

Abstract
Several recent studies have shown that human topoisomerase I (htopoI) can recognize various DNA lesions and thereby form a covalent topoisomerase I–DNA complex, which is known to be detrimental to cells. We have investigated whether htopoI recognizes another htopoI that is covalently trapped on a DNA substrate. For this purpose we created an artificial DNA substrate containing a specific topoisomerase I binding sequence, where the enzyme was trapped in the covalently bound form. We demonstrate that, in vitro, free htopoI stimulates the formation of an additional cleavage complex immediately upstream of the covalently bound topoisomerase I. The predominant distance between the two cleavage sites is 13 nt. In addition we find that these two enzymes may form direct protein–protein contacts and we propose that these may be mediated through the formation of a dimer by domain swapping involving the C-terminal and the core domains. Finally, we discuss the possibility that the double cleavage reaction may be the initial step for the removal of the recognized cleavage complex.
INTRODUCTION
Human topoisomerase I (htopoI) is an abundant enzyme that is involved in several important pathways, such as transcription and replication (1–4), where the removal of positive supercoils is required for ongoing RNA and DNA synthesis. HtopoI catalyses a reversible transesterification reaction by forming a covalent 3′-phosphotyrosyl bond between a DNA strand and an enzyme tyrosine residue while leaving a free 5′-hydroxyl terminus downstream of the cleavage site (5).
The subsequent ligation reaction is inhibited by a family of alkaloids called camptothecins (CPT) that possess an antitumor effect due to a specific interaction with htopoI. This interaction leads to the stabilization of a reversible covalent complex between htopoI and the DNA backbone, thereby introducing a nick into the DNA. The cytotoxic effect of these types of complexes is probably due to fragmentation of the genome that occurs when ongoing replication forks collide with covalently bound topoisomerases (6,7).
It has recently been demonstrated that various DNA lesions can increase the amount of reversible cleavage complexes between htopoI and DNA (8). These lesions include, among others, abasic sites, oxidative damage, gaps, base mismatches and UV-photoproducts (9–13). HtopoI recognizes these lesions and a reversible covalent complex is formed. This may be due to an increased affinity for the DNA or to an indirect inhibition of the ligation process. When present prior to the DNA replication process these complexes block fork migration, and may initiate non-homologous recombination. The complexes may also cause genome fragmentation, as has been shown for complexes formed in the presence of CPT (6,7,14). Wu and Liu (15) found that a collision with the transcription machinery can lead to the formation of irreversible complexes, which pose a great threat to the survival of the cell. Thus, it would be advantageous for cells to have repair mechanisms for the removal of these kinds of DNA–protein crosslinks. However, when a high number of protein–DNA crosslinks are present, it may be beneficial for the organism if these lesions become a signal for the initiation of the apoptotic pathway instead.
A number of studies have searched for a repair mechanism for topoisomerase I–DNA complexes, and several models for such a mechanism have also been suggested (16–19). However, a complete repair mechanism has not yet been detected. Our intention was to investigate if htopoI cleavage complexes would be recognized as a lesion in an in vitro system. A synthetic DNA substrate was designed containing a unique htopoI cleavage site, where htopoI could be trapped; a so called ‘suicide substrate’, L193s. We show here that in an in vitro system a ‘dimer’-like complex can be formed between two htopoI enzymes, where the downstream enzyme is engaged in a suicidal cleavage complex. This was shown with recombinant enzyme expressed in the yeast and the baculovirus system. In addition we show data which suggest that a direct protein–protein interaction takes place and that this interaction may be mediated through a ‘domain-swapping’ mechanism. The formation of such a double cleavage complex is a novel finding which represents an as yet unknown function of htopoI. We speculate that the htopoI enzyme blocked in the cleaved state is specifically recognized by the second htopoI enzyme through putative protein–protein interactions. The cleavage complex could then be excised and a repair pathway initiated. Alternatively, ‘dimer’ formation could act as a signal for apoptosis.
MATERIALS AND METHODS
Preparation of recombinant htopoI
The topoisomerase I expression plasmid pHT143 was transfected into the Saccharomyces cerevisiae strain RS190. Expression and purification of recombinant htopoI was performed as described (20). The final protein concentration was ∼250 µg/ml as measured according to Bradford (21). Storage was at –20°C in 5 mM Tris–HCl pH 7.5, 170 mM NaCl, 50% glycerol, 0.5 mM DTT, 0.5 mM EDTA. Three different batches were used during the course of our investigations.
Topoisomerase I expressed in the baculovirus system was purified in the following way. HiV insect cells were infected with a wild-type htopoI baculovirus clone (a generous gift from Dr James Champoux, University of Washington, Seattle, WA) for 48 h at 27°C. Cells were harvested by centrifugation and resuspended in 4 packed cell volumes (PCV) lysis buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 5 mM DTT, 1 mM PMSF, 5 mM leupeptin, 1% aprotinin). The cells were lysed using a Dounce Teflon homogenizor. Afterwards 4 PCV sucrose/glycerol buffer (50 mM Tris–HCl pH 8.0, 10 mM MgCl2, 25% sucrose, 50% glycerol, 2 mM DTT) was added. One PCV saturated ammonium sulfate (pH 7.0 at 4°C) was added drop-wise. After 30 min on ice the solution was centrifuged for 3 h at 35 000 r.p.m. using a Beckman SW40-Ti rotor. The pellet was discarded and 4 vol saturated ammonium sulfate was added to the supernatant and the solution was incubated on ice for 30 min. The precipitate was centrifuged and redissolved in 42 vol Ni-wash buffer (30 mM HEPES pH 7.9, 150 mM NaCl, 20 mM imidazole pH 8.0, 10% glycerol, 1 mM mercaptoethanol, 1 mM PMSF). The solution was loaded on a 1 ml pre-equilibrated Ni-NTA Agarose (Qiagen) and subsequently washed with Ni-wash buffer. Topoisomerase I was eluted with Ni-elution buffer (30 mM HEPES pH 7.9, 150 mM NaCl, 250 mM imidazole pH 8.0, 10% glycerol, 1 mM mercaptoethanol, 1 mM PMSF) and collected in 0.5 ml fractions. The fractions were analyzed according to Bradford (21) and by SDS–PAGE. HtopoI-containing fractions were pooled and loaded on a pre-equilibrated 1 ml Resource Q column using an FPLC apparatus (Pharmacia). HtopoI did not bind to the column material and was collected in the run-through fractions. These were stored at –20°C in storage buffer (15 mM HEPES–KOH pH 7.8, 170 mM NaCl, 0.5 mM DTT, 0.5 mM EDTA, 50% glycerol). The concentration was ∼250 µg/ml.
Synthetic DNA substrates
For the substrate L193s three different oligonucleotides (DNA Technology, Aarhus, Denmark) were used: OL1, 5′-AAAAAAAGACTTAGA-3′; OL2, 5′-TTTTTTTTTTTTTTTTTTCTAAGTCTTTTTTTGCCTTCGCCCGGATCCCCGCCAAGCTTACCTGCCCTTTGGGCAGGTAAGCTTGGCGGGGATCCGGGCGAAGGC-3′ and OL3, 5′-AGAAAAAAAAAAAAAAAAAAGGATCCCCGGAGTGAATTCGGCCCCTTTGGGCCGAATTCACTCCGGGGATCC-3′. The oligonucleotides were purified by HPLC using a reverse phase column. OL1 was labeled with 100 µCi [γ-32P]ATP (3000 Ci/mmol; Amersham Pharmacia), while OL2 was phosphorylated with non-radioactive ATP. OL1 and OL2 were precipitated, redissolved in ddH2O, and mixed with OL3 in a total volume of 30 µl. Then 1200 U T4 polynucleotide ligase was added (BioLabs) and the mixture was incubated for 3–4 days at 4°C. The ligation mixture was subsequently loaded on a 1 mm 7% denaturing polyacrylamide gel (SequaGel Sequencing System, National Diagnostics). After electrophoresis the full-length band was identified by autoradiography, excised, and eluted overnight in TE buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA) at 37°C. The eluted oligonucleotide was precipitated, redissolved and phosphorylated with ATP for 30 min at 37°C. The full-length oligonucleotide was precipitated and redissolved in 30 µl TE.
In the following experiments between 555 and 925 Bq L193s were used for each reaction. This is equivalent to a molar ratio between L193s and htopoI ranging from 1:500 to 1:1000.
Titration with htopoI using L193s as substrate
L193s was incubated with increasing amounts of htopoI for 40 min at 37°C in 20 µl buffer I (12.5 mM HEPES–KOH pH 7.9, 6 mM MgCl2, 50 mM KCl, 42.5 mM NaCl, 21% glycerol, 0.5 mM EDTA, 1 mM DTT). The reactions were terminated by the addition of 0.4% SDS and 125 µg/ml proteinase K; htopoI was degraded by a further incubation at 37°C for 60 min. The DNA molecules were precipitated by ethanol/LiCl (0.6 M). After 30 min on ice the mixture was centrifuged; the pellet was redissolved in sequence loading buffer, heated to 95°C for 5 min and analyzed on a 20% denaturing polyacrylamide gel. The gels were analyzed using a PhosphorImager (Molecular Dynamics, Storm 860) and quantified with the software ImageQuant.
Comparison of proteinase K with trypsin digestion
L193s was incubated with 0 or 500 ng htopoI in a 10 µl reaction volume in buffer I as described above. The reactions were terminated by the addition of either a final concentration of 0.4% SDS without protease, 125 µg/ml proteinase K or 80 µg/ml trypsin. The samples were incubated at 37°C for 60 min, then the DNA was precipitated by ethanol/LiCl and analyzed on a 14% denaturing polyacrylamide gel.
Investigation of the effect of camptothecin
L193s was incubated with 0 or 500 ng htopoI in a 13 µl reaction volume in buffer I for 5 min at 37°C. Subsequently 2 µl DMSO or CPT dissolved in DMSO was added at the indicated concentrations and samples were incubated for an additional 15 min at 37°C. The reactions were terminated as described. Samples were removed for SDS–PAGE analysis prior to protease digestion in order to quantify the degree of suicide cleavage in each reaction. The remainder of the sample was analyzed as described.
Proteolysis of htopoI with subtilisin
An aliquot of 2 µg htopoI (4 µl) was incubated with TE or various concentrations of subtilisin (as indicated in the text) diluted in TE in a total volume of 10 µl 6 mM HEPES pH 7.9, 6 mM Tris–HCl pH 7.5, 68 mM NaCl and 20% glycerol for 20 min at 25°C. The reaction was terminated by adding 5 mM PMSF then placed on ice. Half of the reaction mixture was analyzed by SDS–PAGE and Coomassie brilliant blue staining. The rest was incubated with L193s in a reaction volume of 15 µl 13 mM HEPES–KOH pH 7.9, 30 mM NaCl, 43 mM KCl, 5 mM MgCl2 and 16% glycerol for 40 min at 37°C. The reactions were terminated with SDS and a sample was taken for analysis of suicide cleavage on a 7.5% SDS–PAGE. The rest was incubated with proteinase K and analyzed as described.
RESULTS
Sequence, two-dimensional structure, and function of the DNA substrates
A so-called suicide substrate, L193s, was designed, which efficiently trapped a htopoI cleavage complex at a defined position. The complete sequence and two-dimensional structure of the DNA substrate is depicted in Figure 1A. L193s consisted of a 193 nt oligonucleotide that folded back on itself, thereby forming a 92 bp double-stranded region with a 3 nt loop at both ends. In the center of the substrate, there was a 3 nt 5′ overhang at a nick. The sequences of the last three nucleotides at each end of the oligonucleotide were identical. Furthermore, a specific 16 nt-long htopoI binding and cleavage sequence (22,23) was included in such a way, that the nick was located 3 nt 3′ to the htopoI cleavage site. Cleavage by htopoI at this site became suicidal due to the presence of a 5′ phosphate next to the cleavage site (Fig. 1). Thus, the presence of the 5′ phosphate prevents ligation and thereby prevented the release of htopoI after the cleavage reaction had taken place. This stable complex was designated as a cleavage complex.
Figure 1.


Two-dimensional structure and complete sequence of the substrate L193s and schematic outline of the function. (A) The htopoI cleavage site is indicated with an arrow. The radioactive label (32P) is located on nucleotide 15 (indicated with an asterisk; base shown in bold). BamHI restriction sites are indicated. (B) HtopoI binds to the hexadecameric binding sequence and cleaves the upper strand forming a covalent phosphotyrosine linkage with the fourth nucleotide (numbered as in A) and thus produces an oligomer consisting of three bases. The overhang is identical with those three bases, therefore, when the trimer is liberated by htopoI, the 5′ end can hybridize to the complementary strand and close the gap. Because the 5′ end is phosphorylated, htopoI is irreversibly trapped. The position of the hexadecameric binding sequence is shown in bold.
Correct substrate folding was verified by the existence of BamHI cleavage sites at the positions indicated in Figure 1A (data not shown). It was also verified that the suicide cleavage of htopoI took place at the correct position. The suicide cleavage reaction catalyzed by htopoI on L193s is schematically outlined in Figure 1B. The utilized recombinant htopoI preparations were either expressed in yeast or baculovirus and purified to near homogeneity (Fig. 2A and B).
Figure 2.
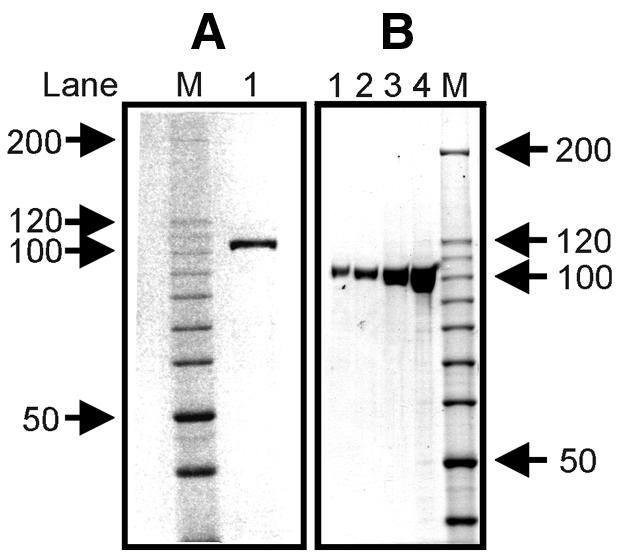
Coomassie staining of yeast and baculovirus-expressed recombinant htopoI. Coomassie staining of a 7.5% polyacrylamide gel. (A) Recombinant htopoI expressed in S.cerevisiae. Lane 1, ∼3 µg yeast-expressed htopoI. (B) Recombinant htopoI expressed by baculoviruses. Lane 1, 200 ng; lane 2, 400 ng; lane 3, 1000 ng; lane 4, 1500 ng. Marker is a 10 kDa ladder and the size of some of the bands are indicated in kDa.
Titration with human topoisomerase I
To investigate whether htopoI could cleave L193s at positions other than at the suicide cleavage site, L193s was incubated with increasing amounts of yeast-expressed htopoI (Fig. 3A). The reactions were terminated by the addition of 0.4% SDS and htopoI was digested with proteinase K. With increasing concentrations of htopoI a cleavage pattern consisting of four double bands occurred. The double bands were designated by the letters A, b, c and d, where A represented the most predominant cleavage site. None of the bands migrated to a position that precisely fitted the sequence identical marker; instead they had an intermediate mobility. Because highly pure recombinant htopoI was used (Fig 2A, lane 1) we assumed that these bands were due to additional htopoI cleavage of substrates that already had been cleaved suicidally.
Figure 3.
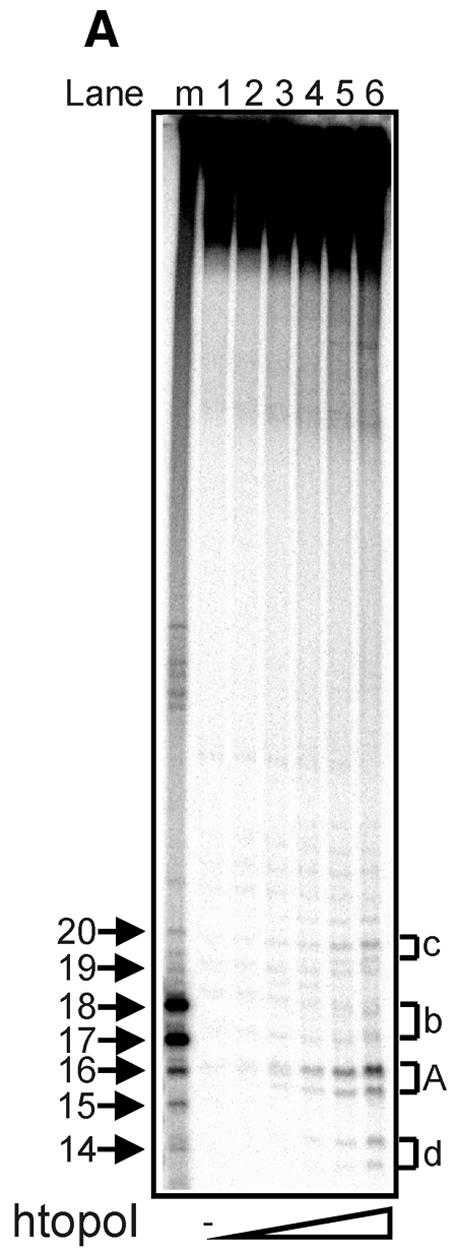
Titration with recombinant yeast-expressed htopoI. (A) Autoradiogram of a 20% sequencing gel. L193s was incubated with: lane 1, no htopoI; lane 2, 125 ng; lane 3, 250 ng; lane 4, 500 ng; lane 5, 750 ng; lane 6, 1250 ng htopoI for 40 min at 37°C. The buffer conditions were identical in all samples. Thereafter all reactions were terminated by the addition of 0.4% SDS and the protein was digested with proteinase K. (B) Autoradiogram of a 20% sequencing gel. L193s was incubated with: lane 1, no htopoI; lanes 2 and 3, 250 ng; lanes 4 and 5, 750 ng; lanes 6–9, 1250 ng yeast-expressed htopoI for 40 min at 37°C. For each concentration of htopoI used the reaction mixtures were split into two aliquots and stopped by the addition of SDS. The samples shown in lanes 1,2,4 and 6 were incubated with proteinase K. The samples shown in lanes 8 and 9 were terminated by the addition of 300 mM NaCl for 10 min at 37°C, divided into two aliquots. One sample was additionally incubated with proteinase K (lane 8). Letters A, b, c and d indicate double bands.
DNA fragments are protein linked
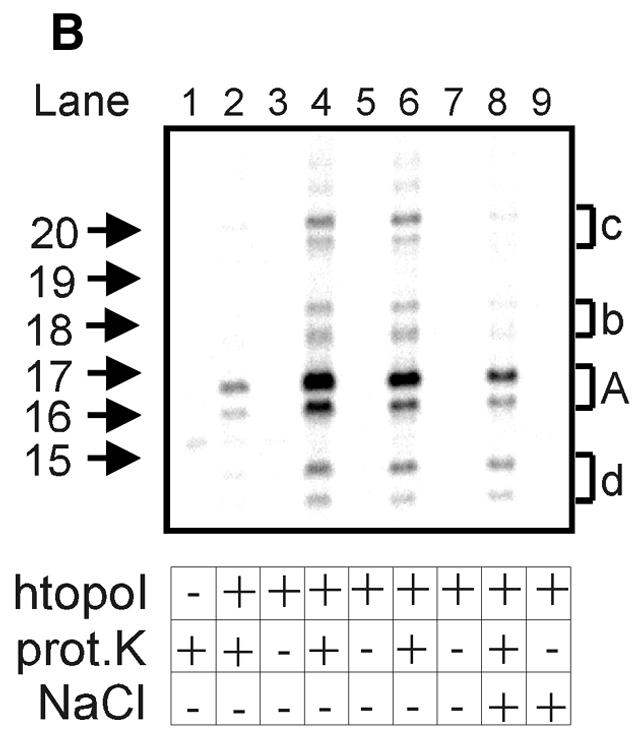
In order to investigate further if the observed bands represented DNA fragments with proteinase K-resistant peptides bound to the 3′ end, three parallel experiments with varying concentrations of yeast-expressed htopoI were performed. We hypothesised that without proteinase K digestion, the DNA from the four double bands should still enter the gel if they were DNA fragments without covalently attached protein moieties. In contrast, if htopoI was attached to the 3′ end, the DNA would no longer be able to enter the gel, but rather remain in the slot. As shown in Figure 3B (lanes 3, 5 and 7) none of the four double bands were detected when the proteinase K digest was omitted, indicating that a large molecule was bound to the DNA fragments.
The cleavage frequency depended on the amount of htopoI added (Fig. 3B, lanes 2 and 4; Fig. 3A, lanes 2–6), indicating that htopoI was indeed responsible for the formation of the double bands. Furthermore, because four double bands were produced, htopoI must have cleaved the substrate at several distinct positions just upstream from the radioactively labeled nucleotide. It is well known that a high concentration of salt shifts the equilibrium of htopoI from the cleaving to the non-bound state. Thus, if cleavage was due to htopoI, it should be possible to reverse the cleavage reactions by the addition of high concentrations of salt. Therefore, one reaction was stopped with 300 mM NaCl and incubated for another 10 min at 37°C prior to the addition of SDS. The reaction mixture was then divided into two halves, and one portion was treated with proteinase K (Fig. 3B, lane 8) whereas the other aliquot was kept untreated (Fig. 3B, lane 9). Salt treatment provoked a clear reduction of the band intensities (Fig. 3B, lanes 6 and 8). For bands A and c, product formation was diminished by 60–70%, while for bands b and d, a reduction of ∼40% was observed. When the samples were incubated for longer than 40 min after the addition of htopoI, the intensities of the cleavage products A, b, c and d declined, but did not completely disappear (data not shown). A similar effect was seen with the salt reversal shown in Figure 3B (lane 8). This strongly suggests that htopoI was responsible for these cleavage reactions, because a putative contaminating endonuclease would be expected to produce an accumulating number of cuts with increasing incubation time.
Position of the second cleavage site on L193s
The results from Figure 3A and B were further supported by comparing digests performed with either proteinase K (Fig. 4, lanes 1 and 2), trypsin (Fig. 4, lane 3), or without protease treatment (Fig. 4, lane 4). In these experiments only band A was clearly seen, because cleavages b, c and d were too weak to be detected. Trypsin produced a different digestion pattern (Fig. 4, lanes 2 and 3). Thus, the double bands produced by proteinase K treatment became a single band when trypsin digestion was employed. It has been previously shown that a complete trypsin digestion of DNA-bound htopoI leaves seven amino acids bound to the DNA (12,24). This supports the results shown in Figure 3B where cleavage by a second htopoI only occurred on substrates that already contained a suicidally attached htopoI molecule. The cleavage product, named A*, represented the same cleavage site as the proteinase K double band A. This shows that proteinase K digestion yielded a protease-resistant peptide with two different lengths most likely corresponding to 5 and 6 amino acids, respectively.
Figure 4.

Position of the second cleavage. Autoradiogram of a 14% sequencing gel. L193s was incubated with: lane 1, no htopoI; lanes 2–4, 500 ng htopoI for 40 min at 37°C. The reactions shown in lanes 1, 2 and 4 were terminated by the addition of SDS and subsequently aliquots 1 and 2 were incubated with proteinase K. The reaction shown in lane 3 was stopped by the addition of trypsin. Lane 4, no protease was added.
Baculovirus-expressed htopoI was used to produce the results shown in Figure 4. The fact that the cleavage sites from Figure 3A and B were reproduced with a highly purified htopoI from another source (Fig. 2B) strongly supports the view that htopoI was responsible for the cleavage reaction. In order to identify the precise location of the cleavage sites we relied on the experimental finding that trypsin cleavage of htopoI covalently bound to DNA generates a mobility shift of ∼5 nt in a 14% sequencing gel (12,24). Because A* had a mobility of 18 nt, the size of the covalently bound DNA fragment can be estimated to 13 nt. Thus, we assume that the incision occurred ∼13 nt upstream from the suicidally bound htopoI. Comparable mobility shifts were also observed for bands b, c and d (data not shown). Therefore, it can be estimated that these bands resulted from cleavages that took place 15, 17 and 11 nt upstream from the covalently attached htopoI, respectively. Because the radioactive label was placed 12 nt upstream from the suicidal cleavage site, band d may also have resulted from a fraction of the suicidally cleaved substrates that were not cleaved at position three, but instead at position five. It is well known that the majority of htopoI incisions take place at position three, but a minor fraction occurs at position five in the htopoI recognition sequence, which was included in our substrate L193s (25,26).
The second cleavage reaction is sensitive to CPT treatment
To further support the view that the cleavage reaction was caused by a htopoI, L193s and htopoI were incubated in the presence of the topoisomerase I-specific drug CPT. A titration with CPT was performed and the influence on the suicide cleavage as well as the cleavage complex specific incisions was tested. CPT showed no effect on the suicide cleavage (Fig. 5A). This, however, is not surprising because the suicide substrate has the same effect as CPT, namely the inhibition of the religation step. Therefore CPT cannot block ligation at the site of suicide cleavage because religation is already inhibited. The effect of CPT on the second cleavage reaction is shown in Figure 5B and C. In the presence of CPT up to ∼12-fold stimulation was observed. However, because DMSO alone also showed a ∼2-fold stimulation, the stimulatory effect of CPT may have been at least 6-fold.
Figure 5.



CPT sensitivity of the second cleavage. Baculovirus-expressed htopoI (500 ng) was incubated with the suicide substrate in the presence of DMSO or CPT as indicated. C, without htopoI; htopoI, htopoI under standard conditions. (A) The graphics represent a quantification of the percentage of suicide cleavage in relation to the total substrate used. (B) Quantification of the relative effect of DMSO and CPT on the second cleavage site ‘A’. (C) Autoradiogram of a 14% sequencing gel showing the results quantified in (B). The results shown are representative of three independent experiments.
The autoradiogram in Figure 5C indicates that CPT also had a stimulatory effect on cleavage sites b, c and, to a smaller extent, d. These alternate cleavage reactions were rather rare events compared to the cleavage at position A.
Limited digest with subtilisin did not affect the suicide cleavage, but eliminated the second cleavage step
The short distance from the suicidal cleavage site to cleavage sites A, b, c and d suggests that the two htopoI molecules were in extreme proximity of each other on the DNA substrate. Therefore, we wanted to investigate whether protein interactions between the two enzymes were taking place. To investigate this we performed a limited proteolysis of htopoI with subtilisin. Stewart et al. (27) reported that limited proteolysis of htopoI with subtilisin removed the N-terminal domain by specifically cleaving in the linker region so that the core and the C-terminal domain were liberated, but remained associated with each other and sustained catalytic activity. More recently, Ireton et al. (28) generated a mutant htopoI that lacked the N-terminal domain and parts of the linker domain. After purifying this enzyme both monomers and dimers were present. Based on these findings a model for the dimerisation was set up and a ‘domain swapping’ interaction of the dimer was suggested. According to this model the core domain of one enzyme binds to the C-terminal domain of the other one and vice versa. We therefore tested whether such a model could explain our results. HtopoI was digested with subtilisin as described. The course of the reaction was followed by SDS–PAGE and Coomassie staining (Fig. 6A). A minor fraction of htopoI was converted to a 80 kDa form (Fig. 6A, lane 2; marked with a scroll symbol) while the major fraction had a molecular weight of ∼60 kDa (Fig. 6A, lane 2; marked with dollar symbol). With increasing concentrations of subtilisin the 80 kDa form was completely converted to the 60 kDa product (Fig. 2, lanes 3–5). Based upon published data (27) it was known that the 80 kDa fragment had lost the N-terminal domain while the 60 kDa fragment had lost both the N- and C-terminal domains. We treated L193s with these different digestion products and measured the amount of suicide cleavage. The bandshift caused by the suicide cleavage reaction changed drastically after digestion with subtilisin (Fig. 6B, lanes 2 and 3). The bandshift caused by undigested htopoI (marked with an asterisk) is shown in lane 2. In lane 3 the upper band represents the bandshift caused by the 80 kDa fragment (marked with a scroll symbol) which has a slightly faster mobility than the full-length htopoI. The lower migrating band (marked with a hash symbol) is due to the attachment of the ∼15 kDa C-terminal domain of htopoI (containing the active site tyrosine) which associates with the 60 kDa fragment under native conditions. From the data presented in Figure 6B it can be concluded that the 60 kDa core domain and the 15 kDa C-terminal domain retain enough activity to support an effective suicide cleavage reaction although this was slightly less effective than that caused by the full-length enzyme. The ability of these htopoI fragments to perform the double cleavage reaction is shown in Figure 6C. When lanes 2 and 3 are compared, a dramatic effect can be seen. Despite the fact that the digested htopoI still retained the ability to perform the suicide cleavage step (Fig. 6B, lanes 3–5) the ability to carry out the double cleavage reaction was lost (Fig. 6C, lanes 3–6). In Figure 6C (lanes 3 and 4) a residual activity of the cleavage product A was still detectable, but this was probably caused by a small fraction of 80 kDa htopoI in these reactions. The data from Figure 6B and C are presented diagrammatically in Figure 6D. Although the degraded forms of htopoI as shown in lanes 2 and 3 retained ∼60% of the suicide cleavage, the ability to perform the double cleavage reaction was reduced to <10% or even totally lost. Because the enzyme was still active this suggests that a protein interaction may be necessary for the double cleavage to occur. This protein contact could be mediated through the core and C-terminal domain. Thus, when only the 80 kDa fragment was produced by subtilisin digestion this had no effect on the suicide or double cleavage (data not shown). The latter result rules out that the N-terminus was involved in the dimerisation reaction.
Figure 6.
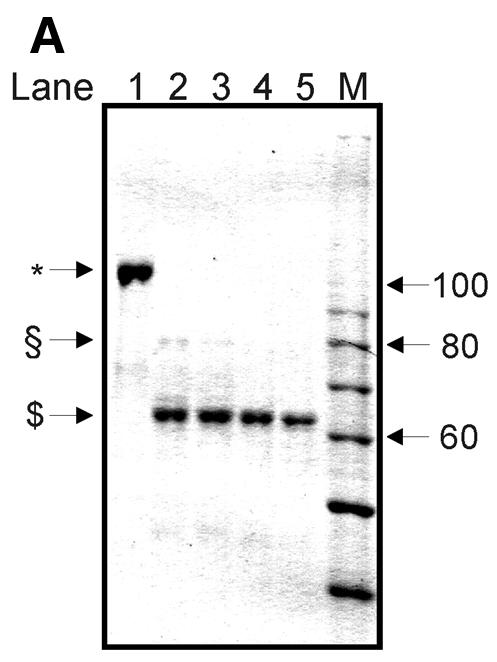
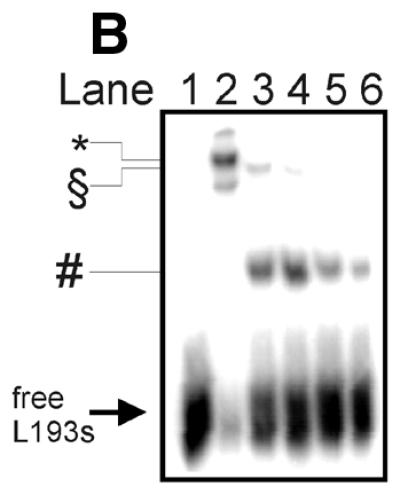


Limited digest of baculovirus-expressed htopoI with subtilisin. (A) Coomassie staining of a 7.5% polyacrylamide gel showing the partial digest of htopoI by subtilisin. HtopoI (2 µg) was incubated with 0, 20, 40, 60 and 120 ng subtilisin for 20 min at room temperature and 800 ng htopoI of each reaction was loaded in lanes 1–5 respectively. Full-length htopoI is indicated with an asterisk; htopoI lacking the N-terminus is indicated with a scroll symbol; core domain lacking the N- and C-terminus is indicated with a dollar symbol; M indicates the 10 kDa protein ladder. (B) Autoradiogram of a 7.5% polyacrylamide gel. L193s was incubated with: lane 1, no htopoI; lanes 2–6, htopoI; and subtilisin at a concentration of , 20 (lane 3),40 (lane 4), 60 (lane 5) and 120 ng (lane 6). The asterisk represents the shift caused by suicide cleavage of full-length htopoI; the scroll symbol represents the suicide cleavage of htopoI missing the N-terminus (80 kDa); the hash symbol represents the suicide cleavage of the C-terminal domain and the core domain; however, only the C-terminal domain is covalently attached to the substrate. (C) Autoradiogram of a 14% sequencing gel. The same numbering is used as in (B). (D) The results from (B) and (C) were quantified and depicted in a column chart. The numbering on the x-axis represents the same as described in (B) and (C). Black bars represent the relative degree of suicide cleavage and quantify the results shown in (B); gray bars represent the relative degree of second cleavage site ‘A’ and quantify the results shown in (C).
DISCUSSION
This study demonstrated that recombinant htopoI expressed in S.cerevisiae or by baculovirus is able to recognize a htopoI cleavage complex and cleave predominantly 13 nt upstream from this complex. Figure 7 is a schematic representation of these cleavage reactions. If only the suicidally cleaved DNA is taken into consideration, the double cleavage efficiency was found to be up to 12% in all experiments. In the presence of CPT this efficiency could be increased by 3–6-fold up to a maximum of 35%. The efficiency was dependent on the quality of the DNA substrate, which was purified fresh for most of the different experiments.
Figure 7.

Schematic representation of the sites of double cleavage. The cleavage sites are indicated by arrows and the same lettering is used as in Figures 3A and B, 4, 5C and 6C. The size of the arrow indicates the cleavage frequency at this site.
The recognition process may have been mediated through dimer formation between the two htopoI molecules on the DNA substrate. Ireton et al. (28) observed that a mutant htopoI (topo70ΔL), which lacked the N-terminus and had a shortened linker, could form dimers in solution. Based on limited digestions of htopoI with subtilisin a model was suggested according to which the C-terminus of one enzyme binds to the core of another enzyme and vice versa. This phenomenon was called domain swapping. Data presented in Figure 6A–D suggest that a similar dimerisation may have taken place. A htopoI enzyme binds and cleaves DNA suicidally. Thus, it is blocked in the catalytic cycle and is ‘frozen’ in the cleavage conformation. This conformation was shown by Stewart et al. (27) to be different to the non-bound state especially in the linker region. It is therefore possible that the interaction between the C-terminus and the core domain is less strong when compared to the non-bound state. This may allow the possibility that the core domain of a second htopoI interacts with the C-terminus of the ‘frozen’ htopoI and simultaneously cleaves the DNA, thus generating the second cleavage seen in our experiments through the formation of a dimer. However, if the linker of htopoI is broken, the second htopoI enzyme may still cleave upstream from the frozen htopoI and also ‘swap’ core domains, but because the linker is broken there are no longer protein contacts between the first and second htopoI that hold the latter in place. Therefore, after religation the second htopoI is released and able to diffuse away. Thus, the second cleavage reaction can no longer be detected. Although still hypothetical this model is nevertheless supported by the data presented in Figure 6C, and by the very close proximity between the two cleavage sites. The distance of 13 nt corresponds to ∼44 Å. Redinbo et al. (29) and Stewart et al. (30) published the crystal structure of a reconstituted htopoI bound or complexed to DNA. They found that htopoI contacted DNA 4 bp upstream and 6 bp downstream from the cleavage site in a htopoI recognition sequence similar to the one used by us. When bound to DNA htopoI covered 60 Å along the DNA. Because the 44 Å we found is considerably less than the 60 Å published for the crystal structure, direct protein–protein interactions between the two adjacent htopoI molecules are highly likely. In addition the longest distance we found between the two cleavage sites was ∼58 Å or 17 nt. Protein–protein interactions should not exceed a distance of 60 Å, which again supports that direct protein interactions could take place leading to dimer formation.
The presence of such dimers in vivo was recently suggested by Mao et al. (31). MCF-7 cells were irradiated with UV and dimethyl suberimidate (DMS) was subsequently added to the media in order to crosslink proteins. The chromosomal DNA was then extracted, bound to a filter, and probed by antibodies for htopoI bound covalently to the DNA. Using this technique there was a significant increase in the amount of htopoI bound covalently to the UV irradiated DNA when DMS was added. This was interpreted as a possible dimer formation between two htopoI molecules that occurred only when the DNA was irradiated with UV (31). Lesions such as abasic sites, UV photoproducts and oxidized bases are preferentially bound by htopoI and all these lesions lead to a stabilization of the enzyme in the cleaved state (9,13,32,33). This situation is mimicked by our suicide substrate. Thus, a recognition of the frozen htopoI as proposed for our in vitro system may also be true for htopoI frozen in the vicinity of the DNA damages in vivo. Such a mechanism would also ensure that a topoI dimer does not form in the absence of damages, because the cleaved state during a normal catalytic cycle of htopoI would be too short to be recognized by another htopoI molecule.
When a htopoI enzyme recognizes a DNA lesion and forms a cleavage complex it is principally able to perform the religation step and diffuse away. However, religation and diffusion is much slower on damaged DNA. The cleavage complex may also be converted into an irreversible lesion (15,34) that effectively blocks transcription and replication. Several groups have searched for a mechanism that would repair such a lesion (17–19), but, until now, very little has been known about the removal of such lesions in human cells. It is possible that the recognition of a cleavage complex is the initiation of a repair event. If, however, there are too many lesions it may also serve as a signal for the induction of apoptosis. If only a few complexes are present a repair pathway could be initiated via recombination with the second htopoI enzyme being used as a ligase. Several groups have shown that htopoI may be involved in recombination events (11,26,35–38). At present this is still a hypothesis which is, however, undergoing further investigation in our laboratory.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Dr Kent Christiansen for important discussions in the beginning of this project and Dr Birgitta R. Knudsen and Dr Michael Lisby (Aarhus) for fruitful discussions and recombinant topoisomerase I expressed in yeast during the experimental work. We are also grateful to Dr Hella Hartmann (Jena) for critical discussions of our data and for helping us to prepare the manuscript. We also wish to thank Dr James Champoux for sending us the wild-type htopoI baculovirus clone. This work was supported by the Deutsche Forschungsgemeinschaft, the Deutsche Krebshilfe and the Danish Research Council.
References
- 1.Stewart A.F. and Schutz,G. (1987) CPT-induced in vivo topoisomerase I cleavages in the transcriptionally active tyrosine aminotransferase gene. Cell, 50, 1109–1117. [DOI] [PubMed] [Google Scholar]
- 2.Stewart A.F., Herrera,R.E. and Nordheim,A. (1990) Rapid induction of c-fos transcription reveals quantitative linkage of RNA polymerase II and DNA topoisomerase I enzyme activities. Cell, 60, 141–149. [DOI] [PubMed] [Google Scholar]
- 3.Snapka R.M., Powelson,M.A. and Strayer,J.M. (1988) Swiveling and decatenation of replicating simian virus 40 genomes in vivo. Mol. Cell. Biol., 8, 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang L., Wold,M.S., Li,J.J., Kelly,T.J. and Liu,L.F. (1987) Roles of DNA topoisomerases in simian virus 40 DNA replication in vitro. Proc. Natl Acad. Sci. USA, 84, 950–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen A.H., Bendixen,C. and Westergaard,O. (1996) DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 6.Ryan A.J., Squires,S., Strutt,H.L. and Johnson,R.T. (1991) Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res., 19, 3295–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Squires S., Ryan,A.J., Strutt,H.L., Smith,P.J. and Johnson,R.T. (1991) Deoxyguanosine enhances the cytotoxicity of the topoisomerase I inhibitor camptothecin by reducing the repair of double-strand breaks induced in replicating DNA. J. Cell Sci., 100, 883–893. [DOI] [PubMed] [Google Scholar]
- 8.Kingma P.S. and Osheroff,N. (1998) The response of eukaryotic topoisomerases to DNA damage. Biochim. Biophys. Acta, 1400, 223–232. [DOI] [PubMed] [Google Scholar]
- 9.Pourquier P., Ueng,L.-M., Fertala,J., Wang,D., Park,H.-J., Essigman,J.M., Bjornsti,M.-A. and Pommier,Y. (1999) Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA-containing oxidative base damages. 7, 8-dihydro-8-oxoguanine and 5-hydroxycytosine. J. Biol. Chem., 274, 8516–8523. [DOI] [PubMed] [Google Scholar]
- 10.Pourquier P., Ueng,L.-M., Kohlhagen,G., Mazumder,A., Gupta,M., Kohn,K.W. and Pommier,Y. (1997) Effects of uracil incorporation, DNA mismatches and abasic sites on cleavage and religation activities of mammalian topoisomerase I. J. Biol. Chem., 272, 7792–7796. [DOI] [PubMed] [Google Scholar]
- 11.Pourquier P., Pilon,A.A., Kohlhagen,G., Mazumder,A., Sharma,A. and Pommier,Y. (1997) Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps. Importance of DNA end phosphorylation and camptothecin effects. J. Biol. Chem., 272, 26441–26447. [DOI] [PubMed] [Google Scholar]
- 12.Christiansen K. and Westergaard,O. (1999) Mapping of eukaryotic DNA topoisomerase I catalyzed cleavage without concomitant religation in the vicinity of DNA structural anomalies. Biochim. Biophys. Acta, 1489, 249–262. [DOI] [PubMed] [Google Scholar]
- 13.Lanza A., Tornaletti,S., Rodolfo,C., Scanavini,M.C. and Pedrini,A.M. (1996) Mapping of eukaryotic DNA topoisomerase I catalyzed cleavage without concomitant religation in the vicinity of DNA structural anomalies. J. Biol. Chem., 271, 6978–6986. [DOI] [PubMed] [Google Scholar]
- 14.Shcherbakova O.G. and Filatov,M.V. (2000) Campothecin enhances random integration of transfected DNA into the genome of mammalian cells. Biochim. Biophys. Acta, 1495, 1–3. [DOI] [PubMed] [Google Scholar]
- 15.Wu J. and Liu,L.F. (1997) Processing of topoisomerase I cleavable complexes into DNA damage by transcription. Nucleic Acids Res., 25, 4181–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai S.D., Liu,L.F., Vazquez-Abad,D. and D’Arpa,P. (1997) Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J. Biol. Chem., 272, 24159–24164. [DOI] [PubMed] [Google Scholar]
- 17.Sastry S. and Ross,B.M. (1998) Mechanisms for the processing of a frozen topoisomerase-DNA conjugate by human cell-free extracts. J. Biol. Chem., 273, 9942–9950. [DOI] [PubMed] [Google Scholar]
- 18.Yang S.-W., Burgin,A.B., Huizenga,B.N., Robertson,C.A., Yao,K.C. and Nash,H.A. (1996) A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl Acad. Sci. USA, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pouliot J.J., Yao,K.C., Robertson,C.A. and Nash,H.A. (1999) Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science, 286, 552–555. [DOI] [PubMed] [Google Scholar]
- 20.Lisby M., Krogh,B.O., Boege,F., Westergaard,O. and Knudsen,B.R. (1998) Camptothecins inhibit the utilization of hydrogen peroxide in the ligation step of topoisomerase I catalysis. Biochemistry, 37, 10815–10827. [DOI] [PubMed] [Google Scholar]
- 21.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 22.Busk H., Thomsen,B., Bonven,B.J., Kjeldsen,E., Nielsen,O.F. and Westergaard,O. (1987) Preferential relaxation of supercoiled DNA containing a hexadecameric recognition sequence for topoisomerase I. Nature, 327, 638–640. [DOI] [PubMed] [Google Scholar]
- 23.Stevnsner T., Mortensen,U.H., Westergaard,O. and Bonven,B.J. (1989) Interactions between eukaryotic DNA topoisomerase I and a specific binding sequence. J. Biol. Chem., 264, 10110–10113. [PubMed] [Google Scholar]
- 24.Svejstrup J.Q., Christiansen,K., Andersen,A.H., Lund,K. and Westergaard,O. (1990) Minimal DNA duplex requirements for topoisomerase I-mediated cleavage in vitro. J. Biol. Chem., 265, 12529–12535. [PubMed] [Google Scholar]
- 25.Christiansen K., Svejstrup,A.B.D., Andersen,A.H. and Westergaard,O. (1993) Eukaryotic topoisomerase I-mediated cleavage requires bipartite DNA interaction. Cleavage of DNA substrates containing strand interruptions implicates a role for topoisomerase I in illegitimate recombination. J. Biol. Chem., 268, 9690–9701. [PubMed] [Google Scholar]
- 26.Christiansen K. and Westergaard,O. (1994) Characterization of intra- and intermolecular DNA ligation mediated by eukaryotic topoisomerase I. Role of bipartite DNA interaction in the ligation process. J. Biol. Chem., 269, 721–729. [PubMed] [Google Scholar]
- 27.Stewart L., Ireton,G.C. and Champoux,J.J. (1996) The domain organization of human topoisomerase I. J. Biol. Chem., 271, 7602–7608. [DOI] [PubMed] [Google Scholar]
- 28.Ireton G.C., Stewart,L., Parker,L.H. and Champoux,J.J. (2000) Expression of human topoisomerase I with a partial deletion of the linker region yields monomeric and dimeric enzymes that respond differently to camptothecin. J. Biol. Chem., 275, 25820–25830. [DOI] [PubMed] [Google Scholar]
- 29.Redinbo M.R., Stewart,L., Kuhn,P., Champoux,J.J. and Hol,W.G.J. (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- 30.Stewart L., Redinbo,M.R., Qiu,X., Hol,W.G.J. and Champoux,J.J. (1998) A model for the mechanism of human topoisomerase I. Science, 279, 1534–1541. [DOI] [PubMed] [Google Scholar]
- 31.Mao Y., Okada,S., Chang,L.-S. and Muller,M.T. (2000) p53 dependence of topoisomerase I recruitment in vivo. Cancer Res., 60, 4538–4543. [PubMed] [Google Scholar]
- 32.Kjeldsen E., Mollerup,S., Thomsen,B., Bonven,B.J., Bolund,L. and Westergaard,O. (1988) Sequence-dependent effect of camptothecin on human topoisomerase I DNA cleavage. J. Mol. Biol., 202, 333–342. [DOI] [PubMed] [Google Scholar]
- 33.Subraminian D., Rosenstein,B.S. and Muller,M.T. (1998) Ultraviolet-induced DNA damage stimulates topoisomerase I–DNA complex formation in vivo: possible relationship with DNA repair. Cancer Res., 58, 976–984. [PubMed] [Google Scholar]
- 34.Tsao Y.-P., Russo,A., Nyamuswa,G., Silber,R. and Liu,L.F. (1993) Interaction between replication forks and topoisomerase I–DNA cleavable complexes: studies in a cell-free SV40 DNA replication system. Cancer Res., 53, 5908–5914. [PubMed] [Google Scholar]
- 35.Zhu J. and Schiestl,R.H. (1996) Topoisomerase I involvement in illegitimate recombination in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Megonigal M.D., Fertala,J. and Bjornsti,M.-A. (1997) Alterations in the catalytic activity of yeast DNA topoisomerase I result in cell cycle arrest and cell death. J. Biol. Chem., 272, 12801–12808. [DOI] [PubMed] [Google Scholar]
- 37.Cheng C., Kussie,P., Pavletic,N. and Shuman,S. (1998) Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell, 92, 841–850. [DOI] [PubMed] [Google Scholar]
- 38.Pourquier P., Jensen,A.D., Gong,S.S., Pommier,Y. and Rogler,C.E. (1999) Human DNA topoisomerase I-mediated cleavage and recombination of duck hepatitis B virus DNA in vitro. Nucleic Acids Res. ,27, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]