

Abstract
Pyroptosis is a form of cell death important in defenses against pathogens that can also result in a potent and sometimes pathological inflammatory response. During pyroptosis, GSDMD (gasdermin D), the pore-forming effector protein, is cleaved, forms oligomers, and inserts into the membranes of the cell, resulting in rapid cell death. However, the potent cell death induction caused by GSDMD has complicated our ability to understand the biology of this protein. Studies aimed at visualizing GSDMD have relied on expression of GSDMD fragments in epithelial cell lines that naturally lack GSDMD expression and also lack the proteases necessary to cleave GSDMD. In this work, we performed mutagenesis and molecular modeling to strategically place tags and fluorescent proteins within GSDMD that support native pyroptosis and facilitate live-cell imaging of pyroptotic cell death. Here, we demonstrate that these fusion proteins are cleaved by caspases-1 and -11 at Asp-276. Mutations that disrupted the predicted p30-p20 autoinhibitory interface resulted in GSDMD aggregation, supporting the oligomerizing activity of these mutations. Furthermore, we show that these novel GSDMD fusions execute inflammasome-dependent pyroptotic cell death in response to multiple stimuli and allow for visualization of the morphological changes associated with pyroptotic cell death in real time. This work therefore provides new tools that not only expand the molecular understanding of pyroptosis but also enable its direct visualization.
Keywords: caspase 1 (CASP1), cell death, inflammasome, inflammation, innate immunity, inflammasome, inflammation, pore, pyroptosis, Gasdermin D, cell death, caspase
Introduction
The innate immune system maintains a delicate balance between pathogen defense and inflammatory disease. Nowhere is this more evident than in pyroptotic cell death, a form of cell death that is important in pathogen defense but that can also result in a robust and sometimes pathological, inflammatory response (1). Pyroptosis was initially characterized as caspase-1-dependent death in cells of monocytic lineage following activation of the inflammasome (2, 3). Activation of caspase-1 was also known to lead to maturation of pro-IL-1β and pro-IL-18, although it was apparent that the cytokine production and maturation could be decoupled from pyroptotic cell death (4). This previous conception of pyroptosis was redefined with the discovery of the non-canonical inflammasome causing pyroptosis in a caspase-11 (caspase-4/5 in humans)–dependent manner (5–8). Pyroptosis was yet again redefined with the discovery of the inflammatory caspase substrate GSDMD (gasdermin D) as the crucial pore-forming protein mediating pyroptotic cell death (9–11). Collectively, these findings thus identified the critical effector protein common to both canonical and non-canonical inflammasome-driven cell death.
The discovery of GSDMD and its role in the pyroptotic cascade provides an essential, unifying protein for both canonical and non-canonical inflammasome directed pyroptosis. Although the exact mechanism by which GSDMD induces cell death is still under investigation, it is thought that GSDMD folds back on itself intramolecularly to achieve an autoinhibited state. Upon stimulation of caspase-1/4/5/11, GSDMD is cleaved. This releases the autoinhibited state and allows the N-terminal p30 GSDMD fragment to form oligomers and insert in lipid membranes, ultimately resulting in the formation of the pyroptotic pore (12–15). Formation of this pyroptotic pore can be kinetically measured by monitoring propidium iodide (PI)2 uptake, because PI moves through the pore and fluoresces with a distinct excitation/emission upon intercalation with DNA (16). The specific mechanism by which p30 GSDMD structurally changes following cleavage, binds to lipid membranes, creates oligomers, and forms mature pores has yet to be delineated, and its study has been complicated by the fact that the rapid induction of pyroptosis has precluded molecular visualization in relevant cell types (17).
Although some studies attempted to overcome this limitation through the use of fluorescent tags and the use of less potent forms of GSDMD, efforts to visualize the key effector protein, GSDMD, have been complicated by the fact that adding tags to the N terminus of the protein interferes with pyroptotic activity (11, 14, 15). Additionally, mutations designed to slow cell death do not allow visualization of real-time kinetics, and overexpression studies have only been done in cell types that do not natively express GSDMD (14, 15). Critically, visualization of GSDMD during and after cleavage by an inflammatory caspase in a relevant cell of myeloid lineage has not been demonstrated. In this work, through molecular and structural biological analysis, we define the molecular parameters by which GSDMD becomes cleaved, oligomerizes, and inserts into cellular membranes in HEK-293T cells and murine bone marrow-derived macrophages. This analysis provided a pathway to internally tag GSDMD such that both biochemical experiments and live-cell imaging can be performed in real time. We further couple CRISPR-Cas9 technology with these novel GSDMD tools to allow direct, real-time visualization of pyroptotic pore formation upon inflammasome activation.
Results
Design of a fluorescent protein-tagged GSDMD
To study GSDMD in macrophages, Gsdmd was genetically deleted in immortalized bone marrow–derived macrophages (iBMDM) through the use of CRISPR-Cas9 as previously described (Fig. 1A) (16, 18). The p30 fragment of murine GSDMD (AA 1–276) has been shown to be cytotoxic via formation of the pyroptotic pore, whereas the cleaved p20 fragment (AA 277–487) is thought to be located diffusely in the cytosol following caspase cleavage (12–15). Shi et al. (9) analyzed truncated versions of p30 GSDMD, demonstrating that residues 1–243 of human GSDMD (1–244 in mouse) were sufficient to cause cell death (Fig. 1B), indicating that residues 245–276 of the p30 GSDMD fragment can likely tolerate alteration without impacting function. Previous work determined the crystal structure of the GSDMD family member gasdermin A3 and demonstrated that a flexible loop lies within gasdermin A3 corresponding to the region of GSDMD near 245–276 (14). To circumvent previously identified issues including a lack of pyroptotic cell death with N-terminal tagging of the p30 fragment of GSDMD (11), a FLAG tag was added following residue 248 in this flexible linker region between the end of the fragment required for pyroptosis (AA 1–244) and before the caspase-1/11 cleavage site located N-terminal to residue 277 (Fig. 1C). This tag allows monitoring of the p30 GSDMD fragment before, during, and after cleavage. Murine GSDMD was cloned into a novel lentiviral expression system. In this system, puromycin resistance and GSDMD expression are linked because they are both constitutively driven by the EFS promoter but are cleaved into separate proteins by the P2A self-cleaving peptide. Gsdmd−/− iBMDM cells were stably reconstituted using this lentiviral expression system optimized for the myeloid lineage (18) resulting in expression of WT-GSDMD or FLAG-GSDMD (Fig. 1, C and D). WT-GSDMD and FLAG-GSDMD reconstituted iBMDM cells demonstrated similar cytotoxicity in response to LPS priming and activation of the NLRP3 inflammasome with the potassium ionophore nigericin (Fig. 1E). Neither WT-GSDMD nor FLAG-GSDMD allowed PI uptake in response to LPS alone (Fig. 1F) but demonstrated similar kinetics in pyroptotic pore formation as measured by PI uptake when LPS-primed and then stimulated with nigericin or ATP, an additional activator of the NLRP3 inflammasome that acts in a manner distinct from nigericin (Fig. 1, G and H).
Figure 1.

Knock-out and reconstitution of gasdermin D in macrophages. A, CRISPR-Cas9 KO of Gsdmd in iBMDM cells. Cells transduced with each CRISPR guide were cloned out, individually verified for loss of GSDMD, and combined to form clonal pools of each guide. B, fragments of p30 GSDMD able to cause cell death identified by Shi et al. (9). C, design of the internal FLAG tag GSDMD construct. GSDMD is separated from the neomycin resistance marker by the P2A self-cleaving peptide, both under the control of an EFS promoter. The lentiviral expression construct allows for stable expression in multiple cell types. D, reconstitution of WT- and FLAG-GSDMD in Gsdmd−/− iBMDM cells. E, LDH release assay in reconstituted iBMDM cell lines primed with 200 ng/ml LPS and unstimulated or stimulated with 10 μm nigericin. F–H, PI uptake assay in WT- and FLAG- GSDMD iBMDM cells with LPS priming alone (F) and in combination with 10 μm nigericin (G) or 5 mm ATP (H). E represents means with S.E. of nine technical replicates of three experiments. F and H represent the means ± S.E. of four technical replicates of two independent experiments. G represents the means ± S.E. of six technical replicates of three independent experiments.
Given that we could successfully recapitulate WT-GSDMD behavior with an internal FLAG tag, we next attempted to insert a much larger fluorescent tag. Because of the size difference between a small 9-AA FLAG tag and a 26-kDa fluorescent tag, we first generated a molecular model of the GSDMD pore based on the crystal structure of the homologous protein GSDMA3 (Protein Data Bank code 5B5R) (19). The crystal structure of mNeonGreen from B. lanceolatum (Protein Data Bank code 5LTR) was modeled between residues 248 and 249 of GSDMD (Fig. 2A). Because reports indicate that p30 oligomerizes with ∼16 p30 GSDMD units per pore (14), the mNeonGreen GSDMD pore was modeled as a 16-mer structure. Although molecular modeling suggests that mNeon-GSDMD is flexible enough to allow for cleavage and pore formation, because mNeon is located in the flexible linker region which could not be crystallized, the actual position of mNeon in relation to p20 or p30 GSDMD could vary greatly from the model. Nonetheless, structural modeling based on the crystal structure of murine GSDMA3 suggests that this placement may allow visualization of near-native pore formation. Additionally, sequence comparison with other members of the gasdermin superfamily demonstrates low sequence homology in this region (supplemental Fig. S1), suggesting that a larger insertion might be tolerated in this region. Based on this, Gibson directed cloning allowed the insertion of the fluorescent mNeon protein into the lentiviral expression system (Fig. 2B).
Figure 2.

Design and modeling of mNeon-gasdermin D. A, structural model of mNeon-GSDMD and the mNeon-GSDMD pyroptotic pore. mNeonGreen from B. lanceolatum (Protein Data Bank code 5LTR) was modeled between residues 248 and 249 of murine GSDMD. The structural model of GSDMD was based on the crystal structure of GSDMA3 (Protein Data Bank code 5B5R). B, design of the internal mNeon-GSDMD lentiviral expression construct for stable reconstitution/expression of mNeon-GSDMD.
mNeon-GSDMD activity in HEK-293T cells
To test whether mNeon-GSDMD retains function, we first assessed cleavage by caspase-1/11. mNeon-GSDMD is cleaved by caspases 1 and 11 when co-expressed in HEK-293T cells (Fig. 3A). Based on the preference of caspase 1 and 11 for aspartate at the cleavage site, mutation of Asp-276 should decrease cleavage of GSDMD and cell death (9, 11). Indeed, mutation of Asp-276 to alanine resulted in a loss of mNeon-GSDMD cleavage when expressed with caspase-1 or -11 (Fig. 3A). Expression of mNeon-GSDMD in HEK-293T cells demonstrated diffuse, cytosolic GSDMD which spared the nucleus consistent with previous imaging studies (Fig. 3B) (14, 15). To ensure that our findings were not mNeon-dependent, the mNeonGreen tag was exchanged for the mRuby2 fluorescent protein (20) (Fig. 3C). mRuby-GSDMD expressed in HEK-293T cells demonstrated diffuse, cytosolic expression of GSDMD confirming an mNeonGreen-independent expression pattern (Fig. 3D and supplemental Fig. S2A). Transfection of mNeon-GSDMD and mRuby-GSDMD in HEK-293T demonstrated analogous distribution patterns, although relative expression levels varied among cells leading to varying coloration from green to red (Fig. 3E and supplemental Fig. S2B). To determine the effect of caspase-11 on mNeon-GSDMD, the catalytic domain of caspase-11 was expressed with mNeon-GSDMD in HEK-293T cells. Expression of the catalytic domain of caspase-11 caused mNeon-GSDMD aggregation and cell death in a caspase-11 dose-dependent manner (Fig. 3F).
Figure 3.

Expression of mRuby and mNeon-gasdermin D in HEK-293T cells. A, Western blot analysis of mNeon-GSDMD or D276A mNeon-GSDMD cleavage by caspase-1 or -11 co-expressed in HEK-293T cells. B, mNeon-GSDMD expression in HEK-293T cells using Lipofectamine transfection on 8-well chamber glass. C, design of the mRuby-GSDMD lentiviral expression construct. D and E, mRuby-GSDMD expression (D) and co-expression of mNeon-GSDMD and mRuby-GSDMD (E) in HEK-293T cells. F, co-expression of mNeon-GSDMD with increasing amounts of the catalytic domain of caspase-11 (left to right: 0.38, 0.56, and 1.14 μg) with DAPI staining in HEK-293T cells.
Probing the p30-p20 interface with mNeon-GSDMD
Modeling of GSDMD based on the crystal structure of gasdermin A3 demonstrated that key residues in the p20 fragment of GSDMD bind and inhibit residues in the p30 fragment (14). Functional studies of gasdermin A3 and GSDMD then demonstrated that mutation of three conserved residues in the p20 fragment of GSDMD (Leu-290, Tyr-373, or Ala-377) to aspartate results in a release of autoinhibition leading to pyroptotic cell death (14). These three conserved residues are present in murine GSDMD as Leu-292, Tyr-376, and Ala-380. The crystal structure of gasdermin A3 further demonstrated that these three residues are likely key for binding to a phenylalanine-tryptophan sequence in the p30 fragment (14). If these novel fluorescent GSDMD tools recapitulate wild-type function, mutation of these residues should release autoinhibition and promote aggregation of the fluorescently tagged GSDMD. Therefore, to investigate the p30-p20 autoinhibitory interface using mNeon-GSDMD, activating mutants (L292D, Y376D, and A380D) and inactivating mutants (D276A) were expressed in HEK-293T cells. As predicted mRuby-GSDMD, as well as non-mutated, L292D, Y376D, and A380D mNeon-GSDMD, demonstrated cleavage in the presence of caspase-11, whereas no cleavage of D276A mNeon-GSDMD was observed (Fig. 4A). Lower levels of mNeon-GSDMD were seen with activating mutants because of their cytotoxic effects. Using confocal microscopy, each of the mNeon-GSDMD variants were expressed in HEK-293T cells and imaged. Expression of L292D, Y376D, and A380D mutations of mNeon-GSDMD caused spontaneous formation of large aggregate structures, suggesting that these mutations spontaneously relieved GSDMD autoinhibition, resulting in oligomerization/aggregation of full-length GSDMD (Fig. 4B). The aggregates were absent from wild type mNeon-GSDMD and D276A mNeon-GSDMD cell lines and present in all three activating mutants, demonstrating that mNeon-GSDMD retains the same p30-p20 autoinhibitory interface as WT-GSDMD. The morphology and abundance of these activating aggregates of GSDMD in overexpression assays do not mean that this finding would be retained in cells naturally harboring an activating mutation. Indeed, we found that stably transduced Gsdmd−/− iBMDM cell lines harboring activating mutations (Y376H/D) expressed significantly lower amounts of GSDMD than WT or a separate point mutant (Y38F), even while retaining the same ability to form pyroptotic pores as the WT (supplemental Fig. S3, A and B). Notably, Y376H or Y376D GSDMD expression decreased with extended time in culture, a phenomenon not seen in wild type or non-activating mutants (data not shown). This suggests that the presence of activating mutations confers a selective disadvantage to high expressing cells. Together, these data suggest that this internally fluorescent GSDMD retains the function of the non-tagged GSDMD.
Figure 4.
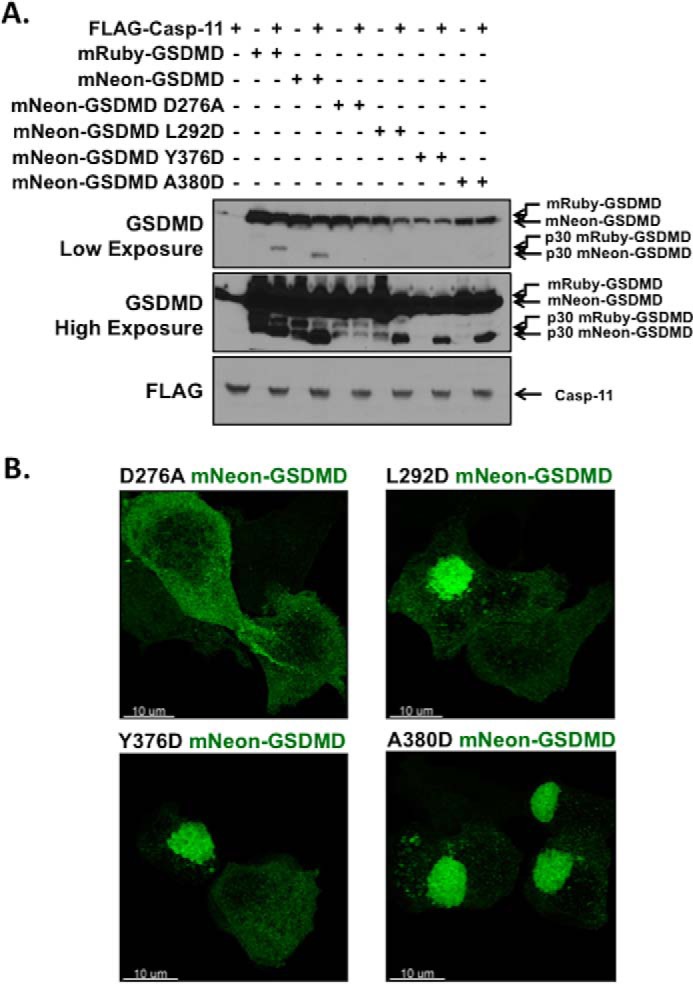
Disruption of the mNeon-gasdermin D autoinhibition. A, Western blot analysis of caspase-11 cleavage of mRuby-GSDMD, mNeon-GSDMD, predicted deactivating mNeon-GSDMD mutation (D276A), and predicted activating mNeon-GSDMD mutations (L292D, Y376D, and A380D). Constructs were co-expressed in HEK-293T cells using calcium phosphate transfection with the catalytic domain of caspase-11. Samples were harvested 24 h post-transfection and analyzed via Western blot. B, representative confocal microscopy images of deactivating and activating mNeon-GSDMD mutations expressed in HEK-293T cells.
Visualizing GSDMD activity during pyroptosis in reconstituted iBMDM cells
Although helpful in probing the mechanics of GSDMD autoinhibition and caspase cleavage, our initial studies employed overexpression in HEK-293T cells and could be subject to overexpression artifacts. Because pyroptotic activity in a cell of myeloid lineages is most relevant to human physiology and pathophysiology, the Gsdmd−/− iBMDM cells were reconstituted with WT-GSDMD and mNeon-GSDMD (Fig. 5A). Expression levels of reconstituted mNeon-GSDMD were similar to that of the reconstituted WT-GSDMD. Stimulation of the NLRP3 inflammasome (LPS with nigericin) resulted in cleavage of mNeon-GSDMD, which was largely absent in unstimulated cells (LPS alone) (Fig. 5B). mNeon-GSDMD cells demonstrated IL-1β release in response to LPS with nigericin, although with slower kinetics than WT-GSDMD reconstituted cells (Fig. 5C). Pyroptotic pore formation and cell death, measured by PI uptake and LDH release respectively, were measured in GSDMD and mNeon-GSDMD reconstituted cells following stimulation with LPS and nigericin. mNeon-GSDMD demonstrated cell death following treatment with LPS and nigericin, although to a lesser degree than WT-GSDMD at 1 h (Fig. 5D). As expected mNeon-GSDMD iBMDM cells demonstrated no pore formation with only LPS stimulation (Fig. 5E). Following treatment with LPS and nigericin or LPS and ATP, mNeon-GSDMD iBMDM cells demonstrated pyroptotic pore formation, albeit at a slower rate than WT-GSDMD iBMDM cells (Fig. 5, F and G).
Figure 5.

Reconstitution of mNeon-GSDMD in macrophages. A, Western blot of wild-type GSDMD or mNEON-GSDMD reconstituted in Gsdmd−/− BMDM cell lines. B, cleavage of mNeon-GSDMD with LPS or LPS with nigericin (LPS + NG) in reconstituted cells (asterisk denotes nonspecific band). The cells were stimulated with 200 ng/ml LPS for 4 h with or without 10 μm nigericin stimulation for 1 h. C, IL-1β release from reconstituted iBMDM cells as measured by sandwich ELISA following stimulation with 200 ng/ml LPS and 10 μm nigericin. D, cytotoxicity as measured by LDH release in reconstituted cell lines in response to LPS priming with or without nigericin stimulation. E–G, pore formation as measured by PI uptake in reconstituted cell lines with LPS priming with no inflammasome activation (E), nigericin stimulation (F), or ATP stimulation (G). Concentrations were LPS, 200 ng/ml; nigericin, 10 μm; and ATP, 5 mm. D represents the mean with S.E. of 15 technical replicates from six experiments. F represents the mean with S.E. of six technical replicates of three independent experiments. E and G represent the mean with S.E. of four technical replicates of two independent experiments.
Live-cell imaging of mNeon-GSDMD reconstituted iBMDM cells demonstrated diffuse mNeon-GSDMD prior to nigericin stimulation, which was followed by membrane blebbing and GSDMD redistribution visible on phase contrast and confocal microscopy following nigericin stimulation (Fig. 6A). A video of GSDMD-induced pyroptotic cell death was then taken of the mNeon-GSDMD cells, providing visualization of the key pyroptotic effector in real time. Consistent with the kinetics of pyroptotic cell death as demonstrated by PI uptake assays, membrane blebbing began 15 min after the addition of nigericin. At this time point, mNeon-GSDMD began to redistribute from a diffusely cytoplasmic localization to form multiple aggregates in the cell that appear to localize at the plasma membrane, as well as various internal membranes (Fig. 6B and supplemental Movie S1). The formation of these bubble-like projections and the loss of membrane integrity are both phenomena that have been previously shown to be characteristic of pyroptosis (15, 21). Moreover, macrophages with extended processes prior to inflammasome activation demonstrated a rapid retraction of the processes following stimulation. Importantly, mNeon-GSDMD provides an important advance over the fixed staining or overexpression of the p30 GSDMD fragment alone in epithelial cell lines. The method presented here allows for the visualization of pyroptotic pore formation in real time before, during, and after inflammasome activation in the myeloid lineage.
Figure 6.

Live-cell imaging of pyroptosis in macrophages. A, mNeon-GSDMD iBMDM cells were primed with 200 ng/ml LPS and stimulated with 10 μm nigericin (NG). Confocal and phase contrast microscopy was used to visualize mNeon-GSDMD during pyroptosis. B, confocal microscopy enabled live-cell imaging of pyroptotic cell death before, during, and after activation of the NLRP3 inflammasome with nigericin. A video of cell death is available in supplemental Movie S1.
Discussion
One of the hurdles in the study of GSDMD has been the disruption of normal pyroptotic activity resulting from N-terminal tagging (11). Here, we have described a novel method for tagging and visualizing GSDMD and demonstrate the applicability of this system in the myeloid lineage, a lineage where inflammasome activation is an important, normal function. We then used mNeon-GSDMD to demonstrate that mutations that disrupt the p30-p20 GSDMD interaction result in the formation of oligomers and aggregates. Cleavage of mNeon-GSDMD results in pyroptotic cell death downstream. Upon encountering activators of the NLRP3 inflammasome, mNeon-GSDMD is cleaved, resulting in pyroptosis, allowing live-cell visualization of pyroptotic cell death across inflammasomes.
In contrast to previous studies of pyroptotic cell death that relied on proxy measurements of pyroptosis such as PI positivity, caspase-1 cleavage, or ASC oligomerization, mNeon-GSDMD allows for direct monitoring of the pyroptotic effector protein. Previous methods were constrained either in that they measured events early in the inflammasome-pyroptosis cascade and not the effector of membrane failure or they could not measure the kinetics or gradations of pyroptotic cell death. mNeon-GSDMD addresses both of these constraints by allowing live-cell imaging of pyroptotic cell death at the level of single cells or small populations of cells. Whereas measuring ASC oligomerization or caspase-1 cleavage only measures the activation of the canonical inflammasome, mNeon-GSDMD provides detailed information about the downstream pyroptotic effector protein. Whereas markers such as PI positivity are binary, mNeon-GSDMD allows gradations of pyroptotic cell death to be visualized. Although mNeon-GSDMD displays slower kinetics in pore formation following activation with LPS and nigericin, this feature is serendipitous in that it allows for more subtle changes in GSDMD mediated death to be studied.
Although the study of cell death and inflammatory disease has been dramatically advanced with the discovery of GSDMD as the protein forming the pyroptotic pore, many important questions remain outstanding. Future investigations are necessary to provide details on the mechanisms by which GSDMD creates oligomers, binds membranes, and forms mature pores. Although GSDMD is known to be present in the intestinal tract, the expression and activity of GSDMD in specific cells and tissues throughout the body remain largely unknown. Exploring GSDMD-induced cell death in a tissue- and disease-specific manner will be important in leveraging the current knowledge of pyroptotic cell death for clinical applications. To that end, the lentiviral delivery system allows for the use of mNeon-GSDMD or mRuby-GSDMD in a wide range of cell types and in combination with other fluorescent markers. Further, because the inflammasome and pyroptotic cell death have been implicated in a myriad of disease processes including sepsis (22), inflammatory bowel disease (23, 24), and immunodeficiencies (25), the role of GSDMD in disease-specific contexts must be further investigated. These are studies in which mNeon-GSDMD could prove a valuable tool. Through building a framework of novel tools and assays, a greater knowledge of pyroptotic cell death can be achieved to solve some of the key outstanding questions and address these serious diseases. mNeon-GSDMD has been used to probe the p30-p20 interface and expand the knowledge of pyroptotic cell death in epithelial and macrophage cells, enabling wider applicability of these novel reagents to address pyroptotic cell death in disease-specific contexts.
Experimental procedures
Cells, plasmids, reagents, antibodies, and Western blotting
Calcium phosphate transfection of HEK-293T cells grown in DMEM (Corning) with 15% SuperCalf Serum (Gemini), 1% penicillin/streptomycin (Gibco), and supplemented with non-essential amino acids (Gibco), l-glutamine (Gibco), and HEPES was used for overexpression assays. Nlrp3−/− iBMDM cells overexpressing NLRP3-FLAG and ASC-mCerulean were a gift from Eicke Latz (University of Bonn, Bonn, Germany) and were cultured in DMEM (Corning) with 10% heat-inactivated SuperCalf Serum, 1% penicillin/streptomycin, and 3 μg/ml puromycin (Invitrogen). Gsdmd−/− cells were generated using CRISPR-Cas9 as previously published (16) under the same culture conditions as iBMDM cells. Knock-out cell lines were verified using Western blot. iBMDM and HEK-293T cell lines were lysed in modified radioimmune precipitation assay buffer with 1 mmol of PMSF (Acros Organics), a protease inhibitor mixture (Sigma), and calyculin (LC Laboratories). The GSDMD monoclonal antibody (SC-393656; Santa Cruz Biotechnology) is designed against an epitope located within the p30 fragment of murine GSDMD. Other primary antibody used were anti-FLAG (F7415; Sigma), caspase-1 p10 (SC-514; Santa Cruz Biotechnology), and GAPDH (60004; Proteintech). All primary antibodies were used according to the manufacturer's instructions. HRP-conjugated secondary antibodies recognizing mouse and rabbit IgG (Cell Signaling Technology) were used at 1:10,000.
PCMV-FLAG-Caspase-11 was a gift from Junying Yuan (Addgene plasmid no. 21145) (26). The GSDMD expression/reconstitution vectors described in Figs. 1B and 2A were Gibson subcloned into the lentiviral expression plasmid previously described (27), which originally utilized LentiCRISPRv2 (Addgene) as a template with GSDMD separated from the neomycin resistance marker by a P2A self-cleaving peptide. GSDMD expression/reconstitution vectors were made CRISPR-Cas9 cut resistant through mutation of the PAM sequence via site-directed mutagenesis while conserving the amino acid sequence. mNeonGreen or mRuby2 were inserted using Gibson cloning following amino acid 248 in the murine gasdermin lentiviral expression construct. L293D, Y376D, A380D, and D276A mutants of GSDMD were created using site-directed mutagenesis on the GSDMD lentiviral reconstitution vector. GSDMD lentivirus was produced in HEK-293T cells using calcium phosphate transfection of the wild-type or mutant GsdmD lentiviral plasmid co-transfected with PsPAX (Addgene) and PMD.2 (Addgene). Lentivirus was harvested, filtered through a 0.45-μm filter, and added to Gsdmd−/− iBMDM cells with Polybrene infection/transfection reagent (Millipore). After 2 days of transduction with the lentivirus, the cells were selected with 1.0 mg/ml of G418 (InvivoGen) for 4 days. Reconstitution of GSDMD into Gsdmd−/− cell lines was verified using Western blot. Following selection, the cells were maintained in 1.0 mg/ml G418.
Propidium iodide, cytotoxicity, and IL-1β release assays
For the propidium iodide assay, iBMDM cells were plated out at 300,000 cells/well in 24-well plate format. 4 h prior to the start of the assay, the cells were stimulated with 200 ng/ml LPS (InvivoGen). At the start of the assay, the cells were washed once with PBS, and an imaging buffer containing 120 mm NaCl, 4 mm KCl, 1.5 mm CaCl2, 1 mm magnesium chloride, 25 mm HEPES, 5 mm glucose, and 0.1% BSA at pH 7.4 with 1 μg/ml propidium iodide (Molecular Probes) was added to the cells. The propidium iodide fluorescence (excitation/emission, 533/617) in each well was measured on Spectramax i3x Multi Mode micro plate reader (Molecular Devices). Following background readings, cells were stimulated with 10 μm nigericin (Sigma) or 5 mm ATP (Fisher) and read kinetically at intervals of 1 min. Maximum PI fluorescence was obtained using 1% Triton X-100 in each well. Relative PI uptake was calculated for each well as (fluorescence at time x − background fluorescence)/(maximum fluorescence − background fluorescence). For the cytotoxicity assay, the cells were plated at 100,000 cells/well in 24-well plate format. The cells were stimulated with 200 ng/ml LPS (Sigma). 4 h after LPS stimulation, the cells were treated with no stimulus (LPS only) or with nigericin (LPS + NG). Background and maximum LDH release were calculated from unlysed and lysed wells, respectively, with LDH release from all wells measured using the Pierce LDH assay (Invitrogen). The percentage of cell death was calculated per the manufacturer's instructions based on LDH release of experimental wells compared with maximal and minimal LDH release control wells.
Alignment and structural modeling of mNeon-GSDMD
Alignment of gasdermin family members was done using Clustal Omega multiple sequence alignment, and supplemental Fig. S1 was prepared using ESPript 3.0 (28). A structural model for the full-length mGSDMD was produced based on the structure of murine GSDMA3 (Protein Data Bank code 5B5R) using the MMM server (29). The crystal structure of an mNeonGreen fluorescent protein from Branchiostoma lanceolatum (Protein Data Bank code 5LTR) was linked to the mGSDMD model between residues Arg-248 and Lys-249. The resulting model was energy-minimized using program Phenix (30). The p30 GSDMD domain is colored gold, the p20 GSDMD domain is silver, and mNeon is green. The caspase cleavage site Asp-276 is marked with a magenta sphere. Based on published data on gasdermin pores (14), a 16-mer symmetric complex assembled by the GSDMD-N domain was modeled. The position and orientation of Neon relative to GSDMD-N is arbitrary.
Cell imaging and analysis
Epifluorescent imaging was conducted on a Leica epifluorescent microscope. HEK-293T cells split onto 6-cm culture plates. The following day, cells were calcium phosphate–transfected with the respective plasmids, incubated for 24 h, and imaged. Confocal imaging was conducted on a Leica SP5 DMI 6000B inverted confocal microscope. For transfected, fixed images, 8-well chamber glass was coated with poly-l-lysine and plated with HEK-293T cells. Transfections were conducted using Lipofectamine 2000 (Invitrogen) in opti-MEM medium. 18 h after transfection, the cells were fixed with 4% paraformaldehyde, washed twice in PBS, and stored in ProLong Antifade Mountant (Molecular Probes) for imaging. For live-cell imaging of mNeon-GSDMD iBMDM cells, macrophages were plated on collagen-coated chamber glass. The following day, the cells were stimulated with 200 ng/ml LPS (Invivogen) followed by stimulation with 10 μm nigericin.
IL-1β release was measured from iBMDM cells primed with 200 ng/ml LPS and stimulated with 10 μm nigericin for the specified amount of time. Supernatants were collected, spun at 13,000 rpm for 30 s to clear any cells, and then assayed for IL-1β concentration using sandwich ELISA (BioLegend).
Confocal live-cell imaging
Live cell confocal imaging was taken on a Leica SP5 DMI 6000B confocal using argon 488-nm and helium–neon 633-nm lasers using a Leica 506375 HC FLUOTAR 25×/0.95 water immersion objective with 0.17-mm glass correction. The cells were cultured in an imaging buffer containing 120 mm NaCl, 4 mm KCl, 1.5 mm CaCl2, 1 mm magnesium chloride, 25 mm HEPES, 5 mm glucose, and 0.1% BSA at pH 7.4. Live-cell imaging was conducted at 37 °C within a custom made environment box utilizing an Airtherm ATX heater (World Precision Instruments). Detection utilized 12 bit Photomultiplier tubes with Leica LAS AF acquisition software. Deconvolution was done with Huygens Professional 16.10 using CMLE with SNR 3 and visualized using maximum intensity projections in Imaris (Bitplane).
Fig. 3 images
Microscopy images were taken on a Leica SP5 DMI 6000B confocal using an argon 488-nm laser with a Leica 506192 HCX PL APO lambda blue 63×/1.4 oil objective with 0.17-mm glass correction. For detection, a 12-bit Photomultiplier tubes were used with Leica LAS AF acquisition software. Deconvolution was done with Huygens Professional 16.10 using CMLE with SNR 5 and visualized using maximum intensity projections in Huygens Professional.
Fig. 4 images
Microscopy images were taken on a Leica SP8 DMI 6000 confocal using a Diode 405-nm laser and white light laser set to 506 nm, with acquisition in sequential mode. A Leica HC PL APO 100×/1.40 Oil STED WHITE lens was used. Detection utilized 12-bit hybrid detectors. Leica LAS X acquisition software and deconvolution with Huygens Professional 16.10 using CMLE with SNR 5 were used. Maximum intensity projections were visualized in Huygens Professional.
Author contributions
J. K. R. and B. L. B. designed and performed experiments, analyzed and interpreted data, and prepared the manuscript. T. S. X. performed the modeling of mNeon-GSDMD and helped prepare the manuscript. S. M. C., J. Y., G. R. D., A. Y. H., and D. W. A. contributed to the conception and design of experiments, the interpretation of data, and the writing of the manuscript.
Supplementary Material
Acknowledgments
We thank Parameswaran Ramakrishnan and Xiaoxia Li for lending helpful comments and expertise. We thank the Abbott, Huang, Dubyak, Xiao, and Ramakrishnan laboratories for insightful discussions and comments on the project and manuscript. We also thank the Case Western Reserve University (CWRU) Imaging Core. Confocal microscopy was supported in part by National Institutes of Health–Office of Research Infrastructure Programs (NIH-ORIP) Grant S10ODO16164 to the CWRU Imaging Facility.
This work was supported by National Institutes of Health Grant T32 GM007250 (to J. K. R., B. L. B., and S. M. C.); Grants TL1 RR024991, T32NS077888, and F31 NS096857 (to B. L. B.); and Grants PO1091222 and RO1086550 (to D. W. A). This work was also supported by the Theresia G. & Stuart F. Kline Family Foundation and the Errol's Cancer Discovery Fund (to A. Y. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S3 and Movie S1.
- PI
- propidium iodide
- iBMDM
- immortalized bone marrow–derived macrophages
- AA
- amino acid(s)
- LDH
- lactate dehydrogenase.
References
- 1. Jorgensen I., Rayamajhi M., and Miao E. A. (2017) Programmed cell death as a defence against infection. Nat. Rev. Immunol. 17, 151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bergsbaken T., Fink S. L., and Cookson B. T. (2009) Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martinon F., Burns K., and Tschopp J. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell. 10, 417–426 [DOI] [PubMed] [Google Scholar]
- 4. Chen K. W., Gross C. J., Sotomayor F. V., Stacey K. J., Tschopp J., Sweet M. J., and Schroder K. (2014) The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 8, 570–582 [DOI] [PubMed] [Google Scholar]
- 5. Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S., Zhang J., Lee W. P., Roose-Girma M., and Dixit V. M. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 [DOI] [PubMed] [Google Scholar]
- 6. Shi J., Zhao Y., Wang Y., Gao W., Ding J., Li P., Hu L., and Shao F. (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 [DOI] [PubMed] [Google Scholar]
- 7. Schmid-Burgk J. L., Gaidt M. M., Schmidt T., Ebert T. S., Bartok E., and Hornung V. (2015) Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 45, 2911–2917 [DOI] [PubMed] [Google Scholar]
- 8. Viganò E., Diamond C. E., Spreafico R., Balachander A., Sobota R. M., and Mortellaro A. (2015) Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 6, 8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., and Shao F. (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 [DOI] [PubMed] [Google Scholar]
- 10. Kayagaki N., Stowe I. B., Lee B. L., O'Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q. T., Liu P. S., Lill J. R., Li H., Wu J., Kummerfeld S., et al. (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 [DOI] [PubMed] [Google Scholar]
- 11. He W., Wan H., Hu L., Chen P., Wang X., Huang Z., Yang Z.-H., Zhong C.-Q., and Han J. (2015) Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25, 1285–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aglietti R. A., Estevez A., Gupta A., Ramirez M. G., Liu P. S., Kayagaki N., Ciferri C., Dixit V. M., and Dueber E. C. (2016) GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. U.S.A. 113, 7858–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sborgi L., Rühl S., Mulvihill E., Pipercevic J., Heilig R., Stahlberg H., Farady C. J., Müller D. J., Broz P., and Hiller S. (2016) GSDMD pore formation in the plasma membrane constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., Sun H., Wang D.-C., and Shao F. (2016) Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 [DOI] [PubMed] [Google Scholar]
- 15. Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V. G., Wu H., and Lieberman J. (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Russo H. M., Rathkey J., Boyd-Tressler A., Katsnelson M. A., Abbott D. W., and Dubyak G. R. (2016) Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J. Immunol. 197, 1353–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aglietti R. A., and Dueber E. C. (2017) Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 38, 261–271 [DOI] [PubMed] [Google Scholar]
- 18. Chirieleison S. M., Marsh R. A., Kumar P., Rathkey J. K., Dubyak G. R., and Abbott D. W. (2017) Nucleotide-binding oligomerization domain (NOD) signaling defects and cell death susceptibility cannot be uncoupled in X-linked inhibitor of apoptosis (XIAP)-driven inflammatory disease. J. Biol. Chem. 292, 9666–9679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shaner N. C., Lambert G. G., Chammas A., Ni Y., Cranfill P. J., Baird M. A., Sell B. R., Allen J. R., Day R. N., Israelsson M., Davidson M. W., and Wang J. (2013) A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lam A. J., St-Pierre F., Gong Y., Marshall J. D., Cranfill P. J., Baird M. A., McKeown M. R., Wiedenmann J., Davidson M. W., Schnitzer M. J., Tsien R. Y., and Lin M. Z. (2012) Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 9, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen X., He W. T., Hu L., Li J., Fang Y., Wang X., Xu X., Wang Z., Huang K., and Han J. (2016) Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deutschman C. S., and Tracey K. J. (2014) Sepsis: current dogma and new perspectives. Immunity 40, 463–475 [DOI] [PubMed] [Google Scholar]
- 23. Zaki M. H., Lamkanfi M., and Kanneganti T. D. (2011) The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 32, 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bauer C., Duewell P., Mayer C., Lehr H. A., Fitzgerald K. A., Dauer M., Tschopp J., Endres S., Latz E., and Schnurr M. (2010) Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 [DOI] [PubMed] [Google Scholar]
- 25. Doitsh G., Galloway N. L., Geng X., Yang Z., Monroe K. M., Zepeda O., Hunt P. W., Hatano H., Sowinski S., Muñoz-Arias I., and Greene W. C. (2014) Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505, 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J., Brieher W. M., Scimone M. L., Kang S. J., Zhu H., Yin H., von Andrian U. H., Mitchison T., and Yuan J. (2007) Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat. Cell Biol. 9, 276–286 [DOI] [PubMed] [Google Scholar]
- 27. Chirieleison S. M., Kertesy S. B., and Abbott D. W. (2016) Synthetic biology reveals the uniqueness of the RIP kinase domain. J. Immunol. 196, 4291–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rai B. K., and Fiser A. (2006) Multiple mapping method: A novel approach to the sequence-to-structure alignment problem in comparative protein structure modeling. Proteins 63, 644–661 [DOI] [PubMed] [Google Scholar]
- 30. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.