

Abstract
ANG II has many biological effects in renal physiology, particularly in Ca2+ handling in the regulation of fluid and solute reabsorption. It involves the systemic endocrine renin-angiotensin system (RAS), but tissue and intracrine ANG II are also known. We have shown that ANG II induces heterodimerization of its AT1 and AT2 receptors (AT1R and AT2R) to stimulate sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) activity. Thus, we investigated whether ANG II-AT1R/AT2R complex is formed and internalized, and also examined the intracellular localization of this complex to determine how its effect might be exerted on renal intracrine RAS. Living cell imaging of LLC-PK1 cells, quantification of extracellular ANG II, and use of the receptor antagonists, losartan and PD123319, showed that ANG II is internalized with AT1R/AT2R heterodimers as a complex in a microtubule-dependent and clathrin-independent manner, since colchicine—but not Pitstop2—blocked this process. This result was confirmed by an increase of β-arrestin phosphorylation after ANG II treatment, clathrin-mediated endocytosis being dependent on dephosphorylation of β-arrestin. Internalized ANG II colocalized with an endoplasmic reticulum (ER) marker and increased levels of AT1R, AT2R, and PKCα in ER-enriched membrane fractions. This novel evidence suggests the internalization of an ANG II-AT1/AT2 complex to target ER, where it might trigger intracellular Ca2+ responses.
Keywords: ANG II-AT1R/AT2R complex, LLC-PK1 luminal membranes, ANG II-AT1R/AT2R internalization, endoplasmic reticulum, microtubule-dependent clathrin-independent endocytosis
the renin-angiotensin system (RAS) is a key hormonal signaling system involved in the homeostasis of body salt and fluid, operating mainly at the kidney level, where the best-described active peptide, ANG II, is the main regulator of Na+ and water reabsorption, a function that depends on finely tuned Ca2+ transport in the cell (6, 15).
RAS has been classically involved in the vasoactive system, acting as an endocrine system and a local tissue paracrine system (20, 47, 66). In the endocrine system, each RAS component is synthesized in different tissues to produce the renal and extrarenal ANG II effects on arterial blood pressure. Briefly, the liver is the main source of angiotensinogen (AGT), the only known precursor in the synthesis of ANG II, which is the main biologically active peptide for RAS. To produce ANG II, kidney-derived renin cleaves AGT to produce ANG I, which is rapidly hydrolyzed by the endothelial angiotensin-converting enzyme (ACE) to ANG II. Several tissues, including the brain, cardiovascular, adipose, gonad, pancreas, placenta, adrenal gland, liver, and kidney tissues, synthesize all of the RAS components, characterizing the local tissue paracrine RAS (7, 18, 43, 44, 54, 57). The paracrine system seems to mediate the physiological and pathophysiological responses of the cell, which includes effects on proliferation, cell growth, and metabolism (reviewed in Ref. 47).
More recently, an intracrine or intracellular RAS (50) was revealed when ANG II was bound to nuclear receptors and triggered gene transcription in kidney proximal tubule cell nuclei (37) and cardiomyocytes (60). Intracrine RAS depends on an intracellular source of ANG II and synthesis of RAS components, which involves different classes of receptors in the nucleus (37) and mitochondria (1, 67).
Intracellular ANG II comes from two sources: 1) cells that synthesize all of the RAS components and 2) extracellular ANG II internalized with its receptors (66). Indeed, ANG II is known to bind to its AT1R, the complex being internalized. A cellular response is triggered, followed by receptor recycling to the plasma membrane or endosome (23). However, the ANG II receptor AT2R may be resistant to internalization and trafficking (27, 31).
ANG II receptors occur in organelles from different cell types, e.g., mitochondria and nuclei (2, 34). In the kidney, mitochondrial AT2R appears to be involved in the regulation of NO formation and O2 consumption (1). In the nucleus, AT1R stimulates ROS, and one possible functional consequence is the modulation of a redox-sensitive signaling pathway (48). Moreover, this class of receptors is involved in Ca2+ homeostasis and function in the nucleus (69). Although it is plausible that ANG II binding to organelle receptors may participate in Ca2+ homeostasis, this has not been properly established, and it is also unknown whether intracrine RAS acts in organelles other than the mitochondria and nuclei.
The role of intracellular RAS has been explored in different tissues; in the kidney, overexpression of intracellular ANG II in a transgenic mouse increases blood pressure and thrombotic microangiopathy (51). In cardiomyocytes, intracellular levels of ANG II are 4−5-fold higher in diabetic (streptozotocin-treated) rats (56) and diabetic hypertensive patients (22) than in nondiabetic patients and were associated with gene expression, oxidative damage, apoptosis, and necrosis.
Although there is no consensus that ANG II always promotes the dimerization of receptors, several reports have demonstrated that in both endocrine and paracrine RAS, ANG II binding to any of its receptors induces their homodimerization and/or heterodimerization, which, in turn, induces different cellular responses (3, 4, 9, 19). For instance, ANG II-induced receptor heterodimerization is required to modulate different ATPase activities involved in ion reabsorption (6, 14). We have previously shown that luminal ANG II induces the heterodimerization of AT1 and AT2 receptors to stimulate the sarco(endo)plasmic reticulum calcium ATPase (SERCA) activity, without an effect on plasma membrane Ca2+-ATPase (PMCA) (19).
However, the specific effect of AT1R/AT2R-induced ANG II on SERCA activity remains unclear in terms of its targets. Our working hypothesis is that the luminal ANG II-AT1R/AT2R complex has to have an intracellular target to regulate Ca2+ storage in renal cells. We show here that ANG II accessing the luminal side is internalized as a complex with AT1R/AT2R heterodimer, and targets the ER to stimulate SERCA activity.
METHODS
Materials.
LLC-PK1 cells and Hanks solution were purchased from American Type Culture Collection (CL-101) (Manassas, VA). Culture medium 199, FBS, trypsin solution (0.25% trypsin plus 0.04% EDTA-tetrasodium salt), ER Tracker Red (E34250) and ANG II-Alexa Fluor 488 (A13439) were obtained from Invitrogen Life Technologies (Carlsbad, CA). Fluorescent carboxyfluorescein-labeled ANG II (FAM-ANG II) (AS-60275) was obtained from Anaspec (Fremont, CA). Antibodies against p-β-arrestin (sc-16639-R, lot F0904), AT1R (sc-57036, lot B1210), AT2R (sc-9040, lot F2513), and PKCα (sc-208, lot E0412) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), and antibodies against Na+/K+-ATPase (A276, lot SLBN0461V), SERCA (S1439, lot SLBL0170V) and β-actin (A5441, lot 014M4759) were obtained from Sigma-Aldrich (St. Louis, MO). Nitrocellulose membranes, secondary antibodies, and ECL system were obtained from GE Healthcare (São Paulo, Brazil). Losartan was obtained from Merck Sharp & Dohme (Whitehouse Station, NJ), PD123319, protease inhibitor cocktail, and trypsin inhibitor (type II-S soybean) from Sigma-Aldrich (St. Louis, MO). Pitstop 2, recombinant AT1R protein (ab157870, lot 1010313–759) and recombinant AT2R protein (ab157871, lot FA261-6226) were purchased from Abcam Biochemicals (Cambridge, UK). All other reagents were of the highest purity available.
Cell culture.
Immortalized porcine proximal tubule epithelial cell line (LLC-PK1) was cultured in medium 199 with 3% FBS in an atmosphere of 5% CO2 in air at 37°C. Approximately 5 × 105 cells were seeded in 25 cm2 culture flasks and incubated until ~90% confluent (~3 days). Cells were washed in PBS at pH 7.5 (10 mM Na2HPO4 + 13.7 mM NaCl + 1.7 mM KH2PO4 + 2.7 mM KCl) and incubated with 1 ml trypsin/EDTA to detach the cells. The cells were centrifuged at 525 g for 2 min, and the pellets were resuspended in 5 ml medium 199 with 3% FBS and distributed into new flasks.
Living cell fluorescent microscopy.
Cultures of 104 cells were grown in 96-well plates in medium 199 with 3% FBS in an atmosphere of 5% CO2 in air at 37°C for 2 days. Cells were incubated with 0.1 μM FAM-ANG II in medium (200 μl) without FBS for 30 min at 37°C in a 5% CO2 atmosphere, as previously described, with modifications (36). Cells were treated with 1 μM losartan and/or PD123319 20 min before incubation with 0.1 μM FAM-ANG II for 30 min to analyze the involvement of AT1R/AT2R heterodimers in ANG II internalization. Cells were washed with PBS, and the cells were observed in fluorescence microscope (ImageXpress Micro; Molecular Devices, Sunnyvale, CA). The concentration of ANG II used in this experiment intended to mimic the tubular ANG II concentration (53).
To analyze ANG II subcellular location, LLC-PK1 cells were grown on coverslips in eight-well plates in medium with 3% FBS in an atmosphere of 5% CO2 at 37°C for 2 days. The cells were incubated with 0.3 μM ANG II-Alexa Fluor 488 for 1 h in 500 μl medium without serum before being washed with Hanks solution and incubated with 0.1 μM ER Tracker Red for 15 min still under culture conditions. Cells were washed in Hanks solution and examined by confocal fluorescence microscopy (TCS SP8, Leica Microsystems, Exton, PA). Images were scanned under identical conditions, and prepared for presentation with PhotoShop CS6 software.
Quantification of ANG II by HPLC.
Cultures of 5 × 105 cells were seeded in 25 cm2 culture flasks for 3 days before being incubated with 8 µM ANG II in 2 ml PBS for 2 h in a 5% CO2 atmosphere at 37°C or 4°C. Supernatants were collected and concentrated in speedvac (Thermo Savant, Holbrook, NY), and analyzed by HPLC (model LC10AS; Shimadzu, Kyoto, Japan), using a C-18 reverse-phase column (Rexcrom, 25 cm × 4.6 mm; Regis Technologies, Morton Grove, IL). To observe the receptors involved in ANG II internalization, cells were treated with 0.1 nM losartan or 0.1 µM PD123319 (or both) 15 min before adding 8 µM ANG II for 2 h at 37°C. To analyze the endocytic pathway, cells were incubated with 30 μM Pitstop 2 or 1 μM colchicine before incubation with 8 µM ANG II for 2 h at 37°C. This concentration of ANG II was used to ensure proper and reproducible UV detection because at <5 μM, the signal is progressively much noisier.
Vesicular membrane fraction.
Preparations enriched with vesicles derived from plasma membranes and ER from LLC-PK1 cells were obtained after growing cells in 12 flasks (150 cm2) under both conditions (with or without 0.1 nM ANG II for 30 min). The methodology for cell fractionation was carried out following Parys et al. (46), with modifications. Briefly, cells were resuspended in 20 ml homogenization buffer containing 250 mM sucrose, 10 mM Tris·HCl (pH 7.6), 0.1 mM PMSF, 1 mg/ml trypsin inhibitor, and protease inhibitor cocktail 1:400, and then lysed with a Potter-Elvejhem homogenizer with a Teflon pestle. Differential centrifugation was used to collect the vesicles derived from the plasma and ER membranes. After recovery of the first fraction, the following were obtained from the supernatants obtained after each centrifugation step: the total homogenate was centrifuged using a Sorvall SS-34 rotor at 2,400 g for 15 min to obtain the p1 fraction. The p2 fraction was obtained from the first supernatant after 15 min at 9,800 g. The next steps used a Beckman-Coulter 70 Ti rotor to collect p3 fraction (17,400 g for 20 min), p4 (23,600 g for 20 min), p5 (36,900 g for 40 min), and finally p6 (105,000 g for 60 min). All of the pellets were resuspended in the same buffer at 1:1 vol/vol. Total protein concentration of pellets was determined by the method of Lowry et al. (39) and ranged from 3 to 6 mg/ml.
Sarco(endo)plasmic reticulum Ca2+-ATPase activity.
Cultures of LLC-PK1 cells (5 × 105) were grown in 25 cm2 culture flasks in medium 199 supplemented with 3% FBS in an atmosphere of 5% CO2 in air at 37°C for 3 days. Cells were starved and preincubated for 20 min at 37°C with 0.1 nM losartan, 0.1 µM PD123319 or in the presence of both antagonists. Then, 0.1 nM ANG II was added, and after 30 min at 37°C in a 5% CO2 atmosphere, the cells were harvested and lysed as described in Ref. 19. SERCA activity was measured in the experimental conditions previously described (19) and expressed as the difference between total Ca2+-ATPase and that measured in the presence of 1 μM thapsigargin, the specific SERCA inhibitor.
Western blot analysis.
To analyze the involvement of β-arrestin in ANG II internalization, cells were incubated with or without 1 µM ANG II for 30 min. They were washed with PBS (pH 7.5) and incubated with medium in the absence of FBS in 5% CO2 at 37°C. Cells were harvested and lysed in a buffer containing 250 mM sucrose, 20 mM HEPES-Tris (pH 7.0), 1 mM EDTA, and 0.15 mg/ml trypsin inhibitor, using a Potter-Elvejhem homogenizer. Cell lysates (50 and 100 μg total protein for p-β-arrestin and 40 μg for Na+/K+-ATPase, SERCA, AT1R, AT2R, and PKC assays) were electrophoresed in a 10% SDS-PAGE and transferred to nitrocellulose membranes. These were probed with primary antibodies, followed by incubation with a horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature, and finally developed with the ECL system. Membrane stripping was done with 0.2 M glycine (pH 2.2) for 1 h at room temperature. Primary antibody incubation was carried out at room temperature for 1 h, at dilutions of anti-p-β-arrestin (1:200), β-actin (1:5,000), Na+/K+-ATPase (1:500), SERCA (1:750), AT1R (1:200), PKCα (1:500). AT2R was probed at 1:400 overnight at 4°C. Secondary antibodies were five times further diluted than their respective primary antibodies.
Despite concerns regarding antibodies specificity, we validated the Santa Cruz Biotechnology antibodies AT1R and AT2R by using adsorption experiments as described before (55), but now using samples from LLC-PK1 cell culture. Recombinant AT1R protein (amino acids 250–359) and AT2R (amino acids 1–363) were used as templates in experiments to see whether they adsorb or not the antibodies against AT1R (raised against amino acids 297–356) and AT2R (raised against amino acids 221–363), thereby checking the specificity of each antibody. A three-fold mass of each recombinant protein was incubated with the two antibodies separately for 72 h at 4°C under gentle shaking. The resulting mixture was centrifuged at 18,000 g for 1 min, and the supernatant was diluted 1:400 with respect to the original antibody. Electrophoresis of LLC-PK1 ER-enriched membrane preparations (obtained as described in “Vesicular Membrane Fraction”) was carried out using 40 µg of total protein in 12% acrylamide SDS-PAGE and transferred to nitrocellulose membranes. Standard molecular weight of 38 kDa was used to confirm the expected band for ANG II receptors (~40 kDa). The membranes were probed with the antibodies preincubated with recombinant AT1R or AT2R proteins. After stripping with 0.2 M glycine (pH 2.2), the membranes were reprobed with anti-AT1R or anti-AT2R antibodies alone (i.e., without the recombinant proteins) at the same dilution (1:400). Probing different membranes in parallel (without stripping) using the same antibodies (alone or preincubated) gave identical results.
Statistical analysis.
Student’s t-test was used to compare two groups, and one-way ANOVA with Newman-Keuls post hoc test was used to compare three or more groups. Statistical significance was set at P < 0.05. Data were analyzed with GraphPad Prism 5.0 software (San Diego, CA).
RESULTS
ANG II is internalized via luminal membranes of LLC-PK1 cells with AT1R/AT2R heterodimers.
In LLC-PK1 cells incubated with 0.1 μM FAM-labeled ANG II, intracellular localization of ANG II was seen after 30 min, indicating that the peptide had been internalized (Fig. 1A). If the antagonists of AT1R (1 μM losartan) or AT2R (1 μM PD123319) receptors were given before FAM-ANG II, they prevented the internalization of FAM-ANG II and, thereby, caused the retention of ANG II in the plasma membrane (Fig. 1, B and C). When both antagonists were added simultaneously before FAM-ANG II incubation (Fig. 1D), the result was unexpectedly the same as without the receptors’ antagonists, i.e., showing the internalization of the peptide after 30 min. Absence of FAM-ANG II was used as the negative control (Fig. 1E).
Fig. 1.

ANG II is internalized through the luminal membranes of LLC-PK1 cells with AT1R/AT2R heterodimers. A: cells incubated with FAM-ANG II (0.1 μM) for 30 min at 37°C. B–D: cells treated with losartan (1 μM) (LOS) or PD123319 (1 μM) (PD), or both antagonists for 20 min before FAM-ANG II incubation. Arrows indicate ANG II intracellular localization (A, D) or plasma membrane retention (B, C). E: negative control (cells only, no FAM-ANG II).
This result was confirmed by the quantification of ANG II levels in the culture medium by HPLC. Figure 2 shows that ANG II is also internalized at the higher concentration of 8 µM—which is required for HPLC analysis—after 2 h of incubation with the cells. This internalization was again partially prevented when losartan or PD123319 was used separately, with no significant difference between them. Incubation with both antagonists did not prevent ANG II internalization, also confirming the observation in Fig. 1D.
Fig. 2.
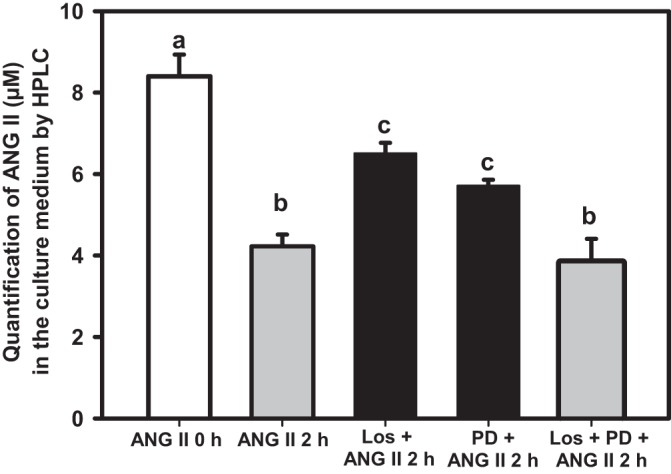
HPLC quantification of ANG II in culture medium containing LLC-PK1 cells preincubated or not with losartan (0.1 nM), PD123319 (0.1 μM), or with both antagonists, as shown on the abscissa, for 15 min at 37°C before adding 8 µM ANG II. The assayed material was further incubated for 2 h at 37°C. Data bars indicate means ± SE (n = 3−7). The empty bar corresponds to ANG II at zero time. Different lowercase letters above the bars indicate statistical difference between the mean values (P < 0.05).
Figure 3 shows 1) the stimulation of SERCA activity after the ANG II internalization, 2) the consequence of the partial internalization blockade by losartan or PD123319, and 3) the recovery of the stimulation profile when both antagonists were assayed together. As previously shown (19), ANG II stimulated SERCA activity by >90%, and preincubation with losartan or PD123319 totally blocked this stimulation. The return of SERCA activity to control levels matches the inhibition of ANG II internalization (∼50%). Interestingly—and in line with Fig. 2—ANG II stimulated SERCA activity to the same extent as ANG II alone when the cells were preincubated with both antagonists.
Fig. 3.
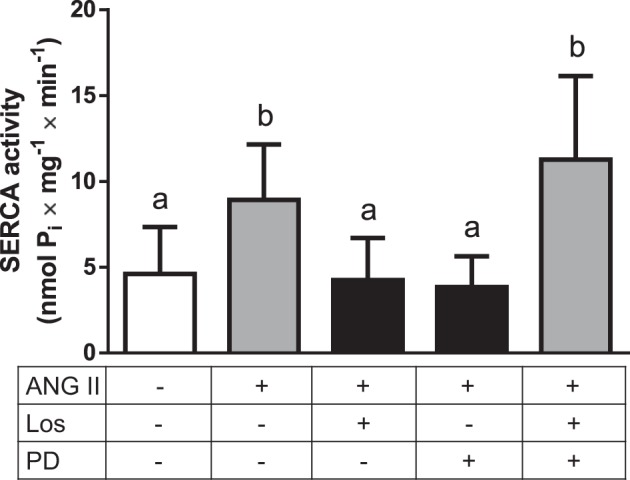
ANG II-induced stimulation of SERCA activity from LLC-PK1 cells is preserved when the AT1R and AT2R antagonists are both added. Cells were preincubated with losartan (0.1 nM) (Los) or PD123319 (0.1 μM) (PD), or with both antagonists, as shown on the abscissa, for 20 min before adding 0.1 nM ANG II. The cells were further incubated at 37°C for 30 min; longer times imply loss of activation, as described in Ref. 19. Data bars indicate means ± SE (n = 3–7). Different lowercase letters above the bars indicate statistical difference between the mean values (P < 0.05). Following the Committee on Publication Ethics (COPE) guidelines, we disclose that we have previously reported internal controls using losartan or PD123319 alone (19).
ANG II-AT1/AT2 complex via luminal membranes of LLC-PK1 cells is internalized through microtubule-dependent and clathrin-independent endocytic pathway.
Different methods were used to investigate the endocytic pathway involved in ANG II internalization in renal epithelial cells. Culturing cells at low temperatures is known to block membrane trafficking and inhibit receptor endocytosis (36). When 8 µM ANG II was incubated with the cells at 4°C, the peptide concentration in the medium decreased significantly (Fig. 4A). Because it is not internalized at this temperature (36), it is clear that the decrease in medium ANG II is due to binding to the membrane surface. The influence of microtubule disruption on ANG II internalization was examined by using 1 µM colchicine treatment of LLC-PK1 cells for 2 h at 37°C; this totally prevented internalization, i.e., ANG II was endocytosed in a microtubule-dependent manner (Fig. 4B). To examine the classical β-arrestin/clathrin endocytic pathway, the cells were incubated with Pitstop 2—which prevents clathrin-mediated endocytosis—for 30 min before ANG II treatment. Pitstop 2 was ineffective in blocking ANG II endocytosis, indicating a clathrin-independent internalization mechanism (Fig. 4C). Indeed, LLC-PK1 cell incubation with 1 μM ANG II for 30 min induced phosphorylation of β-arrestin at Ser-412 (Fig. 5). Dephosphorylation of β-arrestin is required to target clathrin-coated pits for receptor endocytosis, which confirms that this protein is not involved in the endocytic pathway of ANG II-AT1R/AT2R complex internalization.
Fig. 4.
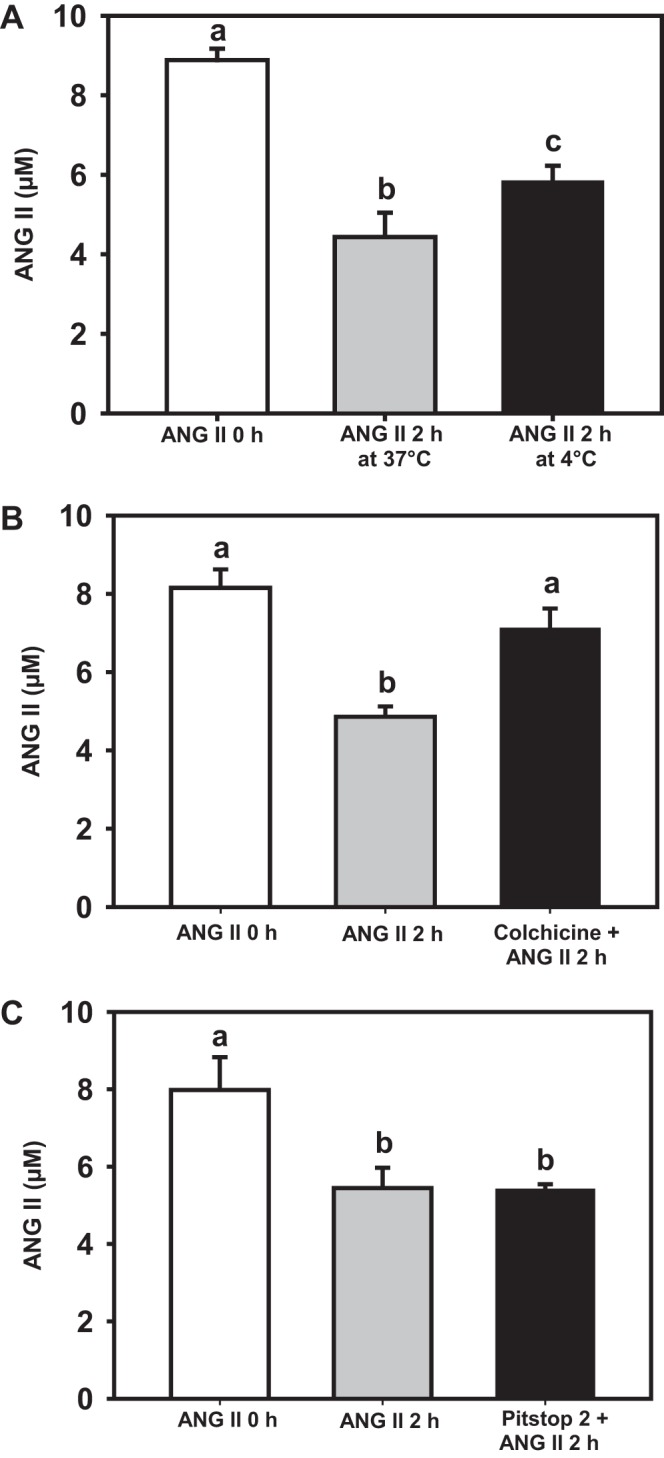
Internalization of ANG II-AT1R/AT2R complex is a microtubule-dependent and clathrin-independent endocytic pathway. The empty column in each panel represents medium ANG II before HPLC development (zero time). A: gray bar shows that 50% of the ANG II incubated for 2 h at 37°C was internalized, while the black bar shows binding of ANG II to the luminal membranes after 2 h at 4°C, since low temperature prevents internalization (27). B: cells incubated with 1 µM colchicine (microtubule polymerization inhibitor) for 2 h before 8 µM ANG II was added followed by incubation for 2 h at 37°C. C: cells incubated with 30 µM Pitstop 2 (a clathrin inhibitor) for 30 min before 8 µM ANG II was added and incubated for 2 h at 37°C. After the last incubation, culture media were carefully collected and analyzed by HPLC. The graphs represent ANG II in the culture media. Data bars indicate means ± SE (n = 3 or 4). Different lowercase letters above the bars indicate statistical differences among the mean values (P < 0.05).
Fig. 5.
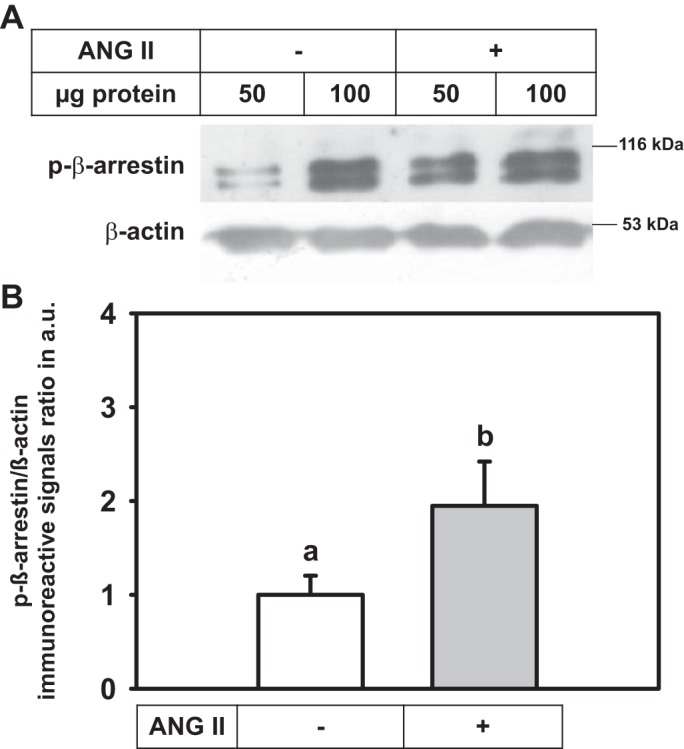
ANG II stimulates β-arrestin phosphorylation in LLC-PK1 cells. A: cells incubated with or without 1 μM ANG II for 30 min. Cell lysates (50 and 100 μg protein) were Western blotted using a polyclonal anti-p-β-arrestin antibody (top blot). After being stripped, membranes were probed with monoclonal anti-β-actin (bottom blot). B: densitometric representation of the immunoreactive signal ratio p-β-arrestin/β-actin. Data bars indicate means ± SE using different cell lysate preparations (n = 7 or 8). Different lowercase letters indicate statistical differences (P < 0.05).
Internalized ANG II via luminal membrane of LLC-PK1 is partially located in the endoplasmic reticulum.
Knowing that luminal ANG II specifically stimulates SERCA activity in LLC-PK1 cells with no effect on PMCA activity (19), this suggests an intracellular ANG II effect, probably in the ER. Living-cell fluorescent imaging was used by incubating LLC-PK1 cells with 0.3 μM ANG II-Alexa Fluor 488 for 1 h and subsequently adding 0.1 μM ER Tracker Red for 15 min. Internalized ANG II was present at high density in the ER (Fig. 6), a physical association that may culminate in the previously described activation of SERCA (19).
Fig. 6.

ANG II internalized via luminal membrane of LLC-PK1 is localized in the ER. LLC-PK1 cells were incubated with ANG II-Alexa Fluor 488 (0.3 μM) for 1 h at 37°C and then with ER Tracker Red (0.1 μM) for 15 min at 37°C. Living cell fluorescent imaging using confocal microscopy was obtained for ANG II-Alexa Fluor 488 (A, D), ER Tracker (B, E), and merge (C, F). A: negative control for ANG II (cells only). B, C: cells incubated with only ER Tracker, i.e., without ANG II.
AT1 and AT2 receptor content is increased in the endoplasmic reticulum of LLC-PK1 cells after ANG II-AT1R/AT2R complex internalization.
To investigate whether the ANG II-AT1R/AT2R complex, rather than the peptide alone, is directed to the ER, LLC-PK1, cells were fractionated by differential centrifugation to obtain plasma membrane and ER-enriched vesicles (Fig. 7). Western blotting was undertaken to characterize membrane vesicles using Na+/K+-ATPase and SERCA as plasma membrane and ER markers, respectively. Fractions p2–p4 contain plasma membrane, while fractions p3–p6 correspond to ER-enriched vesicles (Fig. 7A), showing higher amounts of SERCA. Therefore, p3–p6 fractions were used in Western blotting for AT1R, AT2R, and PKCα—the PKC isoform that is coupled to the luminal effects of ANG II in proximal tubules (15, 19)—in the absence and presence of 0.1 nM ANG II for 30 min, i.e., at a concentration that maximally activates SERCA (19). AT1R and AT2R content clearly increased in p4 to p6 fractions (Fig. 7B). The densitometric analysis of p5, which had the highest SERCA-positive profile and lower Na+/K+-ATPase levels, show 1) an increase of ~40 and ~100% of the AT1R and AT2R immunosignals, respectively, and 2) an increase of 150% in the PKCα immunosignal, compared with nontreated cells, using SERCA content as the loading control (Fig. 7C). Given concerns regarding antibody specificity (28, 32), we validated both AT1R and AT2R antibodies and confirmed the nonexistence of cross-reactivity between them in LLC-PK1 cells (Fig. 8). Preadsorption of AT1R antibody with AT1R recombinant protein abolished the corresponding immunosignal for AT1R (Fig. 8A, left); when the AT2R recombinant protein was used as a template, there was no difference in the AT1R immunosignal (Fig. 8A, right). The preadsorption of AT2R antibody with AT2R recombinant protein partially blocked the immunosignal (Fig. 8B, right), but there was no significant difference when the antibody was incubated with the AT1R recombinant protein (Fig. 8B, left).
Fig. 7.
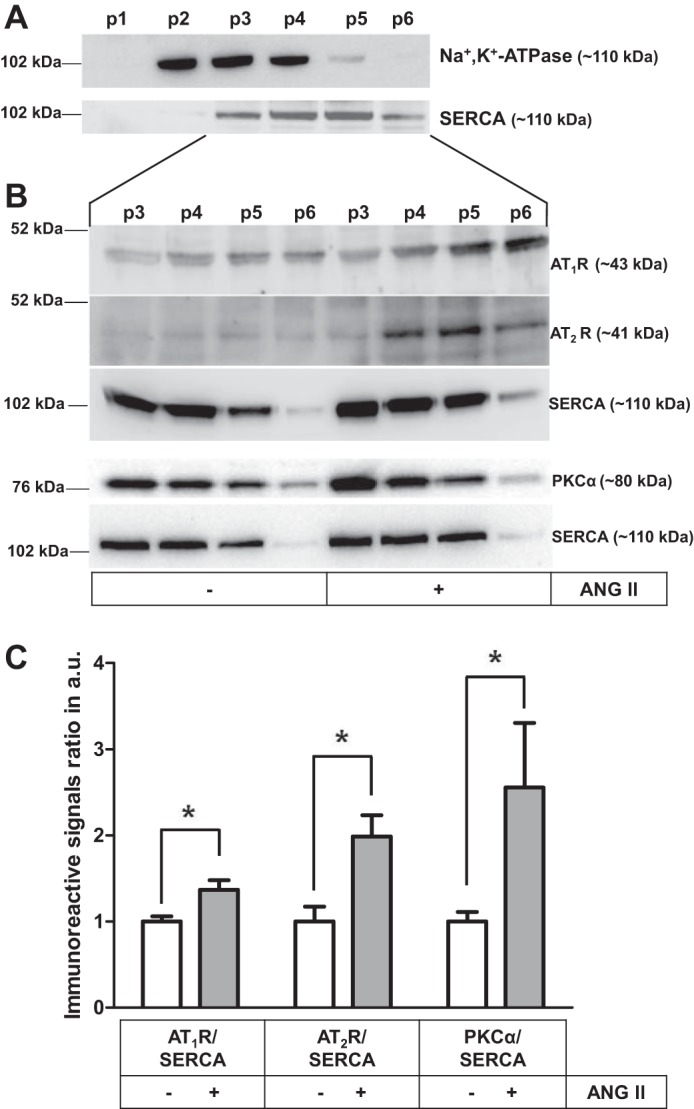
AT1R and AT2R and PKCα content increased in the ER of LLC-PK1 cells after ANG II-AT1R/AT2R internalization. A: cell fractions p1−p6 obtained by differential centrifugation were Western blotted using monoclonal anti-Na+/K+-ATPase and anti-SERCA antibodies (upper and lower blots, respectively). B: cells incubated with or without 0.1 nM ANG II for 30 min, as indicated on the abscissa. Cell fractions p3−p6 were Western blotted using monoclonal anti-AT1R, polyclonal anti-AT2R, monoclonal anti-SERCA, and polyclonal anti-PKCα antibodies. C: densitometric representation of the immunoreactive signal ratio primary antibodies (AT1R, AT2R, or PKCα) and SERCA antibody in p5. Data bars indicate means ± SE (n = 2 or 3). *Statistically different from the control without ANG II (P < 0.05).
Fig. 8.

Specificity of the ANG II AT1R and AT2R antibodies. ER-enriched membrane preparations were probed against AT1R monoclonal (A) or AT2R polyclonal (B) antibodies with or without preincubation with recombinant AT1R or AT2R protein (as indicated). Preincubation of each antibody with the corresponding immunogenic peptide decreased the immunosignal, whereas preincubation with the other recombinant protein did not. 38 kDa corresponds to standard molecular weight used in all conditions.
DISCUSSION
The present study shows that ANG II is internalized as part of a complex with AT1R/AT2R heterodimers, with the ER being a new target organelle for intracrine RAS. This mechanism may explain the specificity of luminal ANG II to stimulate SERCA activity in LLC-PK1 proximal tubule cells (19).
G protein-coupled receptor (GPCR) heterodimerization has been extensively studied, including the functional properties in physiological and pathological contexts (45). AT1R/AT2R heterodimers are not internalized after ANG II induction (49), and AT2R is not internalized after stimulation with ANG II (31). However, both AT1 and AT2 receptor antagonists in our cellular proximal tubule model impeded ANG II internalization when assayed separately, indicating that AT1R and AT2R are involved in luminal ANG II endocytosis (Fig. 1). An explanation for this apparent discrepancy may rest on the cells used. Indeed, Porrello et al. (49) and Hein et al. (31) used HEK293, a nonpolarized human embryonic kidney cell line of epithelial origin, which does not have membrane domains with differentiated “sidedness” (asymmetry and polarity) and functions. In contrast, the LLC-PK1 cells preserve vectorial ion transport, with well-demarcated luminal and basolateral membranes, and, therefore, with architecture similar to that observed in renal tissues in vivo. The data suggest that cell polarity, essential for renal epithelial cell physiology, is important for luminal ANG II to induce AT1R/AT2R heterodimerization. Immunoprecipitation assays clearly indicate that AT1R/AT2R heterodimers in the plasma membrane are essential for luminal ANG II internalization and SERCA stimulation in response to luminal ANG II (19). AT1R/AT2R heterodimers have also been described in membrane fractions from sheep proximal tubules (6) and rat renal cells (14). Unexpectedly, the combination of losartan and PD123319 did not prevent ANG II internalization in LLC-PK1 cells, and, as will be discussed below, in the case of the same effect for AT1R and AT2R, this indicates that simultaneous binding of the antagonists to AT1R/AT2R heterodimers is mutually exclusive.
ANG II internalization was confirmed by another approach: there was a decrease in medium ANG II concentration in HPLC assays when LLC-PK1 cells were incubated with the peptide, a phenomenon that was partially blocked when losartan or PD123319 was present (Fig. 2). At first sight, partial blockade of ANG II internalization (at 8 μM) by 0.1 nM losartan or 0.1 μM PD123319 seems to be unlikely. Both antagonists are considered low-affinity inhibitors, and their effects on ANG II internalization are, therefore, difficult to understand in terms of simple competitiveness. LLC-PK1 cells need to be preincubated with the antagonists for 15 min at 37°C before the addition of ANG II for the blockade of internalization to occur. Thus, the reversible interaction of the antagonists with their respective receptors could be followed by irreversible inactivation in the absence of ANG II. Again, and as encountered for FAM-ANG II (Fig. 1), the simultaneous presence of the antagonists resulted in ANG II internalization at levels similar to those of controls without additions, confirming that interactions with peptide and antagonists at receptor level are the same, irrespective of the ANG II/receptors antagonists’ ratio.
The involvement of β-arrestin-/clathrin-coated pits is well known as an endocytic pathway for GPCRs (62). Internalization of the ANG II-AT1R/AT2R complex occurs by a clathrin-independent pathway, as shown in Fig. 4C, which is supported by the induction of β-arrestin phosphorylation by ANG II in this cell line (Fig. 5). Receptor endocytosis mediated by clathrin binding requires dephosphorylation of β-arrestin (38). Thus, since internalization of the ANG II-AT1R/AT2R complex in LLC-PK1 cells does not involve the classic clathrin-dependent pathway, it may be occurring through the nonclassical receptor endocytic pathway involving the caveolar-type membrane/lipid rafts (30). Indeed, AT1R moves to caveolin-enriched fractions through interaction with caveolin-1 following vascular smooth muscle cell exposure to ANG II (35).
Microtubules are important for membrane traffic, maintenance of epithelial cell polarity, and luminal endocytosis in polarized cell types (5, 17). GPCR interacts with tubulin, suggesting that GPCR membrane trafficking is microtubule-dependent (16), and here, we have shown that internalization of the ANG II-AT1R/AT2R complex in LLC-PK1 cells is microtubule-dependent (Fig. 4B), a view supported by its complete reversal by colchicine. The high specificity of colchicine for tubulin is due to binding site in the tubulin (40), but other independent-microtubule effects of colchicine could not be ruled out, as described by Marques-da-Silva et al. (42). Furthermore, involvement of actin in internalization should be considered, since cytochalasin D blocks endocytosis of high molecular weight complexes in renal cell lines of distal origin (25).
It is tempting to suppose that a caveolar-type membrane/lipid raft microtubule-dependency is the principal mechanism of ANG II-AT1R/AT2R internalization. However, this mechanism itself may be not be sufficient to explain the removal of 4 nmol ANG II/ml from the medium in 2 h in the HPLC assays—crossing a 25 cm2-cell layer—because 1) there are ~120,000 AT1R receptors per LLC-PK1 cell (68) and 2) the 30-s time lapse required by Ca2+ mobilization (19). Considering that megalin in the brush-border membranes of proximal tubule cells is involved in the uptake of ANG II (24), as well as its sensitivity to colchicine (8), we speculate that a colchicine-sensitive, megalin- and microtubule-dependent mechanism participates in ANG II-AT1R/AT2R internalization (17, 26, 36). Moreover, a fast colchicine-sensitive PLC-mediated IP3 release occurs in primary culture of proximal tubule cells after 30 s of luminal ANG II stimulation (52), confirming the fast response that we had observed.
The main source for renal intracellular ANG II is extracellular ANG II internalized with its receptors (65, 66). Nuclear and mitochondrial membranes also have AT1 and AT2 receptors responsive to intracrine ANG II (reviewed in Ref. 2). It is well established that these organelles are involved in intracellular Ca2+ storage (33, 41); however, the ER cannot be neglected because 1) this organelle expresses different proteins involved in Ca2+ storage, and 2) SERCA has a pivotal role in Ca2+ movements and signaling in polarized cells (10, 46). Since ANG II stimulates SERCA activity that induces Ca2+ mobilization (19), it is reasonable to propose that ER can also be an intracellular target of ANG II. An intracrine ANG II effect on Ca2+ homeostasis occurs, but these studies did not indicate the presence of ANG II receptors in the ER (13, 69). Thus, ANG II-associated signaling upon SERCA may be due to the simultaneous endocytosis of the receptors (Figs. 6 and 7).
Assuming a similar t1/2 (~2.3 min) in LLC-PK1 cells like Chinese hamster ovary cells and HEK293 cells (59, 61), we can estimate that just over 10% of the receptors could be internalized by the time Ca2+-ATPase is maximally activated. This estimation is in agreement with the pharmacological principle of the “receptor reserve” theory, which states that a full agonist (e.g., ANG II) only requires binding to a small amount of receptors to elicit its maximal effect with a great efficacy (21, 58). Therefore, a low binding level should be enough to sustain Ca2+ mobilization, as long as internalization continues over 30 min before this process starts to drop (19).
Live cell imaging data confirm that the ER is the target organelle of luminal ANG II, which induces internalization of AT1R/AT2R heterodimers (Fig. 6). The data were confirmed by Western blot analysis of the sequential fractions obtained by differential centrifugation (Fig. 7), which suggests the existence of an intracellular signaling pathway triggered by ANG II in the ER, which accounts for the specific effect of luminal ANG II on SERCA activity to enhance Ca2+ storage (19), a process essential for regulation of Ca2+-modulated fluid reabsorption in proximal tubule cells (15). The ER can form signaling intermediates of the downstream AT1 receptors in the ANG II pathway (63). PKC, a central effector protein in ANG II signaling (15, 19), is not only activated (19), but its content is increased in the ER fraction after ANG II treatment (Fig. 7, B and C), indicating that a full preexisting G protein transduction system is enhanced in response to luminal ANG II. This point deserves special discussion because of the observation that ANG II internalization occurs at control levels when losartan and PD123319 are added together (Figs. 1 and 2). Binding of the antagonists to their corresponding target during preincubation might be mutually exclusive. In other words, with both compounds present, the dynamics of the individual AT1R with losartan and AT2R with PD123319 in the heterodimer is sterically hindered. As a consequence, the heterodimer permits internalization, as under control conditions without the blockers. This phenomenon is in line with the observation that ANG II-stimulated SERCA activity is maintained in the presence of both losartan and PD123319 (Fig. 3), confirming that both antagonists are ineffective in blocking heterodimerization and/or ANG II internalization when assayed together (Figs. 1 and 2). The ligand-specific conformational changes induced in ligand-binding domains, and the long-range intramolecular communications required for ANG II receptor blocking (64), cannot occur simultaneously in the heterodimer.
In conclusion, we have found new evidence for the formation and internalization of a proximal tubule luminal ANG II-AT1R/AT2R complex to target the ER, where it triggers specific intracellular Ca2+ responses. The role of ANG II-AT1R/AT2R signaling in physiological and pathological aspects of Ca2+ homeostasis in the renal system requires further investigation. GPCR heterodimers are known to trigger different signaling pathways compared with homodimers and monomers (9, 12, 45). Understanding the signaling pathways triggered by internalized ANG II heterodimer receptors directed to the ER may help the development of new therapeutic approaches in renal pathophysiological conditions in which imbalance of Ca2+ is a central event (11, 29).
GRANTS
This work was supported by the following agencies: the National Council for Scientific and Technological Development (CNPq), the Carlos Chagas Filho Research Support Foundation, the National Institutes of Science and Technology, and the Coordination for the Improvement of Higher Education Personnel (Brazil); the National Institute of Diabetes & Digestive & Kidney Diseases and the American Heart Association in Aid (USA) (2R01DK-067299 and 1R01DK-102429-01 award to J. L. Zhuo). F. M. Ferrão was supported by a fellowship grant from the Brazilian CNPq to support her stay at the University of Mississippi Medical Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.M.F., A.V., and J.L. conceived and designed research; F.M.F., L.H.C., H.A.D., X.C.L., J.L.Z., D.S.G., L.S.L., and J.L. performed experiments; F.M.F., L.H.C., H.A.D., X.C.L., J.L.Z., D.S.G., L.S.L., A.V., and J.L. analyzed data; F.M.F., L.H.C., H.A.D., X.C.L., J.L.Z., D.S.G., L.S.L., A.V., and J.L. interpreted results of experiments; F.M.F., L.H.C., H.A.D., X.C.L., J.L.Z., L.S.L., A.V., and J.L. prepared figures; F.M.F., L.H.C., L.S.L., A.V., and J.L. drafted manuscript; F.M.F., L.H.C., H.A.D., X.C.L., J.L.Z., D.S.G., L.S.L., A.V., and J.L. edited and revised manuscript; F.M.F., L.H.C., H.A.D., X.C.L., J.L.Z., D.S.G., L.S.L., A.V., and J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
Technical support by Glória Costa-Sarmento is acknowledged. We thank Dr. Richard Roman (Department of Pharmacology & Toxicology, University of Mississippi Medical Center at Jackson, MS) for kindly providing ER Tracker Red. We acknowledge the final English style corrections of the revised manuscript by BioMedES, UK (www.biomedes.co.uk). Publicase (Brazil) performed the English corrections of the original version.
Present address of F. M. Ferrão: Núcleo Multidisciplinar de Pesquisa em Biologia (NUMPEX-BIO), Universidade Federal do Rio de Janeiro, campus Xerém, 25245-390.
REFERENCES
- 1.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O’Rourke B, Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA 108: 14849–14854, 2011. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abadir PM, Walston JD, Carey RM. Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides 38: 437–445, 2012. doi: 10.1016/j.peptides.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem 276: 39721–39726, 2001. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 4.AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 407: 94–98, 2000. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 5.Apodaca G. Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic 2: 149–159, 2001. doi: 10.1034/j.1600-0854.2001.020301.x. [DOI] [PubMed] [Google Scholar]
- 6.Axelband F, Assunção-Miranda I, de Paula IR, Ferrão FM, Dias J, Miranda A, Miranda F, Lara LS, Vieyra A. Ang-(3-4) suppresses inhibition of renal plasma membrane calcium pump by Ang II. Regul Pept 155: 81–90, 2009. doi: 10.1016/j.regpep.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Bader M, Peters J, Baltatu O, Müller DN, Luft FC, Ganten D. Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research. J Mol Med (Berl) 79: 76–102, 2001. doi: 10.1007/s001090100210. [DOI] [PubMed] [Google Scholar]
- 8.Baus M, Medjugorac-Popovski M, Brown D, Sabolic I. In colchicine-treated rats, cellular distribution of AQP-1 in convoluted and straight proximal tubule segments is differently affected. Pflügers Arch 439: 321–330, 2000. doi: 10.1007/s004249900187. [DOI] [PubMed] [Google Scholar]
- 9.Bellot M, Galandrin S, Boularan C, Matthies HJ, Despas F, Denis C, Javitch J, Mazères S, Sanni SJ, Pons V, Seguelas MH, Hansen JL, Pathak A, Galli A, Sénard JM, Galés C. Dual agonist occupancy of AT1-R-α2C-AR heterodimers results in atypical Gs-PKA signaling. Nat Chem Biol 11: 271–279, 2015. doi: 10.1038/nchembio.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caroppo R, Gerbino A, Debellis L, Kifor O, Soybel DI, Brown EM, Hofer AM, Curci S. Asymmetrical, agonist-induced fluctuations in local extracellular [Ca(2+)] in intact polarized epithelia. EMBO J 20: 6316–6326, 2001. doi: 10.1093/emboj/20.22.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chebib FT, Sussman CR, Wang X, Harris PC, Torres VE. Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat Rev Nephrol 11: 451–464, 2015. doi: 10.1038/nrneph.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalrymple MB, Pfleger KD, Eidne KA. G protein-coupled receptor dimers: functional consequences, disease states and drug targets. Pharmacol Ther 118: 359–371, 2008. doi: 10.1016/j.pharmthera.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Deliu E, Tica AA, Motoc D, Brailoiu GC, Brailoiu E. Intracellular angiotensin II activates rat myometrium. Am J Physiol Cell Physiol 301: C559–C565, 2011. doi: 10.1152/ajpcell.00123.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dias J, Ferrão FM, Axelband F, Carmona AK, Lara LS, Vieyra A. ANG-(3-4) inhibits renal Na+-ATPase in hypertensive rats through a mechanism that involves dissociation of ANG II receptors, heterodimers, and PKA. Am J Physiol Renal Physiol 306: F855–F863, 2014. doi: 10.1152/ajprenal.00488.2013. [DOI] [PubMed] [Google Scholar]
- 15.Du Z, Ferguson W, Wang T. Role of PKC and calcium in modulation of effects of angiotensin II on sodium transport in proximal tubule. Am J Physiol Renal Physiol 284: F688–F692, 2003. doi: 10.1152/ajprenal.00261.2002. [DOI] [PubMed] [Google Scholar]
- 16.Duvernay MT, Wang H, Dong C, Guidry JJ, Sackett DL, Wu G. α2B-adrenergic receptor interaction with tubulin controls its transport from the endoplasmic reticulum to the cell surface. J Biol Chem 286: 14080–14089, 2011. doi: 10.1074/jbc.M111.222323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elkjaer ML, Birn H, Agre P, Christensen EI, Nielsen S. Effects of microtubule disruption on endocytosis, membrane recycling and polarized distribution of Aquaporin-1 and gp330 in proximal tubule cells. Eur J Cell Biol 67: 57–72, 1995. [PubMed] [Google Scholar]
- 18.Engeli S, Negrel R, Sharma AM. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension 35: 1270–1277, 2000. doi: 10.1161/01.HYP.35.6.1270. [DOI] [PubMed] [Google Scholar]
- 19.Ferrão FM, Lara LS, Axelband F, Dias J, Carmona AK, Reis RI, Costa-Neto CM, Vieyra A, Lowe J. Exposure of luminal membranes of LLC-PK1 cells to ANG II induces dimerization of AT1/AT2 receptors to activate SERCA and to promote Ca2+ mobilization. Am J Physiol Renal Physiol 302: F875–F883, 2012. doi: 10.1152/ajprenal.00381.2011. [DOI] [PubMed] [Google Scholar]
- 20.Ferrão FM, Lara LS, Lowe J. Renin-angiotensin system in the kidney: What is new? World J Nephrol 3: 64–76, 2014. doi: 10.5527/wjn.v3.i3.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furchgott RF. The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. Adv Drug Res 3: 21–55, 1966. [Google Scholar]
- 22.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes. Circ Res 87: 1123–1132, 2000. doi: 10.1161/01.RES.87.12.1123. [DOI] [PubMed] [Google Scholar]
- 23.Gáborik Z, Hunyady L. Intracellular trafficking of hormone receptors. Trends Endocrinol Metab 15: 286–293, 2004. doi: 10.1016/j.tem.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Villalobos R, Klassen RB, Allen PL, Navar LG, Hammond TG. Megalin binds and internalizes angiotensin II. Am J Physiol Renal Physiol 288: F420–F427, 2005. doi: 10.1152/ajprenal.00243.2004. [DOI] [PubMed] [Google Scholar]
- 25.Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J Cell Biol 120: 695–710, 1993. doi: 10.1083/jcb.120.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutmann EJ, Niles JL, McCluskey RT, Brown D. Colchicine-induced redistribution of an apical membrane glycoprotein (gp330) in proximal tubules. Am J Physiol Cell Physiol 257: C397–C407, 1989. [DOI] [PubMed] [Google Scholar]
- 27.Gwathmey TM, Shaltout HA, Pendergrass KD, Pirro NT, Figueroa JP, Rose JC, Diz DI, Chappell MC. Nuclear angiotensin II type 2 (AT2) receptors are functionally linked to nitric oxide production. Am J Physiol Renal Physiol 296: F1484–F1493, 2009. doi: 10.1152/ajprenal.90766.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hafko R, Villapol S, Nostramo R, Symes A, Sabban EL, Inagami T, Saavedra JM. Commercially available angiotensin II AT2 receptor antibodies are nonspecific. PLoS One 8: e69234, 2013. doi: 10.1371/journal.pone.0069234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart P, Bakris GL. Calcium antagonists: Do they equally protect against kidney injury? Kidney Int 73: 795–796, 2008. doi: 10.1038/sj.ki.5002773. [DOI] [PubMed] [Google Scholar]
- 30.Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta 1838: 532–545, 2014. doi: 10.1016/j.bbamem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hein L, Meinel L, Pratt RE, Dzau VJ, Kobilka BK. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol Endocrinol 11: 1266–1277, 1997. doi: 10.1210/mend.11.9.9975. [DOI] [PubMed] [Google Scholar]
- 32.Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension 61: 253–258, 2013. doi: 10.1161/HYPERTENSIONAHA.112.203679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89: 1145–1153, 1997. doi: 10.1016/S0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 34.Inagami T. Mitochondrial angiotensin receptors and aging. Circ Res 109: 1323–1324, 2011. doi: 10.1161/RES.0b013e31823f05e0. [DOI] [PubMed] [Google Scholar]
- 35.Ishizaka N, Griendling KK, Lassègue B, Alexander RW. Angiotensin II type 1 receptor: relationship with caveolae and caveolin after initial agonist stimulation. Hypertension 32: 459–466, 1998. doi: 10.1161/01.HYP.32.3.459. [DOI] [PubMed] [Google Scholar]
- 36.Li XC, Hopfer U, Zhuo JL. AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through a microtubule-dependent endocytic pathway. Am J Physiol Renal Physiol 297: F1342–F1352, 2009. doi: 10.1152/ajprenal.90734.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li XC, Zhuo JL. Intracellular ANG II directly induces in vitro transcription of TGF-β1, MCP-1, and NHE-3 mRNAs in isolated rat renal cortical nuclei via activation of nuclear AT1a receptors. Am J Physiol Cell Physiol 294: C1034–C1045, 2008. doi: 10.1152/ajpcell.00432.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, Lefkowitz RJ. Clathrin-mediated endocytosis of the β-adrenergic receptor is regulated by phosphorylation/dephosphorylation of β-arrestin1. J Biol Chem 272: 31051–31057, 1997. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 39.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275, 1951. [PubMed] [Google Scholar]
- 40.Lu Y, Chen J, Xiao M, Li W, Miller DD. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 29: 2943–2971, 2012. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marius P, Guerra MT, Nathanson MH, Ehrlich BE, Leite MF. Calcium release from ryanodine receptors in the nucleoplasmic reticulum. Cell Calcium 39: 65–73, 2006. doi: 10.1016/j.ceca.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Marques-da-Silva C, Chaves MM, Castro NG, Coutinho-Silva R, Guimaraes MZ. Colchicine inhibits cationic dye uptake induced by ATP in P2X2 and P2X7 receptor-expressing cells: implications for its therapeutic action. Br J Pharmacol 163: 912–926, 2011. doi: 10.1111/j.1476-5381.2011.01254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morimoto S, Sigmund CD. Angiotensin mutant mice: a focus on the brain renin-angiotensin system. Neuropeptides 36: 194–200, 2002. doi: 10.1054/npep.2002.0894. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen AH, Schauser KH, Poulsen K. Current topic: the uteroplacental renin-angiotensin system. Placenta 21: 468–477, 2000. doi: 10.1053/plac.2000.0535. [DOI] [PubMed] [Google Scholar]
- 45.Parmentier M. GPCRs: Heterodimer-specific signaling. Nat Chem Biol 11: 244–245, 2015. doi: 10.1038/nchembio.1772. [DOI] [PubMed] [Google Scholar]
- 46.Parys JB, De Smedt H, Borghgraef R. Calcium transport systems in the LLC-PK1 renal epithelial established cell line. Biochim Biophys Acta 888: 70–81, 1986. doi: 10.1016/0167-4889(86)90072-8. [DOI] [PubMed] [Google Scholar]
- 47.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 86: 747–803, 2006. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 48.Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, Chappell MC. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun 384: 149–154, 2009. doi: 10.1016/j.bbrc.2009.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porrello ER, Pfleger KD, Seeber RM, Qian H, Oro C, Abogadie F, Delbridge LM, Thomas WG. Heteromerization of angiotensin receptors changes trafficking and arrestin recruitment profiles. Cell Signal 23: 1767–1776, 2011. doi: 10.1016/j.cellsig.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 50.Re RN. The intracrine hypothesis and intracellular peptide hormone action. BioEssays 25: 401–409, 2003. doi: 10.1002/bies.10248. [DOI] [PubMed] [Google Scholar]
- 51.Redding KM, Chen BL, Singh A, Re RN, Navar LG, Seth DM, Sigmund CD, Tang WW, Cook JL. Transgenic mice expressing an intracellular fluorescent fusion of angiotensin II demonstrate renal thrombotic microangiopathy and elevated blood pressure. Am J Physiol Heart Circ Physiol 298: H1807–H1818, 2010. doi: 10.1152/ajpheart.00027.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schelling JR, Hanson AS, Marzec R, Linas SL. Cytoskeleton-dependent endocytosis is required for apical type 1 angiotensin II receptor-mediated phospholipase C activation in cultured rat proximal tubule cells. J Clin Invest 90: 2472–2480, 1992. doi: 10.1172/JCI116139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seikaly MG, Arant BS Jr, Seney FD Jr. Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clin Invest 86: 1352–1357, 1990. doi: 10.1172/JCI114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sernia C. A critical appraisal of the intrinsic pancreatic angiotensin-generating system. JOP 2: 50–55, 2001. [PubMed] [Google Scholar]
- 55.Silva PA, Monnerat-Cahli G, Pereira-Acácio A, Luzardo R, Sampaio LS, Luna-Leite MA, Lara LS, Einicker-Lamas M, Panizzutti R, Madeira C, Vieira-Filho LD, Castro-Chaves C, Ribeiro VS, Paixão AD, Medei E, Vieyra A. Mechanisms involving Ang II and MAPK/ERK1/2 signaling pathways underlie cardiac and renal alterations during chronic undernutrition. PLoS One 9: e100410, 2014. doi: 10.1371/journal.pone.0100410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 57: 3297–3306, 2008. doi: 10.2337/db08-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Speth RC, Daubert DL, Grove KL. Angiotensin II: a reproductive hormone too? Regul Pept 79: 25–40, 1999. doi: 10.1016/S0167-0115(98)00141-4. [DOI] [PubMed] [Google Scholar]
- 58.Stephenson RP. A modification of receptor theory. Br J Pharmacol Chemother 11: 379–393, 1956. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szakadáti G, Tóth AD, Oláh I, Erdélyi LS, Balla T, Várnai P, Hunyady L, Balla A. Investigation of the fate of type I angiotensin receptor after biased activation. Mol Pharmacol 87: 972–981, 2015. doi: 10.1124/mol.114.097030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tadevosyan A, Maguy A, Villeneuve LR, Babin J, Bonnefoy A, Allen BG, Nattel S. Nuclear-delimited angiotensin receptor-mediated signaling regulates cardiomyocyte gene expression. J Biol Chem 285: 22338–22349, 2010. doi: 10.1074/jbc.M110.121749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas WG, Motel TJ, Kule CE, Karoor V, Baker KM. Phosphorylation of the angiotensin II (AT1A) receptor carboxyl terminus: a role in receptor endocytosis. Mol Endocrinol 12: 1513–1524, 1998. doi: 10.1210/mend.12.10.0179. [DOI] [PubMed] [Google Scholar]
- 62.Tian X, Kang DS, Benovic JL. β-arrestins and G protein-coupled receptor trafficking. Handb Exp Pharmacol 219: 173–186, 2014. doi: 10.1007/978-3-642-41199-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ueda Y, Ogiso H, Sato M, Umezawa Y, Okazaki T, Kobayashi T. Asymmetrical diacylglycerol dynamics on the cytosolic and lumenal sides of a single endomembrane in living cells. Sci Rep 5: 12960, 2015. doi: 10.1038/srep12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Unal H, Jagannathan R, Bhatnagar A, Tirupula K, Desnoyer R, Karnik SS. Long range effect of mutations on specific conformational changes in the extracellular loop 2 of angiotensin II type 1 receptor. J Biol Chem 288: 540–551, 2013. doi: 10.1074/jbc.M112.392514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Kats JP, de Lannoy LM, Jan Danser AH, van Meegen JR, Verdouw PD, Schalekamp MA. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension 30: 42–49, 1997. doi: 10.1161/01.HYP.30.1.42. [DOI] [PubMed] [Google Scholar]
- 66.Van Kats JP, Schalekamp MA, Verdouw PD, Duncker DJ, Danser AH. Intrarenal angiotensin II: interstitial and cellular levels and site of production. Kidney Int 60: 2311–2317, 2001. doi: 10.1046/j.1523-1755.2001.00049.x. [DOI] [PubMed] [Google Scholar]
- 67.Wilson BA, Nautiyal M, Gwathmey TM, Rose JC, Chappell MC. Evidence for a mitochondrial angiotensin-(1-7) system in the kidney. Am J Physiol Renal Physiol 310: F637–F645, 2016. doi: 10.1152/ajprenal.00479.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolf G, Zahner G, Mondorf U, Schoeppe W, Stahl RA. Angiotensin II stimulates cellular hypertrophy of LLC-PK1 cells through the AT1 receptor. Nephrol Dial Transplant 8: 128–133, 1993. [PubMed] [Google Scholar]
- 69.Zhuo JL, Li XC, Garvin JL, Navar LG, Carretero OA. Intracellular ANG II induces cytosolic Ca2+ mobilization by stimulating intracellular AT1 receptors in proximal tubule cells. Am J Physiol Renal Physiol 290: F1382–F1390, 2006. doi: 10.1152/ajprenal.00269.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]