

Abstract
The mechanisms underpinning decreased skeletal muscle strength and slowing of movement during aging are ill-defined. “Inflammaging,” increased inflammation with advancing age, may contribute to aspects of sarcopenia, but little is known about the participatory immune components. We discovered that aging was associated with increased caspase-1 activity in mouse skeletal muscle. We hypothesized that the caspase-1-containing NLRP3 inflammasome contributes to sarcopenia in mice. Male C57BL/6J wild-type (WT) and NLRP3−/− mice were aged to 10 (adult) and 24 mo (old). NLRP3−/− mice were protected from decreased muscle mass (relative to body mass) and decreased size of type IIB and IIA myofibers, which occurred between 10 and 24 mo of age in WT mice. Old NLRP3−/− mice also had increased relative muscle strength and endurance and were protected from age-related increases in the number of myopathic fibers. We found no evidence of age-related or NLRP3-dependent changes in markers of systemic inflammation. Increased caspase-1 activity was associated with GAPDH proteolysis and reduced GAPDH enzymatic activity in skeletal muscles from old WT mice. Aging did not alter caspase-1 activity, GAPDH proteolysis, or GAPDH activity in skeletal muscles of NLRP3−/− mice. Our results show that the NLRP3 inflammasome participates in age-related loss of muscle glycolytic potential. Deletion of NLRP3 mitigates both the decline in glycolytic myofiber size and the reduced activity of glycolytic enzymes in muscle during aging. We propose that the etiology of sarcopenia involves direct communication between immune responses and metabolic flux in skeletal muscle.
Keywords: NOD-like receptor family pyrin domain containing 3, muscle, aging, inflammation, caspase, glycolysis
impaired muscle contractile function is one of the most serious consequences of progressive muscle mass loss during sarcopenia. The age-related decline in muscle contractile parameters includes reduced force production and a slowing of contraction and relaxation (24, 29). Reduced skeletal muscle force production is correlated with lower muscle mass and is thought to be derived from a reduction in individual myofiber area as well as myofiber loss during aging (28). The age-related slowing of muscle/myofiber contractile parameters can occur before overt loss of muscle mass or force production (21, 24). Intrinsic slowing of myofiber contractile parameters and consequent slowing of movement during aging is hypothesized to lead to increased risk of falls in the elderly (40). It is possible that age-related mechanisms related to slowing of contraction set the stage for reduced muscle mass. There is a dearth of therapeutic interventions that are safe and effective in preventing these aspects of sarcopenia (19). Surprisingly, even exercise cannot avert aspects of sarcopenia. For example, life-long exercise in elite athletes did not sufficiently attenuate the age-related loss of muscle mass or slowing of contractile parameters in muscle fibers (8, 25).
Candidate strategies to combat sarcopenia have been proposed based on targeting the hormonal regulation of muscle growth or retasking of neurodegenerative drugs (19). However, a better understanding of the stimuli and cellular responses that promote intrinsic changes to muscle during aging may prompt new therapeutic approaches for sarcopenia. Local changes in muscle cell bioenergetics, mitochondrial metabolism, and oxidative damage have all been implicated in the development of sarcopenia (13). Long-lived, differentiated muscle fibers are susceptible to oxidative damage (22), which is a potential trigger for metabolic dysfunction in aged myofibers. Metabolic dysfunction in muscle may limit substrate availability for contractile performance, but glycolytic enzymes can also alter protein synthesis (14). Age-related changes in muscle metabolism can come from external cues. For example, age-related loss of motor unit innervation can promote mitochondrial dysfunction (34). Growth factors such as insulin-like growth factor (IGF-I) can increase oxidative metabolism in muscle (6, 30). In addition to external neural or hormonal cues, we were interested in determining whether immune pathways participate in aspects of sarcopenia. A state of chronic low-grade inflammation often prevalent with advancing age, termed “inflammaging,” may contribute to the development of sarcopenia.
Inflammaging has garnered attention because of the potential for low-level, chronic inflammation to alter the risk of morbidity and mortality during aging (9). There are several ways through which chronic inflammation could contribute to sarcopenia. Inflammatory mediators such as pro-nflammatory cytokines can engage muscle cell-resident receptors and promote wasting and atrophy of muscle tissue under various disease conditions (42). For example, tumor necrosis factor (TNF) promoted myocyte apoptosis via caspase-8 and increased muscle-specific ubiquitin ligase-mediated protein degradation (42). Increased proinflammatory tone can also disrupt hormonal regulation of muscle growth and maintenance of muscle mass. Inflammatory mediators can disrupt postreceptor signaling of insulin and IGF-I cascades that promote protein synthesis and myogenesis (41). Aged muscle has lower myogenic markers associated with protracted inflammatory mediators within regenerating skeletal muscle (37). Muscle catabolic responses can also be elicited by proinflammatory factors such as IL-1β (17), which is regulated by the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome. In fact, deletion of NLRP3 protects mice from multiple aspects of age-related dysfunction, including dysglycemia, bone loss, and thymic involution (39). Compared with wild-type (WT) mice, 18-mo-old NLRP3−/− mice have improved balance and coordination and 24-mo-old NLRP3−/− mice have improved exercise capacity (39). It is important to understand muscle-intrinsic changes vs. circulating factors since elevated serum levels of inflammatory mediators are associated with reduced muscle strength, but many of these circulating factors may not be altered by age in humans (1).
We aimed to test the role of the NLRP3 inflammasome in the functional, structural, and biochemical features of skeletal muscle in old mice. This inflammasome is an assembly of (at least) NLRP3, caspase-1, and apoptosis-associated speck-like protein Ccntaining CARD (ASC). The NLRP3 inflammasome responds to diverse stimuli such as reactive oxygen species (ROS), potassium efflux, lysosomal rupture, metabolic stress, and even therapeutic drugs such as statins (11, 12, 18, 32, 43). This inflammasome can propagate cellular metabolic dysfunction into immune responses (32). Therefore, NLRP3 is positioned as a node for detecting age-related metabolic dysfunction relevant to sarcopenia. NLRP3-mediated processing of caspase-1 can cleave and activate the proinflammatory cytokines IL-1β and IL-18. It is possible that old NLRP3−/− mice would have less circulating IL-1β (and IL-18) and attenuation of inflammation-related sarcopenia. It is not clear whethert the NLRP3 inflammasome regulation of circulating inflammation or local muscle inflammation is relevant to sarcopenia. Furthermore, caspase-1 has other target proteins, including rate-limiting enzymes of glycolysis such as GAPDH (33).
We found that aging increased NLRP3-dependent caspase-1 activity within mouse skeletal muscle. We also found that NLRP3 was required for proteolysis and inactivation of GAPDH in skeletal muscle and decreased glycolytic myofiber size during aging. We found no evidence that systemic inflammation was associated with the NLRP3-dependent decrease in glycolytic potential and myofiber size during aging.
MATERIALS AND METHODS
Animal protocols.
All procedures were approved by the McMaster University Animal Ethics Review Board (McMaster University, Hamilton, ON) and adhered to the guidelines from the Canadian Council on Animal Care. C57Bl/6J (WT) mice were originally purchased from JAX (Bar Harbor, ME) and bred at McMaster University. NLRP3−/− mice were bred at McMaster University, as described previously (11). Breeding colonies of NLRP3−/− mice were kindly provided by Prof. Nicolas Fasel and the Institute of Arthritis Research (Université de Lausanne, Lausanne, Switzerland) and originally supplied by Dr. Dana Philpott (University of Toronto, Toronto, ON). Male WT and NLRP3−/− mice were aged to 10 and 24 mo with ad libitum access to standard chow and water. The numbers of mice in each group were as follows: n = 11 for 10-mo-old WT mice, n = 10 for 24-mo-old WT mice, n = 12 for 10-mo-old NLRP3−/− mice, and n = 9 for 24-mo-old NLRP3−/− mice. Relative muscle strength and endurance were assessed using a wire hang test, as previously done (10). Latency to fall was analyzed from the average of the two best wire hang tests. Mice were euthanized, and serum was prepared from whole blood obtained by cardiac puncture. Hindlimb muscles were excised, trimmed of visible tendons, massed using an analytical balance, frozen, and stored at −80°C. The tibialis anterior (TA) muscle was mounted in tissue-freezing media (Tissue-Tek) and frozen in (thawing) isopentane cooled by liquid nitrogen, as described (30).
Muscle histology and immunofluorescence.
Transverse muscle sections (10 μm) were cryosectioned (Leica CM 1850) from the midbelly of the TA muscle. Muscle architecture was visualized after hematoxylin and eosin (H & E) staining of muscle sections. Muscle fiber cross-sectional area was calculated from ≥200 individual fibers per H & E-stained TA muscle section. The number of myopathic fibers was also quantified from H & E-stained TA sections as the sum of the number of centrally located nuclei per millimeter squared and the number of necrotic fibers per millimeter squared within the entire TA muscle cross-sectional area, as we have published previously (26). Muscle fiber types in TA cross-sections were determined by immunofluorescence using primary antibodies for myosin heavy chain I, IIa, and IIb (Developmental Studies Hybridoma Bank, University of Iowa), as described (2). At least 50 individual fibers from each fiber type per muscle section were used to determine cross-sectional area of specific fiber types. All images were captured with a Nikon 90i Eclipse microscope and analyzed using the Nikon Elements Advanced Research Analysis tool (Nikon, Melville, NY).
Enzymatic assays, immunoblotting, ELISAs, and quantitative PCR.
TA muscles were homogenized with ceramic beads using a Precellys 24 homogenizer at 5 m/s for 60 s in buffer containing 50 mM KH2PO4, 5 mM EDTA, 0.5 mM DTT, and 1.15% KCl with cOmplete protease inhibitor tablets (Roche). Protein concentration was determined using the bicinchoninic acid (BCA) method in the supernatant after homogenates were centrifuged for 10 min at 10,000 g at 4°C. Caspase-1 activity was determined by cleavage of a fluorescent substrate, YVAD-AFC, according to the manufacturer’s instructions (K110; Biovision) using 75 μg of protein lysate and 400-nm excitation and 505-nm emission filters. GAPDH activity was assessed according to the manufacturer’s instructions (K680; Biovision) using 5 μg of protein lysate and a 450-nm emission filter.
For immunoblotting, TA muscle was homogenized using ceramic bead-beating (as above) in buffer containing 50 mM HEPES, 150 mM NaCl, 100 mM NaF, 10 mM NaP2O7, 5 mM EDTA 2H2O, 250 mM sucrose, 20% Triton-X, and 200 mM Na3VO4 2H2O, with cOmplete protease inhibitor tablets (Roche). Protein concentration (BCA method) was determined in the supernatant after homogenates were centrifuged for 15 min at 14,000 g at 4°C and SDS-PAGE/immunoblotting was done using 20 μg of lysate, as described previously (31). The primary antibodies used for immunoblotting were total OxPhos (no. ab110413, 1:10,000; Abcam) and GAPDH (no. 5174, 1:5,000; Cell Signaling Technology). Protein loading for each sample was confirmed by Ponceau staining of polyvinylidene difluoride membranes.
For detection of cytokines/chemokines, a multiplex ELISA kit (no. M60-009RDPD; Bio-Rad) was used according to the manufacturer’s instructions. All 23 targets were detected in a single well for each sample tested. Samples were read on a 96-well plate using a BioRad Bioplex 200 system with Luminex technology. Transcripts were detected using TaqMa- based quantitative PCR in TA muscle after Trizol extraction and cDNA synthesis, as described (7).
Data analysis.
Data are expressed as means ± SE. Unpaired, two-tailed Student’s t-test was used to compare two variables. Comparison of more than two variables in binned cross-sectional area (CSA) data was done by two-way ANOVA and Sidak’s multiple-comparison test when significant interactions were initially detected (Prism 6–7; GraphPad Software).
RESULTS
Deletion of NLRP3 attenuates the progression of sarcopenia in mice.
We first determined whether whole body deletion of NLRP3 altered the progression of sarcopenia by measuring aspects of skeletal muscle structure and function in 10- and 24-mo-old WT and NLRP3−/− mice. NLRP3 transcript was expressed in TA muscles (and gonadal adipose tissue) to ∼30% of the level in the spleen in WT mice but absent in tissues of NLRP3−/− mice (data not shown). Body, muscle, and heart mass of all mice are shown in Fig. 1. Gonadal adipose tissue mass was significantly lower in 24-mo-old NLRP3−/− mice compared with 24-mo-old WT mice (data not shown). We found that the mass of TA, extensor digitorum longus (EDL), and soleus muscles (relative to body mass) decreased by ∼10–17% between 10 and 24 mo of age in WT mice but not in NLRP3−/− mice (Fig. 2A). The changes in the mass of TA, EDL, and soleus muscles between 10 and 24 mo were significantly different between WT and NLRP3−/− mice relative to body mass, whereas the changes in the mass of EDL and soleus muscles were significantly different between WT and NLRP3−/− mice relative to heart mass (P < 0.05; Fig. 2A). Subsequent analysis focused on the TA muscle. We found that the mean CSA of individual myofibers in the TA muscle decreased by ∼150 μm2 between 10 and 24 mo of age in WT mice, but myofiber CSA was similar in TA muscles from adult and old NLRP3−/− mice. Hence, the change in mean myofiber CSA between 10 and 24 mo was significantly different between WT and NLRP3−/− mice (P < 0.05; Fig. 2, B and C). We next assessed changes in the CSA of different fiber types in the TA muscles of mice. We found that changes in mean CSA of type IIB and Ttpe IIA fibers between 10 and 24 mo were significantly different between WT and NLRP3−/− mice (P < 0.05; Fig. 2, D and E).
Fig. 1.

Body and tissue masses for wild-type (WT) and NOD-like receptor family pyrin domain containing 3 (NLPR3−/−) mice at 10 and 24 mo of age. A–H: body mass and tissues masses of 10- and 24-mo-old WT mice. I–P: body mass and tissue masses of 10- and 24-mo-old NLRP3−/− mice. All data are means ± SE, and individual data points represent separate mice; n = 10–11 WT mice; n = 9–12 NLRP3−/− mice. Statistical analyses were performed using Student’s t-test. *P < 0.05.
Fig. 2.

Deletion of NLRP3 attenuates muscle loss, reduces myopathy, and improves muscle function in mice. WT and NLRP3−/− mice were aged to 10 (adult) and 24 mo (old). A: %change in mass of tibialis anterior (TA), extensor digitorum longus (EDL), and soleus muscles between adult and old WT and NLRP3−/− mice were calculated relative to body mass and heart mass. B: representative micrographs of hematoxylin and eosin-stained transverse sections from TA muscle in adult and old WT and NLRP3−/− mice. Scale bar, 250 μm. Inset images of micrographs from 24-mo-old mice shown on the right. C: quantification of changes in mean cross-sectional area (CSA) of TA muscles between 10- and 24-mo-old WT and NLRP3−/− mice. D: representative immunofluorescence micrographs of TA muscles labeled for type IIB (red), type IIX (unstained), and type IIA (green) myofibers. Scale bar, 200 μm. Inset images of micrographs from 24-mo-old mice shown on the right. E: quantification of the mean CSA changes between 10- and 24-mo-old WT and NLRP3−/− mice within each fiber type. F: quantification of myopathic fibers in adult and old WT mice. G: quantification of myopathic fibers in adult and old NLRP3−/−mice. H: quantification of relative muscle strength and endurance using latency to fall during a wire hang test. All data are means ± SE, and individual data points represent separate mice; n = 10–11 WT mice; n = 9–12 NLRP3−/− mice. Statistical analyses were performed using Student’s t-test. *P < 0.05.
Binning the fiber-type CSA data revealed a leftward shift toward smaller type IIB and type IIA fibers in old WT mice (Fig. 3A–C). Specifically, we found an increased abundance of type IIB myofibers that were <1,900 to 2,500 μm2 in TA muscles from 24-mo-old WT mice compared with 10-mo-old WT mice (P < 0.05; Fig. 3A). This increase in smaller type IIB myofibers came at the expense of type IIB myofibers that had a CSA of <3,400 to 4,000 μm2 in WT mice (P < 0.05; Fig. 3A). Similarly, there was increased abundance of type IIA myofibers that were <700 to 800 μm2 (and lower abundance of larger <1,000 to 1,100 μm2 type IIA myofibers) in TA muscles from 24-mo-old WT mice compared with 10-mo-old WT mice (P < 0.05, Fig. 3C). The age-related changes in type IIB and IIA myofiber CSA did not occur in TA muscles from adult and old NLRP3−/− mice (Fig. 3, A–C).
Fig. 3.
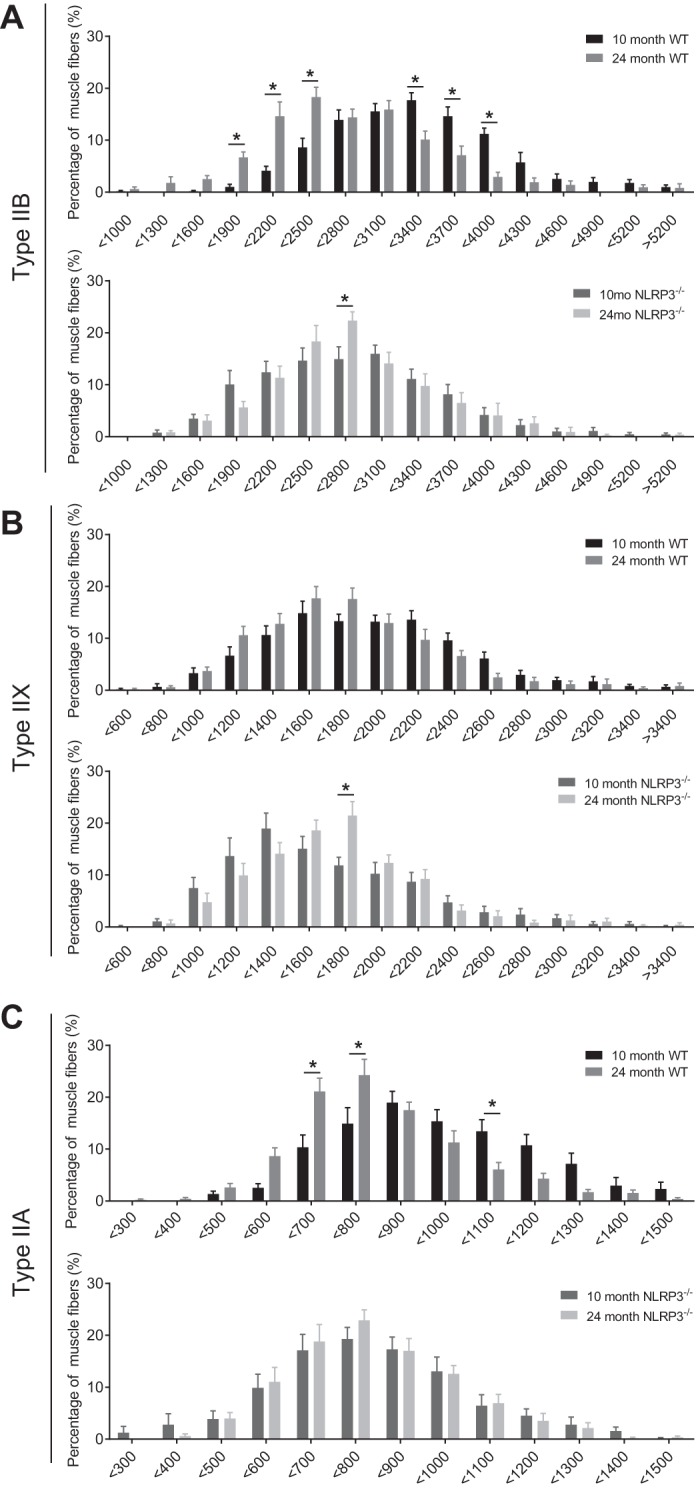
Cross-sectional area of specific fiber types in TA muscle from adult and old WT and NLRP3−/− mice. Frequency histograms of the distribution of fiber cross-sectional areas (μm2) in 10- and 24-mo-old WT and NLRP3−/− mice for type IIB (A), type IIX (B), and type IIA fibers (C). All data are means ± SE; n = 10–11 WT mice; n = 9–12 NLRP3−/− mice. Statistical analyses were performed using 2-way ANOVA. *P < 0.05.
Deletion of NLRP3 reduces myopathy and improves muscle function in old mice.
We next determined whether whole body deletion of NLRP3 altered low-level myopathy and the functional decline that occurs in old mice. We found that TA muscles of 24-mo-old WT mice had higher numbers of myopathic myofibers compared with 10-mo-old WT mice (P < 0.05; Fig. 2F). However, the prevalence of myopathic myofibers was similar in TA muscles from 10- and 24-mo-old NLRP3−/− mice (Fig. 2G). We also found that 24-mo-old NLRP3−/− mice have a longer latency to fall during a wire hang test compared with 24-mo-old WT mice (Fig. 2H). This test is a gross measurement of whole body muscle strength, endurance, and coordination that is dependent on supporting body mass. This direct comparison of mouse genotypes may be confounded by the fact that 24-mo-old NLRP3−/− mice have a lower body mass compared with 10-mo-old NLRP3−/− mice, but body mass is similar in 24- and 10-mo-old WT mice (Fig. 1).
Aging increases NLRP3-dependent caspase-1 activity in mouse muscle.
We found that enzymatic activity of caspase-1 was increased in TA muscle homogenates from 24-mo-old WT mice compared with 10-mo-old WT mice (P < 0.05; Fig. 4A). However, caspase-1 activity was similar in TA muscle homogenates from 10- and 24-mo-old NLRP3−/− mice (Fig. 4B). We next investigated key substrates of caspase-1. We found that IL-1β was below the level of detection in TA muscle (data not shown). Enzymatic activity of GAPDH was lower in TA muscle homogenates from 24-mo-old WT mice compared with 10-mo-old WT mice (P < 0.05; Fig. 4C). GAPDH activity was similar in TA muscle homogenates from 10- and 24-mo-old NLRP3−/− mice (Fig. 4D).
Fig. 4.

Aging increases caspase-1 activity and decreases GAPDH activity in mouse skeletal muscle via NLRP3. A: caspase-1 enzymatic activity in TA muscles from 10- and 24-mo-old WT mice. B: caspase-1 enzymatic activity in TA muscles from 10- and 24-mo-old NLRP3−/− mice. C: GAPDH enzymatic activity in TA muscles from 10- and 24-mo-old WT mice. D: GAPDH enzymatic activity in TA muscles from 10- and 24-mo-old NLRP3−/− mice. All data are means ± SE, and individual data points represent separate mice; n = 10–11 WT mice; n = 8–10 NLRP3−/− mice for caspase-1; n = 9–12 NLRP3−/− mice for GAPDH activity. Statistical analyses were performed using Student’s t-test. *P < 0.05.
Aging increases NLRP3-dependent proteolysis of GAPDH in mouse muscle.
Based on the age-related decrease in muscle GAPDH enzymatic activity, we next investigated GAPDH protein levels. We found no difference in the protein levels of full-length (∼37 kDa) GAPDH in TA muscle homogenates from 10- or 24-mo-old WT or NLRP3−/− mice (Fig. 5, A–C). However, caspase-1 has been shown to cleave GAPDH and generate a ∼25-kDa fragment (33). Here, we found higher levels of GAPDH that was immunoreactive at a size of ∼25 kDa in TA muscle from 24-mo-old WT mice compared with 10-mo-old WT mice (P < 0.05; Fig. 5, D and E). This proteolysis of GAPDH was NLRP3 dependent since there was no change in levels of the ∼25-kDa form of GAPDH in TA muscle from 24- and 10-mo-old NLRP3−/− mice (Fig. 5, D–F).
Fig. 5.

Aging increases NLRP3-dependent proteolysis of GAPDH in mouse TA muscle. A: representative immunoblot for GAPDH using TA muscle lysates prepared from 10- and 24-mo-old WT and NLRP3−/− mice and from TA muscles that were an internal loading control (CON). B and C: quantification of immunoreactive bands at ∼37 kDa predicted to be full-length GAPDH in TA muscle lysates prepared from 10- and 24-mo-old WT and NLRP3−/− mice. D: representative immunoblot for GAPDH after longer exposure times (i.e., higher exposure) using TA muscle lysates prepared from 10- and 24-mo-old WT and NLRP3−/− mice and from TA muscles that were an internal loading CON. E and F: quantification of immunoreactive bands at ∼25 kDa predicted to be a fragment of GAPDH in TA muscle lysates prepared from 10- and 24-mo-old WT and NLRP3−/− mice. G: Ponceau stain of the representative immunoblot showing equal loading of protein between samples; n = 10–11 WT mice; n = 9–10 NLRP3−/− mice. All data are means ± SE, and individual data points represent separate mice. Statistical analyses were performed using Student’s t-test. *P < 0.05. KO, knockout.
Aging did not alter muscle mitochondrial capacity or circulating inflammatory mediators.
We next measured age-related changes in muscle mitochondrial capacity and markers of muscle and systemic inflammation. We found that neither aging nor NLRP3 deletion altered the relative abundance of mitochondrial complexes I–V in TA muscles (Fig. 6, A–C). We also found that neither aging nor NLRP3 deletion altered the relative abundance of transcripts of inflammatory mediators or immune cell markers in the TA muscles (Fig. 6D). Finally, we found no statistical differences in a panel of 23 cytokines/chemokines in the serum of WT or NLRP3−/− mice that were 10 and 24 mo old (Fig. 7, A and B).
Fig. 6.

Aging does not alter muscle mitochondrial capacity or markers of inflammation in WT or NLRP3−/− mice. A: representative immunoblot for mitochondrial complexes I–V using TA muscle lysates prepared from 10- and 24-mo-old WT and NLRP3−/− mice. B: quantification of mitochondrial complexes I–V in TA muscles from 10- and 24-mo-old WT and NLRP3−/− mice. C: Ponceau stain of the representative immunoblot showing equal loading of protein between samples. D: TaqMan-based quantitative PCR detection of transcript levels of selected inflammatory targets in TA muscles of 10- and 24-mo-old WT and NLRP3−/− mice. All data are means ± SE; n = 8–9 WT mice, n = 8–9 NLRP3−/− mice for mitochondrial assessments, n = 8–9 WT mice, and n = 6–11 NLRP3−/− mice for detection of transcripts based on certain transcripts reporting lower than the limit of detection by quantitative PCR in a subset of mice. Statistical analyses were performed using Student’s t-test to compare 10- and 24-mo-old mice within each genotype for each measure.
Fig. 7.

Systemic inflammation in adult and old WT and NLRP3−/− mice. A panel of 23 cytokines/chemokines was analyzed in the serum of WT (A) and NLRP3−/− (B) 10- and 24-mo-old mice. All data are means ± SE; n = 6–9 WT mice and 5–9 NLRP3−/− mice for reported values upon detection of analytes based on certain analytes being lower than the limit of detection by ELISA in a subset of mice. Statistical analyses were performed using Student’s t-test within each analyte.
DISCUSSION
The components of the immune system that link inflammaging to sarcopenia are not well defined. We found increased activity of an inflammatory caspase (i.e., caspase-1) within skeletal muscles of old mice. We then showed that the NLRP3/caspase-1 inflammasome contributes to the age-related decline in muscle mass and function (relative to body mass) in mice. Surprisingly, we found no evidence for NLRP3-mediated alterations in systemic inflammatory markers during aging. Rather, we found that deletion of NLRP3 attenuated muscle-intrinsic responses related to glycolytic metabolism. NLRP3 was required for the age-related decrease in the size of glycolytic myofibers and proteolysis/inactivation of GAPDH, a rate-limiting enzyme in glycolysis.
A two-step process involving priming and activation can regulate the NLRP3 inflammasome. In general, diverse stimuli that engage NF-κB signaling prime the inflammasome. We did not define the source or level of NLRP3 inflammasome priming (i.e., step 1) in our mouse model of aging. NF-κB levels have been reported to be higher in skeletal muscle of ∼70-yr-old vs. ∼28-yr-old men (5). An interesting future goal would be to characterize whether NLRP3 inflammasome priming in skeletal muscle is modifiable during aging. It would also be important to define age-related compartmentalized immune responses in different tissues (20) and know whether inflammasome priming is due to muscle-intrinsic, endogenous triggers of NF-κB activity or external sources such as components of the microbiota (3). We also did not define the source of NLRP3 activation (i.e., step 2) in skeletal muscles of aged mice. Increased caspase-1 activity in aged muscle represents an immune response, but we found no evidence of changes in cytokine-related muscle inflammation or changes in mitochondrial capacity in muscle. However, an important future goal is to determine whether age-related changes in skeletal muscle ROS, ion homeostasis, ATP, or other metabolic stress responses are triggers for NLRP3 inflammasome activation during aging.
Our results have identified NLRP3 as a contributor to age-related decline in myofiber size despite no detectable changes in circulating mediators of inflammation. Our work is consistent with the concept that inflammaging does not necessarily need to be an increase in circulating proinflammatory factors to contribute to sarcopenia. We used 10- and 24-mo-old mice to define aspects of sarcopenia between adulthood and old age. Previous reports have shown that many proinflammatory cytokines are increased in older mice (i.e., 18–22 mo of age) compared with mice that are a couple months old (i.e., 10–16 wk of age) (36). Hence, one factor that may explain our findings of no change in circulating inflammatory mediators is the age of “adult” mice. Our results showing no apparent correlation of circulating inflammation with the onset of sarcopenia are consistent with results in aged humans. Higher serum levels of proinflammatory cytokines are associated with reduced muscle strength and mass in humans, but aging does not necessarily increase human circulating cytokines that can promote muscle wasting (1). Our results also show that NLRP3 inflammasome-mediated inflammaging in the local muscle environment can contribute to aspects of sarcopenia through caspase-1 regulation of cytokine-independent pathways. We have found that NLRP3-dependent reductions in skeletal muscle glycolytic potential may be a link between immunity and sarcopenia through caspase-1 proteolysis of GAPDH.
Intriguingly, attenuation of myofiber atrophy during aging in NLRP3−/− mice may be directly linked to preservation of glycolytic potential in old NLRP3−/− mice. Our results show that GAPDH activity is lower in muscles of old WT mice compared with middle-aged WT mice. GAPDH can be a rate-limiting step in glycolysis, which can affect muscle protein synthesis because lower glycolytic flux increases the interaction between GAPDH and Rheb (16). The consequence of increased GAPDH association with Rheb is suppression of mammalian target of rapamycin complex 1 (mTORC1)-mediated protein synthesis, which has been expertly reviewed (14). Here, we have shown that NLRP3 deletion prevents increased caspase-1 activity in muscle and proteolysis of GAPDH in old muscle. The interactions of full-length and cleaved forms of GAPDH with Rheb/mTORC1 and consequent changes in protein synthesis in aged muscle warrant investigation. In addition, glycolytic enzymes such as GAPDH and hexokinase are associated with the mitochondria. This is also important to consider since GAPDH can promote apoptosis by interacting with mitochondrial voltage-dependent anion channel (35), whereas dissociation of hexokinase from the mitochondria can activate the NLRP3 inflammasome (38). The role of GAPDH in apoptosis warrants investigation given the NLRP3-dependent protection from age-related increase in myopathic fibers we observed in mouse TA muscles.
We used whole body NLRP3−/− mice that were back-crossed >10 generations but compared with nonlittermate WT C57 Bl/6J mice. Mouse substrain differences can alter metabolic responses (23), but the relevance to sarcopenia is not yet clear. We cannot discount mouse substrain differences in our measurements, but the focus in our experiments was to compare each mouse strain over time. We have not yet determined the cell type(s) involved in NLRP3-mediated contributions to sarcopenia. The role of NLRP3 in metabolic programming of muscle precursor cells (15, 27) and immune cells during sarcopenia should be determined in littermate controls and cell-specific deletion of NLRP3 in muscle progenitors, differentiated muscle fibers, and myeloid cells. Also, from a therapeutic standpoint, NLRP3 inflammasome inhibitors (such as MCC950) should be tested in models of sarcopenia (4). Furthermore, we have not identified the cellular source of increased caspase-1 activity in aged muscle. Caspase-1 is a secreted protein, and it is possible that muscle-resident immune cells (such as macrophages) are a source of active caspase-1. Immune cell-derived active caspase-1 should be considered in a model of paracrine signaling to muscle fibers, since transcript levels of caspase-1 in whole TA muscle are much lower (i.e., 10–15%; data not shown) compared with the spleen. We found that NLRP3 deletion influenced age-related muscle fiber intrinsic changes in glycolytic potential, where is it probable that the majority of GAPDH is derived from muscle fibers. However, determining whether caspase-1 acts in an autocrine or paracrine manner to alter muscle GAPDH requires direct testing.
In summary, we have found that NLRP3 contributes to the age-related decline in myofiber size and lower glycolytic potential within skeletal muscles of mice. NLRP3 was required for the age-related increase in caspase-1 activity, which corresponded with proteolysis and reduced activity of GAPDH in skeletal muscle. We propose that the NLRP3 inflammasome propagates age-related danger/damage-associated molecular patterns into metabolic flux alterations that can regulate myofiber size and contractility.
GRANTS
This work was supported by an operating grant to J. D. Schertzer and T. J. Hawke from the Natural Sciences and Engineering Research Council (NSERC) though a discovery grant and an early career researcher supplement (J. D. Schertzer). J. D. Schertzer holds Canadian Diabetes Association Scholar (SC-5-12-3891-JS) and Canadidan Institutes of Health Research (CIHR) New Investigator awards (MSH-136665). CIHR supported Y. E. Li with a Canada Graduate Scholarship and T. C. Lau and D. M. D’Souza with a Frederick Banting and Charles Best Canada Graduate Scholarships. K. P. Foley was supported by an NSERC fellowship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.J.M., K.P.F., D.M.D., Y.E.L., T.C.L., and J.D.S. performed experiments; M.J.M., K.P.F., D.M.D., Y.E.L., T.C.L., and J.D.S. analyzed data; M.J.M., K.P.F., T.J.H., and J.D.S. interpreted results of experiments; M.J.M., K.P.F., and J.D.S. prepared figures; M.J.M., K.P.F., D.M.D., Y.E.L., T.C.L., T.J.H., and J.D.S. approved final version of manuscript; K.P.F. and J.D.S. conceived and designed research; K.P.F. and J.D.S. drafted manuscript; K.P.F., D.M.D., Y.E.L., T.C.L., T.J.H., and J.D.S. edited and revised manuscript.
REFERENCES
- 1.Bano G, Trevisan C, Carraro S, Solmi M, Luchini C, Stubbs B, Manzato E, Sergi G, Veronese N. Inflammation and sarcopenia: A systematic review and meta-analysis. Maturitas 96: 10–15, 2017. doi: 10.1016/j.maturitas.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Bloemberg D, Quadrilatero J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One 7: e35273, 2012. doi: 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chi W, Dao D, Lau TC, Henriksbo BD, Cavallari JF, Foley KP, Schertzer JD. Bacterial peptidoglycan stimulates adipocyte lipolysis via NOD1. PLoS One 9: e97675, 2014. doi: 10.1371/journal.pone.0097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KHG, Masters SL, Schroder K, Cooper MA, O’Neill LAJ. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255, 2015. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J 19: 422–424, 2005. doi: 10.1096/fj.04-2640fje. [DOI] [PubMed] [Google Scholar]
- 6.Das S, Morvan F, Jourde B, Meier V, Kahle P, Brebbia P, Toussaint G, Glass DJ, Fornaro M. ATP citrate lyase improves mitochondrial function in skeletal muscle. Cell Metab 21: 868–876, 2015. doi: 10.1016/j.cmet.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Denou E, Lolmède K, Garidou L, Pomie C, Chabo C, Lau TC, Fullerton MD, Nigro G, Zakaroff-Girard A, Luche E, Garret C, Serino M, Amar J, Courtney M, Cavallari JF, Henriksbo BD, Barra NG, Foley KP, McPhee JB, Duggan BM, O’Neill HM, Lee AJ, Sansonetti P, Ashkar AA, Khan WI, Surette MG, Bouloumié A, Steinberg GR, Burcelin R, Schertzer JD. Defective NOD2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol Med 7: 259–274, 2015. doi: 10.15252/emmm.201404169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faulkner JA, Davis CS, Mendias CL, Brooks SV. The aging of elite male athletes: age-related changes in performance and skeletal muscle structure and function. Clin J Sport Med 18: 501–507, 2008. doi: 10.1097/JSM.0b013e3181845f1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69, Suppl 1: S4–S9, 2014. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 10.Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA, Lynch GS. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 484: 394–398, 2012. doi: 10.1038/nature10980. [DOI] [PubMed] [Google Scholar]
- 11.Henriksbo BD, Lau TC, Cavallari JF, Denou E, Chi W, Lally JS, Crane JD, Duggan BM, Foley KP, Fullerton MD, Tarnopolsky MA, Steinberg GR, Schertzer JD. Fluvastatin causes NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes 63: 3742–3747, 2014. doi: 10.2337/db13-1398. [DOI] [PubMed] [Google Scholar]
- 12.Henriksbo BD, Schertzer JD. Is immunity a mechanism contributing to statin-induced diabetes? Adipocyte 4: 232–238, 2015. doi: 10.1080/21623945.2015.1024394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hepple RT. Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci 6: 211, 2014. doi: 10.3389/fnagi.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koopman R, Ly CH, Ryall JG. A metabolic link to skeletal muscle wasting and regeneration. Front Physiol 5: 32, 2014. doi: 10.3389/fphys.2014.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laker RC, Ryall JG. DNA methylation in skeletal muscle stem cell specification, proliferation, and differentiation. Stem Cells Int 2016: 5725927, 2016. doi: 10.1155/2016/5725927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MN, Ha SH, Kim J, Koh A, Lee CS, Kim JH, Jeon H, Kim DH, Suh PG, Ryu SH. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol Cell Biol 29: 3991–4001, 2009. doi: 10.1128/MCB.00165-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W, Moylan JS, Chambers MA, Smith J, Reid MB. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 297: C706–C714, 2009. doi: 10.1152/ajpcell.00626.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao YH, Lin YC, Tsao ST, Lin YC, Yang AJ, Huang CT, Huang KC, Lin WW. HMG-CoA reductase inhibitors activate caspase-1 in human monocytes depending on ATP release and P2X7 activation. J Leukoc Biol 93: 289–299, 2013. doi: 10.1189/jlb.0812409. [DOI] [PubMed] [Google Scholar]
- 19.Lynch GS. Update on emerging drugs for sarcopenia - age-related muscle wasting. Expert Opin Emerg Drugs 13: 655–673, 2008. doi: 10.1517/14728210802544476. [DOI] [PubMed] [Google Scholar]
- 20.McPhee JB, Schertzer JD. Immunometabolism of obesity and diabetes: microbiota link compartmentalized immunity in the gut to metabolic tissue inflammation. Clin Sci (Lond) 129: 1083–1096, 2015. doi: 10.1042/CS20150431. [DOI] [PubMed] [Google Scholar]
- 21.Narayanan N, Jones DL, Xu A, Yu JC. Effects of aging on sarcoplasmic reticulum function and contraction duration in skeletal muscles of the rat. Am J Physiol Cell Physiol 271: C1032–C1040, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Narciso L, Fortini P, Pajalunga D, Franchitto A, Liu P, Degan P, Frechet M, Demple B, Crescenzi M, Dogliotti E. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc Natl Acad Sci USA 104: 17010–17015, 2007. doi: 10.1073/pnas.0701743104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicholson A, Reifsnyder PC, Malcolm RD, Lucas CA, MacGregor GR, Zhang W, Leiter EH. Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity (Silver Spring) 18: 1902–1905, 2010. doi: 10.1038/oby.2009.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plant DR, Lynch GS. Excitation-contraction coupling and sarcoplasmic reticulum function in mechanically skinned fibres from fast skeletal muscles of aged mice. J Physiol 543: 169–176, 2002. doi: 10.1113/jphysiol.2002.022418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Power GA, Minozzo FC, Spendiff S, Filion ME, Konokhova Y, Purves-Smith MF, Pion C, Aubertin-Leheudre M, Morais JA, Herzog W, Hepple RT, Taivassalo T, Rassier DE. Reduction in single muscle fiber rate of force development with aging is not attenuated in world class older masters athletes. Am J Physiol Cell Physiol 310: C318–C327, 2016. doi: 10.1152/ajpcell.00289.2015. [DOI] [PubMed] [Google Scholar]
- 26.Rebalka IA, Raleigh MJ, Snook LA, Rebalka AN, MacPherson REK, Wright DC, Schertzer JD, Hawke TJ. Statin therapy alters lipid storage in diabetic skeletal muscle. Front Endocrinol (Lausanne) 7: 95, 2016. doi: 10.3389/fendo.2016.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryall JG, Cliff T, Dalton S, Sartorelli V. Metabolic reprogramming of stem cell epigenetics. Cell Stem Cell 17: 651–662, 2015. doi: 10.1016/j.stem.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryall JG, Schertzer JD, Lynch GS. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 9: 213–228, 2008. doi: 10.1007/s10522-008-9131-0. [DOI] [PubMed] [Google Scholar]
- 29.Schertzer JD, Plant DR, Ryall JG, Beitzel F, Stupka N, Lynch GS. β2-agonist administration increases sarcoplasmic reticulum Ca2+-ATPase activity in aged rat skeletal muscle. Am J Physiol Endocrinol Metab 288: E526–E533, 2005. doi: 10.1152/ajpendo.00399.2004. [DOI] [PubMed] [Google Scholar]
- 30.Schertzer JD, Ryall JG, Lynch GS. Systemic administration of IGF-I enhances oxidative status and reduces contraction-induced injury in skeletal muscles of mdx dystrophic mice. Am J Physiol Endocrinol Metab 291: E499–E505, 2006. doi: 10.1152/ajpendo.00101.2006. [DOI] [PubMed] [Google Scholar]
- 31.Schertzer JD, Tamrakar AK, Magalhães JG, Pereira S, Bilan PJ, Fullerton MD, Liu Z, Steinberg GR, Giacca A, Philpott DJ, Klip A. NOD1 activators link innate immunity to insulin resistance. Diabetes 60: 2206–2215, 2011. doi: 10.2337/db11-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science 327: 296–300, 2010. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 33.Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 282: 36321–36329, 2007. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- 34.Spendiff S, Vuda M, Gouspillou G, Aare S, Perez A, Morais JA, Jagoe RT, Filion M-E, Glicksman R, Kapchinsky S, MacMillan NJ, Pion CH, Aubertin-Leheudre M, Hettwer S, Correa JA, Taivassalo T, Hepple RT. Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J Physiol 594: 7361–7379, 2016. doi: 10.1113/JP272487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, Zamzami N, Jan G, Kroemer G, Brenner C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 26: 2606–2620, 2007. doi: 10.1038/sj.onc.1210074. [DOI] [PubMed] [Google Scholar]
- 36.Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, Schertzer JD, Larché MJ, Davidson DJ, Verdú EF, Surette MG, Bowdish DME. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21: 455–466.e4, 2017. doi: 10.1016/j.chom.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Poel C, Gosselin LE, Schertzer JD, Ryall JG, Swiderski K, Wondemaghen M, Lynch GS. Ageing prolongs inflammatory marker expression in regenerating rat skeletal muscles after injury. J Inflamm (Lond) 8: 41, 2011. doi: 10.1186/1476-9255-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, Underhill DM. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166: 624–636, 2016. doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Münzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 18: 519–532, 2013. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu F, Hedström M, Cristea A, Dalén N, Larsson L. Effects of ageing and gender on contractile properties in human skeletal muscle and single fibres. Acta Physiol (Oxf) 190: 229–241, 2007. doi: 10.1111/j.1748-1716.2007.01699.x. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Q, Yang ST, Wang JJ, Zhou J, Xing SS, Shen CC, Wang XX, Yue YX, Song J, Chen M, Wei YY, Zhou QP, Dai T, Song YH. TNF alpha inhibits myogenic differentiation of C2C12 cells through NF-κB activation and impairment of IGF-1 signaling pathway. Biochem Biophys Res Commun 458: 790–795, 2015. doi: 10.1016/j.bbrc.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 42.Zhou J, Liu B, Liang C, Li Y, Song YH. Cytokine signaling in skeletal muscle wasting. Trends Endocrinol Metab 27: 335–347, 2016. doi: 10.1016/j.tem.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]