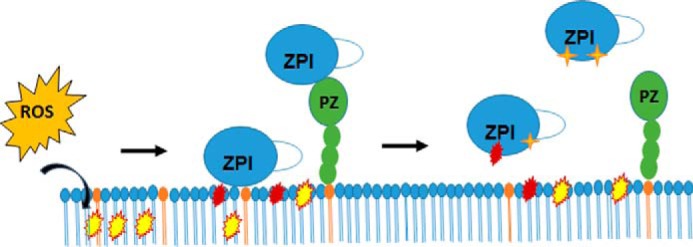
Scheme illustrating the proposed mechanism of oxidative inactivation of free or PZ-complexed ZPI by oxidized PS/PC lipid vesicles. Reactive oxygen species (ROS) generated by oxidative stress oxidize polyunsaturated fatty acids in the vesicles to hydroperoxides (yellow). Further oxidation causes fragmentation of the fatty acid chains and results in reactive aldehydes at the ends of these chains (red) and reactive hydroperoxides to be exposed on or near the vesicle surface. Binding of the ZPI-PZ complex through protein Z (green) or free ZPI (blue) through the heparin-binding site to the vesicle surface brings the serpin in close proximity to the reactive groups on the lipid surface. The latter then modify amino acids of ZPI (red and orange symbols) that weaken PZ binding and cause ZPI release from PZ.
The nature of the reactive oxidants generated by oxidation of the polyunsaturated 18:2 and 18:3 fatty acids in PS/PC vesicles is suggested by our finding that the potency of OxPS/PC vesicles to inactivate ZPI or the ZPI-PZ complex was greatly enhanced by prolonged oxidation of the vesicles even after the conjugated diene markers of lipid peroxidation had maximally formed. Fatty acid lipid hydroperoxides are known to undergo further oxidation that causes fragmentation of the fatty acid chains, resulting in shorter chains terminating in polar aldehyde and carboxylate functional groups (5). The increased polarity of these truncated fatty acid chains, both free and covalently linked to phospholipid, result in their exposure on the vesicle surface and a consequent reduction in vesicle size (36). We in fact observed that prolonged oxidation of vesicles resulted in a reduction in their size relative to non-oxidized vesicles. Prolonged oxidation of PS/PC vesicles may thus be required to shrink the vesicles and place reactive aldehydes and fatty acid hydroperoxides in close proximity to bound ZPI to promote rapid ZPI inactivation (Fig. 11). The involvement of peroxides would be consistent with the partial protection of ZPI from inactivation by the peroxide and radical scavenger, KI (24, 25).
Oxidized PS/PC vesicles were found to retain their ability to function as a membrane surface to promote ZPI-PZ complex inhibition of membrane-associated FXa in the presence of calcium both in vitro and ex vivo in plasma. However, their efficacy in supporting ZPI-PZ anticoagulant function was reduced relative to non-oxidized vesicles due fully or in part to the concomitant inactivation of the bound ZPI-PZ complex. Interestingly, other studies have found that oxidized membranes possess an enhanced ability to support coagulation reactions. Oxidation of PS/PC vesicles containing 20:4 fatty acids was found to enhance the activity of the prothrombinase complex 6-fold (6) and oxidation of natural brain or synthetic PS/PE/PC but not PS/PC phospholipid vesicles containing 18:2 or 20:4 unsaturated fatty acids was reported to enhance both the protein S-dependent and -independent ability of activated protein C to inactivate factor Va (7). More recently, non-oxidized PS or oxidized PS or PE vesicles with 20:4 fatty acid chains were reported to promote the binding of the serpin, protein C inhibitor (PCI), to lipid and stimulate the inhibition of aPC in the presence of calcium (8). Such effects may result from the increased negative charge of the vesicle surface resulting from the exposure of carboxylate groups produced by oxidation of polyunsaturated fatty acids (36). Notably, PCI was shown to bind lipid through the heparin-binding site of PCI, which mediates heparin promotion of PCI inhibition of aPC. Such findings parallel our present finding that free ZPI binds to PS containing lipid vesicles through the heparin-binding site of the serpin and may be related to the structural similarities of these serpins, both of which belong to the A clade.
Our findings have implications for the pathogenesis of chronic diseases such as atherosclerosis in which oxidative stress and lipid peroxidation are present continuously as a low strength stimulus (3). In such situations, the gradual loss of ZPI together with the reported loss of TFPI anticoagulant activity (9) and gain of procoagulant activities associated with lipid peroxidation (5) could contribute to a prothrombotic state as a result of the inability to regulate FXa procoagulant activity.
Experimental procedures
Materials
Plasma-derived human FXa and protein Z were purchased from Enzyme Research Laboratories. Recombinant human ZPI (rZPI) was expressed in baculovirus-infected insect cells and purified as described (16). Plasma ZPI (pZPI) was kindly provided by Professor George Broze, Jr. (Washington University) or purified as described (12). rZPI and pZPI behaved indistinguishably as cofactor-dependent inhibitors of FXa (16). ZPI refers to the recombinant protein unless otherwise indicated. Human recombinant α1-antitrypsin was expressed and purified as reported (37). Plasma-derived human α-antithrombin was purified as described (38). Protein concentrations were determined from the absorbance at 280 nm based on published extinction coefficients (16, 17, 37, 38). The phospholipids, 1,2-dioleoyl-sn-glycero-3-phosphocholine (18:1 PC), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (18:2 PC), 1,2-dilinolenoyl-sn-glycero-3-phosphocholine (18:3 PC), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (18:1 PS) with or without an NBD fluorescent label, natural porcine brain polar lipids, or purified natural PE and PC phospholipids from bovine heart were purchased from Avanti Polar Lipids. Phospholipid concentrations were determined by colorimetric assay (39). 50-Saccharide heparin was purified from unfractionated heparin as described (38).
Preparation of lipid vesicles
Phospholipid vesicles containing the synthetic lipids, 100% 18:3 PC or 30% 18:1 PS plus 70% of 18:1 PC, 18:2 PC, or 18:3 PC (by weight) (designated 18:1 PS/PC, 18:2 PS/PC, 18:3 PS/PC, respectively), as well as the natural porcine brain lipid mixture or synthetic 18:1 PS (20%) plus bovine heart PE (40%) and PC (40%) were prepared by sonication of dried phospholipids after suspension in 50 mm Tris, 0.1 m NaCl, pH 7.4, essentially as described except that the centrifugation step was omitted (40).
Experimental conditions
Experiments were performed in 50 mm Tris, 0.1 m NaCl, pH 7.4, at 25 °C unless specified otherwise.
Lipid promotion of ZPI-PZ anti-FXa activity
Kinetic studies comparing the ability of oxidized and non-oxidized 18:3 PS/PC vesicles to accelerate ZPI-PZ complex inhibition of FXa were done under pseudo-first order conditions ([ZPI-PZ]o ≫ [FXa]o). 0.6 nm FXa and 25–50 μm lipid were mixed in buffer containing 5 mm calcium and reactions were initiated by adding 12–25 nm rZPI and equimolar PZ. Residual FXa activity was measured after increasing reaction times by dilution into fluorogenic substrate as described below. Control reactions omitted either ZPI-PZ complex or lipid.
Clotting assays
To 100 μl of citrated normal human plasma (BIOPHEN) was added 900 μl of buffer containing 1 mg/ml of ovalbumin, 6 mm Ca2+, 50 μm 18:3 OxPS/PC or PS/PC, 0.7–2 pm tissue factor (TF) (Innovin, Dade-Behring), and 0–50 nm rZPI and equimolar PZ at 25 °C with rZPI/PZ added last. Clot formation was continuously monitored from the sigmoidal increase in absorbance at 350 nm due to light scattering until a plateau absorbance was reached. The clotting time was determined as the time corresponding to the intersection of a line drawn through the steepest part of the clotting curve with the baseline absorbance (41). To block ZPI/PZ activity, goat anti-PZ polyclonal antibody (1.7 mg/ml) (Cedarlane Lab) was preincubated with an equimolar mixture of ZPI and PZ at a 10-fold higher concentration at 25 °C before adding to clotting reactions.
Effect of oxidized lipid vesicles on ZPI activity
rZPI or pZPI (40–150 nm) was mixed with oxidized or non-oxidized PS/PC or PC vesicles with different fatty acid compositions in the absence or presence of equimolar PZ, 5 mm Ca2+ and heparin as indicated. To ensure consistency and reproducibility, a standardized vortex mixing procedure was adopted (2.5 rpm for 20 s). At different times, an aliquot was diluted 4–6-fold into buffer containing 5 mm calcium, 25 μm nonoxidized 18:1 PS/PC, 0.1–0.5 nm FXa and PZ to yield 10–23 nm ZPI and PZ in 100 μl. 0.1% PEG and 0.1 mg/ml of ovalbumin were included in the dilution buffer. After reaction for a fixed time of 40–90 s, 1 ml of 40 μm fluorogenic FXa substrate (Pefafluor FXa, Sekisui Diagnostics) in 20 mm sodium phosphate, 0.1 m NaCl, 50 μg/ml of Polybrene, pH 6.7, was added and the residual FXa activity was measured by monitoring the linear increase in fluorescence due to FXa hydrolysis of the substrate (λex 360 nm, λem 440 nm). Low pH buffer minimized ZPI-FXa complex dissociation during the 2–3 min required to monitor substrate hydrolysis (16). The reaction of ZPI-PZ complex with FXa under these conditions is pseudo-first order with the observed rate constant (kobs) given by the equation,
| (Eq. 1) |
where vo and vt are the initial velocities of FXa hydrolysis of substrate measured for reactions in the absence of inhibitor and in the presence of inhibitor after the fixed reaction time t, respectively. Velocities were corrected for a minor fraction of degraded FXa that is resistant to inhibition by the ZPI-PZ complex (∼5%). kobs is proportional to the concentration of ZPI-PZ complex over this range of inhibitor concentrations and thus provides a measure of the active ZPI concentration remaining after preincubation with lipid (16).
Analysis of ZPI binding to lipid
rZPI (1.5 μm) with or without equimolar PZ was mixed with (i) buffer, (ii) PC or PS/PC vesicles (0.25–1 mm), (iii) heparin (30–170 μm), or (iv) both PS/PC vesicles and heparin in 50 mm Tris, 0.1 m NaCl, pH 7.1, with or without 5 mm Ca2+. Mixtures (200 μl) were then applied to a Superdex 200 (1 × 30 cm) gel filtration column and eluted with buffer at 0.4 ml/min. Protein peaks were detected by fluorescence (λex 280 nm, λem 340 nm) and quantitated with Waters HPLC software. Similar experiments were performed with the serpins, ovalbumin, α1-antitrypsin, and antithrombin.
PAGE and immunoblotting
rZPI or pZPI with or without equimolar PZ were mixed with OxPS/PC or PS/PC vesicles in buffer with or without 5 mm Ca2+. Samples were analyzed by native PAGE at 4 °C with 5.5% gels or by non-reducing SDS-PAGE with 10% polyacrylamide gels as described (16). Protein bands were either stained with Coomassie Blue or transferred to a PVDF membrane and detected by immunoblotting with a rabbit anti-ZPI antibody (Sigma) and chemiluminescence.
Author contributions
X. H., Y. W., and S. T. O. designed the study; X. H., B. L., R. B., and H. Y. performed experiments and analyzed data; X. H. and S. T. O. wrote the manuscript.
Acknowledgment
We thank Dr. Peter Gettins of the Department of Biochemistry and Molecular Genetics, University of Illinois at Chicago, for critical comments on the manuscript.
Note added in proof
In the version of this article that was published as a Paper in Press on July 17, 2017, Fig. 10, A and B, did not indicate the borders between different sections of a gel. These errors have now been corrected and do not affect the results or conclusions of this work.
This work was supported by American Heart Association Scientist Development Grant SDG 48880022 (to X. H.), National Institutes of Health Grant R37 HL39888 (to S. T. O.), International Exchange Program for Graduate Student, Shanghai Tongji University Grants 2015020040 (to B. L.) and 2015020041 (to H. Y.), National Natural Science Foundation of China Grants 30800466 and 81270193 (to Y. W.), and the MOST Summer Undergraduate Program, University of Illinois College of Dentistry (to R. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- aPC
- activated protein C
- PZ
- protein Z
- FXa
- factor Xa
- PS
- phosphatidylserine
- PC
- phosphatidylcholine
- PS/PC
- phosphatidylserine/phosphatidylcholine vesicles
- OxPS/PC
- oxidized phosphatidylserine/phosphatidylcholine vesicles
- NBD
- N,N′-dimethyl-N-(acetyl)-N′-(7-nitrobenz-3-oxa-1,3-diazol-4-yl)ethylenediamine
- TFPI
- tissue factor pathway inhibitor
- TF
- tissue factor
- BHT
- butylated hydroxytoluene
- PE
- phosphatidylethanolamine
- PCI
- protein C inhibitor.
References
- 1. Steinberg D. (1997) Low density lipoprotein oxidation and its pathobiological significance. J. Biol. Chem. 272, 20963–20966 [DOI] [PubMed] [Google Scholar]
- 2. Lee S., Birukov K. G., Romanoski C. E., Springstead J. R., Lusis A. J., and Berliner J. A. (2012) Role of phospholipid oxidation products in atherosclerosis. Circ. Res. 111, 778–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ellulu M. S., Patimah I., Khaza'ai H., Rahmat A., Abed Y., and Ali F. (2016) Atherosclerotic cardiovascular disease: a review of initiators and protective factors. Inflammopharmacology 24, 1–10 [DOI] [PubMed] [Google Scholar]
- 4. Fruhwirth G. O., Loidl A., and Hermetter A. (2007) Oxidized phospholipids: from molecular properties to disease. Biochim. Biophys. Acta 1772, 718–736 [DOI] [PubMed] [Google Scholar]
- 5. Salomon R. G. (2012) Structural identification and cardiovascular activities of oxidized phospholipids. Circ. Res. 111, 930–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weinstein E. A., Li H., Lawson J. A., Rokach J., FitzGerald G. A., and Axelsen P. H. (2000) Prothrombinase acceleration by oxidatively damaged phospholipids. J. Biol. Chem. 275, 22925–22930 [DOI] [PubMed] [Google Scholar]
- 7. Safa O., Hensley K., Smirnov M. D., Esmon C. T., and Esmon N. L. (2001) Lipid oxidation enhances the function of activated protein C. J. Biol. Chem. 276, 1829–1836 [DOI] [PubMed] [Google Scholar]
- 8. Malleier J. M., Oskolkova O., Bochkov V., Jerabek I., Sokolikova B., Perkmann T., Breuss J., Binder B. R., and Geiger M. (2007) Regulation of protein C inhibitor (PCI) activity by specific oxidized and negatively charged phospholipids. Blood 109, 4769–4776 [DOI] [PubMed] [Google Scholar]
- 9. Hiraishi S., Horie S., and Seyama Y. (2002) Oxidation products of phospholipid-containing Δ-9 fatty acids specifically impair the activity of tissue factor pathway inhibitor. Biochem. Biophys. Res. Commun. 298, 468–473 [DOI] [PubMed] [Google Scholar]
- 10. Horie S., Hiraishi S., Hamuro T., Kamikubo Y., and Matsuda J. (2002) Oxidized low-density lipoprotein associates strongly with carboxy-terminal domain of tissue factor pathway inhibitor and reduces the catalytic activity of the protein. Thromb. Haemost. 87, 80–85 [PubMed] [Google Scholar]
- 11. Becatti M., Marcucci R., Bruschi G., Taddei N., Bani D., Gori A. M., Giusti B., Gensini G. F., Abbate R., and Fiorillo C. (2014) Oxidative modification of fibrinogen is associated with altered function and structure in the subacute phase of myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 34, 1355–1361 [DOI] [PubMed] [Google Scholar]
- 12. Han X., Fiehler R., and Broze G. J. Jr. (1998) Isolation of a protein Z-dependent plasma protease inhibitor. Proc. Natl. Acad. Sci. U.S.A. 95, 9250–9255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han X., Huang Z. F., Fiehler R., and Broze G. J. Jr. (1999) The protein Z-dependent protease inhibitor is a serpin. Biochemistry 38, 11073–11078 [DOI] [PubMed] [Google Scholar]
- 14. Tabatabai A., Fiehler R., and Broze G. J. Jr. (2001) Protein Z circulates in plasma in a complex with protein Z-dependent protease inhibitor. Thromb. Haemost. 85, 655–660 [PubMed] [Google Scholar]
- 15. Han X., Fiehler R., and Broze G. J. Jr. (2000) Characterization of the protein Z-dependent protease inhibitor. Blood 96, 3049–3055 [PubMed] [Google Scholar]
- 16. Huang X., Swanson R., Broze G. J. Jr., and Olson S. T. (2008) Kinetic characterization of the protein Z-dependent protease inhibitor reaction with blood coagulation factor Xa. J. Biol. Chem. 283, 29770–29783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang X., Dementiev A., Olson S. T., and Gettins P. G. (2010) Basis for the specificity and activation of the serpin protein Z-dependent proteinase inhibitor (ZPI) as an inhibitor of membrane-associated factor Xa. J. Biol. Chem. 285, 20399–20409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang X., Yan Y., Tu Y., Gatti J., Broze G. J. Jr., Zhou A., and Olson S. T. (2012) Structural basis for catalytic activation of protein Z-dependent protease inhibitor (ZPI) by protein Z. Blood 120, 1726–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yin Z. F., Huang Z. F., Cui J., Fiehler R., Lasky N., Ginsburg D., and Broze G. J. Jr. (2000) Prothrombotic phenotype of protein Z deficiency. Proc. Natl. Acad. Sci. U.S.A. 97, 6734–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J., Tu Y., Lu L., Lasky N., and Broze G. J. Jr. (2008) Protein Z dependent protease inhibitor deficiency produces a more severe murine phenotype than protein Z deficiency. Blood 111, 4973–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vossen R. C., van Dam-Mieras M. C., Hornstra G., and Zwaal R. F. (1993) Continuous monitoring of lipid peroxidation by measuring conjugated diene formation in an aqueous liposome suspension. Lipids 28, 857–861 [DOI] [PubMed] [Google Scholar]
- 22. Patel R. P., Svistunenko D., Wilson M. T., and Darley-Usmar V. M. (1997) Reduction of Cu(II) by lipid hydroperoxides: implications for the copper-dependent oxidation of low-density lipoprotein. Biochem. J. 322, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Esterbauer H., Striegl G., Puhl H., and Rotheneder M. (1989) Continuous monitoring of in vitro oxidation of human low density lipoprotein. Free Radic. Res. Commun. 6, 67–75 [DOI] [PubMed] [Google Scholar]
- 24. Hicks M., and Gebicki J. M. (1979) A spectrophotometric method for the determination of lipid hydroperoxides. Anal. Biochem. 99, 249–253 [DOI] [PubMed] [Google Scholar]
- 25. Blok J., and Verhey W. S. (1968) The attack of free radicals on biologically active DNA in irradiated aqueous solutions. Radiat. Res. 34, 689–703 [PubMed] [Google Scholar]
- 26. Huang X., Rezaie A. R., Broze G. J. Jr., and Olson S. T. (2011) Heparin is a major activator of the anticoagulant serpin, protein Z-dependent protease inhibitor. J. Biol. Chem. 286, 8740–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang L., Ding Q., Huang X., Olson S. T., and Rezaie A. R. (2012) Characterization of the heparin-binding site of the protein Z-dependent protease inhibitor. Biochemistry 51, 4078–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Porter N. A., Caldwell S. E., and Mills K. A. (1995) Mechanisms of free radical oxidation of unsaturated lipids. Lipids 30, 277–290 [DOI] [PubMed] [Google Scholar]
- 29. Vossen R. C., van Dam-Mieras M. C., Lemmens P. J., Hornstra G., and Zwaal R. F. (1991) Membrane fatty acid composition and endothelial cell functional properties. Biochim. Biophys. Acta 1083, 243–251 [DOI] [PubMed] [Google Scholar]
- 30. Bochkov V. N., Oskolkova O. V., Birukov K. G., Levonen A. L., Binder C. J., and Stöckl J. (2010) Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 12, 1009–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jain S. K. (1985) In vivo externalization of phosphatidylserine and phosphatidylethanolamine in the membrane bilayer and hypercoagulability by the lipid peroxidation of erythrocytes in rats. J. Clin. Invest. 76, 281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McDonald J. F., Shah A. M., Schwalbe R. A., Kisiel W., Dahlbäck B., and Nelsestuen G. L. (1997) Comparison of naturally occuring vitamin K-dependent proteins: correlation of amino acid sequences and membrane binding properties suggests a membrane contact site. Biochemistry 36, 5120–5127 [DOI] [PubMed] [Google Scholar]
- 33. Gettins P. (2002) Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–4804 [DOI] [PubMed] [Google Scholar]
- 34. Powell L. M., and Pain R. H. (1992) Effects of glycosylation on the folding and stability of human, recombinant and cleaved α1-antitrypsin. J. Mol. Biol. 224, 241–252 [DOI] [PubMed] [Google Scholar]
- 35. Berlett B. S., and Stadtman E. R. (1997) Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272, 20313–20316 [DOI] [PubMed] [Google Scholar]
- 36. Greenberg M. E., Li X. M., Gugiu B. G., Gu X., Qin J., Salomon R. G., and Hazen S. L. (2008) The lipid whisker model of the structure of oxidized cell membranes. J. Biol. Chem. 283, 2385–2396 [DOI] [PubMed] [Google Scholar]
- 37. Stratikos E., and Gettins P. G. (1998) Mapping the serpin-proteinase complex using single cysteine variants of α1-proteinase inhibitor Pittsburgh. J. Biol. Chem. 273, 15582–15589 [DOI] [PubMed] [Google Scholar]
- 38. Olson S. T., Björk I., and Shore J. D. (1993) Kinetic characterization of heparin-catalyzed and uncatalyzed inhibition of blood coagulation proteinases by antithrombin. Methods Enzymol. 222, 525–559 [DOI] [PubMed] [Google Scholar]
- 39. Stewart J. C. (1980) Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 104, 10–14 [DOI] [PubMed] [Google Scholar]
- 40. Anderson P. J., Nesset A., Dharmawardana K. R., and Bock P. E. (2000) Role of proexosite I in factor Va-dependent substrate interactions of prothrombin activation. J. Biol. Chem. 275, 16435–16442 [DOI] [PubMed] [Google Scholar]
- 41. De Cristofaro R., and Di Cera E. (1991) Phenomenological analysis of the clotting curve. J. Protein Chem. 10, 455–468 [DOI] [PubMed] [Google Scholar]