

Abstract
Chemical RNA modifications are central features of epitranscriptomics, highlighted by the discovery of modified ribonucleosides in mRNA and exemplified by the critical roles of RNA modifications in normal physiology and disease. Despite a resurgent interest in these modifications, the biochemistry of 3-methylcytidine (m3C) formation in mammalian RNAs is still poorly understood. However, the recent discovery of trm141 as the second gene responsible for m3C presence in RNA in fission yeast raises the possibility that multiple enzymes are involved in m3C formation in mammals as well. Here, we report the discovery and characterization of three distinct m3C-contributing enzymes in mice and humans. We found that methyltransferase-like (METTL) 2 and 6 contribute m3C in specific tRNAs and that METTL8 only contributes m3C to mRNA. MS analysis revealed that there is an ∼30–40% and ∼10–15% reduction, respectively, in METTL2 and -6 null-mutant cells, of m3C in total tRNA, and primer extension analysis located METTL2-modified m3C at position 32 of tRNAThr isoacceptors and tRNAArg(CCU). We also noted that METTL6 interacts with seryl-tRNA synthetase in an RNA-dependent manner, suggesting a role for METTL6 in modifying serine tRNA isoacceptors. METTL8, however, modified only mRNA, as determined by biochemical and genetic analyses in Mettl8 null-mutant mice and two human METTL8 mutant cell lines. Our findings provide the first evidence of the existence of m3C modification in mRNA, and the discovery of METTL8 as an mRNA m3C writer enzyme opens the door to future studies of other m3C epitranscriptomic reader and eraser functions.
Keywords: human, mouse, mRNA, RNA methyltransferase, transfer RNA (tRNA), Mettl2, Mettl6, Mettl8, m3C, mice
Introduction
Building on decades of study of transfer and ribosomal RNA (tRNA and rRNA, respectively), the resurgent interest in RNA modifications as central features of epitranscriptomics has been sparked by the discovery of modified ribonucleosides in messenger RNA (mRNA) (1, 2) and by the discovery of novel functions for non-coding RNA modifications in normal physiology and disease (3–6). Although tRNA and rRNA are the most extensively modified RNA species in terms of the variety and abundance of modifications (7, 8), all known classes of RNA are subject to enzymatic modification. This point is illustrated by the recent discoveries of 6-methyladenosine (m6A),4 pseudouridine, 5-methylcytidine (m5C), inosine (I), 2′-O-ribose methylation, and 1-methyladenosine (m1A) in mammalian mRNA (2, 9–11). 3-Methylcytidine (m3C) (Fig. 1A), first discovered in Saccharomyces cerevisiae total RNA (12) and later found in other eukaryotic tRNA (13, 14), occurs most frequently at position 32 in several cytoplasmic and mitochondrial tRNA isoacceptors, at position 47d on the long variable loop of several cytoplasmic tRNASer, and at position 20 of tRNAMet-e (15–19). Maraia and co-workers (18) recently used a sequencing-based polymerase misincorporation assay to map methylation modifications in tRNAs in mouse and human cells and found nearly identical m3C distributions: tRNAArgCCU and tRNAArgUCU and all tRNASer and tRNAThr. Here, we shed light on the landscape of m3C-catalyzing activities in mouse and human cells with the discovery of m3C “writer” functions for three methyltransferase-like (METTL) proteins: METTLs 2, 6, and 8.
Figure 1.

Contribution of METTL2 and METTL6 to tRNA m3C in mouse and human cell lines. A, structure of m3C. Positions 2–4 are involved in Watson-Crick base pairing, with the N3-methylation serving as a block to polymerases. This polymerase-blocking phenomenon was exploited to map the location of methylations in tRNA. B and C, relative m3C levels in tRNA isolated liver and brain tissues, respectively. Samples from wild-type (WT), Mettl2 KO (M2 KO), Mettl6 mutant (M6 KO), and Mettl8 KO (M8 KO) mice were analyzed. D, relative m3C levels in HEK293T wild-type and METTL2 KO cell lines (loss of both METTL2A and -2B). E, relative m3C levels in HCT116 METTL8 wild-type and knock-out sample. m3C modification levels were normalized against levels of canonical cytidine. Data represent mean ± S.D. for at least three biological replicates, with asterisks denoting significant differences by Student's t test; **, p < 0.01. ns, not significant.
RNA methyltransferases (MTases) represent a diverse family of enzymes that transfer a methyl group from S-adenosylmethionine (SAM) to a variety of positions in RNA either directly, such as the 2′-O position of ribose, and the carbons, exocyclic nitrogens, and heterocyclic nitrogens of nucleobases, or as steps in the synthesis of hypermodified nucleobases, such as queuosine (7, 17, 20, 21). TRM140 (also known as ABP140) of S. cerevisiae is the first enzyme recognized to synthesize m3C in RNA, specifically in threonine and serine tRNAs (22, 23). The situation is more complicated in fission yeast (Saccharomyces pombe) in which Trm140 and Trm141 catalyze m3C at position 32 in tRNAThr and tRNASer, respectively (24). In mammals, several methyltransferase-like (METTL) proteins have been well characterized, including the formation of m6A in mRNA by a complex of METTL3 and METTL14 in human cells (25, 26, 36). Because human Mettl2B and Mettl6 have been proposed to be homologs of yeast trm140 and trm141, respectively (18, 19, 23), it is not surprising that sequence similarity analyses have revealed a unique clustering of METTL2A, -2B, -6, and -8 in humans and the homologous METTL2, -6, and -8 in mice (18, 27). The sequence alignments shown in supplemental Fig. S1 demonstrate this strong similarity.
Apart from the observation of a reduction in m3C in Mettl2B knockdown studies in human cells (23), little is known about the enzymology of m3C or the function of METTL proteins in mammals. Here, we have systematically analyzed the role of METTL2, -6, and -8 in RNA modification in mice and humans, with the discovery that both METTL2 and -6 contribute to m3C in specific tRNAs, and m3C is a METTL8-dependent modification in mRNA.
Results
METTL2 and METTL6, but not METTL8, contribute to m3C formation in tRNA
To define the catalytic activity of METTL2, -6, and -8 with tRNA, we generated null-mutant mice and cell lines by CRISPR/Cas9. All mutant mice were born with normal Mendel ratio without observable developmental defects (supplemental material and supplemental Figs. S2 and S3). We then used an established size-exclusion chromatography method to purify tRNA from total RNA (supplemental Fig. S4) (28). Using HPLC-coupled triple quadrupole mass spectrometry (LC-MS/MS), we both identified and quantified m3C in tRNA fractions in brain and liver tissues from wild-type (WT) mice and Mettl2, -6, and -8 mutants. tRNA from both tissues of Mettl2 KO mice showed an ∼35% reduction of m3C compared with WT tissues, whereas the Mettl6 mutants showed an ∼12% m3C reduction (Fig. 1, B and C). Loss of Mettl8 did not produce a significant change in m3C levels in tRNA in either tissue (Fig. 1, B and C). Importantly, the levels of 18 other RNA modifications were not significantly affected in any of the mutant mice in total RNA (supplemental Fig. S5). Similar to mice, loss of human METTL2A and -2B (M2 KO in Fig. 1D) contributes ∼35% of the m3C in tRNA in HEK293T cells, whereas loss of METTL8 in HCT116 cells again did not affect m3C levels (Fig. 1E). Importantly, m3C was significantly elevated in METTL2A and -2B KO cells overexpressing ectopic METTL2B, with m3C increasing 15 and 21% in transfected cells after 6 and 10 days, respectively (supplemental Fig. S6E). Unfortunately, we were not able to obtain METTL6 KO clones in the human cell lines. Nevertheless, these data support the conclusion that METTL2 and -6 catalyze m3C in tRNAs, whereas METTL8 has no effect on tRNA m3C levels.
METTL2 targets tRNAThr(UGU) and tRNAArg(CCU)
Having established METTL2- and METTL6-catalyzed m3C formation in tRNA fractions, we next defined the location of the METTL-dependent m3C in specific tRNAs. Here, we used primer extension assays to identify polymerase-blocking modifications in specific tRNAs predicted or known to contain m3C at position 32 (18, 19, 22, 23). For threonine tRNA(UGU) isoacceptors, primer extension on total tRNA substrates from WT mice generated RNA bands consistent with blockage of the reverse transcriptase at position 32 (Fig. 2, A and C, lanes 1 and 3; bands 2 nt longer than the 22-nt probe). In contrast, the intensity of the 24-nt extension cDNA was greatly reduced in RNA from Mettl2 KO mice, indicating a significant reduction of m3C at position 32. Instead, an even longer cDNA band was generated (Fig. 2C, lane 2), consistent with read-through past C32 but blockage at another unknown modification. Similarly, Mettl2 is required for C32 modification of tRNAArg(CCU) (Fig. 2, B and D), with loss of Mettl2 leading to several longer RNA fragments perhaps caused by the presence of polymerase-blocking m1G and m2G as inferred from previous studies at position 10 (19, 21, 23). Moreover, there is no RT stop at C32 in tRNAArg(ACG) from either WT, Mettl2 or -6 mutant mouse tissues (supplemental Fig. S8E). These discoveries are in agreement with the recent observation that m3C32 is present in tRNAArg with U at position 36 (e.g. tRNAArg(CCU)), but not in tRNAArg with G at position 36 (tRNAArg(ACG)) (19). Like mouse Mettl2, human METTL2A and -2B double mutants showed reduced C32 modification in both tRNAThr(AGU) and tRNAThr(UGU)(supplemental Fig. S6, A–C), with longer cDNA bands in tRNAThr(AGU) most likely caused by N2-dimethylguanosine (m2,2G) for tRNAThr(AGU), as inferred from previous studies (23). Complementation of METTL2 KO cells with ectopic METTL2B restored a polymerase-blocking modification, as indicated by the loss of longer read-through cDNA bands observed in the METTL2 KO cells (supplemental Fig. S6D). Further evidence comes from primer extension analyses in tRNA from METTL2 KO cells incubated with recombinant WT METTL2B or mutants in which the GXGXG SAM-binding motif is replaced with three alanines (G3A). The longer read-through cDNA is absent with WT METTL2B but is present in lanes with G3A mutants (supplemental Figs. S6, F and G, and S9).
Figure 2.
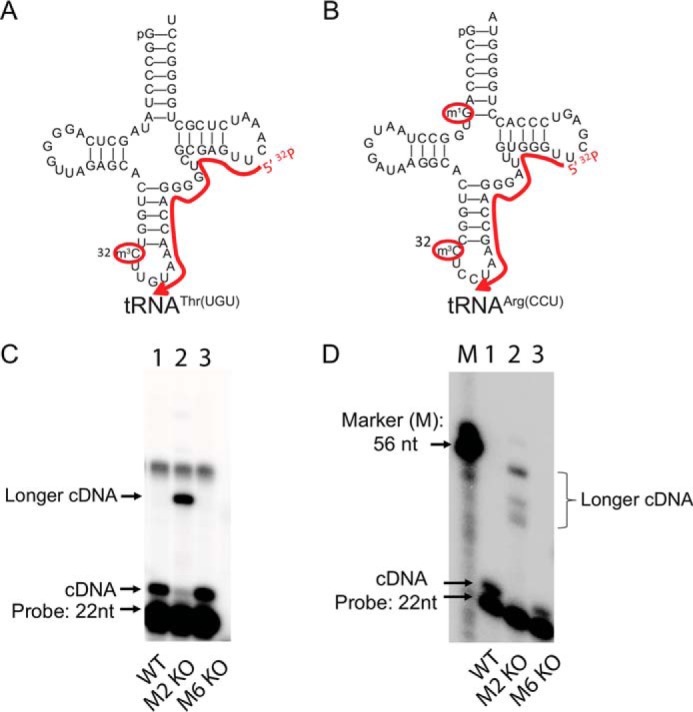
Mouse METTL2 is responsible for position 32 modification of tRNAThr(UGU) and tRNAArg(CCU). A and B, schematic showing one tRNAThr(UGU) and one tRNAArg(CCU) isoacceptor from mice, with red curved lines denoting the coverage of probes used in primer extension assay to map polymerase-blocking modification at position C32. C, primer extension assay using probes targeting tRNAThr(UGU) using RNA from wild type (WT), Mettl2 KO (M2 KO), and Mettl6 (M6 KO) mutants. Free probes are 22 nt long. D, primer extension for tRNAArg(CCU). Marker lane (M) contains a 56-nt-long ssDNA probe.
Human METTL6 interacts with seryl-tRNA synthetase in an RNA-dependent manner
Although METTL2 contributed 35% of the m3C level in total tRNA, METTL6 accounted for 12%, so we next assessed METTL6's role in tRNA modification. METTL6 bears homology with Trm141 in S. pombe, which targets serine tRNAs, so we assessed the presence of m3C32 in tRNASer(AGA) and tRNASer(GCU) using the primer extension assay. This revealed the presence of read-through bands consistent with loss of RT-blocking modification(s) near position 32 on tRNASer(AGA) and tRNASer(GCU) in Mettl6 KO cells (supplemental Fig. S8, C and D). It was recently revealed that yeast seryl-tRNA synthetase (SARS) interacts with TRM140 and greatly stimulates m3C modification of tRNASer in vitro (24). We also performed immunoprecipitation (IP) and verified this interaction in human HEK293T cell lines. Analysis of immunoprecipitates from lysates of cells stably overexpressing FLAG-METTL6 revealed the presence of SARS as the protein associated with FLAG-METTL6, with SARS absent in the pulldowns from FLAG-METTL6-ΔSAM (GXGXG motif deletion) mutant cells or pulldowns from FLAG-METTL6 cells using a nonspecific IgG control (Fig. 3). Because both SARS and METTL6 target tRNA, we assessed the potential for RNA-dependent SARS-METTL6 interactions by digesting cell pulldowns with RNase A or DNase I before the IP washing steps and then resolving the proteins by SDS-PAGE. The binding between SARS and METTL6 was disrupted by RNase treatment, but not DNase treatment (Fig. 3), which points to an RNA-dependent interaction between SARS and METTL6, possibly involving the serine tRNA substrates of both enzymes.
Figure 3.
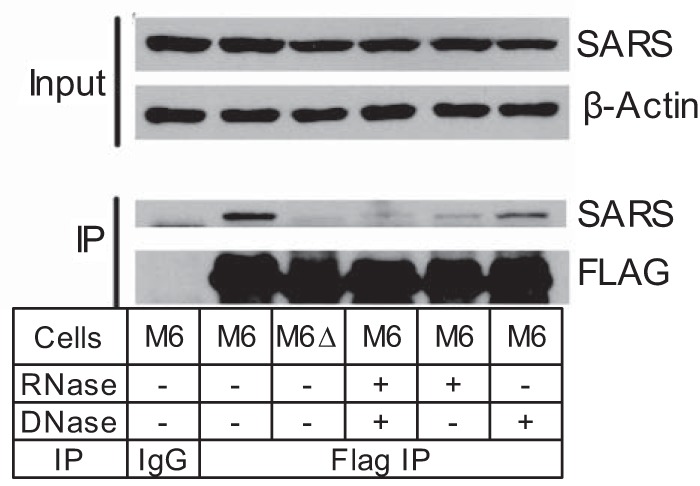
RNA-dependent interaction between METTL6 and SARS. HEK293T cells were transfected with either FLAG-METTL6 (M6) or FLAG-METTL6-ΔSAM (M6Δ). Equal amounts (protein level) of cell extracts were immunoprecipitated (IP) with either nonspecific IgG as a control or FLAG antibody, and treated with either buffer (−), RNase (+), or DNase (+), followed by SDS-PAGE, blotting, and probing with SARS or FLAG antibodies. Blots with Input cell extracts were probed with SARS and β-actin antibodies.
METTL8 catalyzes m3C formation in mRNA
Although both METTL2 and -6 contribute to m3C formation in tRNA, METTL8 showed no such activity (Fig. 1), which motivated us to analyze other potential RNA substrates for METTL2, -6, and -8. Here, we provide definitive proof that m3C is a modification in mRNA and that METTL8 in mice and humans catalyzes its formation. We first established the presence of m3C in mRNA. tRNA is the most abundant source of m3C, with levels about 4 orders-of-magnitude higher than large RNA species (Fig. 4C). To prevent contamination of mRNA by tRNA and other small RNAs, we systematically and progressively purified the mRNA following the strategy shown in Fig. 4A, with assessment of purity at each stage by Bioanalyzer analysis (Fig. 4B and supplemental Fig. S4) and quantification of m3C by LC-MS/MS analysis. First, we used size-exclusion chromatography to purify RNA larger than 5.8S rRNA in size (“input RNA” in Fig. 4C) (28), and Bioanalyzer electropherograms showed no detectable small RNAs in this RNA fraction (Fig. 4B). mRNA in this fraction was then enriched by two rounds of oligo(dT) affinity purification, which resulted in an ∼5–7-fold enrichment of m3C compared with input RNA (Fig. 4C). Some 28S and 18S rRNA was still present in the poly(A)-enriched samples (Fig. 4B). The residual rRNA was further removed using a commercial rRNA depletion kit (supplemental Materials and Methods and Fig. 4B), which resulted in an additional ∼1.5-fold increase in the m3C level (Fig. 4C). Importantly, we also purified individual 28S and 18S rRNA fractions and showed extremely low m3C levels (Fig. 4C). The progressive enrichment of m3C as the mRNA fraction was progressively purified establishes poly(A)-containing mRNA as the source of the m3C and rules out contamination with all small RNA species (5.8S rRNA and other RNA smaller than 5.8S in size), 18S or 28S rRNA. We also monitored m1A, an established mRNA modification during the purification process. It is not surprising to see higher m1A levels in 28S rRNA than that in mRNA, because m1A is known to be present in 28S rRNA (supplemental Fig. S10A). We also failed to observe N6-threonylcarbamoyladenosine (t6A) modification, which is only known to exist in tRNA, in the fully purified mRNA (supplemental Fig. S10B), which further excludes tRNA contamination. To place the level of m3C in perspective, we quantified m1A, m6A, and m5C levels in the purified mRNA (after rRNA removal step) (Fig. 4D), with m6A being the most abundant about the four, and other three showed similar percentages at the same order of magnitude. The comparison with other published results will be discussed below.
Figure 4.

Poly(A) mRNA enrichment and m3C levels monitored during purification. A, schematics of poly(A) mRNA enrichment and separation of 28S and 18S rRNA. B, typical electropherogram for the following: 1. total RNA; 2. RNA >5.8S isolated by SEC; 3. poly(A) RNA enriched by oligo(dT) selection; 4. poly(A)-enriched mRNA after rRNA depletion; 5. 28S; and 6. 18S rRNA purified by SEC. C, LC-MS/MS quantitation of m3C level in tRNA, RNA larger than 5.8S rRNA, oligo(dT)-enriched mRNA, ribo-zero-treated mRNA, 18S rRNA, 28S rRNA. D, LC-MS/MS quantitation of m3C, m5C, m1A, and m6A in human HCT116 mRNA purified by SEC, poly(A)-enrichment, and rRNA depletion. (The level of each modified ribonucleoside is expressed relative to its unmodified form. Data represent mean ± S.D. for n > = 3.)
We next sought to establish which gene is responsible for m3C in mRNA: Mettl2, -6, or -8. Here, we observed dramatic reduction in m3C in mRNA isolated from liver tissue from Mettl8 KO mice compared with WT mice littermate, and no reduction of m3C levels in liver tissue from Mettl2 or Mettl6 KO mice (Fig. 5, A and B). Similar reductions in m3C signal were observed in METTL8 KO human HCT116 and HeLa S3 cells but not in METTL2 KO HEK293T cells (Fig. 5, C–E, and supplemental Fig. S11). Furthermore, ectopic expression of METTL8 in HeLa S3 METTL8 KO cells for 3 days restores the m3C level in mRNA to ∼59%, whereas complementation with a SAM-binding site mutant of METTL8 (METTL8-ΔSAM) failed to increase m3C (Fig. 5E). These results establish METTL8 as the m3C writer in mRNA in mouse liver tissue and two human cell lines.
Figure 5.

METTL8 catalyzes formation of m3C in mRNA in mouse liver tissues and human cell lines. mRNA was isolated from mouse liver tissue, human HCT116, and HeLa S3 cells using the purification scheme shown in Fig. 4A. A, LC-MS/MS chromatogram for m3C, m4C, and m5C in wild-type mice (WT) and mice lacking Mettl2 (M2 KO), Mettl6 (M6 KO), or Mettl8 (M8 KO), as well as a double knock-out for METTL6 and -8 (M6,8 KO). Peak areas are noted numerically above each peak. B, LC-MS/MS chromatogram for m3C, m4C, and m5C in HeLa S3 METTL8 KO cells transfected with a vector expressing either wild-type (M8(WT)) or a SAM-binding mutant METTL8 (M8(ΔSAM)). Peak areas are noted numerically above each peak. C–E, LC-MS/MS quantification of m3C levels in mouse liver tissue (C), HCT116 cells (D), and HeLa S3 cells (E). Abbreviations are the same as in A and B. (The level of m3C is expressed relative to canonical cytidine. Data with error bars represent mean ± S.D. for at least three biological replicates.)
METTL2, -6, and -8 family in cell line growth rate and global translation
Although no observable developmental defects are observed in METTL2, -6, and -8 null-mutant mice, we explored the effects of this METTL family enzymes on growth rate and global translation in cell lines. For HEK293T, there was no significant change in growth rate in METTL2 and -8 mutant cell lines or METTL6 knockdown cell lines, compared with control cell lines (from which those mutant or knockdown cell lines are derived) (supplemental Figs. S12 and S13). Next, we assess the effects of m3C on global translation by polysome profiling, comparing wild-type and null-mutant cells. HEK293T WT and METTL2 KO cell lines showed a similar ratio of polysomes to monosomes (supplemental Fig. S14), whereas METTL8 KO in HCT116 cell lines showed slightly lower polysomes to monosomes. This suggests a possible increased ribosomal stalling when METTL8 is absent in HCT116 (supplemental Fig. S15). Nevertheless, more work such as ribosome profiling is needed to reveal unrevealed biological functions of m3C.
Discussion
As a renewal of interest in the functional consequences of RNA modification, the emerging field of epitranscriptomics is in the early stages of expanding the “catalogue” of modifications and modifying enzymes from precedent with tRNA and rRNA to all other forms of non-coding and coding RNA. Mammalian cells have seen the most significant advances in the discovery of mRNA modifications, such as m6A, 2′-O-methylations, pseudouridine, m5C, and m1A, whereas our understanding of the enzymology of RNA modifications has lagged behind that of prokaryotes. Here we have used a combination of bioanalytical chemistry and genetics to expand the repertoire of RNA-modifying enzymes in mammalian cells with the definitive assignment of function to three homologous METTL MTases and the discovery of m3C as a new epitranscriptomic mark in mRNA.
Of the over 25 METTL proteins and other MTases, METTL2, -6, and -8 stand out in terms of homology, with the suggestion that they may be related to m3C from the observation by Suzuki and co-workers (23) of reduced m3C levels in Mettl2B knockdown studies in human cells. We pursued a systematic and quantitative analysis of m3C in the major forms of RNA in mouse and human cells lacking METTL2, -6, and -8. These studies revealed that METTL2 and -6 are tRNA-specific enzymes that account for a total of 45–55% of the m3C in tRNA, with METTL8 having no activity with tRNA. This leaves nearly half of the m3C in tRNA to other yet-to-be discovered MTases. Some care must be exercised in assigning the proportion of m3C synthesis to each enzyme, as the METTL6 mutant mice used in this study could conceivably use an alternative splicing strategy that skips the deleted region in our construct and produces a functional variant of METTL6, as we could see from the strongest band in supplemental Fig. S2E. A targeted proteomic analysis of METTL enzymes in each of our human and mouse mutants would resolve this problem.
Beyond m3C catalysis, we were also able to assign the specific function of C32 methylation to METTL2 and -6. Although the primer extension assays for METTL6 precluded observation of the actual polymerase blockage site, the evidence for polymerase read-through in mutant cells, the RNA-dependent interaction between METTL6 with seryl-tRNA synthetase, and the conservation of function across eukaryotes points to serine tRNA as the likely substrate. It is likely that the range of substrates for both METTL2 and METTL6 exceeds the tRNAs analyzed in this study. Indeed, there are 15 m3C sites in mammalian cytoplasmic tRNAs detected or predicted so far at the isoacceptor level (supplemental Table S5) (19).
A systematic purification and analysis of m3C in individual RNA species from WT and mutant cells also led to the discovery of m3C in mRNA and of METTL8 activity as the major if not the only m3C writer in mRNA. Contamination with m3C-containing tRNA or rRNA species could not account for the m3C observed in poly(A)-purified RNA given the progressive enrichment of m3C at each stage of mRNA purification, including size-exclusion chromatography to remove all small RNAs, oligo(dT)-based affinity purification, and removal of rRNAs by affinity purification. LC-MS/MS analysis revealed that m3C is present in mRNA at a level of ∼5 m3C per 105 cytidine. This is lower than the level of m6A in mRNA at ∼1–2 per 103 adenosines but of similar level as m1A at ∼7 per 105 adenosines and m5C at ∼9 per 105 cytidines in mRNA (Fig. 4D). In 28S rRNA, m1A in only known to be present at position 1322, and if this is the real case, the frequency of m1A in pure 28S rRNA should be around 1.25 m1A per 1000 adenosines (∼800 adenosines per 28S rRNA molecule). Our measured frequency is ∼1.7 per 1000 adenosines (supplemental Fig. S10A). Future absolute quantification using isotope-labeled internal standards is needed to get more accurate m1A and m3C levels in mRNA. Notably, the purified 18S rRNA has a signal of 0.5 m1A per 10,000 adenosine nucleotides. It is most likely due to the contamination of 28S rRNA (supplemental Fig. S4, G and K).
The possible contamination from small RNA species (<200 nt) could be excluded because METTL2 or -6 mutant cells failed to show any reduction of m3C in purified mRNA (Fig. 5). It is clear from the studies with the METTL mutant cells (Fig. 5) that METTL8, but not METTL2 or METTL6, is responsible for m3C in mRNA.
More work is needed to reveal the distribution and dynamics of m3C in mRNA and at which stage m3C is inserted. Affinity pulldown studies could be done similarly as with m1A studies (9, 10). There are already commercial antibodies targeting against 2′-deoxy-3-methylcytidine (m3dC) (29) but no antibodies targeting m3C was reported. Future work is needed to characterize those m3dC-targeting antibodies and to check their specificity and affinity to m3C-containing RNA. Otherwise, new m3C-targeting antibodies needed to be developed. The m3C-targeting antibodies could be used to pull down m3C-containing RNA fragments and proceed to RNA-sequencing. In this way, m3C-containing transcripts could be discovered. Deep sequencing-based methods provide another approach to identify nucleobase methylations (19, 30, 31) and could shed light on the locations of m3C in mRNA. Future work like ribosome profiling and quantitative proteomics would be to decipher more biochemical and biological functions of m3C on specific tRNA and mRNA. It was also recently reported that m3C is a pre-requisite for cytidine to uridine deamination in Trypanosoma brucei (32). It opens a lot of testable hypotheses, for example, whether m3C is required for C-to-U deamination in mammalian tRNA, mRNA, or even DNA.
Experimental procedures
Materials
All plasmids and oligos used in the present studies are listed in supplemental Tables S1–S4.
Cell culture
The human cell lines HEK293T, HeLa S3, and HCT116 were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Sigma or Gibco) and antibiotics (100 units/ml penicillin, 100 μg/ml streptomycin) unless otherwise specified.
Generation of METTL2, -6, and -8 mutants by CRISPR/Cas9
Mettl2, -6, and -8 mutant mice were generated at Model Animal Research Centre, Nanjing University. gRNA for D10A CAS9 was inserted into a U6 promoter-driven expression vector (pUC57), and targeting efficiency was tested in 3T3 cells first before injecting into fertilized eggs. All mutant mice were verified by Sanger sequencing of targeted loci (supplemental Fig. S2). Mettl6 and Mettl8 wild-type and KO tissues were checked by Western blotting (anti-mouse Mettl6, Proteintech, catalogue no. 16527-1-AP, Mettl8 antibody (Prestige Antibodies® Sigma)) (supplemental Fig. S2G). Mice were bred in comparative medicine, National University of Singapore, following the polices and guidelines of National University of Singapore Institutional Animal Care and Use Committee (IACUC). Vector pST1374-NLS-FLAG-linker-Cas9 containing human codon-optimized Cas9 and human-gRNA-expression vector MLM3636 were from Addgene and used for generation of mutants in human cell lines (supplemental Table S1). MISSION shRNA targeting METTL6 was purchased from Sigma.
Primer extension analysis of m3C in RNA
Primer extension assays were performed as described previously (22, 33). The high sequence similarity among some tRNA isoacceptors (supplemental Fig. S7) makes it possible to use only one 32P-labeled probe to differentiate among different isoacceptors, and when it is not possible, we designed probes that target the most frequent tRNA isodecoder in that group. To label oligos, 10–30 pmol of the primers were labeled with twice this amount of [γ-32P]ATP (20–60 pmol) (PerkinElmer Life Sciences BLU002Z500UC) using T4 polynucleotide kinase (New England Biolabs), followed by removal of excess ATP by G-25 spin columns (GE Healthcare). Oligos for primer extension assay are listed in supplemental Table S2. 1–2 pmol of 5′-end-labeled primers were annealed to 2 μg of total RNA or 500 ng to 1 μg of tRNA in 0.5 μl of Superscript III buffer (Invitrogen) by heating at 80 °C for 5 min, with slow cooling (turning off heat block) to 25 °C for 20–30 min. The annealed reaction was then extended using 1 μl of Superscript III (Invitrogen), 0.1 mm of each dNTP (A, C, G, and T) in Superscript III buffer at 55 °C for 30 min, and stopped by adding RNA loading dye (formamide containing 0.1% bromphenol blue), heated at 85 °C for 5 min, and resolved on a 20-cm-long 15% polyacrylamide gel containing 8 m urea. The gel was dried under vacuum (Bio-Rad) and exposed in a cassette with a piece of X-ray film at −80 °C or a phosphor screen (GE Healthcare) at room temperature overnight or longer.
Size-exclusion chromatography purification of various RNA species
Samples with RIN >9 (Nano 6000 Chips, Agilent) were used for different RNA isolation by size-exclusion chromatography as described previously (28, 34). For tRNA isolation and the collection of large RNA species (Depletion of tRNA, 5.8S, and 5S rRNA), total RNA was separated by Bio SEC-3 column (Agilent; inner diameter, 7.8 mm; length, 300 mm; particle size, 3 μm; and pore size, 100 Å). For separation of 28S and 18S, Bio SEC-5 (Agilent; 7.8 mm; length, 300 mm; particle size, 5 μm; pore size, 2000 Å) column was used. All separation was run under 100 mm ammonium acetate isocratic elution, at flow rate of 0.5 ml/min at 60 °C.
Poly(A)-tailed mRNA enrichment
Cell lines or mouse liver tissues were lysed and purified by TRIzol reagent (Invitrogen/Thermo Fisher Scientific). Small RNA (5.8S rRNA and smaller RNAs) was depleted using size-exclusion HPLC, as described above. 20 μg of small RNA-depleted RNAs were used as input for two successive rounds of poly(A)+ selection using either Dynabeads® oligo(dT)25 beads or NEBNext® poly(A) mRNA Magnetic Isolation Module (New England Biolabs) according to the manufacturer's instructions. For the New England Biolabs kit, the reactions are scaled up 5-fold and performed in 1.5-ml tubes instead of 0.2-ml PCR tubes. 85 μl of elution buffer provided by the kits were used to elute RNA from the magnetic beads. Ribo-Zero gold rRNA removal kits (Illumina) were used according to the manufacturer's instructions to further deplete rRNAs before LC-MS/MS analysis. 1 μl of the elution was reserved for bioanalyzer pico kit analysis. All the remaining eluted RNAs were digested and analyzed by LC-MS/MS.
Quantification of RNA modifications by LC-MS/MS
RNA fractions were concentrated with a 10-kDa MWCO spin filter (Millipore), spin-dialyzed against ultra-pure water. Isolated RNA was checked by Bioanalyzer nano (for 28S and 18S rRNA and tRNAs), pico (for poly(A)-tailed mRNA), to ensure no contamination from other cellular RNAs. An equal amount of RNA (500 ng or 1 μg of tRNA and 100 or 200 ng of mRNA calculated by Bioanalyzer) was enzymatically hydrolyzed to a single ribonucleoside using Ultrapure Benzonase (Sigma), bacterial alkaline phosphatase (Invitrogen), and phosphodiesterase I (Affymetrix) in the presence of antioxidants and deaminase inhibitors as described previously (35), cleaned up by spin dialysis (10 kDa, Millipore). The ribonucleosides were resolved by HPLC (Agilent 1290) using a Hypersil Gold aQ column (Thermo Fisher Scientific, inner diameter, 3 mm; length, 50 mm; and particle size, 1.9 μm) with a gradient elution starting with 0.1% (v/v) formic acid in water and adding acetonitrile (LC-MS grade) with 0.1% (v/v) formic acid at 25 °C. The HPLC eluent was analyzed on Agilent 6460 or 6490 triple quadrupole mass spectrometer using multiple-reaction monitoring in positive ion mode. Transitions for adenosine (A) 268.1→136.1 m/z, guanosine (G) 284.1→152.1 m/z, uridine (U) 245.1→113.1 m/z, cytidine (C) 244.1→112.1 m/z, m3C, m5C and 4-methylcytidine (m4C), 258.1→126.1 m/z, m1A, m6A, 282.1→150.1 m/z, t6A, 413.1→281.1 were used for quantification. A detailed procedure and instrumentation for the LC-MS/MS analyses are described elsewhere (35). For relative comparison of one modification among the same batch of samples, the signal intensity is normalized against the intensity of canonical cytidine. For quantifications, a series of concentrations of nucleoside standards, m3C (Sigma, M5753), m5C (Berry and Associates, PY7637), m4C (CambridgeChem, B246277), m1A (Cayman Chemical, 16937), m6A (Berry and Associates, PR 3732), adenosine (Sigma, 01890), cytidine (Sigma, C122106), and t6A (Biolog, Bremen, Germany, C022), were run for every batch of experiments to obtain their corresponding standard curves. Concentrations of nucleosides in mRNA samples were interpolated by fitting the signal intensities onto calibration curves. The ratios of m3C/C, m5C/C, m1A/A, and m6A/A were subsequently calculated. Data were analyzed using Microsoft Excel and Prism 6 software.
Immunoprecipitation
Cell lysates were extracted in lysis buffer consisting of 50 mm Tris-HCl, pH 7.5, 0.1 mm EGTA, 1% Triton X-100, 5 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 50 mm sodium fluoride, 0.27 m sucrose, 0.1% (v/v) 2-mercaptoethanol, and 1 tablet/50 ml of EDTA-free complete protease inhibitor mixture (Roche Applied Science). Protein lysates from cells were centrifuged at 13,000 × g for 5 min at 4 °C, and the insoluble debris was discarded. FLAG M2 antibody (10 μl; for FLAG IP, Sigma) or 2 μg of nonspecific IgG antibody (Santa Cruz Biotechnology) was coupled to protein G-agarose beads (10 μl; Thermo Fisher Scientific) and washed with 1 ml of lysis buffer before incubation with 1 mg of total lysate for 1 h at 4 °C on an orbital shaker at 1000 rpm. Proteins bound to the beads were separated from the cell lysate by centrifugation at 10,000 × g for 1 min, washed twice with 1 ml of lysis buffer containing 0.5 m NaCl, and twice with 1 ml of Buffer B (50 mm Tris-HCl, 0.27 m sucrose, and 0.1% v/v 2-mercaptoethanol, pH 7.5). After removing the remaining supernatant, SDS-Laemmli buffer was added to the beads to release the immunoprecipitated protein. The samples were boiled for 10 min before being resolved by SDS-PAGE and blotted for subsequent Western analysis. SARS antibody is from Abcam (ab154825). β-Actin is from Santa Cruz Biotechnology (SC-47778). FLAG antibody is from Sigma (F3165).
Author contributions
L. X. conceived the study, performed the experiments, and wrote the paper; X. L. generated human Mettl8 KO cell lines and performed the experiments; K. S. O. performed Mettl6 IP with SARS; J. L., F. Y., and Y. H. C. provided technical assistance on cell line culture, protein purification, and LC-MS/MS, respectively; N. S. and J. X. generated all mutant mice strains; Y. G. G. provided technical guidance and advice to the paper; P. C. D. designed LC-MS/MS experiments and wrote the paper; X. Y. F. conceived the study and wrote the paper.
Supplementary Material
Acknowledgments
We thank Vonny Leo and Dr. Leah Vardy (IMB, Singapore) and Nah Jiemin and Dr. Guo Huili (IMCB, Singapore) for technical advice on polysome profiling. We thank Prof. Chuan He (University of Chicago) for advice on this study.
This work was supported by the National Research Foundation Singapore, the Singapore Ministry of Education under its Research Centres of Excellence Initiative, Cancer Science Institute of Singapore (CSI, Singapore) Grant 713001010271 (to X.-Y. F.), funds from the Office of Deputy President of National University of Singapore, partly under the Singapore-MIT Alliance for Research and Technology (to P. C. D.), and by National Institutes of Health Grant ES026856 from the NIEHS (to P. C. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Materials and Methods, Tables S1–S5 and Figs. S1–S15.
This article was selected as one of our Editors' Picks.
- m6A
- 6-methyladenosine
- m3C
- 3-methylcytidine
- m5C
- 5-methylcytidine
- m1A
- 1-methyladenosine
- MTase
- methyltransferase
- SAM
- S-adenosylmethionine
- METTL
- methyltransferase-like
- IP
- immunoprecipitation
- SARS
- seryl-tRNA synthetase
- SEC
- size-exclusion chromatography
- nt
- nucleotide
- oligo
- oligonucleotide
- t6A
- N6-threonylcarbamoyladenosine
- m3dC
- 2′-deoxy-3-methylcytidine.
References
- 1. O'Connell M. (2015) RNA modification and the epitranscriptome; the next frontier. RNA 21, 703–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao B. S., Roundtree I. A., and He C. (2017) Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan C. T., Deng W., Li F., DeMott M. S., Babu I. R., Begley T. J., and Dedon P. C. (2015) Highly predictive reprogramming of tRNA modifications is linked to selective expression of codon-biased genes. Chem. Res. Toxicol. 28, 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chionh Y. H., McBee M., Babu I. R., Hia F., Lin W., Zhao W., Cao J., Dziergowska A., Malkiewicz A., Begley T. J., Alonso S., and Dedon P. C. (2016) tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat. Commun. 7, 13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lewis C. J., Pan T., and Kalsotra A. (2017) RNA modifications and structures cooperate to guide RNA-protein interactions. Nat. Rev. Mol. Cell Biol. 18, 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Helm M., and Motorin Y. (2017) Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 18, 275–291 [DOI] [PubMed] [Google Scholar]
- 7. Phizicky E. M., and Hopper A. K. (2010) tRNA biology charges to the front. Genes Dev. 24, 1832–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jackman J. E., and Alfonzo J. D. (2013) Transfer RNA modifications: nature's combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 4, 35–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dominissini D., Nachtergaele S., Moshitch-Moshkovitz S., Peer E., Kol N., Ben-Haim M. S., Dai Q., Di Segni A., Salmon-Divon M., Clark W. C., Zheng G., Pan T., Solomon O., Eyal E., Hershkovitz V., et al. (2016) The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li X., Xiong X., Wang K., Wang L., Shu X., Ma S., and Yi C. (2016) Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 12, 311–316 [DOI] [PubMed] [Google Scholar]
- 11. Gilbert W. V., Bell T. A., and Schaening C. (2016) Messenger RNA modifications: form, distribution, and function. Science 352, 1408–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hall R. H. (1963) Isolation of 3-methyluridine and 3-methylcytidine from soluble ribonucleic acid. Biochem. Biophys. Res. Commun. 12, 361–364 [DOI] [PubMed] [Google Scholar]
- 13. Iwanami Y., and Brown G. (1968) Methylated bases of ribosomal ribonucleic acid from HeLa cells. Arch. Biochem. Biophys. 126, 8–15 [DOI] [PubMed] [Google Scholar]
- 14. Maden B. E., and Salim M. (1974) The methylated nucleotide sequences in HELA cell ribosomal RNA and its precursors. J. Mol. Biol. 88, 133–152 [DOI] [PubMed] [Google Scholar]
- 15. Weissenbach J., Kiraly I., and Dirheimer G. (1977) Structure primaire des tRNAThr la et b de levure de bière. Biochimie 59, 381–391 [DOI] [PubMed] [Google Scholar]
- 16. Olson M. V., Page G. S., Sentenac A., Piper P. W., Worthington M., Weiss R. B., and Hall B. D. (1981) Only one of two closely related yeast suppressor tRNA genes contains an intervening sequence. Nature 291, 464–469 [DOI] [PubMed] [Google Scholar]
- 17. Jühling F., Mörl M., Hartmann R. K., Sprinzl M., Stadler P. F., and Pütz J. (2009) tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 37, D159–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arimbasseri A. G., Iben J., Wei F. Y., Rijal K., Tomizawa K., Hafner M., and Maraia R. J. (2016) Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA 22, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clark W. C., Evans M. E., Dominissini D., Zheng G., and Pan T. (2016) tRNA base methylation identification and quantification via high-throughput sequencing. RNA 22, 1771–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. El Yacoubi B., Bailly M., and de Crécy-Lagard V. (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 46, 69–95 [DOI] [PubMed] [Google Scholar]
- 21. Machnicka M. A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K. M., Helm M., Bujnicki J. M., and Grosjean H. (2013) MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 41, D262–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. D'Silva S., Haider S. J., and Phizicky E. M. (2011) A domain of the actin binding protein Abp140 is the yeast methyltransferase responsible for 3-methylcytidine modification in the tRNA anti-codon loop. RNA 17, 1100–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noma A., Yi S., Katoh T., Takai Y., Suzuki T., and Suzuki T. (2011) Actin-binding protein ABP140 is a methyltransferase for 3-methylcytidine at position 32 of tRNAs in Saccharomyces cerevisiae. RNA 17, 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Han L., Marcus E., D'Silva S., and Phizicky E. M. (2017) S. cerevisiae Trm140 has two recognition modes for 3-methylcytidine modification of the anticodon loop of tRNA substrates. RNA 23, 406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., Dai Q., Chen W., and He C. (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang X., Feng J., Xue Y., Guan Z., Zhang D., Liu Z., Gong Z., Wang Q., Huang J., Tang C., Zou T., and Yin P. (2016) Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578 [DOI] [PubMed] [Google Scholar]
- 27. Richon V. M., Johnston D., Sneeringer C. J., Jin L., Majer C. R., Elliston K., Jerva L. F., Scott M. P., and Copeland R. A. (2011) Chemogenetic analysis of human protein methyltransferases. Chem. Biol. Drug Des. 78, 199–210 [DOI] [PubMed] [Google Scholar]
- 28. Chionh Y. H., Ho C. H., Pruksakorn D., Ramesh Babu I., Ng C. S., Hia F., McBee M. E., Su D., Pang Y. L., Gu C., Dong H., Prestwich E. G., Shi P. Y., Preiser P. R., Alonso S., and Dedon P. C. (2013) A multidimensional platform for the purification of non-coding RNA species. Nucleic Acids Res. 41, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dango S., Mosammaparast N., Sowa M. E., Xiong L. J., Wu F., Park K., Rubin M., Gygi S., Harper J. W., and Shi Y. (2011) DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol. Cell 44, 373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tserovski L., Marchand V., Hauenschild R., Blanloeil-Oillo F., Helm M., and Motorin Y. (2016) High-throughput sequencing for 1-methyladenosine (m(1)A) mapping in RNA. Methods 107, 110–121 [DOI] [PubMed] [Google Scholar]
- 31. Hauenschild R., Tserovski L., Schmid K., Thüring K., Winz M. L., Sharma S., Entian K. D., Wacheul L., Lafontaine D. L., Anderson J., Alfonzo J., Hildebrandt A., Jäschke A., Motorin Y., and Helm M. (2015) The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence-dependent. Nucleic Acids Res. 43, 9950–9964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiang Y., Laurent B., Hsu C. H., Nachtergaele S., Lu Z., Sheng W., Xu C., Chen H., Ouyang J., Wang S., Ling D., Hsu P. H., Zou L., Jambhekar A., He C., and Shi Y. (2017) RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chan P. P., and Lowe T. M. (2009) GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan C. T., Dyavaiah M., DeMott M. S., Taghizadeh K., Dedon P. C., and Begley T. J. (2010) A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 6, e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Su D., Chan C. T., Gu C., Lim K. S., Chionh Y. H., McBee M. E., Russell B. S., Babu I. R., Begley T. J., and Dedon P. C. (2014) Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat. Protoc. 9, 828–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ping X. L., Sun B. F., Wang L., Xiao W., Yang X., Wang W. J., Adhikari S., Shi Y., Lv Y., Chen Y. S., Zhao X., Li A., Yang Y., Dahal U., Lou X. M., et al. (2014) Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.