

Abstract
In utero hypoxia influences the structure and function of most fetal arteries, including those of the developing cerebral circulation. Whereas the signals that initiate this hypoxic remodeling remain uncertain, these appear to be distinct from the mechanisms that maintain the remodeled vascular state. The present study explores the hypothesis that chronic hypoxia elicits sustained changes in fetal cerebrovascular reactivity to endothelin-1 (ET-1), a potent vascular contractant and mitogen. In fetal lambs, chronic hypoxia (3,820-m altitude for the last 110 days of gestation) had no significant effect on plasma ET-1 levels or ETA receptor density in cerebral arteries but enhanced contractile responses to ET-1 in an ETA-dependent manner. In organ culture (24 h), 10 nM ET-1 increased medial thicknesses less in hypoxic than in normoxic arteries, and these increases were ablated by inhibition of PKC (chelerythrine) in both normoxic and hypoxic arteries but were attenuated by inhibition of CaMKII (KN93) and p38 (SB203580) in normoxic but not hypoxic arteries. As indicated by Ki-67 immunostaining, ET-1 increased medial thicknesses via hypertrophy. Measurements of colocalization between MLCK and SMαA revealed that organ culture with ET-1 also promoted contractile dedifferentiation in normoxic, but not hypoxic, arteries through mechanisms attenuated by inhibitors of PKC, CaMKII, and p38. These results support the hypothesis that chronic hypoxia elicits sustained changes in fetal cerebrovascular reactivity to ET-1 through pathways dependent upon PKC, CaMKII, and p38 that cause increased ET-1-mediated contractility, decreased ET-1-mediated smooth muscle hypertrophy, and a depressed ability of ET-1 to promote contractile dedifferentiation.
Keywords: calcium-calmodulin-dependent protein kinase II, endothelin receptors, myosin light chain kinase, p38 MAP kinase, protein kinase C
for most mammals, the final weeks of gestation are a period of rapid change, particularly for the fetal cardiovascular system. This brisk pace of change renders the immature vasculature vulnerable to many stresses common during the perinatal period, including hypoxia, which can be secondary to compromised placental flow, maternal pulmonary disease, diabetes, or drug abuse (32). Hypoxic stresses, in turn, typically promote vascular remodeling that alters vascular structure and function in the short term (51) and increases the risk of later onset cardiovascular disease in adulthood (75). Within this context, our previous work has shown that hypoxia modulates fetal vascular structure and function, and that vascular endothelial growth factor (VEGF) is involved in this modulation (1). In the present study, we examine the possible involvement of another vasotrophic molecule, endothelin-1 (ET-1), which, like VEGF, is also upregulated by hypoxia through the actions of the transcription factor hypoxia-inducible factor-1α (HIF-1α) (30), as summarized in our recent review of hypoxic cerebrovascular remodeling in the fetal cerebral circulation (56).
ET-1 is a potent contractant in many arteries (17) and also exerts marked mitogenic effects in both vascular and nonvascular tissues, due in large part to the broad expression of endothelin receptors in many different cell types (17). ET-1 binds and activates two main G protein-coupled receptors, ETA and ETB, which are responsible for vasoconstriction and vasodilation, respectively (17). These receptors also can activate multiple signaling pathways, including those involving protein kinase C (PKC) (58), Ca2+-calmodulin-dependent protein kinase II (CaMKII) (57, 76), and p38 MAP kinase (54). Through these pathways, ET-1 and its receptors influence proliferation in neonatal pulmonary arteries (58), cardiomyocyte hypertrophy (76), vascular smooth muscle growth, proliferation, and migration (57), and many other effects.
To test our hypothesis that chronic hypoxia modulates the influence of ET-1 signaling on the fetal cerebral circulation, we measured by immunoassay the circulating levels of ET-1 in normoxic and chronically hypoxic term fetal lambs. In these two groups, we also measured and compared the abundances of ETA and ETB receptors in fetal cerebral arteries, and the contractile effects of ET-1 in the presence and absence of the ETA antagonist PD-156707. To assay the effects of chronic hypoxia on ET-1 signaling through mitogenic pathways, we studied the effects of organ culture with ET-1 in the presence and absence of chelerythrine (a PKC inhibitor), KN93 (a CaMKII inhibitor), and SB203580 (a p38 MAP kinase inhibitor) in normoxic and hypoxic fetal cerebral arteries. In these organ culture experiments, multiple end points were examined, including the medial thickness within the artery wall, the extent of smooth muscle proliferation as indicated by positive staining for Ki-67, and the organization of contractile proteins as revealed by confocal colocalization between myosin light chain kinase (MLCK) and smooth muscle α-actin (SMαA). Together, these experiments revealed that in the fetal cerebral circulation, ET-1 signaling has multiple important and diverse effects that are discretely modulated by chronic hypoxia.
MATERIALS AND METHODS
The protocols used in these studies were approved by the Animal Research Committee of Loma Linda University and complied with all policies in the National Institutes for Health Guide for the Care and Use of Laboratory Animals. Tissue harvesting and preparation have been previously described in detail (1, 12).
All tissues used in these experiments were obtained from normoxic and chronically hypoxic term fetal (139–142 days gestation) and young (18–24 mo old) nulliparous adult sheep. Normoxic animals were maintained at the LLU animal care facility (353-m altitude), where arterial oxygen tensions () averaged 23 ± 1 Torr and 102 ± 2 Torr in fetal and adult sheep, respectively (33). Chronically hypoxic sheep were maintained for the final 110 days of gestation at the Barcroft Laboratory, White Mountain Research Station, Bishop, CA (altitude 3,820 m). At high altitude, values averaged 19 ± 1 and 64 ± 2 Torr for fetal and adult sheep, respectively (33). Although chronic hypoxia decreased arterial oxygen tension by only ~4 Torr in the fetus, the corresponding decrease in hemoglobin oxygen saturation could fall as much as 30% or more (5, 50, 69), owing to the steep slope of oxygen dissociation curve for fetal hemoglobin.
Ewes were anesthetized with 10 mg/kg ketamine and 5 mg/kg midazolam (iv), intubated, and ventilated on 1–2% isoflurane with balance % O2. Term fetuses (between 139 and 142 days gestation) were accessed via a midline incision, after which whole blood was collected from the umbilical vein of normoxic and hypoxic term fetal sheep, into heparinized syringes. After blood collection, the umbilical cord was cut and the fetus weighed before immediate exsanguination by rapid removal of the heart. Ewes were also euthanized by rapid exsanguination.
Tissue harvest.
Brains were collected from fetuses gestated at sea level (FN) or at 3,820 m (FH) for the last 110 days of gestation. As previously described (1), harvested brains and arteries were continuously bubbled in a HEPES buffer solution (pH 7.4, 122.1 mM NaCl, 25 mM HEPES, 5.16 mM KCl, 2.4 mM MgSO4, 11.1 mM dextrose, 1.6 mM CaCl2, and 50 μM EDTA) with 95% O2-5% CO2. All arteries were dissected and cleaned of loose connective tissue and blood. Middle cerebral arteries (MCAs) designated for contractility and organ culture experiments were mechanically denuded of endothelium, then cut into 3-mm segments.
Measurement of plasma ET-1.
The hematocrit of collected whole blood was quantified within 30 min of blood collection using the HemataSTAT-II Microhematocrit System. Plasma isolated by centrifugation at 2,500 rpm (~1800 rcf) for 15 min at 4°C was aliquoted and frozen at −20°C until further processing. Plasma ET-1 levels were quantified with an ET-1 ELISA kit (R&D Systems, Minneapolis, MN) with 90–100% recovery efficiency. All samples were analyzed in duplicate.
Western blotting for ETA and ETB receptor levels.
In light of evidence demonstrating that multiple mRNA splice variants can be produced for both ETA and ETB receptors (17), and that certain microRNAs can attenuate endothelin receptor expression (55), we chose to directly measure endothelin receptor expression via Western blotting. Endothelium-intact MCAs designated for Western blot experiments were weighed, fast frozen in liquid N2, and kept at −80°C until tissue homogenization and protein extraction. MCAs first were equilibrated briefly at a 1:20 tissue-to-buffer ratio in ice-cold RIPA buffer solution containing 150 mM NaCl, 50 mM Tris, 10 mM EDTA, 5 mM EGTA, 1% Triton X-100, 0.05% sodium deoxycholate, 0.10% SDS, 10% glycerol, 20 mM DTT, and 5 μl/ml of buffer protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, no. M1745), all at a pH of 8.0. Proteins were extracted from the MCA samples via water bath sonication for 1 h at 4°C. Thereafter, the samples were then shaken for 90 min at 4°C before centrifugation at 10,000 g for 10 min. Supernatants were aliquoted and frozen at −80°C until protein assay and Western blotting.
Supernatant protein contents were quantified with the Bio-Rad DC Protein Assay using BSA as a standard, according to manufacturer’s directions. Supernatant from the protein homogenates were then separated by SDS-PAGE on 4–10% acrylamide gels. Increasing known amounts of common carotid protein homogenates were used as a relative standard. β-Mercaptoethanol (150 μl) was added to the top reservoir and gels were run at 35-mA constant current. Separated proteins were transferred onto nitrocellulose membranes at a constant 30 V overnight (16 h) in Bjerrum buffer (40 mM Tris, 39 mM glycine, and 0.01% SDS) with 20% methanol at 4°C.
The membranes were blocked with 5% milk in PBS at pH 7.45 (m-PBS) for 1 h at room temperature with gentle agitation. All subsequent incubations and washes were performed with 0.1% Tween-20 in 5% m-PBS (m-PBS-T20). After blocking, membranes were incubated with primary antibodies (anti-ETA at 1 μg/ml, Abcam, ab85163; and anti-ETB at 1:2,000, Abcam, ab117529) overnight (16–18 h) at 4°C with gentle agitation. Membranes were then washed 5 min for a total of 6 times with m-PBS-T20 before application of secondary antibody (GAR Dylight 800; Pierce Chemical, Rockford, IL, no. 46422) for 90 min. Membranes were again washed 6 × 5 min each in m-PBS-T20 followed by 2 × 5 min in PBS pH 7.45. Membranes were imaged with the LI-COR Bioscience Odyssey System.
Contractility.
As previously described (1, 12), MCAs designated for contractility measurements were mechanically denuded of endothelium with a small tungsten rod and then sectioned into 3-mm segments. The endothelium-denuded fetal MCA segments were mounted onto tungsten wires suspended between isometric force transducers, adjusted to a resting tension of 0.5 g, and equilibrated for 30 min in a Na+-Krebs buffer solution containing (in mM) 122.1 NaCl, 25.6 NaHCO3, 11.1 dextrose, 5.16 KCl, 2.5 MgSO4, 1.60 CaCl2, 0.114 ascorbic acid, 0.1 l-NAME, 0.1 l-NNA, and 0.027 EDTA, all at pH 7.4. The arteries were equilibrated at normal ovine temperature (38°C) and continuously bubbled with 95% O2-5% CO2. Contractions were induced with a K+-Krebs buffer solution containing (in mM) 122.1 KCl, 25.6 NaHCO3, 11.1 dextrose, 5.16 NaCl, 2.5 MgSO4, 1.60 CaCl2, 0.114 ascorbic acid, 0.1 l-NAME, 0.1 l-NNA, and 0.027 EDTA at pH 7.4. Following contraction in K+-Krebs buffer, the arteries were returned to resting conditions in Na+-Krebs buffer.
To confirm endothelial denudation, the MCA segments were incubated in Na+-Krebs buffer with 10 µM 8-phenyltheophylline (8-PT) (Sigma-Aldrich, no. P2278) for 20 min at 0.75 g, then contracted with 1 µM serotonin hydrochloride (5-HT) (Sigma-Aldrich, no. H9523). The addition of 1 µM ADP (Sigma-Aldrich, no. A5285) following 5-HT contraction was used to validate endothelium removal. The 8-PT was present during exposure to ADP to minimize any relaxation due to activation of vascular P1 receptors by adenosine released via ADP degradation. Na+-Krebs buffer without 8-PT was used to wash the arteries and return them to basal resting conditions. The contractile response to K+-Krebs buffer was then recorded once more to fully load all intracellular calcium stores, after which the arteries were returned to basal resting conditions. At this point, 10 nM PD-156707 (Sigma-Aldrich, no. PZ0141), an ETA antagonist, was added to the Na+-Krebs solution and the arteries were incubated in the solution for 30 min. The concentration used (10 nM) was approximately the IC90 concentration based on published results (40). Control arteries received the same amount of diluent as arteries treated with PD-156707 (0.02% DMSO in 0.02% 100 mM NaOH). A contractile dose-response relation was then determined for ET-1 (Sigma-Aldrich, no. E7764) starting at 10−12 M and increasing in half-log increments to a final concentration of 3.16 × 10−7 M.
Organ culture.
Endothelium-denuded MCA segments were organ-cultured at pH 7.45 in sterile DMEM (Sigma-Aldrich, no. M56469C) without FBS, to which was added 3.7 g/l of NaHCO3, 0.5% amino acid solution (Sigma-Aldrich, no. M5550), 1% non-essential amino acid solution (Sigma-Aldrich, no. M7145), 4 mM glutamine (Sigma-Aldrich, no. G7513), 2% antibiotic-antimycotic solution (GIBCO, Carlsbad, CA, no. 15240–096), and 70 μg/ml of gentamycin (GIBCO, no. 15750–060). The artery segments were cultured in untreated 12-well plates and maintained in a humidified incubator with 5% CO2 in room air at 37°C for 24 h.
After the first 24 h of culture, DMSO was added to all culture wells at 0.01875%, which was the final concentration used to solubilize all inhibitors. Matched sets of five adjacent segments were then treated as follows: 1) starved controls, 2) ET-1 at a physiological concentration of 10 nM, 3) 10 nM ET-1 plus 6.6 μM chelerythrine (Santa Cruz Biotechnology, Santa Cruz, CA, SC-3547), 4) 10 nM ET-1 plus 10 μM KN93 (Cayman Chemical, San Diego, CA, no. 13319), and 5) 10 nM ET-1 plus 10 μM SB203580 (Tocris Bioscience, Bristol, UK, no. 1402). Chelerythrine, KN93, and SB203580 were added to inhibit PKC, CaMKII, and p38 pathways, respectively, at approximately the EC90 concentration for each inhibitor (26, 31, 67). Owing to the limited availability of only 6 MCA segments from a single fetus, additional animals were used to prepare appropriate negative controls. Arteries were prepared for organ culture as already described, but received the following four treatments: 1) starved controls, 2) 6.6 μM chelerythrine, 3) 10 μM KN93, and 4) 10 μM SB203580.
Fluorescent immunohistochemistry and confocal imaging.
As previously described (1), arteries designated for imaging were fixed for 24 h in 4% neutral buffered formaldehyde (Electron Microscopy Sciences, Hatfield, PA, no. 15713S), paraffin embedded, and sectioned at 5 μm. Histoclear solution (National Diagnostic, Atlanta, GA, no. HS-200) was used to deparaffinize the slides before rehydration in decreasing concentrations of alcohol. Slides were then incubated in 100 mM glycine in PBS, pH 7.45, for 10 min to decrease background staining, then rinsed with gentle agitation in PBS at pH 7.45 for 5 min. Antigen retrieval was performed by microwave irradiation in citrate buffer at pH 6.03 for 5 min. Following antigen retrieval, the artery sections were washed with gentle agitation in PBS at pH 7.45, 3 times for 5 min each, and then permeabilized. Nonspecific blocking was achieved by exposure to 5% normal goat serum (Pierce Biotechnology, no. 31873) with 1% bovine serum albumin (Santa Cruz Biotechnology, no. SC-2323) and 0.3% Triton X-100 (Sigma-Aldrich, no. T-8787) in PBS at pH 7.45 for 30 min.
Slides carrying the MCA sections were double stained with three marker-pair combinations: 1) polyclonal anti-Ki-67 (Abcam, ab15580) at 1:100 and anti-SMαA at 1:400; 2) polyclonal anti-MLCK (Santa Cruz Biotechnology, SC-25428) at 1:50 and monoclonal anti-SMαA (Sigma-Aldrich, no. A5228) at 1:400; and 3) monoclonal anti-MLC20 (Sigma-Aldrich, no. M4401) at 1:100 and anti-MLCK at 1:50. Slides were incubated in primary antibodies overnight at 4°C in incubation chambers with slight agitation. The following morning the slides were washed in PBS + 0.1% Tween-20 at pH 7.45, 3 times for 5 min each, after which the second antibodies (DyLight 488 conjugate and DyLight 633 conjugate, Pierce Chemical) were applied for 2 h in the dark at room temperature with slight agitation. Slides then were washed in PBS + 0.1% Tween-20 at pH 7.45, 2 times for 5 min each followed by a wash in PBS at pH 7.45 for 5 min and a final rinse in 50% PBS. Finally, mounting medium (SlowFade Gold Antifade Mountant, S36937) was applied and the slides were coverslipped and stored at 4°C in the dark until imaged with the Olympus FV1000 at 200× for wall thickness measurements and at 600× for quantification of colocalization.
Colocalization between markers was determined using a custom nonparametric quadrant analysis that provided a measure of contractile protein colocalization independent of pixel intensity that identified VSM phenotype (1). The voxel dimensions for this analysis were 146 × 146 × 545 nm for the green channel (488 nm) and 185 × 185 × 693 nm for the red channel (633 nm). For each image, the distribution of pixel intensities was analyzed to determine the numbers of pixels above median intensity for each channel using CoLocalizer Pro version 2.6.1 (Colocalization Research Software). Colocalization of Ki-67 with SMαA was calculated as the ratio of the number of pixels above median intensity for both Ki-67 and SMαA, divided by the total number of pixels above median intensity for just SMαA. Colocalization of MLCK with SMαA was calculated as the ratio of the number of pixels above median intensity for both MLCK and SMαA, divided by the total number of pixels above median intensity for just SMαA. Similarly, colocalization of MLC20 with MLCK was calculated as the ratio of the number of pixels above median intensity for both MLC20 and MLCK divided by the total number of pixels above median intensity for just MLCK.
Data analysis and statistics.
All middle cerebral arteries were analyzed in matched sets within each of the main protocols including contractility, Western blotting, and organ culture. For contractility experiments, paired segments were distributed to the control and PD-156707 treatments. Contractile responses to ET-1 were normalized to the corresponding maximal K+-induced contraction, and arteries exhibiting a maximum contractile response to potassium of <1.5 g, a vasodilator response to ADP of >15%, or a maximum response to ET-1 <20% of the maximum response to potassium were excluded from further analysis. Results from duplicate segments from the same animal were averaged and counted as n = 1. Emax and pD2 values from FN and FH MCAs were determined via nonlinear regression.
Each Western blot gel run to analyze ETA and ETB receptor abundances included 5 lanes with known standards that were used to construct a logistic standard curve from which relative abundances were directly calculated. ETA and ETB standards were prepared from endothelium-intact common carotid arteries harvested from nonpregnant adult ewes. For organ cultured arteries, MCAs stained with SMαA and imaged at 200× were analyzed with ImagePro6 to measure medial wall thicknesses. Colocalization ratios from organ cultured arteries were normalized relative to their corresponding negative controls.
Results obtained for Emax, pD2, relative abundances, medial thicknesses, and colocalization ratios were analyzed using a univariate ANOVA with oxygen (normoxia or hypoxia) and treatment (control, ET, ET + inhibitor, etc.) as factors (SPSS software, version 23). For ANOVA results with at least one significant effect, post hoc comparisons were performed using a least significant difference test. Following elimination of outliers identified using the 2SD rule (SPSS software, version 23), homogeneity of variance and normal distribution were verified for all datasets. Two-group comparisons were analyzed using a Behrens-Fisher test with pooled variance to identify significant differences (P < 0.05). A minimum observed statistical power of 0.8 was routinely obtained for all nonsignificant differences.
RESULTS
This study used a total of 85 fetal lambs. Of these, 14 normoxic fetuses (FN) and 9 hypoxic fetuses (FH) provided endothelium-intact fetal MCAs for Western blots to quantify ETA and ETB receptor abundances. All other protocols used mechanically denuded fetal MCA segments harvested from 19 FN and 27 FH. Throughout the text, “n” indicates the number of animals used in each experiment, not the number of segments. All values represent mean values ± SE, with statistical significance defined as P < 0.05.
Effects of chronic hypoxia on ET-1-induced contractions.
The ETA receptor antagonist PD-156707 caused a right shift in ET-1 concentration-response relations in endothelium-denuded MCAs from both FN and FH groups (Fig. 1). The pD2 values for ET-1 in control normoxic (7.96 ± 0.16, n = 11) and hypoxic (7.97 ± 0.096, n = 7) arteries did not differ significantly, but treatment with PD-156707 significantly decreased pD2 values in both FN (7.44 ± 0.11, n = 7) and FH (7.52 ± 0.09, n = 5) MCAs. These results suggest that ETA receptors mediate the contractile responses to ET-1 in both normoxic and hypoxic fetal MCAs.
Fig. 1.

Chronic hypoxia altered ET-1-induced contractility in fetal MCAs. The ETA receptor antagonist (PD-156707) caused a right shift in the ET-1 concentration-response relation and significantly decreased pD2 in both normoxic (FN) and hypoxic (FH) fetal MCAs. Untreated FH MCAs exhibited a higher Emax than FN MCAs. Additionally, PD-156707 treatment decreased Emax in FH but not FN MCAs. Values shown represent means ± SE for n = 7–11 (FN) and n = 5–7 (FH). ‡P < 0.05, FN vs. FH. *P < 0.05, control vs. PD-156707.
The maximum contractile response to ET-1 (Emax) was significantly greater in untreated (control) chronically hypoxic fetal MCAs (98.3 ± 2.4, n = 7) than in normoxic fetal MCAs (85.3 ± 6.2, n = 11). Treatment with PD-156707 significantly decreased Emax values in FH (72.7 ± 9.0, n = 5) but not FN (81.6 ± 2.8, n = 7) MCAs.
Effects of chronic hypoxia on fetal plasma ET-1 levels.
In our model, chronic hypoxia during the last 110 days of gestation did not affect circulating ET-1 levels at the end of gestation in ovine fetuses. ET-1 levels in fetal plasma at term did not differ significantly between normoxic (1.04 ± 0.06 pg/ml, n = 14) and hypoxic (1.02 ± 0.07 pg/ml, n = 16) fetuses.
Effects of chronic hypoxia on ETA and ETB abundances.
ETA and ETB receptor abundances were calculated relative to levels measured in pooled common carotid arteries from adult nonpregnant ewes. ETA receptor abundances did not differ significantly between normoxic (0.788 ± 0.082, n = 12) and hypoxic (0.852 ± 0.084, n = 9) fetal MCAs (Fig. 2). In contrast, the relative abundances of ETB receptors were significantly greater in normoxic (0.121 ± 0.027, n = 14) than in hypoxic (0.074 ± 0.005, n = 6) fetal MCAs. In addition, the relative abundances were markedly greater for ETA than for ETB receptors, indicating that in term fetuses ETA receptor levels were much closer to adult carotid values than were ETB receptors.
Fig. 2.
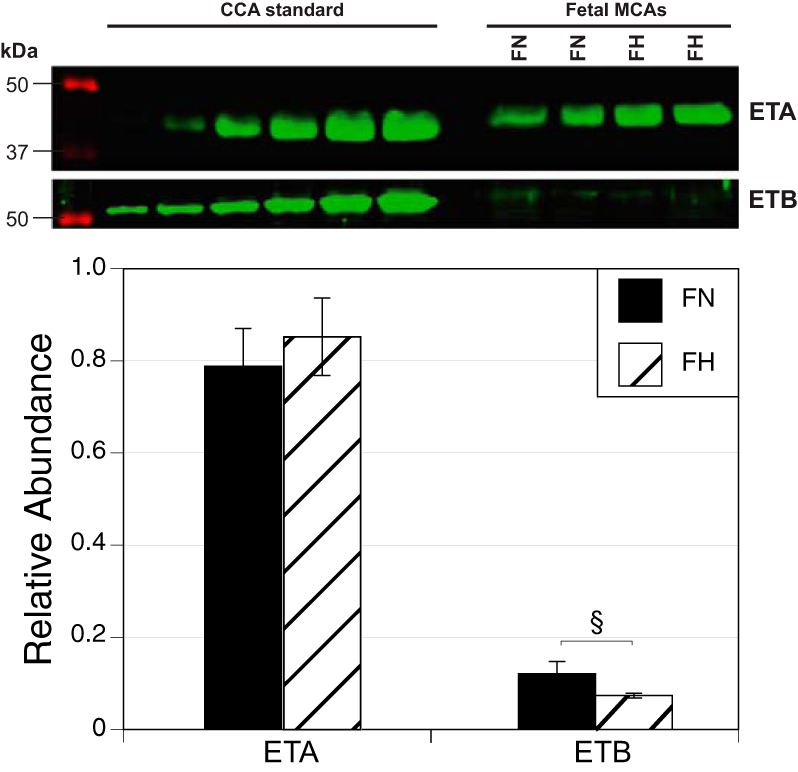
Chronic hypoxia alters ET receptor expression in fetal MCAs. Chronic hypoxia did not alter ETA receptor expression (FN n = 12, FH n = 9) but significantly decreased ETB receptor expression (FN n = 14, FH n = 6) in endothelium-intact MCAs from term ovine fetuses. Results are presented as means ± SE. §P < 0.05. CCA, common carotid artery.
Effects of chronic hypoxia and ET-1 on medial thicknesses.
Organ culture with 10 nM ET-1 for 24 h significantly increased medial thicknesses, and more so in normoxic (146.0 ± 3.4% compared with starved FN controls, n = 7) than in hypoxic (124.9 ± 7.5% compared with starved FH controls, n = 13) fetal MCAs (Fig. 3). Arteries treated with both 10 nM ET-1 and 6.6 µM chelerythrine exhibited no differences in medial thickness for either normoxic (96.0 ± 14.3% of control, n = 9) or hypoxic (101.8 ± 6.5% of control, n = 14) fetal MCAs, indicating that PKC inhibition blocked the effects of organ culture with ET-1 on medial thicknesses. Arteries treated with both 10 nM ET-1 and 10 µM KN93 exhibited medial thicknesses that were significantly less than observed with ET-1 treatment alone for normoxic (110.9 ± 12.5% of control, n = 9) but not for hypoxic (122.4 ± 11.6% of control, n = 12) fetal MCAs, suggesting that CaMKII activity was required for ET-1-induced increases in medial thicknesses in normoxic but not hypoxic arteries. Similarly, arteries treated with both 10 nM ET-1 and 10 µM SB203580 exhibited medial thicknesses that were significantly less than observed with ET-1 treatment alone for normoxic (122.8 ± 4.5% of control, n = 7) but not for hypoxic (120.7 ± 7.0% of control, n = 14) fetal MCAs, suggesting that p38 activity also was required for ET-1-induced increases in medial thicknesses in normoxic but not hypoxic arteries.
Fig. 3.
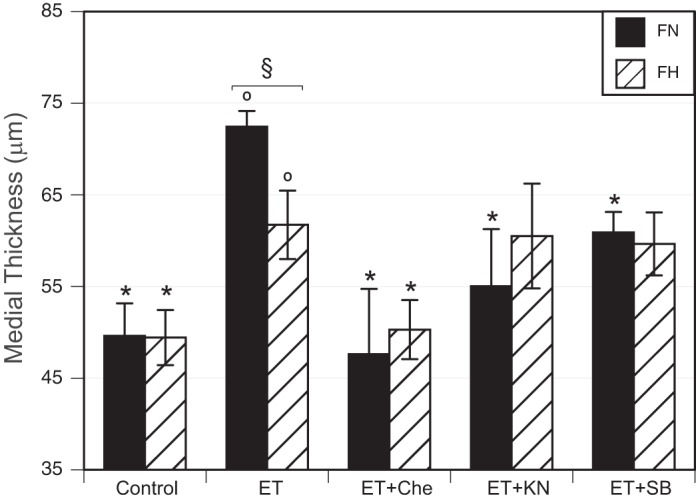
Effects of chronic hypoxia and ET-1 on medial wall thicknesses. Fetal MCAs incubated with 10 nM ET-1 for 24 h exhibited increased medial wall thicknesses (oP < 0.05), an effect significantly attenuated by chronic hypoxia. In both FN and FH arteries, the effect of ET-1 appeared to be PKC dependent because chelerythrine significantly depressed the response to ET-1 in both groups. Both CaMKII (inhibited by KN93) and p38 (inhibited by SB203580) pathways may also play a role in ET-1-stimulated thickening of the medial layer in FN but not FH arteries. Thickness measurements represent mean values ± SE for n = 7–9 (FN) and n = 12–14 (FH). §P < 0.05, FN vs. FH. *P < 0.05, significantly different from ET-1 alone.
Effects of chronic hypoxia and ET-1 on smooth muscle proliferation.
To quantify the separate and combined effects of chronic hypoxia and ET-1 on smooth muscle proliferation, we measured the colocalization of Ki-67 with smooth muscle α-actin (SMαA). Given that our previous work has indicated that smooth muscle characteristics differ between inner regions of the medial layer near the lumen and outer regions near the adventitia (12), the colocalization of Ki-67 with SMαA was measured separately in the inner and outer medial layers of artery wall (Fig. 4).
Fig. 4.
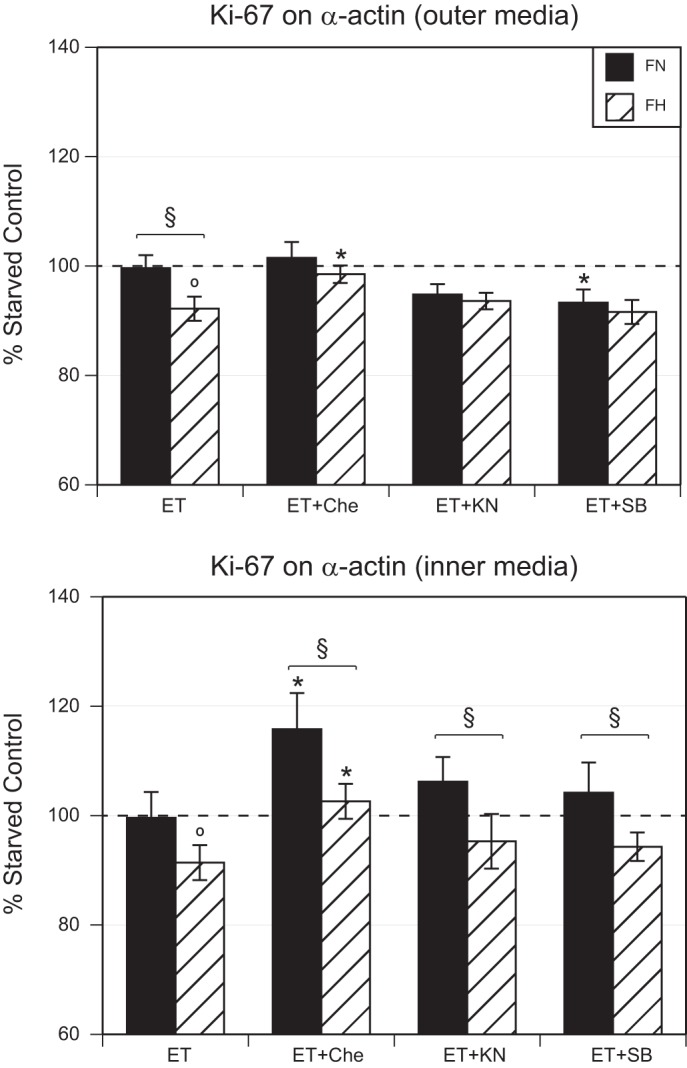
Effects of chronic hypoxia and ET-1 treatment on proliferation. ET-1 treatment significantly decreased colocalization of Ki-67 and SMαA in FH but not FN MCAs in both the inner and outer medial layers. In FN arteries, treatment with the PKC inhibitor chelerythrine was without effect in the outer layer but increased colocalization in the inner layer. In FH arteries, treatment with chelerythrine increased colocalization in both layers. Treatment with the CaMKII inhibitor KN93 was without effect compared with ET-1 alone in both layers of both normoxic and hypoxic arteries. Treatment with the p38 inhibitor SB203580 significantly decreased colocalization only in the outer layer of normoxic arteries. FN and FH values were significantly different for ET-1-treated arteries in the outer layer and were significantly different in the inner layer for all three groups treated with kinase inhibitors. Results are presented as means ± SE for n = 7–9 (FN) and n = 10–13 (FH). §P < 0.05, FN vs. FH. *P < 0.05, ET-1 vs. ET-1 with inhibitor treatments.
In the outer medial layer, organ culture with ET-1 significantly decreased colocalization between Ki-67 and SMαA in hypoxic (92.2 ± 2.2% of control, n = 12) but not normoxic (99.6 ± 2.4% of control, n = 7) fetal MCAs. Organ culture with both ET-1 and chelerythrine did not alter Ki-67/SMαA colocalization in normoxic MCAs (101.5 ± 2.9% of control, n = 8) but the hypoxic value (98.5 ± 1.6% of control, n = 12) was significantly greater than observed with ET-1 treatment alone. Organ culture with both ET-1 and KN93 did not alter Ki-67/SMαA colocalization in either normoxic (94.8 ± 1.9%, n = 8) or hypoxic (93.6 ± 1.5%, n = 11) MCAs when compared with ET-1 treatment alone. Organ culture with both ET-1 and SB203580 significantly decreased Ki-67/SMαA colocalization in normoxic (93.3 ± 2.4, n = 7) but not hypoxic (91.6 ± 2.2, n = 12) MCAs with ET-1 treatment alone. Within each of the treatment groups, normoxic values were significantly different from hypoxic values only in arteries cultured with only ET-1.
In the inner medial layer, organ culture with ET-1 significantly decreased colocalization between Ki-67 and SMαA in hypoxic (91.4 ± 3.2%, n = 10) but not normoxic (99.6 ± 4.7%, n = 9) fetal MCAs. Organ culture with both ET-1 and chelerythrine significantly increased Ki-67/SMαA colocalization in normoxic (115.8 ± 6.6%, n = 7) and hypoxic (102.6 ± 3.2%, n = 13) MCAs compared with that observed with ET-1 treatment alone. Organ culture with both ET-1 and KN93 did not significantly alter Ki-67/SMαA colocalization in normoxic (106.2 ± 4.5%, n = 9) or hypoxic (95.3 ± 5.0%, n = 11) MCAs compared with ET-1 treatment alone. Organ culture with both ET-1 and SB203580 did not significantly affect Ki-67/SMαA colocalization in normoxic (104.2 ± 5.5%, n = 9) or hypoxic MCAs (94.3 ± 2.6, n = 11) compared with ET-1 treatment alone. Within each of the treatment groups, normoxic values differed significantly from hypoxic values for all treatments except in arteries cultured with only ET-1.
Overall, the effects of ET-1 and chronic hypoxia on Ki-67/SMαA colocalization differed markedly in the inner and outer medial layers. Normoxic values were significantly different from hypoxic values for all treatment groups except ET-1 alone in the inner layer, but for no treatment groups except ET-1 alone in the outer layer. In addition, the effects of chelerythrine and SB203580 on responses to ET-1 differed between normoxic and hypoxic arteries only in the outer medial layer.
Effects of chronic hypoxia and ET-1 on contractile protein colocalization.
To explore further the interactive effects of chronic hypoxia and ET-1 on cerebrovascular smooth muscle, serial sections of fetal MCAs were double stained for MLC20 and MLCK, or for MLCK and SMαA for all treatment groups (Fig. 5). For colocalization of MLC20 with MLCK, organ culture with ET-1 did not affect colocalization in either normoxic (94.4 ± 3.5%, n = 8) or hypoxic (95.1 ± 2.9%, n = 13) fetal MCAs. Because there was no significant difference between control and ET-1 treatment, further experiments with kinase inhibitors were irrelevant.
Fig. 5.
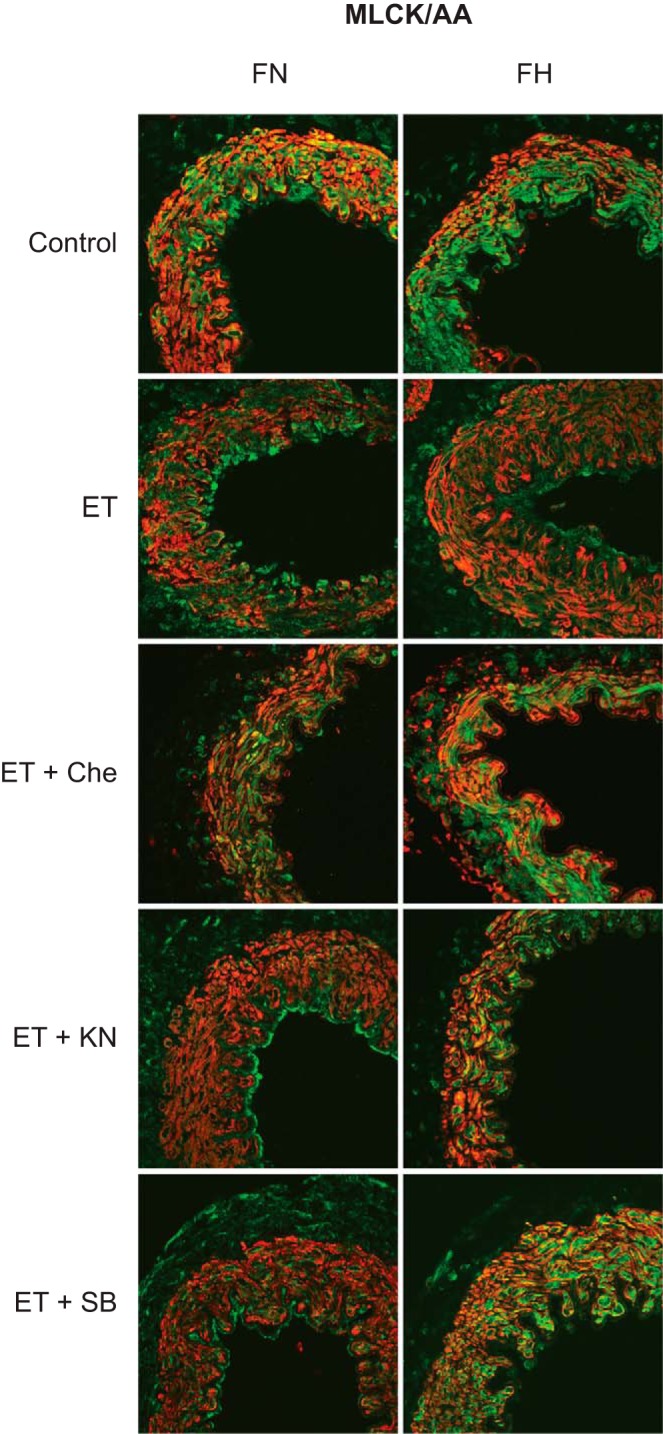
Effect of chronic hypoxia and ET-1 on MLCK and SMαA colocalization. Coronal sections of fetal MCAs were double stained with MLCK (green) and SMαA (red) after treatment with ET-1 and kinase inhibitors for PKC (chelerythrine), CaMKII (KN93), and p38 (SB203580). Yellow indicates areas of colocalization in these merged images.
Analysis of the colocalization of MLCK with SMαA revealed that organ culture with ET-1 significantly depressed colocalization in normoxic (75.1 ± 4.3% of control, n = 9) but not hypoxic (95.6 ± 2.7%, n = 13) fetal MCAs (Fig. 6). Organ culture with both ET-1 and chelerythrine significantly increased MLCK/SMαA colocalization more in normoxic (93.1 ± 2.9%, n = 8) and hypoxic MCAs (103.1 ± 3.3%, n = 13) than with ET-1 treatment alone. Organ culture with both ET-1 and KN93 significantly increased MLCK/SMαA colocalization in normoxic (89.7 ± 1.4%, n = 6) and hypoxic MCAs (110.5 ± 2.7%, n = 11) compared with ET-1 treatment alone. Similarly, organ culture with both ET-1 and SB203580 significantly increased MLCK/SMαA colocalization in normoxic (92.5 ± 2.5%, n = 9) and hypoxic MCAs (103.6 ± 2.7%, n = 13) compared with ET-1 treatment alone. Within each of the treatment groups, normoxic values differed significantly from hypoxic values of MLCK/SMαA colocalization for all treatments.
Fig. 6.
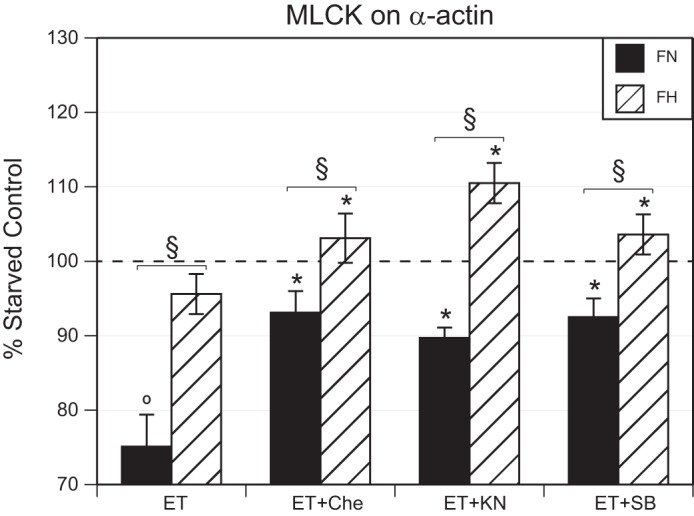
Effect of chronic hypoxia and ET-1 on MLCK and SMαA colocalization. ET-1 significantly decreased the colocalization of MLCK with SMαA in FN but not FH MCAs. In both FN and FH arteries, treatment with ET-1 together with inhibition of PKC (Che), p38 (SB), and CaMKII (KN93) increased MLCK and SMαA colocalization. FN and FH values differed significantly in all treatment groups. Results are presented as means ± SE for n = 6–9 (FN) and n = 11–13 (FH). §P < 0.05, FN vs. FH. *P < 0.05, ET-1 vs. ET-1 with inhibitor treatments.
Together, the results demonstrate that chronic hypoxia significantly altered the influence of ET-1 on contractile protein colocalization. For MLCK/SMαA colocalization, hypoxia attenuated ET-1-induced decreases in colocalization and increased the extent of colocalization observed following combined treatment with ET-1 and any of the three kinase inhibitors tested.
DISCUSSION
Despite numerous studies of hypoxic vascular remodeling in the cardiac and pulmonary circulations, the effects of hypoxic remodeling on the fetal cerebrovasculature remain poorly understood, particularly in relation to ET-1, a potent vascular growth factor (65). The present study evaluates the hypothesis that hypoxic remodeling of fetal cerebral arteries alters ET-1 signaling and offers three original findings: 1) chronic hypoxia did not alter ET-1 levels in fetal plasma or ETA receptor abundance in fetal MCAs, but decreased ETB receptor abundance and increased ET-1-induced contractility; 2) chronic hypoxia attenuated the ability of organ culture with ET-1 to stimulate smooth muscle hypertrophy and increase medial thickness through pathways dependent upon PKC, CaMKII, and p38; and 3) chronic hypoxia also attenuated the ability of organ culture with ET-1 to depress colocalization of MLCK with SMαA via mechanisms dependent upon PKC, CaMKII, and p38 in organ cultured fetal MCAs. Together, these results support the hypothesis that chronic hypoxia modulates the PKC-, CaMKII-, and p38-dependent mechanisms through which ET-1 influences contractile protein organization, wall thickness, and contractility in fetal MCAs.
Hypoxic vascular remodeling in the fetus.
Intrauterine hypoxia is a common but serious condition that typically results in numerous fetal complications and neonatal morbidities. Hypoxic cardiovascular remodeling during fetal development often causes lifelong consequences that compromise cardiopulmonary function and broadly increases susceptibility to ischemia-reperfusion injury (75). The mechanisms involved in hypoxic fetal vascular remodeling remain uncertain, but abundant evidence suggests that HIFs, the transcription factors increased by hypoxia that mediate many hypoxic effects, bind to HREs in multiple gene promoters to directly stimulate the release of vascular growth factors such as VEGF (15) and ET-1 (30) that then alter vascular structure and function. An important feature of these responses is that an initial elevation of HIF-1α typically falls within a few weeks of hypoxic exposure, despite continued hypoxia (13). Correspondingly, hypoxia can initially increase VEGF production in the adult mouse brain, after which VEGF later returns to basal normoxic levels despite continued hypoxia (37). Similarly, previous work from our group has shown that hypoxia causes sustained increases in artery medial wall thickness and VEGF receptor densities in fetal lambs that persist as long as hypoxia continues, even though VEGF levels return to basal normoxic levels (1). Such evidence suggests that hypoxia acts transiently through HIFs to promote the short-term release of vascular growth factors that then produce lasting changes in capillary density, collateralization, and vascular function that together constitute the vascular remodeling response to chronic hypoxia. In this manner, HIF-1α can rise transiently but produce long-term changes in the coupling between cell surface receptors and the contractile apparatus on the one hand, and vascular gene expression on the other. These hypoxic changes in endothelin receptor coupling are a focus of the current study.
Evidence from a broad variety of experimental models demonstrates that chronic hypoxia increases the expression of ET-1 (2) and also alters reactivity to ET-1 in many vascular tissues including guinea pig pulmonary arteries (64), rat pulmonary arteries (2, 68), rat mesenteric arteries (3), and rat carotid body (14). Due perhaps to differences in the duration and severity of chronic hypoxia, however, other studies report that long-term hypoxia does not yield a lasting increase in circulating ET-1 and may even decrease ETA receptor density in rat thoracic aorta (7). At the individual organism level, chronic hypoxia also can exert selective effects on ET-1, ETA, and ETB expression that vary significantly among different tissues (39). Together, this evidence emphasizes that the effects of chronic hypoxia on ET-1 are governed by multiple factors that complicate efforts to generalize the effects of chronic hypoxia on ET-1 signaling.
In contrast to VEGF and PDGF, which modulate vascular smooth muscle proliferation and differentiation through activation of tyrosine kinase receptors, other vascular growth factors act through G protein-coupled receptors to exert both acute and chronic effects on vascular smooth muscle structure and function. As for serotonin and norepinephrine (24, 74), ET-1 also activates G protein-coupled receptors to produce both immediate and potent changes in vascular tone as well as longer-term changes in gene expression that influence wall thickness, smooth muscle phenotype, and contractility (18, 21). Given the findings that hypoxia can increase ET-1 signaling (30) and that ET-1 can influence smooth muscle structure-function relations (18), the present study explored the hypothesis that chronic hypoxia modulates ET-1 signaling in the fetal cerebrovasculature.
Chronic hypoxia increased the maximum response to ET-1 in fetal cerebral arteries.
Whereas most previous studies of interactions between chronic hypoxia and ET-1 signaling have focused on the pulmonary circulation, the present study examined the fetal cerebral circulation, which has not been widely studied despite the high clinical frequency of cerebral morbidities associated with fetal hypoxia (66). Consistent with findings in rat pulmonary (68), mesenteric (3) and carotid (38) arteries, 110 days of hypoxia in our fetal sheep model increased contractile responses to ET-1 (Fig. 1). In contrast to lamb pulmonary (27) and rat mesenteric (3) arteries, however, our fetal lamb cerebral arteries did not exhibit any change in sensitivity to ET-1 following hypoxic acclimatization (Fig. 1), suggesting that the affinity of ET-1 for the ETA receptor(s) probably was not influenced by hypoxia in fetal lamb MCAs.
Hypoxic enhancement of contractile responses to ET-1 was not maintained by increased circulating levels of ET-1 because plasma levels of ET-1 were similar in normoxic and hypoxic fetuses (N: 1.04 ± 0.06; H: 1.02 ± 0.07 pg/ml; see Effects of chronic hypoxia on fetal plasma ET-1 levels). This latter result extends our previous finding that 110 days of chronic hypoxia did not alter circulating levels of VEGF in the fetal lamb (1) and furthers the view that hypoxia produces transient increases in HIFs (13) that drive secondary increases in ET-1 (7, 10) and VEGF (1), which resolve to normoxic levels once hypoxic adaptation is complete, typically within ~3 wk (13). In aggregate, this evidence reinforces the interpretation that hypoxic adaptation produced lasting changes in the structure and function of fetal cerebral arteries, particularly in relation to ET-1 signaling.
Chronic hypoxia did not markedly alter ET-1 receptors in fetal cerebral arteries.
Contractile responses to ET-1 in cerebral arteries are most commonly mediated by ETA receptors (44). Correspondingly, in fetal MCAs, dose-response relations for ET-1 were significantly right-shifted by the ETA antagonist PD-156707 (Fig. 1). ETA receptor abundances, however, were not significantly affected by chronic hypoxia (Fig. 2), implying that hypoxic enhancement of contractile responses to ET-1 was not due to increased ETA receptor levels. Instead, chronic hypoxia appears to have altered coupling of ETA receptors to the contractile response, perhaps through enhanced calcium release or elevated myofilament calcium sensitivity as suggested by other studies (4, 35); separate experiments will be required to confirm the involvement of these mechanisms. In turn, the absence of an effect of chronic hypoxia on ETA receptors has been reported previously in mouse astrocytes (28) and cat carotid body (52), whereas hypoxia decreased ETA receptors in gravid rat uterus (62) but increased ETA abundances in multiple rat tissues including carotid body (14), coronary arteries (3), lungs (3, 68), kidneys (3), and cerebral arteries (59). This pattern of findings emphasizes that the effects of chronic hypoxia on ETA receptor expression are highly species and tissue specific.
As also reported in rat pulmonary arteries (68) and mouse astrocytes (28), chronic hypoxia significantly decreased ETB levels in the fetal lamb MCAs (Fig. 2). In other studies, hypoxia exerted no effect on ETB abundance in a variety of noncerebral rat tissues (3), but increased ETB receptor abundance in cultured hypoxic human pulmonary endothelial cells (34), cat carotid bodies (52), and rat cerebral arteries (59). In fetal lamb MCAs, however, the normoxic and hypoxic abundances of ETB receptors averaged no more than 12% and 7% of the levels found in normoxic adult ovine carotids, respectively. Although hypoxic decreases in ETB receptors, which typically promote cerebral endothelium-dependent vasodilatation (44), could augment contractile responses to ET-1, the very low abundance of ETB receptors compared with ETA receptors minimized their physiological influence on ET-1-induced contractions. Alternatively, the hypoxic increases in contractile responses to ET-1 observed in fetal lamb MCA were more likely due to hypoxic changes in the coupling between ETA receptors and the contractile apparatus in cerebrovascular smooth muscle.
Chronic hypoxia depressed effects of ET-1 on medial thickness through selective modulation of kinase signaling.
Through activation of ETA receptors, ET-1 induces cerebral vasoconstriction via phospholipase C-dependent increases in intracellular Ca2+ (11) and can also potently stimulate DNA synthesis and proliferation in smooth muscle (36). Many of these intracellular effects of ET-1 depend on protein kinase C (PKC) cascades (11), but Ca2+-calmodulin-dependent kinase II (CaMKII) (11) and p38 MAP kinase (71) can also contribute, particularly under pathophysiological conditions. In turn, hypoxia can modulate PKC signaling (58), CaMKII signaling (6), and p38 signaling (8), suggesting that hypoxia could modulate ET-1 signaling through influences on these kinases. In addition, each of these kinases independently can influence smooth muscle proliferation, differentiation, and phenotypic transformation (25, 41, 57). Together, this evidence motivated the hypothesis tested in this study, namely that chronic hypoxia influences the PKC-, CaMKII-, and p38-dependent mechanisms through which ET-1 modulates contractile protein organization, wall thickness, and contractility in fetal MCAs.
In agreement with previous studies in rat pulmonary arteries (10), organ culture with ET-1 significantly increased medial thicknesses by 46% in normoxic fetal MCAs (Fig. 3). Treatment with chelerythrine completely eliminated this effect, and treatment with KN93 and SB203580 significantly attenuated the thickness responses to ET-1 by 76% and 51%, respectively. In contrast, organ culture of hypoxic fetal MCAs with ET-1 significantly increased medial thicknesses by only 25%, which was significantly less than observed in normoxic arteries. In hypoxic fetal MCAs, treatment with chelerythrine again completely eliminated the response of thickness to ET-1, but neither KN93 nor SB203580 exhibited any significant effect. This pattern of results demonstrated that the influence of ET-1 on medial thickness was completely dependent on PKC in both normoxic and hypoxic fetal MCAs, and that CaMKII and p38 significantly contributed to this thickness response in normoxic, but not hypoxic arteries. This finding raises the possibility that hypoxic elimination of contributions from CaMKII and p38 may help explain the reduced magnitude of response to ET-1 in hypoxic arteries; further experiments will be needed to explore this idea. Nonetheless, the data clearly demonstrate that chronic hypoxia attenuated the roles of CaMKII and p38 in coupling between ET-1 and expansion of medial thickness.
An important feature of the thickness measurements summarized in Fig. 3 was that these were restricted to the medial layer of the fetal MCAs. Numerous studies established years ago that hypoxia can increase total wall thickness, but adventitial expansion accounted for much of this increase (23, 45). The present focus on the medial layer enabled assessment of the relative effects of hypoxia on smooth muscle hypertrophy and hyperplasia, which has been a recurrent topic in studies of hypoxic increases in wall thickness (47). To assess hyperplasia, we measured immunoreactivity to Ki-67, an established marker of proliferation (53). To ensure that we measured Ki-67 levels only in smooth muscle cells, we quantified colocalization between Ki-67 and SMαA, a known cytoplasmic marker of smooth muscle (72). In light of our previous work demonstrating a gradient in smooth muscle differentiation between the adventitial and luminal boundaries of the medial layer (12), we quantified Ki-67- SMαA colocalization in both boundary regions (Fig. 4). Using this approach, ET-1 did not increase colocalization of Ki-67 with SMαA in either region, but instead decreased this colocalization, and presumably proliferation. These results suggest that ET-1-induced increases in medial thickness did not involve increased proliferation and instead were mediated by hypertrophy and not hyperplasia. As such, these results were consistent with previous reports that ET-1 can induce a nonproliferative, hypertrophic response in both vascular (19) and nonvascular (22) tissues. More importantly, in the present study, ET-1 significantly decreased apparent proliferation in both the luminal and adventitial boundary regions, but only in hypoxic arteries, again suggesting that hypoxia altered coupling between ET-1 receptors and the intracellular pathways governing hyperplasia.
To explore how hypoxia might alter the effects of ET-1 on smooth muscle proliferation, we assayed the effects of our kinase inhibitors on ET-1-induced changes in Ki-67/SMαA colocalization. In the outer medial layer near the adventitia, apparent proliferation in the presence of ET-1 was influenced only by SB203580 (p38 inhibitor) in normoxic arteries, but was affected only by chelerythrine (PKC inhibitor) in hypoxic arteries. Owing to the small magnitudes of these effects, it is unlikely they were physiologically significant. In the inner medial layer, however, apparent proliferation in the presence of ET-1 was affected only by chelerythrine in both normoxic and hypoxic arteries. Together, these results suggest that hypoxia converted a small (6 ± 2%) pro-proliferative effect of p38 into a small (6 ± 2%) antiproliferative effect of PKC in the outer medial layer; these changes were at best of minor physiological significance. In the inner medial layer, however, hypoxia reduced the magnitude of an antiproliferative effect of PKC from 16 ± 3% (normoxic) to 11 ± 3% (hypoxic). This pattern emphasizes that the (anti)proliferative effects of ET-1 are not only artery specific, but also vary among different regions of the same artery as previously shown for VEGF (12). These results also imply that the previously reported antiproliferative effects of PKC (16) are largely responsible for the antiproliferative effects of ET-1, and that the magnitude of these effects are modulated by hypoxia in fetal cerebral arteries.
Chronic hypoxia depressed effects of ET-1 on MLCK organization through selective modulation of kinase signaling.
One mechanism whereby chronic hypoxia could modulate the ability of ET-1 to elicit contraction and smooth muscle hypertrophy would be to alter the phenotype of smooth muscle in fetal cerebral arteries. A broad variety of evidence, including findings in fetal lamb carotid arteries (1), has demonstrated that chronic hypoxia can modulate smooth muscle differentiation and phenotype (60). In turn, such changes can dramatically alter contractile reactivity and phenotypic responses to numerous receptor agonists and growth factors (60). To assess the extent to which chronic hypoxia influenced the ability of ET-1 to elicit changes in smooth muscle differentiation and phenotype, we measured markers for the contractile phenotype of smooth muscle. First, we examined ET-1-induced changes in MLC20/MLCK colocalization. MLC20 is a component of thick filaments and is typically tightly attached to the cross-bridges of myosin heavy chains (20). Similarly, MLCK is also typically tightly associated with the myosin molecules in thick filaments (29). Due to the nature of this organization, MLC20/MLCK colocalization is not highly dynamic and is closely associated with contractile capacity. Correspondingly, organ culture with 10 nM ET-1 did not change MLC20/MLCK colocalization in either normoxic or hypoxic arteries, indicating that ET-1 did not influence this phenotypic marker.
Previous work from multiple laboratories (9), including our own (1), has shown that the extent of colocalization between MLCK, the rate-limiting enzyme for contraction, and SMαA, the main component of the smooth muscle cytoskeleton, is a reliable marker for the contractile phenotype of smooth muscle. In contrast to MLC20, SMαA can have a highly variable association with MLCK, depending on smooth muscle phenotype. In fetal cerebral arteries, organ culture with a physiological concentration of ET-1 (10 nM) significantly depressed MLCK/SMαA colocalization, suggesting attenuated contractility, in normoxic but not hypoxic arteries (Fig. 6). In light of observations that ET-1 can signal through PKC (11), CaMKII (22, 57), and p38 (71) to modulate phenotype and contractile protein organization, we examined the effects of inhibitors of these kinases on phenotypic responses to ET-1. Coculture of ET-1 with chelerythrine, KN93, or SB203580 all attenuated ET-1-induced decreases in MLCK/SMαA colocalization in normoxic arteries, indicating that PKC (18 ± 3%), CaMKII (15 ± 1%), and p38 (17 ± 3%) all contributed to ET-1-induced contractile dedifferentiation. In hypoxic arteries, inhibitors of all three kinases also significantly increased MLCK/SMαA colocalization compared with culture with ET-1 alone, again indicating that PKC (8 ± 3%), CaMKII (15 ± 3%), and p38 (8 ± 3%) all exerted a modest influence toward contractile dedifferentiation, even in hypoxic arteries. Following treatment with each of the three kinase inhibitors, the extent of MLCK/SMαA colocalization was greater in hypoxic than in normoxic arteries, again suggesting that hypoxia attenuated the ability of ET-1 to influence smooth muscle phenotype through PKC, CaMKII, and p38.
Overview.
The combined results extend previous observations that chronic hypoxia modulates the phenotype of vascular smooth muscle. In contrast to adult arteries, in which structure, contractility, and smooth muscle phenotype are relatively stable, fetal arteries are highly dynamic and undergo near-continuous changes in stiffness (48), calcium handling (42), endothelial function (70), and many other characteristics (49). Importantly, these vascular changes occur simultaneously with age-related increases in cerebral perfusion, oxygen consumption, and sensitivity to hypoxia (61), reinforcing the view that functional demands and regulatory mechanisms are very different in fetal and adult cerebral arteries, particularly with respect to the modulating influences of hypoxia. In freshly dissected fetal cerebral arteries, the effects of chronic hypoxia did not appear to involve changes in ETA abundance but decreased ETB abundance by 40%. Despite these changes, contractile responses to ET-1 were enhanced in fetal cerebral arteries in an ETA-dependent manner, suggesting that coupling between the ETA receptor and the contractile apparatus was enhanced by chronic hypoxia. In organ culture, ET-1-induced increases in medial thicknesses were depressed by chronic hypoxia. These changes were dependent on PKC, CaMKII, and p38 in normoxic arteries, but only upon PKC in hypoxic arteries. Estimates of proliferation based on Ki-67 colocalization with SMαA revealed that ET-1 increased medial thickness through hypertrophy and not hyperplasia in a PKC-dependent manner in both normoxic and hypoxic arteries. Chronic hypoxia ablated the ability of ET-1 to depress colocalization of MLCK with SMαA, a marker for the contractile phenotype of smooth muscle. In both normoxic and hypoxic arteries, inhibition of PKC, CaMKII, and p38 all increased colocalization of MLCK with SMαA, suggesting that each of these kinases exerted a tonic influence toward contractile dedifferentiation in both normoxic and hypoxic arteries. Overall, this pattern of results demonstrates that many of the effects of ET-1 are mediated through PKC to a varying degree in normoxic and hypoxic fetal cerebral arteries. In contrast, CaMKII and p38 help mediate ET-1’s effects on medial hypertrophy in normoxic but not hypoxic arteries. These kinases, along with PKC, variably promote contractile dedifferentiation in both normoxic and hypoxic arteries. Although the abundance of ETA receptors was far greater than for ETB receptors in both normoxic and hypoxic arteries, it remains possible that both receptor types contributed to the observed responses. Equally important, it remains possible that hypoxic attenuation of the roles of PKC, CaMKII, and p38 in ET-1 signaling were generalized to all pathways dependent on these kinases and were not exclusive to ET-1 signaling. It also remains possible that the observed cerebrovascular effects of hypoxia on intracellular kinases may affect smooth muscle migration in a manner similar to that demonstrated in pulmonary arteries (73); further experiments will be needed to evaluate this important possibility. Owing to the instability of the contractile phenotype in cultured smooth muscle (46), ideally such measurements would be conducted in vivo using either genetic labeling or cell fate measurements (63), which have yet to be attempted in a large animal model.
Given that chronic hypoxia decreased fetal arterial oxygen tension by only ~4 Torr, but elicited major changes in fetal artery contractility and kinase signaling, the present results emphasize that the fetal cerebral vasculature is exquisitely sensitive to chronic hypoxia. Maternal mechanisms probably also contribute (43), but the net result is that chronic hypoxia can significantly alter the structure, function, and reactivity of fetal cerebral arteries. In that context, the present results clearly demonstrate that chronic hypoxia significantly influenced ET-1 signaling in fetal cerebral arteries, as indicated by hypoxia’s ability to increase ET-1-mediated contractility, decrease ET-1-mediated smooth muscle hypertrophy, and depress ET-1’s ability to promote contractile dedifferentiation. In turn, these results demonstrate that hypoxia can act not only through classical receptor tyrosine kinase-dependent pathways (e.g., VEGF and PDGF) to modulate smooth muscle phenotype, structure, and function, but can also modulate signaling through G protein-coupled receptors to produce many of the same effects. The extents to which hypoxia achieves these effects through direct action on smooth muscle, as opposed to indirect and whole body effects on cardiac output, arterial pressure, and cerebral perfusion, remain uncertain. Nonetheless, the present results clearly suggest that chronic hypoxia modulates ET-1 signaling, and the effects of ET-1 on structure and function, in fetal cerebral arteries.
GRANTS
This work was supported by National Institutes of Health Grants HL-54120, HD-31266, HL-64867, and NS-076945 and the Loma Linda University School of Medicine (LLUSM).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.S. and W.J.P. conceived and designed the research; J.S. and D.K. performed experiments; J.S., D.K., J.M.W., O.O.A., R.B.T., and W.J.P. analyzed data; J.S., D.K., J.M.W., O.O.A., R.B.T., and W.J.P. interpreted results of experiments; J.S. and W.J.P. prepared figures; J.S. and W.J.P. drafted manuscript; J.S., O.O.A., R.B.T., and W.J.P. edited and revised manuscript; J.S., D.K., J.M.W., O.O.A., R.B.T., and W.J.P. approved final version of manuscript.
ACKNOWLEDGMENTS
Imaging was performed in the LLUSM Advanced Imaging and Microscopy Core with support of National Science Foundation Grant No. MRI-DBI 0923559 (S. M. Wilson) and the Loma Linda University Schools of Medicine and Pharmacy.
REFERENCES
- 1.Adeoye OO, Butler SM, Hubbell MC, Semotiuk A, Williams JM, Pearce WJ. Contribution of increased VEGF receptors to hypoxic changes in fetal ovine carotid artery contractile proteins. Am J Physiol Cell Physiol 304: C656–C665, 2013. doi: 10.1152/ajpcell.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguirre JI, Morrell NW, Long L, Clift P, Upton PD, Polak JM, Wilkins MR. Vascular remodeling and ET-1 expression in rat strains with different responses to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 278: L981–L987, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Allahdadi KJ, Cherng TW, Pai H, Silva AQ, Walker BR, Nelin LD, Kanagy NL. Endothelin type A receptor antagonist normalizes blood pressure in rats exposed to eucapnic intermittent hypoxia. Am J Physiol Heart Circ Physiol 295: H434–H440, 2008. doi: 10.1152/ajpheart.91477.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Artamonov MV, Momotani K, Stevenson A, Trentham DR, Derewenda U, Derewenda ZS, Read PW, Gutkind JS, Somlyo AV. Agonist-induced Ca2+ sensitization in smooth muscle: redundancy of Rho guanine nucleotide exchange factors (RhoGEFs) and response kinetics, a caged compound study. J Biol Chem 288: 34030–34040, 2013. doi: 10.1074/jbc.M113.514596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avni R, Golani O, Akselrod-Ballin A, Cohen Y, Biton I, Garbow JR, Neeman M. MR imaging-derived oxygen-hemoglobin dissociation curves and fetal-placental oxygen-hemoglobin affinities. Radiology 280: 68–77, 2016. doi: 10.1148/radiol.2015150721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balla Z, Hoch B, Karczewski P, Blasig IE. Calcium/calmodulin-dependent protein kinase IIdelta 2 and gamma isoforms regulate potassium currents of rat brain capillary endothelial cells under hypoxic conditions. J Biol Chem 277: 21306–21314, 2002. doi: 10.1074/jbc.M200553200. [DOI] [PubMed] [Google Scholar]
- 7.Barbier J, Reboul C, Goret L, Saïag B, Catheline M, Gibault A, Dauzat M, Obert P, Tanguy S. Aortic vasoconstriction related to smooth muscle cells ET-A and ET-B receptors is not involved in hypoxia-induced sustained systemic arterial hypertension in rats. Vascul Pharmacol 47: 209–214, 2007. doi: 10.1016/j.vph.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Blaschke F, Stawowy P, Goetze S, Hintz O, Gräfe M, Kintscher U, Fleck E, Graf K. Hypoxia activates beta(1)-integrin via ERK 1/2 and p38 MAP kinase in human vascular smooth muscle cells. Biochem Biophys Res Commun 296: 890–896, 2002. doi: 10.1016/S0006-291X(02)02033-8. [DOI] [PubMed] [Google Scholar]
- 9.Blue EK, Goeckeler ZM, Jin Y, Hou L, Dixon SA, Herring BP, Wysolmerski RB, Gallagher PJ. 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am J Physiol Cell Physiol 282: C451–C460, 2002. doi: 10.1152/ajpcell.00333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blumberg FC, Wolf K, Arzt M, Lorenz C, Riegger GA, Pfeifer M. Effects of ET-A receptor blockade on eNOS gene expression in chronic hypoxic rat lungs. J Appl Physiol (1985) 94: 446–452, 2003. doi: 10.1152/japplphysiol.00239.2002. [DOI] [PubMed] [Google Scholar]
- 11.Bouallegue A, Daou GB, Srivastava AK. Endothelin-1-induced signaling pathways in vascular smooth muscle cells. Curr Vasc Pharmacol 5: 45–52, 2007. doi: 10.2174/157016107779317161. [DOI] [PubMed] [Google Scholar]
- 12.Butler SM, Abrassart JM, Hubbell MC, Adeoye O, Semotiuk A, Williams JM, Mata-Greenwood E, Khorram O, Pearce WJ. Contributions of VEGF to age-dependent transmural gradients in contractile protein expression in ovine carotid arteries. Am J Physiol Cell Physiol 301: C653–C666, 2011. doi: 10.1152/ajpcell.00413.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chávez JC, Agani F, Pichiule P, LaManna JC. Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol (1985) 89: 1937–1942, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Chen J, He L, Dinger B, Stensaas L, Fidone S. Role of endothelin and endothelin A-type receptor in adaptation of the carotid body to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 282: L1314–L1323, 2002. doi: 10.1152/ajplung.00454.2001. [DOI] [PubMed] [Google Scholar]
- 15.D’Angelo G, Ladoux A, Frelin C. Hypoxia-induced transcriptional activation of vascular endothelial growth factor is inhibited by serum. Biochem Biophys Res Commun 267: 334–338, 2000. doi: 10.1006/bbrc.1999.1947. [DOI] [PubMed] [Google Scholar]
- 16.Das M, Burns N, Wilson SJ, Zawada WM, Stenmark KR. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of PKCzeta in the pulmonary artery adventitia. Cardiovasc Res 78: 440–448, 2008. doi: 10.1093/cvr/cvn014. [DOI] [PubMed] [Google Scholar]
- 17.Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, Pollock DM, Webb DJ, Maguire JJ. Endothelin. Pharmacol Rev 68: 357–418, 2016. doi: 10.1124/pr.115.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Frutos S, Caldwell E, Nitta CH, Kanagy NL, Wang J, Wang W, Walker MK, Gonzalez Bosc LV. NFATc3 contributes to intermittent hypoxia-induced arterial remodeling in mice. Am J Physiol Heart Circ Physiol 299: H356–H363, 2010. doi: 10.1152/ajpheart.00341.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng H, Hershenson MB, Lei J, Anyanwu AC, Pinsky DJ, Bentley JK. Pulmonary artery smooth muscle hypertrophy: roles of glycogen synthase kinase-3β and p70 ribosomal S6 kinase. Am J Physiol Lung Cell Mol Physiol 298: L793–L803, 2010. doi: 10.1152/ajplung.00108.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddinger TJ, Meer DP. Myosin II isoforms in smooth muscle: heterogeneity and function. Am J Physiol Cell Physiol 293: C493–C508, 2007. doi: 10.1152/ajpcell.00131.2007. [DOI] [PubMed] [Google Scholar]
- 21.Fisher SA, Ikebe M, Brozovich F. Endothelin-1 alters the contractile phenotype of cultured embryonic smooth muscle cells. Circ Res 80: 885–893, 1997. doi: 10.1161/01.RES.80.6.885. [DOI] [PubMed] [Google Scholar]
- 22.Gangopadhyay JP, Ikemoto N. Intracellular translocation of calmodulin and Ca2+/calmodulin-dependent protein kinase II during the development of hypertrophy in neonatal cardiomyocytes. Biochem Biophys Res Commun 396: 515–521, 2010. doi: 10.1016/j.bbrc.2010.04.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffith SL, Rhoades RA, Packer CS. Pulmonary arterial smooth muscle contractility in hypoxia-induced pulmonary hypertension. J Appl Physiol (1985) 77: 406–414, 1994. [DOI] [PubMed] [Google Scholar]
- 24.Han X, Chen C, Cheng G, Liang L, Yao X, Yang G, You P, Shou X. Peroxisome proliferator-activated receptor γ attenuates serotonin-induced pulmonary artery smooth muscle cell proliferation and apoptosis inhibition involving ERK1/2 pathway. Microvasc Res 100: 17–24, 2015. doi: 10.1016/j.mvr.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Hedges JC, Yamboliev IA, Ngo M, Horowitz B, Adam LP, Gerthoffer WT. p38 mitogen-activated protein kinase expression and activation in smooth muscle. Am J Physiol Cell Physiol 275: C527–C534, 1998. [DOI] [PubMed] [Google Scholar]
- 26.Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun 172: 993–999, 1990. doi: 10.1016/0006-291X(90)91544-3. [DOI] [PubMed] [Google Scholar]
- 27.Herrera EA, Riquelme RA, Ebensperger G, Reyes RV, Ulloa CE, Cabello G, Krause BJ, Parer JT, Giussani DA, Llanos AJ. Long-term exposure to high-altitude chronic hypoxia during gestation induces neonatal pulmonary hypertension at sea level. Am J Physiol Regul Integr Comp Physiol 299: R1676–R1684, 2010. doi: 10.1152/ajpregu.00123.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho MC, Lo AC, Kurihara H, Yu AC, Chung SS, Chung SK. Endothelin-1 protects astrocytes from hypoxic/ischemic injury. FASEB J 15: 618–626, 2001. doi: 10.1096/fj.99-1022com. [DOI] [PubMed] [Google Scholar]
- 29.Hong F, Haldeman BD, Jackson D, Carter M, Baker JE, Cremo CR. Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys 510: 135–146, 2011. doi: 10.1016/j.abb.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun 245: 894–899, 1998. doi: 10.1006/bbrc.1998.8543. [DOI] [PubMed] [Google Scholar]
- 31.Huang LH, He JY, Yuan BX, Cao YX. Lipid soluble smoke particles upregulate endothelin receptors in rat basilar artery. Toxicol Lett 197: 243–255, 2010. doi: 10.1016/j.toxlet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Hutter D, Kingdom J, Jaeggi E. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int J Pediatr 2010: 401323, 2010. doi: 10.1155/2010/401323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamitomo M, Longo LD, Gilbert RD. Right and left ventricular function in fetal sheep exposed to long-term high-altitude hypoxemia. Am J Physiol Heart Circ Physiol 262: H399–H405, 1992. [DOI] [PubMed] [Google Scholar]
- 34.Kang BY, Kleinhenz JM, Murphy TC, Hart CM. The PPARγ ligand rosiglitazone attenuates hypoxia-induced endothelin signaling in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 301: L881–L891, 2011. doi: 10.1152/ajplung.00195.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kikkawa Y, Matsuo S, Kameda K, Hirano M, Nakamizo A, Sasaki T, Hirano K. Mechanisms underlying potentiation of endothelin-1-induced myofilament Ca(2+) sensitization after subarachnoid hemorrhage. J Cereb Blood Flow Metab 32: 341–352, 2012. doi: 10.1038/jcbfm.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komuro I, Kurihara H, Sugiyama T, Yoshizumi M, Takaku F, Yazaki Y. Endothelin stimulates c-fos and c-myc expression and proliferation of vascular smooth muscle cells. FEBS Lett 238: 249–252, 1988. doi: 10.1016/0014-5793(88)80489-7. [DOI] [PubMed] [Google Scholar]
- 37.Kuo NT, Benhayon D, Przybylski RJ, Martin RJ, LaManna JC. Prolonged hypoxia increases vascular endothelial growth factor mRNA and protein in adult mouse brain. J Appl Physiol (1985) 86: 260–264, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Lefebvre B, Godin-Ribuot D, Joyeux-Faure M, Caron F, Bessard G, Lévy P, Stanke-Labesque F. Functional assessment of vascular reactivity after chronic intermittent hypoxia in the rat. Respir Physiol Neurobiol 150: 278–286, 2006. doi: 10.1016/j.resp.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S, Elton TS. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J Appl Physiol (1985) 77: 1451–1459, 1994. [DOI] [PubMed] [Google Scholar]
- 40.Lin CL, Jeng AY, Howng SL, Kwan AL. Endothelin and subarachnoid hemorrhage-induced cerebral vasospasm: pathogenesis and treatment. Curr Med Chem 11: 1779–1791, 2004. doi: 10.2174/0929867043364919. [DOI] [PubMed] [Google Scholar]
- 41.Nakashima S. Protein kinase C alpha (PKC alpha): regulation and biological function. J Biochem 132: 669–675, 2002. doi: 10.1093/oxfordjournals.jbchem.a003272. [DOI] [PubMed] [Google Scholar]
- 42.Nauli SM, Williams JM, Akopov SE, Zhang L, Pearce WJ. Developmental changes in ryanodine- and IP3-sensitive Ca2+ pools in ovine basilar artery. Am J Physiol Cell Physiol 281: C1785–C1796, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Newby EA, Myers DA, Ducsay CA. Fetal endocrine and metabolic adaptations to hypoxia: the role of the hypothalamic-pituitary-adrenal axis. Am J Physiol Endocrinol Metab 309: E429–E439, 2015. doi: 10.1152/ajpendo.00126.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nilsson T, Cantera L, Adner M, Edvinsson L. Presence of contractile endothelin-A and dilatory endothelin-B receptors in human cerebral arteries. Neurosurgery 40: 346–351; discussion 351–353, 1997. doi: 10.1097/00006123-199702000-00023. [DOI] [PubMed] [Google Scholar]
- 45.Ono S, Westcott JY, Voelkel NF. PAF antagonists inhibit pulmonary vascular remodeling induced by hypobaric hypoxia in rats. J Appl Physiol (1985) 73: 1084–1092, 1992. [DOI] [PubMed] [Google Scholar]
- 46.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801, 2004. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 47.Packer CS, Roepke JE, Oberlies NH, Rhoades RA. Myosin isoform shifts and decreased reactivity in hypoxia-induced hypertensive pulmonary arterial muscle. Am J Physiol 274: L775–L785, 1998. [DOI] [PubMed] [Google Scholar]
- 48.Pearce WJ, Hull AD, Long DM, Longo LD. Developmental changes in ovine cerebral artery composition and reactivity. Am J Physiol Regul Integr Comp Physiol 261: R458–R465, 1991. [DOI] [PubMed] [Google Scholar]
- 49.Pearce WJ, Khorram O. Maturation and differentiation of the fetal vasculature. Clin Obstet Gynecol 56: 537–548, 2013. doi: 10.1097/GRF.0b013e31829e5bc9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prystowsky H, Blechner JN, Cotter JR. Oxygen dissociation curves of the bloods of maternal and fetal goats as they exist in vivo. Yale J Biol Med 42: 229–234, 1969. [PMC free article] [PubMed] [Google Scholar]
- 51.Ream M, Ray AM, Chandra R, Chikaraishi DM. Early fetal hypoxia leads to growth restriction and myocardial thinning. Am J Physiol Regul Integr Comp Physiol 295: R583–R595, 2008. doi: 10.1152/ajpregu.00771.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rey S, Corthorn J, Chacón C, Iturriaga R. Expression and immunolocalization of endothelin peptides and its receptors, ETA and ETB, in the carotid body exposed to chronic intermittent hypoxia. J Histochem Cytochem 55: 167–174, 2007. doi: 10.1369/jhc.6A7079.2006. [DOI] [PubMed] [Google Scholar]
- 53.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol 182: 311–322, 2000. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 54.Schorlemmer A, Matter ML, Shohet RV. Cardioprotective signaling by endothelin. Trends Cardiovasc Med 18: 233–239, 2008. doi: 10.1016/j.tcm.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sestito R, Cianfrocca R, Rosanò L, Tocci P, Semprucci E, Di Castro V, Caprara V, Ferrandina G, Sacconi A, Blandino G, Bagnato A. miR-30a inhibits endothelin A receptor and chemoresistance in ovarian carcinoma. Oncotarget 7: 4009–4023, 2016. doi: 10.18632/oncotarget.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silpanisong J, Pearce WJ. Vasotrophic regulation of age-dependent hypoxic cerebrovascular remodeling. Curr Vasc Pharmacol 11: 544–563, 2013. doi: 10.2174/1570161111311050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singer HA. Ca2+/calmodulin-dependent protein kinase II function in vascular remodelling. J Physiol 590: 1349–1356, 2012. doi: 10.1113/jphysiol.2011.222232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Snow JB, Gonzalez Bosc LV, Kanagy NL, Walker BR, Resta TC. Role for PKCβ in enhanced endothelin-1-induced pulmonary vasoconstrictor reactivity following intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol 301: L745–L754, 2011. doi: 10.1152/ajplung.00020.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stenman E, Malmsjö M, Uddman E, Gidö G, Wieloch T, Edvinsson L. Cerebral ischemia upregulates vascular endothelin ET(B) receptors in rat. Stroke 33: 2311–2316, 2002. doi: 10.1161/01.STR.0000028183.04277.32. [DOI] [PubMed] [Google Scholar]
- 60.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 61.Szymonowicz W, Walker AM, Cussen L, Cannata J, Yu VY. Developmental changes in regional cerebral blood flow in fetal and newborn lambs. Am J Physiol Heart Circ Physiol 254: H52–H58, 1988. [DOI] [PubMed] [Google Scholar]
- 62.Thaete LG, Jilling T, Synowiec S, Khan S, Neerhof MG. Expression of endothelin 1 and its receptors in the hypoxic pregnant rat. Biol Reprod 77: 526–532, 2007. doi: 10.1095/biolreprod.107.061820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tulis DA, Mnjoyan ZH, Schiesser RL, Shelat HS, Evans AJ, Zoldhelyi P, Fujise K. Adenoviral gene transfer of fortilin attenuates neointima formation through suppression of vascular smooth muscle cell proliferation and migration. Circulation 107: 98–105, 2003. doi: 10.1161/01.CIR.0000047675.86603.EB. [DOI] [PubMed] [Google Scholar]
- 64.Underwood DC, Bochnowicz S, Osborn RR, Luttmann MA, Hay DW. Nonpeptide endothelin receptor antagonists. X. Inhibition of endothelin-1- and hypoxia-induced pulmonary pressor responses in the guinea pig by the endothelin receptor antagonist, SB 217242. J Pharmacol Exp Ther 283: 1130–1137, 1997. [PubMed] [Google Scholar]
- 65.Vignon-Zellweger N, Heiden S, Miyauchi T, Emoto N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci 91: 490–500, 2012. doi: 10.1016/j.lfs.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 66.Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res 50: 553–562, 2001. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Waldsee R, Ahnstedt H, Eftekhari S, Edvinsson L. Involvement of calcium-calmodulin-dependent protein kinase II in endothelin receptor expression in rat cerebral arteries. Am J Physiol Heart Circ Physiol 298: H823–H832, 2010. doi: 10.1152/ajpheart.00759.2009. [DOI] [PubMed] [Google Scholar]
- 68.Wang Z, Li AY, Guo QH, Zhang JP, An Q, Guo YJ, Chu L, Weiss JW, Ji ES. Effects of cyclic intermittent hypoxia on ET-1 responsiveness and endothelial dysfunction of pulmonary arteries in rats. PLoS One 8: e58078, 2013. doi: 10.1371/journal.pone.0058078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wedegärtner U, Tchirikov M, Schäfer S, Priest AN, Kooijman H, Adam G, Schröder HJ. Functional MR imaging: comparison of BOLD signal intensity changes in fetal organs with fetal and maternal oxyhemoglobin saturation during hypoxia in sheep. Radiology 238: 872–880, 2006. doi: 10.1148/radiol.2383042213. [DOI] [PubMed] [Google Scholar]
- 70.Williams JM, Pearce WJ. Age-dependent modulation of endothelium-dependent vasodilatation by chronic hypoxia in ovine cranial arteries. J Appl Physiol (1985) 100: 225–232, 2006. doi: 10.1152/japplphysiol.00221.2005. [DOI] [PubMed] [Google Scholar]
- 71.Yamboliev IA, Hruby A, Gerthoffer WT. Endothelin-1 activates MAP kinases and c-Jun in pulmonary artery smooth muscle. Pulm Pharmacol Ther 11: 205–208, 1998. doi: 10.1006/pupt.1998.0139. [DOI] [PubMed] [Google Scholar]
- 72.Yamin R, Morgan KG. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J Physiol 590: 4145–4154, 2012. doi: 10.1113/jphysiol.2012.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu X, Li T, Liu X, Yu H, Hao Z, Chen Y, Zhang C, Liu Y, Li Q, Mao M, Zhu D. Modulation of pulmonary vascular remodeling in hypoxia: role of 15-LOX-2/15-HETE-MAPKs pathway. Cell Physiol Biochem 35: 2079–2097, 2015. doi: 10.1159/000374015. [DOI] [PubMed] [Google Scholar]
- 74.Zhang C, Shan XL, Liao YL, Zhao P, Guo W, Wei HC, Lu R. Effects of stachydrine on norepinephrine-induced neonatal rat cardiac myocytes hypertrophy and intracellular calcium transients. BMC Complement Altern Med 14: 474, 2014. doi: 10.1186/1472-6882-14-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang L. Prenatal hypoxia and cardiac programming. J Soc Gynecol Investig 12: 2–13, 2005. doi: 10.1016/j.jsgi.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 76.Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, Mizukami M, Toko H, Shibasaki F, Yazaki Y, Nagai R, Komuro I. Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem 275: 15239–15245, 2000. doi: 10.1074/jbc.275.20.15239. [DOI] [PubMed] [Google Scholar]