

Abstract
Forskolin, a selective activator of adenylyl cyclase (AC), commonly is used to establish actions of G protein-coupled receptors (GPCRs) that are initiated primarily through activation of AC/cAMP signaling pathways. In the present study, forskolin was used to evaluate the potential role of AC/cAMP, which is a major signaling mechanism for the pituitary adenylate cyclase-activating polypeptide (PACAP)-selective PAC1 receptor, in the regulation of guinea pig cardiac neuronal excitability. Forskolin (5–10 µM) increases excitability in ~60% of the cardiac neurons. The forskolin-mediated increase in excitability was considered related to cAMP regulation of a cyclic nucleotide gated channel or via protein kinase A (PKA)/ERK signaling, mechanisms that have been linked to PAC1 receptor activation. However, unlike PACAP mechanisms, forskolin enhancement of excitability was not significantly reduced by treatment with cesium to block currents through hyperpolarization-activated nonselective cation channels (Ih) or by treatment with PD98059 to block MEK/ERK signaling. In contrast, treatment with the clathrin inhibitor Pitstop2 or the dynamin inhibitor dynasore eliminated the forskolin-induced increase in excitability; treatments with the inactive Pitstop analog or PP2 treatment to inhibit Src-mediated endocytosis mechanisms were ineffective. The PKA inhibitor KT5702 significantly suppressed the forskolin-induced change in excitability; further, KT5702 and Pitstop2 reduced the forskolin-stimulated MEK/ERK activation in cardiac neurons. Collectively, the present results suggest that forskolin activation of AC/cAMP/PKA signaling leads to the recruitment of clathrin/dynamin-dependent endosomal transduction cascades, including MEK/ERK signaling, and that endosomal signaling is the critical mechanism underlying the forskolin-induced increase in cardiac neuron excitability.
Keywords: autonomic neuron, forskolin, PKA, MAPK signaling, neuronal excitability
g protein-coupled receptors (GPCRs) can activate a number of downstream intracellular cascades, which in turn can regulate multiple cellular functions (14, 16, 17). Recent studies have shown that activation of intracellular signaling occurs not only through well-established plasma membrane delimited mechanisms such as activation of adenylyl cyclase (AC) and phospholipase C (PLC), but also through clathrin- and dynamin-dependent GPCR endocytosis and formation of a signaling endosome (8, 12, 21, 27). Recent evidence also strongly suggests that for some GPCRs, receptor activation leads to AC internalization and incorporation into the signaling endosome. The signaling endosome contributes to sustained cAMP generation distinct from plasma membrane AC-mediated downstream signaling and also can direct cAMP to specific intracellular sites with high spatial and temporal resolution (4, 5, 11, 35).
The neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) enhances excitability of guinea pig cardiac neurons (2, 22, 29). Multiple factors contribute to the peptide effect, some of which, such as enhancement of Ih currents flowing through hyperpolarization-activated nonselective cationic channels (HCN), are mediated by activation of the AC/cAMP signaling cascade and others mediated through PACAP/PAC1 receptor internalization and initiation of endosomal signaling (20, 25). Forskolin, a potent activator of AC, is commonly used to establish potential cellular actions of GPCRs that are specifically mediated by activation of AC/cAMP signaling (10, 15) and distinct from endosomal signaling. Forskolin also enhances cardiac neuron excitability, an observation supporting the hypothesis that the AC/cAMP cascade contributes to the regulation of neuronal excitability by PACAP (30). Our earlier studies have indicated that forskolin and PACAP stimulate an increase in cAMP levels to comparable extents in two different cells types, rat sympathetic neurons and human embryonic kidney cells (19, 22). Consequently, forskolin was used in the present study to quantify the contribution of AC/cAMP signaling, which should be independent of the endosomal signaling mechanisms associated with PAC1 receptor internalization, in the regulation of membrane excitability in guinea pig cardiac neurons.
Our results confirm that forskolin can significantly increase cardiac neuron excitability. However, treatment with cesium to block the cyclic nucleotide gated Ih and with the MEK inhibitor PD98059 to blunt MEK/ERK activation did not appear to significantly reduce the forskolin-induced increase in excitability. By contrast, neuronal exposure to inhibitors of clathrin/dynamin-mediated endocytosis eliminated the forskolin-induced increase in cardiac neuron excitability. Interestingly, pretreatment with a protein kinase A (PKA) inhibitor KT5720, but not the Src kinase family inhibitor PP2, blunted the forskolin-induced increase in excitability. From these observations, rather than direct cAMP effects, forskolin activation of AC/cAMP/PKA signaling appears to lead to the recruitment of endosomal signal transduction cascades that are critical mechanisms underlying the forskolin-induced modulation of cardiac neuron excitability.
METHODS
Animals.
All experiments were performed using cardiac ganglia whole mount preparations from Hartley guinea pigs (either sex, 250–350 g), following animal protocols approved by the Institutional Animal Care and Use Committees of the University of Vermont, the University of California, Los Angeles, and Ithaca College. Approved procedures also followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The guinea pigs were euthanized by isoflurane overdose and exsanguination. Hearts were quickly removed and placed in cold Krebs solution (in mM: 121 NaCl, 5.9 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, 8 glucose; pH 7.4 maintained by 95% O2–5% CO2 aeration). The cardiac ganglia, located on the epicardium of the atria, were then removed to form a whole mount preparation suitable for intracellular recording from visually identified cells and for confocal analysis of immunolabeled neurons (7, 9, 22, 30).
Chemicals.
All drugs were obtained from commercial sources: forskolin, and the MEK inhibitor PD 98059 (2′-amino-3′-methoxyflavone) (Calbiochem, La Jolla, CA); 8-bromo-cAMP (Sigma-Aldrich, St Louis, MO); Pitstop 2 (N-[5-(4-bromobenzylidene)-4-oxo-4,5-dihydro-1,3-thiazol-2-yl]naphthalene-1-sulfonamide), Pitstop inactive analog (5-(4-bromobenzylidene)-2-(naphthalen-1-ylamino)-1,3-thiazol-4(5H)-one) and PP2 (1-tert-butyl-3-(4-chlorophenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine) (Abcam Biochemicals, Cambridge, UK); KT5720 from Tocris (Ellisville MO). Forskolin, Pitstop2, Pitstop inactive analog, PP2, PD98059, and KT5702 were prepared as DMSO stocks, diluted and added directly to the bath solution. The concentration of DMSO was ≤ 0.2%.
Immunocytochemistry and confocal imaging.
The methods used to quantify phosphorylated ERK (pERK) levels in the cardiac neurons followed those reported previously (7, 30). Following different experimental treatments at 37°C, cardiac ganglia whole mounts were fixed in a 2% paraformaldehyde and 0.2% picric acid solution for 2 h at 4°C, washed in blocking solution, and treated with ice-cold methanol for 10 min before incubation overnight in 1:1,000 rabbit anti-phosphoERK1/2 (D13.14.4E, Cell Signaling Technology, Beverly, MA) for visualization with Cy3-conjugated donkey anti-rabbit IgG (1:500; Jackson ImmunoResearch, West Grove, PA). The guinea pig cardiac ganglia whole mounts were then mounted on glass slides, coverslipped and imaged with a Nikon/Yokogawa CSU W1 Spinning Disk confocal microscope using a Nikon Apo LWD 25X/1.10NA objective lens. Excitation was accomplished with a 100 mW 561 nm solid state laser, and emission collected from 590 nm-650 nm as 16-bit Nikon nd2 image files. Z-series (0.4 µm-step) were taken on five random ganglia in each sample, and regions of interest (ROI) were generated for individual neurons at the axial midpoint adjacent to the nucleus and avoiding the cell membrane. Specifically, a single Z-slice was selected through the middle of the cell and a unique circular ROI was generated for each cell cytoplasm depending on cell size. Each ganglion generally contained 3–10 cells that met measurement criteria. All hardware settings were carefully maintained across samples and cross-checked by reviewing data header files. Data collection and analysis were all performed using Nikon Elements 4.30.10 (Build 1021). Data obtained from multiple neurons in different cardiac ganglia whole mounts were averaged.
Intracellular recordings from cardiac ganglia neurons.
Intracellular recordings from cardiac neurons followed methods described previously (2, 22, 29–31). Cardiac ganglia preparations were superfused continuously (6–7 ml/min) with Krebs solution containing 10 mM NaHEPES (32–35°C) and individual neurons impaled using 2 M KCl-filled microelectrodes (60–120 MΩ). Membrane voltage was recorded using an Axoclamp-2A amplifier coupled with a Digidata 1322A data acquisition system and pCLAMP 8 software (Axon Instruments, Foster City, CA). Depolarizing current steps (0.1–0.5 nA, 1 s) were applied to characterize neuron excitability, and excitability curves were generated by plotting the number of action potentials generated at each stimulus intensity. Hyperpolarizing current steps (500 ms) of increasing amplitude were used to test for rectification in the current-induced hyperpolarization, which occurs when the hyperpolarization-activated current Ih is initiated.
A variable amplitude (generally <10 mV), transient depolarization occurred in some cells during the initial exposure to micromolar concentrations of forskolin. Consequently, some intracellular recordings were made with the cells electronically held at ~−60 mV to negate any transient depolarization. There was no noticeable difference in results between cells, which were held at −60 mV electrotonically throughout the recording, or those cells whose membrane potential was not maintained electrotonically.
Statistics.
Statistics were performed using GraphPad Prism statistical software (version 5.4; La Jolla, CA). Data are presented as means ± SE. When changes in excitability curves were evaluated, differences in mean action potential numbers elicited by 1-s depolarizing voltage steps at each current magnitude (0.1 to 0.5 nA) were tested for significance. Depending on the experimental protocol, differences between means were either determined using an unpaired Student’s t-test or by one-way ANOVA followed by a Tukey’s post hoc test. Fisher’s exact test was used to determine differences between percentages of responsive cells in different conditions. The significance of change in mean pERK-immunoreactivity (IR) values were determined using an one-way ANOVA followed by a Tukey’s post hoc test. Values were considered statistically significant at P < 0.05.
RESULTS
Forskolin increases cardiac neuron excitability.
PACAP is an endogenous ligand in vagal preganglionic parasympathetic fibers to the guinea pig heart and exogenous or neurally released PACAP can consistently increase cardiac neuron excitability (2, 29). The PACAP/PAC1 receptor-mediated increase in cardiac neuron excitability is modulated in part by cAMP regulation of a hyperpolarization activated, cyclic nucleotide gated Ih current and pERK, which can enhance currents through Nav1.7 channels (23, 30). As PAC1 receptor mechanisms can initiate multiple signaling pathways (i.e., cAMP/PKA, PKC, ERK) to regulate ionic channel function, we wanted to evaluate the relative contribution of the AC/cAMP/PKA signaling in cardiac neuron excitability using the diterpene AC activator forskolin.
To quantify the percentage of cardiac neurons responding to forskolin, recordings were made in the same phasic cell before and during 5 µM forskolin exposure. Recordings were obtained from 18 neurons in 18 separate cardiac ganglia whole mount preparations. Forskolin produced a shift from phasic to multiple firing (from 1 to 2 to ≥ 5 action potentials elicited by the depolarizing steps) in 10 out of 18 cardiac neurons tested (56%).
An example recording from a forskolin “responsive cell” is shown in Fig. 1. In this case, multiple recordings of the action potentials elicited by 1-s duration, constant current steps of increasing magnitude were obtained before and during 5 µM forskolin application. This cell exhibited a phasic firing pattern before forskolin exposure (Fig. 1A1), but shifted to a multiple firing pattern in forskolin (Fig. 1B1). The data from these 18 cells were used to create averaged excitability curves for all cells before and during 5 µM forskolin exposure (Fig. 1C).
Fig. 1.

Forskolin can increase cardiac neuron excitability and the rectification in hyperpolarizations elicited by constant current injection. A1 and B1: example recording of action potentials elicited by a 1,000 ms, 0.3 nA depolarizing step before (A1) and during 5 µM forskolin exposure (B1). A2 and B2: the slight sag (rectification) in the hyperpolarization induced by the constant current step to ~−90 mV (A2) is due to the activation of the hyperpolarization-activated inward current, Ih. This rectification is more evident during 5 µM forskolin exposure (B2), indicative of an enhanced Ih. In this cell, a rebound depolarization followed the termination of the hyperpolarization, which in A2, but not B2, was sufficient in magnitude to elicit an action potential. The resting membrane potential was initially −60 mV, but over the course of the recording, the cell hyperpolarized by a few millivolts such that the rebound depolarization, although still evident, did not reach the threshold for action potential generation. C: averaged excitability curves for different cells before and during exposure to 5 µM forskolin. ○, Averaged excitability curve for 18 cells before forskolin; ●, averaged excitability curve for these same cells during exposure to 5 µM forskolin; *, number of action potentials elicited were significantly greater in forskolin-treated than in control cells.
From cAMP generation, forskolin, like PACAP/PAC1 receptor Gαs signaling, enhances a cyclic nucleotide gated hyperpolarization-activated Ih current in dissociated cardiac neurons (23) and expectedly, forskolin enhanced the inward rectification noted in hyperpolarizations elicited by 500-ms constant current steps (Fig. 1, A2 and B2). As cesium treatments blocked Ih and reduced the PACAP-induced increase in cardiac neuron excitability (32), we examined whether CsCl could also suppress the forskolin-mediated increase in excitability. Intracellular recordings were obtained from 15 phasic cells pretreated with 2 mM cesium chloride for at least 15 min and then exposed to 5 µM forskolin and cesium. During forskolin treatment, 5 of the 15 cesium-pretreated cells exhibited a multiple firing pattern (≥5 action potentials) when stimulated with 1-s depolarizing constant current steps of increasing magnitude. Although the percentage of cells that exhibited multiple firing when pretreated with cesium and then exposed to forskolin (33%) was less than that for those exposed to forskolin alone (56%), the difference did not reach statistical significance (Fisher’s exact test, P = 0.3). In addition, the averaged excitability curve for the cells exposed to cesium and forskolin (Fig. 2A) was not different from that estimated for cells exposed to forskolin alone (Fig. 1C).
Fig. 2.

Neither cesium nor PD98059 pretreatment significantly decreases the forskolin-induced increase in excitability. A: averaged excitability curves for cells pretreated with 1 mM cesium (Cs) and then exposed to Cs and 5 µM forskolin. □, Averaged excitability curve for 15 cells during treatment with cesium alone; ■, averaged excitability curve for 15 cells during exposure to cesium and forskolin. B: averaged excitability curves for cells pretreated with 50 µM PD98059 (PD) and then exposed to PD98059 and 5 µM forskolin. ∇, Averaged excitability curve for 8 cells during pretreatment with PD98059 alone; ▼, averaged excitability curve for the same 8 cells during exposure to PD98059 and forskolin. From comparison of the excitability curves in A (Cs and forskolin) and B (PD and forskolin), with the excitability curve for forskolin alone in Fig. 1C, it was determined that neither cesium nor PD98059 significantly decreased the number of action potentials elicited by each depolarizing step. Furthermore, the averaged excitability curves determined before forskolin exposure were the same for control cells or cells just exposed to cesium or PD98059.
The results of the above studies suggested additional signaling pathways downstream of cAMP mediated the forskolin-enhanced excitability. Previous PAC1 receptor studies implicated ERK signaling in modulation of excitability; the PACAP-induced increase in cardiac neuronal excitability could be reduced by pretreatment with the MEK inhibitor PD98059 (30, 34). As forskolin can increase cardiac neuron pERK (7), we tested whether treatment with the MEK inhibitor PD98059 to block MEK/ERK signaling would suppress the forskolin-induced increase in excitability. In this series of experiments, cardiac ganglia whole mount preparations were pretreated with the MEK inhibitor PD98059 (50 µM) for at least 15 min and recordings were obtained from the same phasic cell exposed to PD98059 alone and then during exposure to PD98059 and 5 µM forskolin. Excitability shifted from a phasic to multiple firing pattern (≥5 action potentials) in 2 of 8 cells, which is a smaller percentage (25%) than seen with forskolin alone (56%). However, as with CsCl pretreatment, the difference in percentage of forskolin-induced multiple firing cells with and without PD98059 treatment did not reach significance (Fishers exact test, P = 0.2). In addition, even though the number of action potentials elicited by each current step was consistently less for cells exposed to forskolin and PD98059 (Fig. 2B) than that determined for cells exposed to forskolin alone (Fig. 1C), the differences did not reach significance by an unpaired Student’s t-test).
The observation that neither cesium nor PD98059 treatment significantly decreased the forskolin-induced increase in excitability suggested that a direct cAMP action on Ih or activation of ERK signaling may not have been major contributors to the forskolin effect and that other mechanisms drive the forskolin enhancement of excitability.
The forskolin-induced increase in excitability is suppressed by a PKA inhibitor.
Cellular actions of forskolin are mediated by a rise in cytosolic cAMP and one well-established cellular effect of cAMP is the activation of PKA, which in turn phosphorylates downstream targets of the cAMP/PKA signaling cascade. As pretreatment with a PKA inhibitor blunted the PACAP-induced increase in excitability (34), additional experiments tested a potential role of PKA in the forskolin-induced increase in excitability.
For this series of experiments, the concentration of forskolin was increased to 10 µM to potentially increase the number of cells that responded to forskolin with an increase in excitability. Also, to reduce the number of animals needed to quantify the results, and to avoid having to maintain stable recordings during prolonged exposures to forskolin, most recordings were not made in the same cell before and during forskolin treatment. Rather, recordings were made from different groups of neurons in multiple cardiac ganglia preparations before and during forskolin exposure.
The initial recordings were obtained from 14 cells in 7 cardiac ganglia whole mount preparations not exposed to forskolin. All of these control cells were phasic (Fig. 3A). To then establish the effect of 10 µM forskolin on excitability, recordings were obtained from 18 cells from 5 cardiac ganglia whole mounts exposed to forskolin for up to 40 min. Eleven of the 18 cells (61%) exposed to 10 µM forskolin exhibited a multiple firing pattern (≥ 5 action potentials), a percentage comparable to that obtained with 5 µM forskolin. The averaged excitability curves for the control cells and 10 µM forskolin treated cells are shown in Fig. 3A.
Fig. 3.

Pretreatment with KT5702, an inhibitor of PKA, and inhibitors of clathrin/dynamin-mediated endocytosis blunts the forskolin-induced increase in excitability. A: 1 µM KT5702 pretreatment significantly depresses the forskolin-induced increase in excitability. ○, Averaged excitability curve for 17 control cells; □, averaged excitability curve for 18 cells exposed to 10 µM forskolin. △, averaged excitability curve for 9 cells pretreated with 1 µM KT5702 and then exposed to KT5702 and 10 µM forskolin. The number of action potentials generated by each depolarizing step was significantly less (P < 0.05, unpaired t-test) in cells exposed to KT5702 and forskolin than for cells exposed to forskolin alone. B: pretreatment with Pitstop2 and dynasore, but not the inactive analog of Pitstop, reduces the 10 µM forskolin-induced increase in excitability. □, Averaged excitability curve for 12 cells pretreated with the inactive analog of Pitstop (15 µM) and then exposed to the Pitstop inactive analog and 10 µM forskolin; ○, averaged excitability curve for 16 cells pretreated with Pitstop2 and then exposed to Pitstop2 and 10 µM forskolin; ∇, averaged excitability curve for 10 cells pretreated with 20 µM dynasore and then exposed to dynasore and 10 µM forskolin. The number of action potentials generated by each depolarizing step was significantly less (P < 0.05, one-way ANOVA) in cells exposed to Pitstop2 and forskolin or dynasore and forskolin than for cells exposed to the inactive analog of Pitstop.
The next experiments tested whether activation of PKA was a requisite for the forskolin-induced increase in excitability. Three whole mount preparations were pretreated with the PKA inhibitor KT5720 (1 µM) for at least 15 min and then exposed to 10 µM forskolin and the inhibitor. Recordings were obtained from 9 cells; only 2 of the 9 cells exhibited a multiple firing pattern, whereas the remaining 7 were phasic. The averaged excitability curve for the 9 cells exposed to forskolin and KT5702 is shown in Fig. 3A. The averaged number of action potentials elicited by each depolarizing step was significantly less (P < 0.05) in cells exposed to KT5702 and forskolin than for cells exposed to forskolin alone. These results demonstrate that inhibition of PKA significantly depressed the forskolin-induced increase in excitability.
Pitstop2 and dynasore suppress the forskolin-induced increase in cardiac neuron excitability.
PAC1 receptors can engage diverse transduction pathways via AC/cAMP/PKA, PLC/PKC and β-arrestin-mediated endosomal signaling platforms (19). From recent work, PAC1 receptor endosomal signaling appears to represent a key mechanism for long-term cellular activation and function (19). Because the clathrin inhibitor Pitstop2 (36) eliminates the PACAP-induced increase in cardiac neuron excitability (22), experiments were completed to test whether Pitstop2 affected the forskolin-induced increase in excitability. Cardiac ganglia whole mounts were pretreated with 15 µM Pitstop2 and then exposed to 10 µM forskolin and 15 µM Pitstop2. Recordings were obtained from 16 cells for up to 40 min. Only 1 of the 16 cells exposed to Pitstop2 and forskolin exhibited a multiple firing pattern, clearly indicating that Pitstop2 suppressed the forskolin-induced increase in excitability. The averaged excitability curve for the cells in Pitstop2/forskolin is presented in Fig. 3B.
In contrast to that noted for Pitstop2, a 15-min pretreatment with the inactive analog of Pitstop (15 µM) did not significantly suppress the forskolin-induced increase in excitability (Fig. 3, A and B). When cells were pretreated with the Pitstop inactive analog, forskolin increased excitability in 5 of 12 cells (42%) and the averaged excitability curve was significantly greater for cells exposed to the inactive analog plus forskolin than that determined for cells pretreated with Pitstop2 and forskolin (Fig. 3B).
Clathrin can play a role in intracellular signaling that is separate from supporting endocytosis mechanisms (3). Consequently, to test further that the inhibition by Pitstop2 was due to its ability to blunt endocytosis, experiments also were completed using dynasore, a dynamin inhibitor, which also blocks endocytosis (18). Dynasore eliminates the PACAP-induced increase in excitability, but blocks endocytosis by a mechanism distinct from that of Pitstop2 (22). Two whole mounts were pretreated with 20 µM dynasore and then exposed to dynasore along with 10 µM forskolin. Recordings were obtained from 10 cells, none of which exhibited multiple firing. The excitability curve determined for cells exposed to dynasore and forskolin was similar to that noted for cells treated with Pitstop2 along with forskolin (Fig. 3B).
Previously, the cell-permeable analog of cAMP, 8-bromo-cAMP, was determined to increase cardiac neuron excitability to an extent comparable to that of forskolin (34). In separate experiments, we tested whether Pitstop2 also blunted the increase in neuronal excitability by 8-bromo-cAMP. Recordings were obtained from 8 cells in 2 cardiac ganglia preparations that were pretreated with 15 µM Pitstop2 and then exposed to 1 mM 8-bromo-cAMP and Pitstop2. All 8 cells retained a phasic firing pattern during exposure to 1 mM 8-bromo-cAMP and Pitstop2 (data not shown), indicating that Pitstop2 pretreatment can also effectively eliminate the 8-bromo-cAMP-induced increase in excitability.
Inhibition of Src family kinases does not blunt the forskolin-induced increase in excitability.
Src family kinases can play critical roles in recruiting cofactors needed for initiation of trophic factor endocytosis (1). Also, the Src family kinase inhibitor PP2 blocked PACAP-induced PAC1 internalization and reduced pERK generation in a human embryonic kidney (HEK) cell line stably expressing the green fluorescent protein (GFP)-tagged PAC1 receptor (May V and Parsons RL, unpublished observations). Consequently, experiments tested whether pretreatment with PP2 affected the forskolin-induced increase in excitability. For these experiments, two cardiac ganglia whole mounts were pretreated for 15 min with 10 µM PP2 and then exposed to 10 µM forskolin and 10 µM PP2. Recordings were obtained from 16 cells over a 40-min period. Seven of the 16 cells (42%) exposed to PP2 and forskolin exhibited a multiple firing pattern, whereas the remaining nine exhibited a phasic firing pattern. The percentage of cells exhibiting a multiple firing pattern was less in cells exposed to PP2 and forskolin (42%) than those exposed to only forskolin (56%); the difference was not significant (Fisher’s exact test, P = 0.7). Further, the averaged excitability curve for cells exposed to 10 µM forskolin and 10 µM PP2 was not different from the excitability curve for cells exposed to forskolin alone (data not shown).
Pitstop2 and KT5702 decrease the forskolin activation of MEK/ERK signaling.
Forskolin can initiate MEK/ERK signaling in guinea pig cardiac neurons (7). Confirming our earlier observations, treatment with 10 µM forskolin for 20 min significantly increased neuronal cytosolic pERK-IR (Fig. 4, A and B), an effect eliminated by a 15-min pretreatment with the MEK inhibitor PD98059 (50 µM) (Fig. 4C).
Fig. 4.

Pitstop2 and KT5702 decrease the forskolin-induced generation of neuronal pERK in cardiac neurons. A–C: examples of pERK-immunoreactivity in cardiac neurons under control conditions (A), following a 20-min exposure to 10 µM forskolin (B), and following a 20-min exposure to 10 µM forskolin and 50 µM PD98059 that was preceded by a 15-min pretreatment with 50 µM PD98059 (C). Calibration in C equals 20 µm. D: the forskolin-induced increase in pERK generation is eliminated by pretreatment with 50 µM PD98059 (F+PD) and reduced by pretreatment with 15 µM Pitstop2 (F+Pitstop) or 1 µM KT5702 (F+KT). Results are summarized from multiple cells in different cardiac ganglia whole mount preparations: Control, 53 cells in 4 whole mounts; forskolin, 88 cells in 3 whole mounts; PD98059/forskolin, 30 cells in 2 whole mounts; Pitstop2/forskolin, 58 cells in 3 whole mounts, and KT5702/forskolin, 47 cells in 2 whole mounts. Treatment with PD98059, Pitstop2, or KT5702 all significantly reduced the forskolin-induced generation of pERK (*P < 0.05 by one-way ANOVA).
Because Pitstop2 blocked the forskolin-induced increase in excitability, additional experiments were done to explore the possible involvement of a clathrin-mediated mechanism in the forskolin-induced activation of MEK/ERK signaling in the cardiac neurons. Pretreatment with the clathrin inhibitor Pitstop2 (15 µM) reduced the forskolin-induced increase in neuronal pERK-IR by ~50% (Fig. 4D).
Stimulation of AC/cAMP/PKA signaling can lead to activation of the MEK/ERK signaling cascade. Consequently, we also tested whether the forskolin-induced rise in neuronal pERK was depressed by pretreatment with the PKA inhibitor KT5702. Indeed, treatment with 1 µM KT5702 diminished forskolin-induced pERK-IR by ~30% in the cardiac neurons (Fig. 4D. Consequently, as was determined for PACAP (6), the forskolin-induced increase in neuronal pERK involves both activation of endosomal signaling and the AC/cAMP/PKA transduction pathways.
DISCUSSION
Two novel observations obtained in the present study are 1) that Pitstop2 and dynasore, two established inhibitors of clathrin/dynamin-mediated endocytosis, eliminated a forskolin-induced increase in cardiac neuron excitability and 2) that Pitstop2 also significantly diminished the forskolin-induced activation of MEK/ERK signaling.
Our previous work has shown that PACAP/PAC1 receptor signaling can engage diverse and intersecting mechanistic routes to enhance cardiac neuron excitability (25, 30). Some of these mechanisms appear to be canonical plasma membrane delimited signaling pathways including G protein-dependent activation of AC to engage cAMP/PKA-modulation of ionic conductances. Cyclic AMP appears to regulate directly a cyclic nucleotide/hyperpolarization activated Ih current and PKA has been suggested to enhance a T-type calcium current (IT). However, as for many GPCRs, ligand binding to the PAC1 receptor can also initiate β-arrestin-mediated receptor internalization for G protein-independent endosomal signaling (19). The formation of signaling endosomes allows the assembly of components that may generate signals distinct from those at the plasma membrane with spatiotemporal dynamics that can have more long-lasting consequences in neuronal activation and plasticity. PAC1 receptor endosomal signaling has been best studied with respect to ERK, which may regulate a sodium conductance in cardiac neurons (7, 30). While intersecting, the plasma membrane (AC/cAMP/PKA) and endosomal routes to ERK may be functionally distinct.
The diterpene compound forskolin activates AC directly and was used in the current studies to delineate the relative contribution of plasma membrane-initiated cAMP/PKA signaling in cardiac neuron excitability. Similar to PACAP/PAC1 receptor signaling, forskolin-mediated generation of cAMP enhances neuronal excitability and Ih currents in voltage-clamped, dissociated cardiac neurons (23). Consistent with our prior findings, forskolin consistently enhanced the rectification in hyperpolarizing steps. Cesium chloride blocks Ih, and treatments of forskolin-stimulated cells with cesium chloride decreased the percentage of multiple firing cells and reduced the averaged number of action potentials generated by most depolarizing steps. However, these changes did not reach significance, which appeared consistent with prior studies that noted not all cardiac neurons express functional HCN channels (9, 33). Consequently, the contribution of Ih to the regulation of excitability would depend on whether these channels are expressed and level of expression in a given cell. Cesium treatment would only affect the forskolin-induced modulation of excitability in cells expressing HCN channels, even though forskolin can increase excitability in neurons that do not appear to express HCN channels. Thus, the limited effect of cesium on forskolin-stimulated cells could reflect the variable expression of HCN channels in the cardiac neurons.
Previously, we reported that MEK inhibition significantly decreased the PACAP-induced increase in cardiac neuron excitability, decreasing both the number of multiply firing cells and the excitability curve (30). However, some PACAP-stimulated cells pretreated with PD98059 still exhibited an enhanced excitability, indicating that inhibition of MEK/ERK signaling did not diminish the PACAP effect in all cells (30). There are multiple routes of forskolin to ERK activation including PKA and exchange protein activated by cAMP (EPAC). We have previously shown that EPAC signaling is likely not engaged in cardiac neuron signaling, thereby suggesting that, of the two pathways, PKA may be the most direct route for forskolin-mediated ERK signaling (7). PD98059 pretreatment to block MEK/ERK signaling also decreased the number of multiple firing cells in forskolin by 50% and consistently decreased the averaged number of action potentials elicited by depolarizing steps. Although the reduction of the forskolin-induced increase in excitability by PD98059 pretreatment did not reach significance, we suggest that activation of MEK/ERK signaling by forskolin could still be one mechanism contributing, at least in some cells, to the forskolin-induced increase in excitability. However, because PD98059 treatment totally suppressed forskolin-stimulated pERK-immunoreactivity in cardiac neurons, but did not eliminate the increase in excitability by either forskolin or PACAP, MEK/ERK signaling must be only one participatory mechanism regulating cardiac neuron excitability. These observations are consistent with the view that multiple mechanisms can potentially contribute to the peptide modulation of excitability (20, 25).
Pitstop2 and dynasore are distinct pharmacological inhibitors of endocytosis. Pitstop2 is a cell-permeable 1,8-naphthalimide-derived clathrin terminal domain inhibitor (36); dynasore is an inhibitor to dynamin 1/2 GTPase important for endocytic budding and scission (18). Our previous work showed that both inhibitors effectively suppressed PACAP-induced PAC1 receptor vesicular internalization in our stable HEK PAC1R-EGFP cell line, blunted PAC1 receptor-mediated ERK activation and eliminated the PACAP-induced increase in excitability (19, 22). On the basis of these observations, it is proposed that a PACAP-induced internalization of the PAC1 receptor into a signaling endosome is the most critical mechanism underlying the PACAP modulation of excitability and possibly is a mechanism that efficiently regulates function of a number of different ionic conductances (20). Among the compounds evaluated, Pitstop2 and dynasore completely and unexpectedly eliminated the forskolin-induced increase in excitability. The inhibitory actions of Pitstop2 and dynasore are mechanistically different in endocytosis yet yielded comparable results, and the inactive Pitstop2 analog had no effects. Hence, the results with Pitstop2 and dynasore appear to uncover mechanisms by which forskolin can activate clathrin/dynamin to initiate membrane endocytosis and formation of signaling endosomes. This would create a platform for recruitment of multiple transduction cascades, such as MEK/ERK signaling, that potentially contribute to the forskolin modulation of cardiac neuron excitability.
At present, we can only speculate how forskolin treatment might stimulate membrane endocytosis/endosomal signaling. As forskolin is a potent activator of AC, one possible mechanism is that an elevation of cAMP at the inner surface of the plasma membrane or subsequent activation of PKA triggers endocytosis of membrane patches, some of which could contain AC. The fact that the increase in neuronal excitability by 8-bromo-cAMP was eliminated by Pitstop2 provides support for a mechanism driven by a rise in cAMP. Pretreatment with the PKA inhibitor KT5702 also markedly reduced the forskolin-induced increase in excitability, an observation indicating a requirement for downstream PKA phosphorylation in the forskolin enhancement of excitability. In contrast to many trophic factor receptors (1, 26), the studies with PP2 treatment of forskolin-stimulated cells showed that Src family kinase activity was not required for the enhanced excitability.
Prior studies indicate that PKA-mediated phosphorylation of membrane proteins can facilitate processes resulting in their internalization. As example, similar to our results, forskolin-initiated PKA phosphorylation of the UT-A1 urea transporter, which is structurally similar to AC, results in clathrin-dependent UT-A1 endocytosis and downregulation (28). AC has a number of potential PKA phosphorylation sites, and previous publications have shown that AC function is regulated by PKA (6, 13, 24).
As summarized in the schematic in Fig. 5, we suggest that phosphorylation of AC, and potentially scaffolding proteins, by PKA rather than by Src family kinases, is a critical step in initiation of endosomal formation and recruitment of downstream signaling cascades that underlie the forskolin-induced modulation of excitability.
Fig. 5.
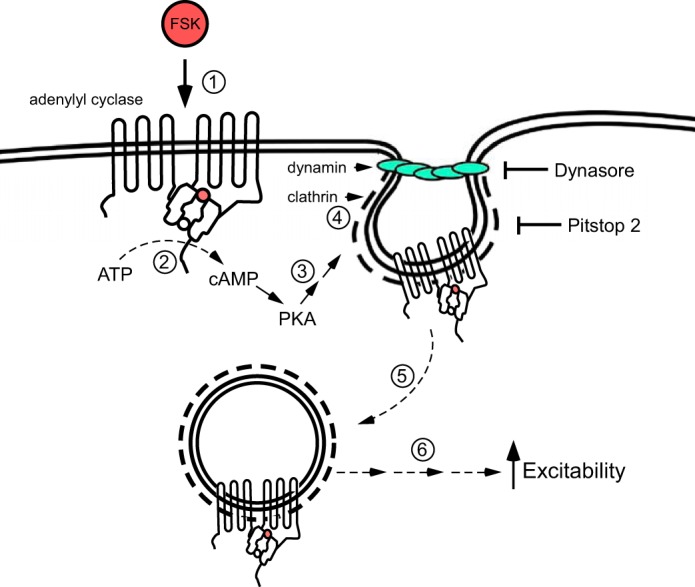
Forskolin (FSK) recruits endosomal signaling to enhance neuronal excitability. This schematic summarizes the results obtained in the present study, which support the hypothesis that forskolin recruits endosomal signaling. Forskolin stimulation of adenylyl cyclase (1) results in the catalysis of ATP to cAMP (2), leading to the activation of PKA (3). PKA facilitates clathrin and dynamin accumulation (4) to initiate plasma membrane endocytosis, creating a signaling endosome (5). The endosome becomes a platform for the recruitment of downstream signaling cascades, including MEK/ERK and other unidentified transduction cascades (6) that modulate ionic conductances regulating neuronal excitability.
PACAP, in nanomolar concentrations, increases excitability in 75–90% of the cardiac neurons (2, 22, 33, 34), whereas forskolin (5–10 µM) only increases excitability in 55–60% of the cardiac neurons. Given this marked difference in effectiveness between forskolin and PACAP on cardiac neuron excitability, we suggest that the PACAP-induced PAC1 internalization/endosome recruitment must be considerably more efficient than the endosomal formation/signaling induced by forskolin.
In conclusion, we postulate that a forskolin-induced membrane endocytosis and formation of signaling endosomes is the critical step in the recruitment of downstream signaling cascades that modulate ionic conductances underlying the enhancement of cardiac neuron excitability by forskolin. The results of the present study provide further support for the hypothesis that recruitment of endosomal signaling is an essential mechanism regulating neuronal excitability (20). Furthermore, AC internalization and incorporation into a signaling endosome may be a mechanism by which forskolin-induced cAMP production is sustained. This mechanism has been suggested for GPCRs, which are coupled to AC. With sustained agonist exposure, both the receptor and associated AC can be internalized into signaling endosomes and this mechanism can sustain prolonged cAMP production (4, 5, 35). To our knowledge this is the first time evidence supporting this novel mechanism of forskolin action has been reported.
GRANTS
This work was supported in part by National Institutes of Health (NIH) National Institute of General Medical Sciences Grant P30 GM103498/National Center for Research Resources P30 RR032135 (R. L. Parsons) and NIH/Department of Health and Human Services Grant S10 OD017969-01 (R. L. Parsons).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.C.H., T.A.C., J.D.T., B.M.G., C.N.B., and L.A.M. performed experiments; J.C.H., T.A.C., J.D.T., B.M.G., C.N.B., L.A.M., V.M., and R.L.P. analyzed data; J.C.H., T.A.C., J.D.T., V.M., and R.L.P. interpreted results of experiments; J.C.H., T.A.C., J.D.T., B.M.G., L.A.M., V.M., and R.L.P. edited and revised manuscript; J.C.H., T.A.C., J.D.T., B.M.G., C.N.B., L.A.M., V.M., and R.L.P. approved final version of manuscript; T.A.C., B.M.G., and L.A.M. prepared figures; V.M. and R.L.P. conceived and designed research; V.M. and R.L.P. drafted manuscript.
REFERENCES
- 1.Auciello G, Cunningham DL, Tatar T, Heath JK, Rappoport JZ. Regulation of fibroblast growth factor receptor signalling and trafficking by Src and Eps8. J Cell Sci 126: 613–624, 2013. doi: 10.1242/jcs.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braas KM, May V, Harakall SA, Hardwick JC, Parsons RL. Pituitary adenylate cyclase-activating polypeptide expression and modulation of neuronal excitability in guinea pig cardiac ganglia. J Neurosci 18: 9766–9779, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodsky FM, Sosa RT, Ybe JA, O’Halloran TJ. Unconventional functions for clathrin ESCRTs, and other endocytic regulators in the cytoskeleton, cell cycle, nucleus, and beyond: links to human disease. Cold Spring Harb Perspect Biol 2014: a0117004, 2016. doi: 10.1101/cshperspect.a017004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 7: e1000172, 2009. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calebiro D, Nikolaev VO, Persani L, Lohse MJ. Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci 31: 221–228, 2010. doi: 10.1016/j.tips.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Harry A, Li J, Smit MJ, Bai X, Magnusson R, Pieroni JP, Weng G, Iyengar R. Adenylyl cyclase 6 is selectively regulated by protein kinase A phosphorylation in a region involved in Gαs stimulation. Proc Natl Acad Sci USA 94: 14100–14104, 1997. doi: 10.1073/pnas.94.25.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clason TA, Girard BM, May V, Parsons RL. Activation of MEK/ERK signaling by PACAP in guinea pig cardiac neurons. J Mol Neurosci 59: 309–316, 2016. doi: 10.1007/s12031-016-0766-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Fiore PP, von Zastrow M. Endocytosis, signaling, and beyond. Cold Spring Harb Perspect Biol 6: a016865, 2014. doi: 10.1101/cshperspect.a016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards FR, Hirst GD, Klemm MF, Steele PA. Different types of ganglion cell in the cardiac plexus of guinea-pigs. J Physiol 486: 453–471, 1995. doi: 10.1113/jphysiol.1995.sp020825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emery AC, Eiden LE. Signaling through the neuropeptide GPCR PAC1 induces neuritogenesis via a single linear cAMP- and ERK-dependent pathway using a novel cAMP sensor. FASEB J 26: 3199–3211, 2012. doi: 10.1096/fj.11-203042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 5: 734–742, 2009. doi: 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495: 534–538, 2013. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwami G, Kawabe J, Ebina T, Cannon PJ, Homcy CJ, Ishikawa Y. Regulation of adenylyl cyclase by protein kinase A. J Biol Chem 270: 12481–12484, 1995. doi: 10.1074/jbc.270.21.12481. [DOI] [PubMed] [Google Scholar]
- 14.Jalink K, Moolenaar WH. G protein-coupled receptors: the inside story. BioEssays 32: 13–16, 2010. doi: 10.1002/bies.200900153. [DOI] [PubMed] [Google Scholar]
- 15.Klinger M, Kudlacek O, Seidel MG, Freissmuth M, Sexl V. MAP kinase stimulation by cAMP does not require RAP1 but SRC family kinases. J Biol Chem 277: 32490–32497, 2002. doi: 10.1074/jbc.M200556200. [DOI] [PubMed] [Google Scholar]
- 16.Luttrell LM, Gesty-Palmer D, Sibley DR. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev 62: 305–330, 2010. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283: 655–661, 1999. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 18.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10: 839–850, 2006. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 19.May V, Buttolph TR, Girard BM, Clason TA, Parsons RL. PACAP-induced ERK activation in HEK cells expressing PAC1 receptors involves both receptor internalization and PKC signaling. Am J Physiol Cell Physiol 306: C1068–C1079, 2014. doi: 10.1152/ajpcell.00001.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May V, Parsons RL. G protein-coupled receptor endosomal signaling and regulation of neuronal excitability and stress responses: signaling options and lessons from the PAC1 receptor. J Cell Physiol 232: 698–706, 2017. doi: 10.1002/jcp.25615. [DOI] [PubMed] [Google Scholar]
- 21.McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12: 517–533, 2011. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 22.Merriam LA, Baran CN, Girard BM, Hardwick JC, May V, Parsons RL. Pituitary adenylate cyclase 1 receptor internalization and endosomal signaling mediate the pituitary adenylate cyclase activating polypeptide-induced increase in guinea pig cardiac neuron excitability. J Neurosci 33: 4614–4622, 2013. doi: 10.1523/JNEUROSCI.4999-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merriam LA, Barstow KL, Parsons RL. Pituitary adenylate cyclase-activating polypeptide enhances the hyperpolarization-activated nonselective cationic conductance, Ih, in dissociated guinea pig intracardiac neurons. Regul Pept 123: 123–133, 2004. doi: 10.1016/j.regpep.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 24.Murphey KS, Zhou H, Makhlouf GM. PKA-dependent activation of PDE3A and PDE4 and inhibition of adenylyl cyclase V/VI in smooth muscle. Am J Physiol Cell Physiol 311: C508–C517, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Parsons RL, Tompkins JD, Hardwick JC, Merriam LA, Girard BM, May V. Multiple mechanisms contribute to the PAC1 modulation of parasympathetic cardiac neuron excitability. In: Pituitary Adenylate Cyclase Activating Polypeptide – PACAP, edited by Reglodi D, Tamas A. New York: Springer Nature, 2016, p. 205–225. doi: 10.1007/978-3-319-35135-3_13. [DOI] [Google Scholar]
- 26.Reinecke J, Caplan S. Endocytosis and the Src family of non-receptor tyrosine kinases. Biomol Concepts 5: 143–155, 2014. doi: 10.1515/bmc-2014-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scita G, Di Fiore PP. The endocytic matrix. Nature 463: 464–473, 2010. doi: 10.1038/nature08910. [DOI] [PubMed] [Google Scholar]
- 28.Su H, Carter CB, Laur O, Sands JM, Chen G. Forskolin stimulation promotes urea transporter UT-A1 ubiquitination, endocytosis, and degradation in MDCK cells. Am J Physiol Renal Physiol 303: F1325–F1332, 2012. doi: 10.1152/ajprenal.00248.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tompkins JD, Ardell JL, Hoover DB, Parsons RL. Neurally released pituitary adenylate cyclase-activating polypeptide enhances guinea pig intrinsic cardiac neurone excitability. J Physiol 582: 87–93, 2007. doi: 10.1113/jphysiol.2007.134965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tompkins JD, Clason TA, Hardwick JC, Girard BM, Merriam LA, May V, Parsons RL. Activation of MEK/ERK signaling contributes to the PACAP-induced increase in guinea pig cardiac neuron excitability. Am J Physiol Cell Physiol 311: C643–C651, 2016. doi: 10.1152/ajpcell.00164.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tompkins JD, Hardwick JC, Locknar SA, Merriam LA, Parsons RL. Ca2+ influx, but not Ca2+ release from internal stores, is required for the PACAP-induced increase in excitability in guinea pig intracardiac neurons. J Neurophysiol 95: 2134–2142, 2006. doi: 10.1152/jn.01077.2005. [DOI] [PubMed] [Google Scholar]
- 32.Tompkins JD, Lawrence YT, Parsons RL. Enhancement of Ih, but not inhibition of IM, is a key mechanism underlying the PACAP-induced increase in excitability of guinea pig intrinsic cardiac neurons. Am J Physiol Regul Integr Comp Physiol 297: R52–R59, 2009. doi: 10.1152/ajpregu.00039.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tompkins JD, Merriam LA, Girard BM, May V, Parsons RL. Nickel suppresses the PACAP-induced increase in guinea pig cardiac neuron excitability. Am J Physiol Cell Physiol 308: C857–C866, 2015. doi: 10.1152/ajpcell.00403.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tompkins JD, Parsons RL. Identification of intracellular signaling cascades mediating the PACAP-induced increase in guinea pig cardiac neuron excitability. J Mol Neurosci 36: 292–298, 2008. doi: 10.1007/s12031-008-9086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vilardaga J-P, Jean-Alphonse FG, Gardella TJ. Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol 10: 700–706, 2014. doi: 10.1038/nchembio.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, Chircop M, Sakoff J, von Kries JP, Saenger W, Kräusslich HG, Shupliakov O, Robinson PJ, McCluskey A, Haucke V. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146: 471–484, 2011. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]