

Abstract
Enhanced expression of cyclooxygenase 2 (COX2) in podocytes contributes to glomerular injury in diabetic kidney disease, but some basal level of podocyte COX2 expression might be required to promote podocyte attachment and/or survival. To investigate the role of podocyte COX2 expression in diabetic kidney disease, we deleted COX2 specifically in podocytes in a mouse model of Type 1 diabetes mellitus (Akita mice). Podocyte-specific knockout (KO) of COX2 did not affect renal morphology or albuminuria in nondiabetic mice. Albuminuria was significantly increased in wild-type (WT) and KO Akita mice compared with nondiabetic controls, and the increase in albuminuria was significantly greater in KO Akita mice compared with WT Akita mice at both 16 and 20 wk of age. At the 20-wk time point, mesangial expansion was also increased in WT and KO Akita mice compared with nondiabetic animals, and these histologic abnormalities were not improved by KO of COX2. Tubular injury was seen only in diabetic mice, but there were no significant differences between groups. Thus, KO of COX2 enhanced albuminuria and did not improve the histopathologic features of diabetic kidney disease. These data suggest that 1) KO of COX2 in podocytes does not ameliorate diabetic kidney disease in Akita mice, and 2) some basal level of podocyte COX2 expression in podocytes is necessary to attenuate the adverse effects of diabetes on glomerular filtration barrier function.
Keywords: diabetes mellitus, diabetic nephropathy, glomerular podocyte, cyclooxygenase, eicosanoids
diabetic nephropathy (DN) is a serious complication of both Type 1 and Type 2 diabetes mellitus (31). The economic consequences of the disease are significant because DN is the most common cause of end-stage kidney disease in developed countries (31), and both the incidence and prevalence of diabetes are increasing worldwide (27, 31). While current therapies slow disease progression (1, 26), ∼20% of patients with overt DN ultimately develop end-stage renal disease, requiring renal replacement therapy to sustain life (27, 31). As a result, much effort has been devoted to identifying new therapeutic targets and treatment strategies.
A large body of evidence suggests that glomerular podocytes play a key role in DN (51). These highly differentiated cells are important for maintaining the integrity of the glomerular filtration barrier (17, 50). In diabetes, podocyte injury is a common feature of both experimental and human diabetic kidney disease (17, 51). In advanced disease, a reduction in podocyte number and/or density is characteristic of both human DN and animal models of diabetic kidney disease (7, 29, 40, 48). Because podocytes are terminally differentiated cells having limited ability to replicate (24, 50), a decrease in podocyte number leads to instability of the glomerular tuft and glomerulosclerosis (50). Although the cause of podocyte loss is multifactorial, apoptosis and detachment likely play prominent roles (39, 51). An important mediator of podocyte injury in diabetes is cyclooxygenase 2 (COX2) (9, 19, 44). In diabetic kidney disease, COX2 is upregulated in glomerular podocytes (21), and treatment with COX2 inhibitors reduces albuminuria in both animal models (5, 36) and humans with DN (42). Moreover, the COX2 metabolites PGE2 and thromboxane A2 (TxA2) promote podocyte apoptosis (3, 44), and overexpression of COX2 specifically in glomerular podocytes in vivo exacerbated diabetic kidney disease in streptozotocin (STZ)-treated mice (3, 4). On the basis of these observations, we hypothesized that inhibition of eicosanoid synthesis specifically in podocytes would attenuate glomerular injury in diabetes. To investigate this hypothesis, we created diabetic Akita mice lacking COX2 specifically in glomerular podocytes using COX2-flox/flox mice (43), and the podocyte-specific podocin promoter to express Cre recombinase specifically in glomerular podocytes in vivo (30). Using this model, we examined the effect of podocyte-specific COX2 deletion on the severity of kidney disease in diabetic Akita mice.
MATERIALS AND METHODS
Experimental protocol.
All experiments were performed using male mice on the 129/SvEv background for >10 generations. Male mice were used for the experiments because female 129/SvEv Akita mice develop only mild hyperglycemia, as well as modest functional and histologic features of diabetic kidney disease (12). In previous studies (2), male 129/SvEv Akita mice have been shown to develop albuminuria and early histopathologic features of DN (13). To create Akita mice lacking COX2 in podocytes, we obtained mice with loxP sites flanking exons 6-8 of the COX2 genetic locus (43) from Dr. Garret S. Fitzgerald at the University of Pennsylvania (COX2-flox/flox mice). These exons are critical for the enzymatic function of COX2 (43). To create mice lacking COX2 specifically in podocytes, COX2-flox/flox mice were crossed with mice expressing Cre recombinase under the regulation of the podocyte-specific podocin promoter (NPHS2-Cre mice) (30) from Jackson Laboratories (stock no. 008205). To determine the effectiveness of Cre-mediated recombination, NPHS2-Cre mice were crossed with reporter mice (32), expressing a two-color fluorescent Cre reporter allele (Jackson Laboratories, stock nos. 007576 and 007676). Genotyping was performed using previously described techniques (13, 30, 32).
For the experiments, the following groups were studied: wild-type (WT) Akita mice, COX2 KO Akita mice (KO Akita mice), nondiabetic WT controls (COX2+/+ mice), and nondiabetic KO mice (COX2−/−POD mice). In the first set of experiments, nondiabetic COX2−/−POD mice were compared with age-matched nondiabetic COX2+/+ mice to examine the functional and histopathologic effects induced by KO of COX2 in podocytes. A subset of these mice was euthanized at 6 wk of age, and kidneys were examined by light microscopy and transmission electron microscopy (TEM), as described below. The remainder of the mice had urine collected at 12 and 20 wk of age for measurement of albuminuria, as described below. In the second set of experiments, WT Akita mice, KO Akita mice, nondiabetic COX2+/+ mice, and nondiabetic COX2−/−POD mice were examined. Blood glucose measurements were made at 8, 12, 16, and 20 wk of age using the AlphaTRAK 2 testing system (Abbott Laboratories, Chicago, IL) calibrated for glucose measurements in mice, according to directions of the manufacturer. Twenty-four-hour urine collections were obtained at 12, 16, and 20 wk of age. Systolic blood pressure (SBP) was measured at 12 and 20 wk of age, as described below. After the last urine collection, mice were given an injection of pentobarbital sodium (250 mg/kg ip). When the mice were unconscious, blood was obtained using a retro-orbital approach, placed in an Eppendorf tube, and allowed to clot at room temperature. After collecting blood, kidneys were rapidly removed and placed in ice-cold saline. The left kidney and most of the right kidney tissue were used to isolate sufficient glomeruli for immunoblotting and collection of mRNA. The remaining tissue was divided and saved in formalin for light microscopy, 8% glutaraldehyde for electron microscopy or frozen in OCT (optimal cutting temperature) compound for immunofluorescence studies. Serum was collected after centrifugation of the clotted blood at 1,500 g at 4°C for 15 min and stored at −70°C until assayed. The experiments conformed to the Guide for the Care and Use of Laboratory Animals (34), and the studies were approved by both the Duke and Durham Veterans Administration Medical Centersʼ Institutional Animal Care and Use Committee.
SBP measurements.
SBP was measured using a computerized tail-cuff system (Hatteras Instruments, Cary, NC) in conscious mice, as previously described (47). This technique has previously been shown to correlate closely with intra-arterial measurements (49).
Histopathology.
Light microscopy sections were stained with hematoxylin & eosin (H&E), as well as periodic acid Schiff and then evaluated by a pathologist (A. F. Buckley) who was blinded to genotype. Mesangial expansion, tubule dilation, and casts, as well as tubulointerstitial inflammation and fibrosis, were graded on a semiquantitative scale of 0–3 (0, normal, 1, mild, 2, moderate, 3, severe), as previously described (2).
Albuminuria and urinary eicosanoid excretion.
Urine collections were performed using metabolic cages specifically designed for collection of urine in mice (Hatteras Instruments). Urinary albumin concentrations were measured using a kit from AssayPro (St. Charles, MO), and urine creatinine levels were measured using a kit from Exocell (Philadelphia, PA). Urinary albumin excretion was expressed as the albumin·mg creatinine ratio−1·30 g body wt−1. Urinary eicosanoids were measured using kits from Cayman Chemical (Ann Arbor, MI), and data were expressed as pg·mg creatinine ratio−1·30 g body wt−1.
Serum cystatin C levels.
Serum levels of cystatin C were measured using a kit from R&D Systems (Minneapolis, MN), according to the manufacturer’s directions.
TEM.
Electron microscopy was performed as previously described (47). Analysis at the ultrastructural level was performed in a qualitative fashion, and areas of interest were selected in semithin sections for preparation of ultrathin sections for examination by a pathologist (A. F. Buckley) blinded to genotype.
Glomerular basement membrane width and number of filtration slits per micrometer of glomerular basement membrane.
Glomerular basement membrane (GBM) width was measured using the orthogonal intercept technique (18) and Photoshop CS6 Extended software after calibrating the measurement tool to convert pixels to a known distance in the digital images. For the actual measurements, the Photoshop software was used to place a grid over the digital images by choosing View>Show>Gridlines. At the intercept of each gridline, the minimal distance between the endothelial and epithelial cell membranes was measured (orthogonal intercept), avoiding the mesangial areas (8). An average of 62 ± 18 measurements was obtained per capillary loop, and an average of 9.4 ± 1.2 capillary loops was examined per mouse. The harmonic mean of these measurements was calculated to reduce the effect of outliers and then corrected for oblique sectioning using the correction factors described by Ramage et al. (37).
The extent of foot process (FP) effacement was quantitated by counting the number of filtrations slit per micrometer of GBM length after calibrating the measurement tool in Photoshop CS6 Extended software (see above) to convert pixels to a known distance in the digital images (47). All images were evaluated without knowledge of either genotype or experimental group.
Quantitation of glomerular volume, podocyte number, and podocyte density.
Podocyte density (podocytes per glomerular volume) [Nv(P/Glom)] was quantitated using the methodology described by Wiggins and colleagues (41), which requires the number of podocyte nuclei per glomerular profile and the section thickness (41). This method has the advantage of using a single histologic section and provides an estimate of podocyte density comparable to other more rigorous methodologies (41). The mean glomerular volume (VGlom) was obtained from the arithmetic mean of the glomerular surface area (AG) using the method of Weibel and the equation described by Gundersen and colleagues (16), as previously described (47). For the studies, podocyte nuclei were stained with a rabbit polyclonal WT1 (Wilms tumor 1) antibody (rhodamine) (Santa Cruz Biotechnology, Santa Cruz, CA; cat. no. sc-192), podocytes in the glomerular tuft were stained with a mouse monoclonal antibody to the podocyte marker synaptopodin (fluorescein) (Progen Biotechnik, cat. no. 65194), and nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI), as previously described (47). Podocyte nuclei were then quantitated by counting nuclei that both colocalized with DAPI and were associated with synaptopodin staining. Using this technique, we found that nuclear WT1 staining was consistently associated with synaptopodin-stained cells. Podocyte density was then quantitated using the methodology described by Wiggins and colleagues (41), as described above, and an Excel spreadsheet supplied as supplemental data to the article (41). An average of 22 ± 1.1 glomeruli (range 16–29) was evaluated per mouse, and the specimens were examined without knowledge of either genotype or experimental group.
The mean glomerular volume (VGlom) was obtained from the harmonic mean of the glomerular surface area using the method of Weibel and the equation described by Gundersen and colleagues (16): VGlom = K/β × (AG)3/2, where AG was obtained as described above, K equals 1.10 and is the distribution coefficient, and β equals 1.38 and is the shape coefficient, which pertains to spheres. For the studies, glomerular cross-sectional area was determined by evaluating digital images using Adobe Photoshop CS6 Extended software after calibrating the measurement tool to convert pixels to a known surface area. The glomerular area in the digital images was then outlined using the “lasso” tool, and the number of pixels in the glomerular area was converted to square micrometers. An estimate of the total number of podocytes per glomerulus was obtained by multiplying podocyte density [Nv(P/Glom] by the glomerular volume (VGlom) (25, 41). All measurements were made without knowledge of either genotype or experimental group.
Podocyte COX2 expression.
Expression of COX2 in podocytes was detected by indirect immunofluorescence using a rabbit monoclonal antibody to COX2 from Cell Signaling Technology (cat. no. 12282) and a mouse monoclonal antibody to the podocyte marker synaptopodin (Progen Biotechnik). For the studies, frozen tissue sections were fixed in 2% paraformaldehyde for 5 min, air-dried, treated with 0.1% Triton-X in Dulbecco’s phosphate buffered saline (D-PBS) for 5 min and then blocked for 1 h in 20 mM Tris·HCl, 137 mM NaCl, pH 7.6 (TBS) with 0.2% Tween 20 (T-TBS) and 5% BSA. The COX2 and synaptopodin primary antibodies were then added at a 1:500 dilution and 1:50, respectively, in T-TBS with 5% BSA. After 1 h, slides were washed three times in D-PBS and then incubated for 1 h with an Alexa Fluor 488-labeled donkey anti-rabbit antibody (Life Technologies, A21206) and a rhodamine-labeled goat anti-mouse antibody (Life Technologies, cat. no. A6393) both at a dilution of 1:1,000 in T-TBS with 5% BSA. Sides were then washed three times in D-PBS and then examined by confocal microscopy using a Zeiss 510 inverted confocal fluorescent microscope.
Immunoblotting of glomerular lysates.
Immunoblotting was performed as previously described (47) using the following antibodies: 1) a goat polyclonal antibody to renin 1 (R&D Systems, cat. no. AF4277) and 2) a mouse monoclonal antibody to β-actin (Chemicon International, cat. no. MAB1501R). For the studies, enriched glomerular pellets were solubilized in NP-40 lysis buffer [50 mM Tris·HCl, 150 mM sodium chloride, 2 mM EDTA, 1% IGEPAL CA-630 (NP-40)] with protease inhibitors (Sigma-Aldrich) by sonication and frozen at −70°C until the time of study. For immunoblotting, proteins were separated using the XCell SureLock Bis-Tris mini-cell electrophoresis system (Thermo Fisher Scientific) and transferred to PVDF (polyvinylidene fluoride) membranes, according to the directions of the manufacturer. Immunoblots were then blocked in 20 mM Tris·HCl, 137 mM NaCl, pH 7.6 (TBS) with 0.2% T-TBS, and 5% nonfat dry milk. The primary antibody was added at a concentration of 0.1 µg/ml in blocking buffer and was incubated overnight. After washing, the horseradish peroxidase (HRP)-linked secondary antibody to goat (Santa Cruz Biotechnology) was added a 1:2,000 in blocking solution and incubated for 1 h prior to washing. Protein detection was performed using enhanced chemiluminescence (Thermo Scientific), according to the directions of the manufacturer. To assess protein loading, immunoblots were stripped according to the directions of the manufacturer, and immunoblotting was performed using the mouse monoclonal antibody to β-actin (0.5 µg/ml) in blocking solution and an HRP-linked anti-mouse secondary antibody (1:2,000) from Santa Cruz Biotechnology. For densitometry, the immunoblots were converted into a digital format using an Epson Perfection scanner 1670 (Seiko Epsom Corporation) and were then analyzed using ScanAnalysis 2.5 software (Biosoft). For the analyses, the densitometric data for the renin protein signal were divided by the matched signal for β-actin. To compare separate immunoblots, densitometric data were normalized to nondiabetic COX2+/+ mice.
Coomassie staining protocol.
Urinary proteins were separated using the XCell SureLock Bis-Tris mini-cell electrophoresis system (Thermo Fisher Scientific). To compensate for the large differences in urine volumes between diabetic and nondiabetic mice, the amount of urine loaded was 1:250 of the total urine volume. Protein gels were then stained using 0.1% Coomassie Brilliant Blue R-250 (Bio-Rad, Hercules, CA) in 10% acetic acid 50% methanol for 20 min and then destained with 50% methanol and 10% acetic acid overnight.
Quantitative RT-PCR.
Enriched glomerular preparations, isolation of glomerular mRNA, and quantitative RT-PCR were performed as previously described (47). Quantitation of renin mRNA was performed using the primer pairs (forward: ATGAAGGGGGTGTCTGTGGGGTC and reverse: ATGCGGGGAGGGTGGGCACCTG), and the amplification signals were normalized to the endogenous β-actin mRNA levels. Quantitation of COX2 mRNA expression was performed using primer pairs described in a previous publication (46).
Statistical analysis.
Data are presented as the means ± SE, and statistical analyses were performed using the Prism 5 computer program (GraphPad Software, San Diego, CA). For comparison of continuous variables, a test of normality was performed (Kolmogorov–Smirnov test) prior to assessing statistical significance using the following statistical methods: 1) two-tailed t-test for variables passing the normality test, or 2) Mann-Whitney U-test for variables that were not normally distributed. For comparisons between more than two groups, statistical analyses included either a one-way ANOVA followed by a Bonferroni multiple-comparisons post hoc test for normally distributed variables, or a Kruskal-Wallis test followed by a Dunn multiple-comparisons post hoc test for variables that were not normally distributed. When more than two categories were present (such as diabetes and genotype), data were analyzed using a two-way ANOVA followed a Bonferroni multiple-comparisons post hoc test.
For noncontinuous variables, data were analyzed using a Fisher’s exact test. Histologic data were analyzed as a noncontinuous variable (categorical data) using the number of mice with the specified histologic abnormality. P values less than 0.05 were considered significant. Graphs of the histologic findings were presented as the percentage of mice with the specified abnormality to permit a more effective comparison of the differences between the experimental manipulations in studies with an imbalance in the number of mice in each group.
RESULTS
Cre-mediated recombination.
To determine the effectiveness of Cre-mediated recombination, mice expressing Cre recombinase specifically in podocytes (NPHS2-Cre mice) were crossed with a reporter animal expressing a two-color fluorescent Cre reporter allele (32). The reporter allele contains a cell membrane-localized, red fluorescence protein (tdTomato) with widespread expression in all tissues and cell types prior to Cre recombinase exposure. Expression of Cre recombinase induces a cell membrane-localized enhanced green fluorescence protein (EGFP). As shown in Fig. 1, green fluorescence is induced specifically in podocytes in transgenic mice expressing both the reporter construct and NPHS2-Cre, with red fluorescence confined to other cell types (Fig. 1A). Little EGFP expression was observed in mice expressing only the reporter construct (Fig. 1B). In data not shown, no fluorescence was observed in mice expressing only Cre recombinase.
Fig. 1.
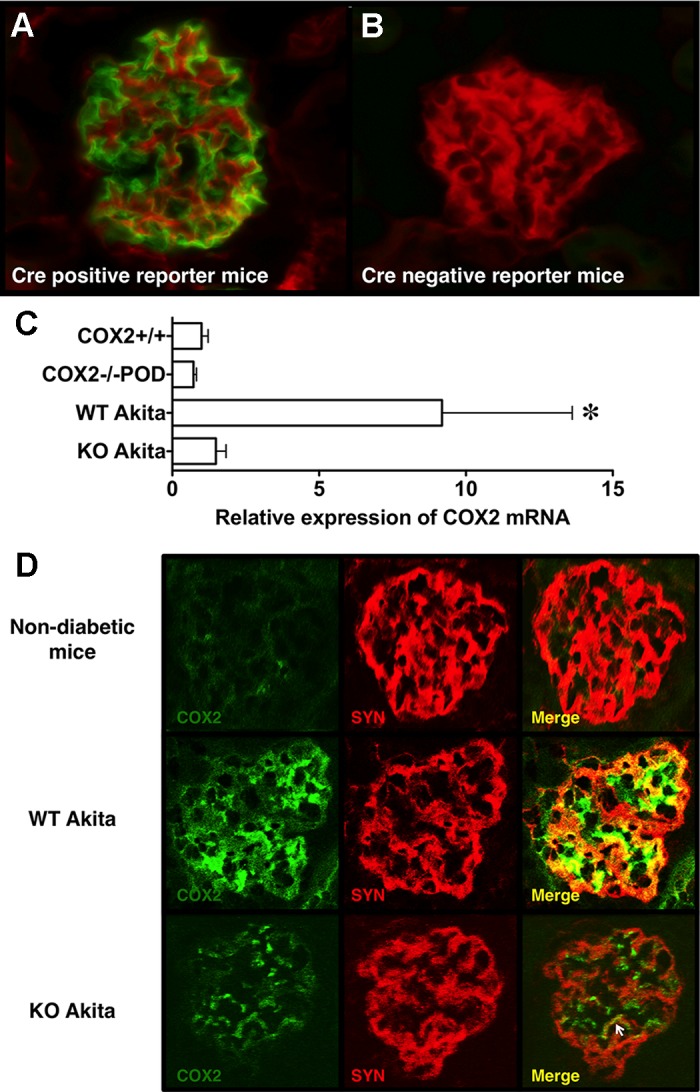
Effectiveness of Cre-mediated recombination in NPHS2-Cre mice. To examine Cre-medicated recombination in vivo, NPHS2-Cre mice were crossed with a transgenic (TG) reporter animal expressing a two-color fluorescent Cre reporter allele (32). The reporter allele contains a cell membrane-localized red fluorescence protein (tdTomato) with widespread expression in all tissues and cell types prior to Cre recombinase exposure. Expression of Cre recombinase induces a cell membrane-localized enhanced green fluorescence protein (EGFP). A: in TG mice expressing both transgenes, green fluorescence is induced specifically in podocytes with red fluorescence confined to other cell types. B: little EGFP expression was observed in mice expressing only the reporter construct. C: we next determined the effect of podocyte-specific knockout (KO) on COX2 mRNA expression using quantitative RT-PCR. The presence of diabetes significantly affected glomerular COX2 mRNA levels. The increase in glomerular COX2 mRNA in wild-type (WT) Akita mice was largely prevented by KO of podocyte COX2 in diabetic animals. D: representative glomerular images stained for COX2 (green) and the podocyte marker synaptopodin (SYN) (red) and examined by confocal microscopy. In nondiabetic COX2+/+ and COX2−/− mice, COX2 staining was faint and largely confined to mesangial areas. In WT Akita mice, COX2 staining was granular and increased in both mesangial areas and podocytes compared to nondiabetic mice. SYN was also granular in both groups of Akita mice, as we have seen in another model of podocyte injury (45). As shown in the merged images, podocyte COX2 expression (yellow) was decreased by ~70%, by KO of COX2 in Akita mice, although focal areas of COX2 expression were still detected in KO Akita amice (arrow). Mesangial COX2 expression was not affected KO of COX2 in podocytes of diabetic mice. For the immunofluorescence studies, three to six mice were examined per group. For the quantitative RT-PCR experiments, we studied 11 KO Akita, 10 WT Akita, 5 COX2+/+, and 5 COX2−/− mice. Statistical analyses were performed using a two-way ANOVA followed by a Bonferroni post-hoc analyses. *P < 0.05 vs nondiabetic mice and KO Akita mice.
To determine whether podocyte-specific Cre activation reduced expression of COX2, we measured COX2 expression in enriched glomerular preparations from both diabetic and nondiabetic mice using primers specific for COX2 (46). As shown in Fig. 1C, COX2 mRNA expression tended to be reduced in nondiabetic mice lacking COX2 specifically in podocytes (COX2−/−POD mice) compared with nondiabetic WT mice (COX2+/+ mice), but this difference was not statistically significant. The presence of diabetes significantly affected glomerular COX2 mRNA levels, as reported by other investigators (4). In knockout (KO) Akita mice, glomerular COX2 mRNA induction was largely prevented, resulting in a significant decrease in COX2 mRNA levels in KO Akita mice compared with WT Akita mice.
We next determined whether immunoreactive COX2 was decreased in podocytes of KO Akita mice. Figure 1D shows representative images of glomerular profiles stained for COX2 (green) and podocyte marker synaptopodin (red), as described in the materials and methods. In both groups of nondiabetic mice, COX2 staining was faint and largely confined to mesangial areas. In WT Akita mice, COX2 staining was increased and granular in both mesangial areas and podocytes compared with nondiabetic mice. Synaptopodin was also granular in both groups of Akita mice, as we have seen in another model of podocyte injury (45). In KO Akita mice, the increase in podocyte COX2 expression was decreased by ~70%, although there were focal areas of COX2 staining that colocalized with synaptopodin. Mesangial COX2 expression was not affected by KO of podocyte COX2 in diabetic mice. Taken together, these findings suggest that COX2 was effectively deleted in podocytes.
Effect of podocyte-specific COX2 KO on kidney pathology.
Systemic KO of COX2 causes renal dysplasia, characterized by smaller kidneys, as well as renal structural abnormalities that develop postnatally (35), including reduced kidney weight, tubular dilation and cyst formation, outer cortical glomerular hypoplasia, periglomerular fibrosis, variable glomerulosclerosis, and diffuse interstitial fibrosis. To determine whether podocyte-specific COX2 KO mice develop renal structural abnormalities, kidneys from male, nondiabetic COX2+/+ mice were compared with kidneys from male, nondiabetic COX2−/−POD mice. As shown in Fig. 2A, kidney weight per gram body weight was similar in nondiabetic COX2+/+ and nondiabetic COX2−/−POD mice. Moreover, body weight was similar in both groups of nondiabetic mice at the time points examined (Table 1). Mild mesangial expansion, as well as mild tubule dilation and casts, were observed in a few nondiabetic mice, but there were no significant differences in either mesangial expansion or tubular dilation and casts between COX2+/+ and COX2−/−POD mice (Fig. 2, B–E). There was also no evidence of cyst formation, glomerular hypoplasia, periglomerular fibrosis, glomerulosclerosis, or interstitial fibrosis in any of the animals studied.
Fig. 2.
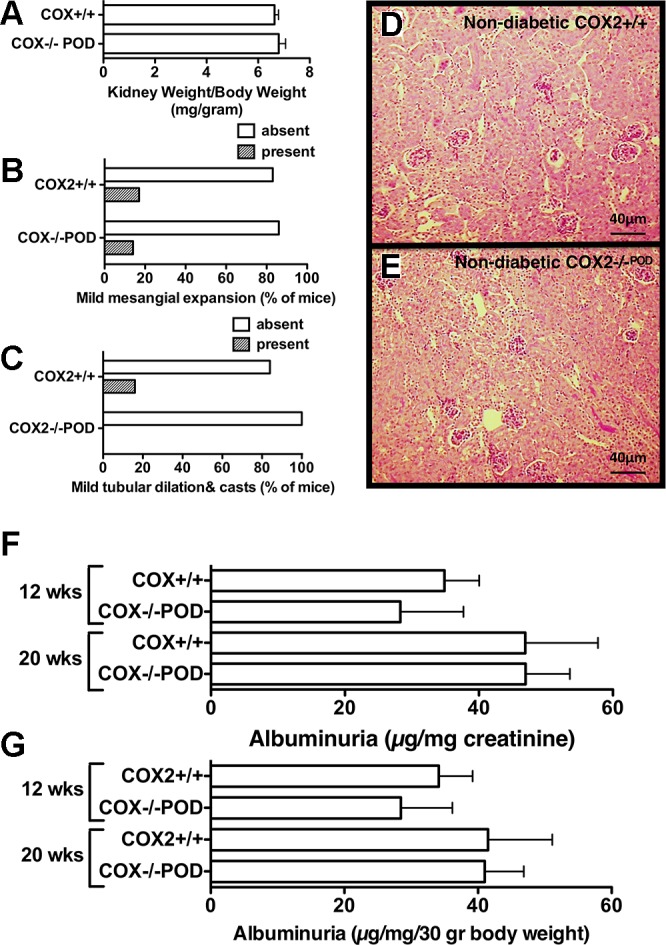
Effect of podocyte-specific COX2 KO on kidney histopathology. A: kidney weights were similar in nondiabetic COX2+/+ mice and nondiabetic COX2−/−POD mice. B: minimal mesangial expansion was observed in either group of nondiabetic mice. C: tubule dilation and casts were similar in nondiabetic COX2+/+ mice and nondiabetic COX2−/−POD mice. D and E: representative low-power views of kidneys from nondiabetic COX2+/+ mice and nondiabetic COX2−/−POD mice. F and G: albuminuria was similar in both groups of nondiabetic mice at 12- and 20-wk of age. For the histologic studies, kidneys from 10 COX2+/+ and 8 COX2−/− mice were examined. Statistical analyses of histologic data were performed using a Fishers Exact test. Statistical analyses of the albuminuria data were performed using an ANOVA followed by a Bonferroni post hoc analysis.
Table 1.
Fasting blood glucose levels, body weights, and urine volume
| COX2+/+ | COX2−/−POD | WT Akita | KO Akita | |
|---|---|---|---|---|
| (n = 5) | (n = 5) | (n = 12) | (n = 7) | |
| 8 wk | ||||
| Glucose, mg/dl | 119 ± 6.9* | 112 ± 2.6† | 416 ± 13.5 | 400 ± 13.63 |
| Urine, ml/24 h | not done | not done | not done | not done |
| Weight, g | not done | not done | not done | not done |
| 12 wk | ||||
| Glucose, mg/dl | 112 ± 2.3 | 111 ± 3.6 | 400 ± 9.0* | 419 ± 5.8† |
| Urine, ml/24 h | 1.4 ± 0.2 | 1.1 ± 0.1 | 6.7 ± 0.5* | 6.7 ± 0.6† |
| Weight, g | 28 ± 2.6 | 28 ± 3.8 | 23 ± 2.1* | 23 ± 0.9† |
| 16 wk | ||||
| Glucose, mg/dl | 117 ± 2.5 | 112 ± 3.9 | 397 ± 9.7* | 443 ± 24.8† |
| Urine, ml/24 h | 0.9 ± 0.1 | 1.0 ± 0.1 | 7.9 ± 0.3* | 7.6 ± 0.1† |
| Weight, g | 33 ± 0.5 | 31 ± 0.4 | 26 ± 0.4* | 26 ± 1.0† |
| 20 wk | ||||
| Glucose, mg/dl | 114 ± 4.5 | 113 ± 3.6 | 434 ± 22.0* | 400 ± 9.0† |
| Urine, ml/24 h | 0.9 ± 0.1 | 1.0 ± 0.1 | 8.8 ± 0.4* | 7.5 ± 0.1† |
| Weight, g | 35 ± 0.5 | 35 ± 0.4 | 28 ± 0.8* | 26 ± 0.7† |
Values are expressed as means ± SE.
P < 0.01 vs. age-matched COX2+/+ mice.
P < 0.01 vs. age-matched COX2−/−POD mice.
To assess the functional effect of podocyte-specific COX2 KO on glomerular permselectivity, we measured albuminuria in nondiabetic COX2+/+ and nondiabetic COX2−/−POD mice at 12 and 20 wk of age. As shown in Fig. 2, F and G, albuminuria was similar in age-matched groups at 12 and 20 wk of age.
To examine podocyte morphology more closely, TEM was performed in COX2+/+ and COX2−/−POD mice at 12 wk of age. As shown in Fig. 3, A and B, podocyte morphology was qualitatively normal in both groups of nondiabetic animals. There were also no qualitative differences in FP effacement in either COX2+/+ or COX2−/−POD mice. To provide a more quantitative assessment of FP effacement, the number of filtration slits per micrometer of GBM was measured. As shown in Fig. 3C, filtration slits per micrometer GBM were similar in both groups.
Fig. 3.

Glomerular ultrastructure in nondiabetic mice. A and B: examination of kidneys by transmission electron microscopy (TEM) revealed no qualitative differences in glomerular ultrastructure in nondiabetic COX2+/+ mice or nondiabetic COX2−/−POD mice. C: number of filtration slits per micrometer (µm) of glomerular basement membrane (GBM) was similar in both groups. For the studies, two mice were studied in each group. Statistical analysis was performed using an unpaired t-test.
Effect of podocyte-specific COX2 KO on blood glucose levels and systemic blood pressure in diabetes.
Hyperglycemia and systemic blood pressure are important determinants of the severity of kidney disease in diabetes mellitus (31). Therefore, we first examined blood glucose levels in the Akita mice. As shown in Table 1, blood glucose levels were similarly elevated in both groups of Akita mice compared to nondiabetic mice at 8, 12, 16, and 20 wk of age. The increase in blood glucose levels was associated with significant polyuria in diabetic mice, which may have accounted for the similar decrease in body weight in the diabetic groups (Table 1).
We next evaluated SBP, as well as serum cystatin C levels, as an index of renal function. As shown in Table 2, there were no significant differences in either SBP or cystatin C levels between the groups.
Table 2.
Effect of COX2 KO on SBP and cystatin C levels
| SBP, mmHg | SBP, mmHg | Cystatin C, ng/ml | |
|---|---|---|---|
| 12 wk of age | 20 wk of age | 20 wk of age | |
| COX2−/− | 129 ± 8.5 | 125 ± 4.1 | 567 ± 31.5 |
| (n = 5) | |||
| COX2−/−POD | 138 ± 7.5 | 126 ± 2.7 | 545 ± 44 |
| (n = 5) | |||
| WT Akita | 133 ± 4.7 | 131 ± 4.1 | 527 ± 54 |
| (n = 12) | |||
| KO Akita | 133 ± 6.0 | 135 ± 6.2 | 538 ± 62 |
| (n = 7) |
Values are expressed as means ± SE.
Effect of diabetes and podocyte specific COX2 KO on albuminuria, renal histopathology and glomerular ultrastructure.
Figure 4A shows a Coomassie-stained gel of urinary protein samples separated by protein electrophoresis, as described in the materials and methods. Prominent bands at the molecular weight of albumin (~66 kDa) were detected in diabetic mice, but these bands were difficult to detect in nondiabetic controls. Figure 4, B and C shows the quantitative effects of podocyte-specific COX2 KO on albuminuria. Because albuminuria was similar in nondiabetic COX2+/+ and COX2−/−POD mice (Fig. 2, F and G), these data were combined for the analyses. Both diabetes and COX2 genotype had a significant effect albuminuria results. There was a significant increase in albuminuria in WT and KO Akita mice compared to nondiabetic mice at the 16- and/or 20-wk time points. The increase in albuminuria was, however, significantly greater in KO Akita mice compared to WT Akita mice at both 16 and 20 wk of age. Figure 4, B–F shows the effect of diabetes and podocyte-specific COX2 KO on renal histology. Because kidney histology was similar in nondiabetic COX2+/+ and nondiabetic COX2−/−POD mice (Figure 2, A–E), these data were combined for the data analyses. As shown in Fig. 4, D–G, mesangial expansion was significantly increased in both groups of diabetic mice compared to nondiabetic controls, but the degree of mesangial expansion was similar in WT and KO Akita mice. Tubular dilation and casts were observed only in diabetic mice, but there were no statistically significant differences in either group of diabetic mice compared to nondiabetic controls (Fig. 4H). No significant tubulointerstitial inflammation or fibrosis was detected in either group of mice (not shown).
Fig. 4.
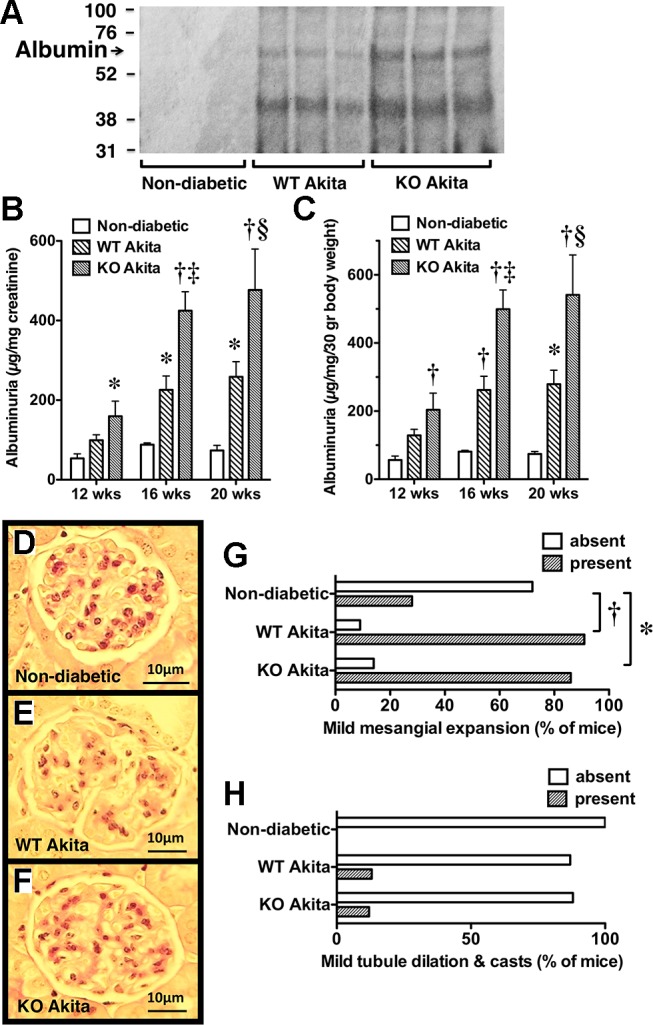
Effect of podocyte-specific COX2 KO on albuminuria and kidney histopathology in diabetic mice. A: as an index of glomerular albuminuria, urinary protein samples were separated by protein electrophoresis, and the gels were stained with Coomassie Brilliant Blue R-250, as described in materials and methods. Prominent bands of the molecular weight of albumin (~66 kDa) were detected in diabetic mice, but these bands were difficult to detect in nondiabetic controls. B and C: both diabetes and genotype (COX2+/+ or COX2−/−) significantly influenced the albuminuria results. Albuminuria was increased in both groups of diabetic mice compared to controls at 16 and 20 wk of age. Podocyte-specific KO of COX2 further enhanced albuminuria at the 16- and 20-wk time points. D–G: mesangial expansion was significantly increased in diabetic WT and KO Akita mice compared to nondiabetic animals, but the extent of mesangial expansion was similar in both groups of Akita mice. H: tubule dilation and casts were observed only in diabetic mice, but there were no significant differences between groups. For the histologic studies, kidneys from seven nondiabetic controls, 14 WT Akita mice, and seven KO Akita mice were examined in each group. For the albuminuria studies, seven nondiabetic controls, 12 WT Akita mice and 8 KO Akita mice were studied in each group. Statistical analysis of the albuminuria data was performed using a two-way ANOVA followed by a Bonferroni post hoc analysis. Categorical data were analyzed using a Fishers exact test. *P < 0.05 or †P < 0.01 vs. nondiabetic mice. ‡P < 0.05 or §P < 0.01 vs. WT Akita mice.
We next evaluated the effect of podocyte injury on glomerular ultrastructure in mice at 20 wk of age. As shown in Fig. 5, A–C, focal areas of foot process (FP) fattening were seen in both diabetic and nondiabetic mice. Quantitation of the filtration slits per micrometer (µm) of GBM tended to decrease in diabetic mice, but these differences were not significantly different between groups (Fig. 5D). The GBM exhibited areas of focal irregularity in all groups. As shown in Fig. 5E, GBM width tended to be increased in both groups of Akita mice compared with controls, but these increases in GBM width were not statistically significant.
Fig. 5.

Glomerular ultrastructure in diabetic and nondiabetic mice. A–C: focal areas of foot process (FP) effacement were seen in both groups of Akita mice compared to nondiabetic controls (CTLs). RBC, red blood cell. D: number of filtration slits per micrometer (µm) of GBM tended to be decreased in both groups of Akita mice, but these differences were not statistically significant. E: glomerular basement membrane (GBM) width also tended to increase in both groups of Akita mice compared to nondiabetic CTLs, but these differences were not statistically significant. For the studies, four or five mice were studied in each group. Statistical analyses were performed using a two-way ANOVA followed by a Bonferroni post hoc analysis.
Effect of diabetes and podocyte-specific KO glomerular volume, podocyte number, and podocyte density.
Table 3 shows the effects of diabetes and podocyte-specific COX2 KO on glomerular volume, podocyte number, and podocyte density. Glomerular volume similarly increased in both groups of Akita mice compared to nondiabetic animals. Podocyte number and density were quantitated using the recently described methodology described by Wiggins and colleagues (41). Podocyte density was significantly decreased in both groups of diabetic mice compared with nondiabetic animals. Podocyte number was similar in all groups.
Table 3.
Effect of COX2 KO on glomerular structure in Akita mice
| Podocytes per | VGlom | NV (P/Glom) | Podocytes per | |
|---|---|---|---|---|
| Glomerular Profile | (×105 µm3) | (×10−5/µm3) | Glomerulus | |
| COX2+/+ | 8.14 ± 0.19 | 2.58 ± 0.09 | 22.6 ± 0.5 | 58.3 ± 1.7 |
| (n = 5) | ||||
| COX2−/−POD | 7.82 ± 0.23 | 2.56 ± 0.06 | 22.6 ± 0.7 | 56.8 ± 1.7 |
| (n = 5) | ||||
| WT Akita | 7.54 ± 0.20 | 3.35 ± 0.14† | 18.1 ± 0.4‡ | 60.5 ± 1.4 |
| (n = 16) | ||||
| KO Akita | 7.31 ± 0.19 | 3.20 ± 0.13* | 18.4 ± 0.4§ | 55.5 ± 1.4 |
| (n = 13) |
Values are expressed as means ± SE.
P < 0.05, vs. nondiabetic COX2−/−.
P < 0.01, vs. nondiabetic COX2+/+.
P < 0.001, vs. nondiabetic COX2+/+.
P < 0.001, vs. nondiabetic COX2−/−.
Effect of podocyte-specific COX2 KO on urinary eicosanoid excretion.
Figure 6 shows the effects of podocyte-specific COX2 KO on urinary eicosanoid excretion. Urinary eicosanoid excretion was similar in nondiabetic COX2+/+ mice and nondiabetic COX2−/−POD mice. Therefore, we combined results for urinary excretion of PGE2 (6.1 ± 0.9 for COX2+/+ vs. 5.7 ± 0.5 for COX2−/−POD pg·mg creatinine−1·30 g body wt−1; P = not significant, NS), as well as urinary excretion the TxA2 metabolite TxB2 (276 ± 95 for COX2+/+ vs. 217 ± 58 for COX2−/−POD pg·mg creatinine−1·30 g body wt−1; P = NS) in nondiabetic mice. As shown in Fig. 6, A and B, the presence of diabetes significantly affected the profile of urinary PGE2 excretion. There was a significant and similar increase in urinary excretion of PGE2 in WT Akita mice compared with nondiabetic animals (Fig. 6A).
Fig. 6.

Effect of podocyte-specific COX2 KO on urinary eicosanoid excretion and glomerular renin expression. A and B: presence of diabetes significantly enhanced urinary eicosanoid excretion. Both urinary PGE2 metabolites and the urinary thromboxane 2 (TxA2) metabolite TxB2 were increased in both groups of diabetic mice compared to nondiabetic controls. This increase in urinary eicosanoid excretion was statistically significant for PGE2 in WT Akita mice compared to nondiabetic animals. C and D: similarly, the presence of diabetes significantly reduced expression of renin mRNA. Relative expression of renin mRNA was decreased in both groups of diabetic mice compared to nondiabetic controls. For the urinary eicosanoid studies, eight nondiabetic controls, 15 WT Akita, and 11 KO Akita mice were studied. For the renin mRNA studies, eight mice were studied in each group. For the immunoblotting studies, eight nondiabetic, six WT Akita, and six KO Akita mice were studied. For the quantitative RT-PCR studies, eight mice were examined in each group. Statistical analyses were performed using a two-way ANOVA followed by a Bonferroni post hoc analysis. *P < 0.05 or †P < 0.01 vs. nondiabetic mice.
Glomerular renin expression.
High glucose is reported to increase renin activity in cultured podocytes, and this increase is prevented by pharmacologic inhibition of COX2 (4). To investigate the effects of diabetes and podocyte COX2 KO on glomerular renin expression, we measured renin mRNA in enriched glomerular preparations using quantitative RT-PCR. Glomerular levels of renin mRNA were similar in nondiabetic COX2+/+ mice and nondiabetic COX2−/−POD mice (1.00 ± 0.26 for COX2+/+ vs. 0.94 ± 0.17 for COX2−/−POD relative expression; P = NS), and these data were combined for the analyses. As shown in Fig. 6C, the presence of diabetes significantly affected the glomerular renin mRNA levels. There was a significant decrease in renin mRNA in both groups of diabetic mice compared to nondiabetic controls.
We next determined whether renin protein levels were altered in enriched glomerular preparations by immunoblotting. Glomerular levels of renin protein were similar in nondiabetic COX2+/+ mice and nondiabetic COX2−/−POD mice (1.00 ± 0.05 for COX2+/+ vs. 1.1 ± 0.13 for COX2−/−POD densitometry units; P = NS), and these data were combined for the analyses. As shown in Fig. 6D, protein levels of renin tended to decrease in glomerular preparations from diabetic mice compared with nondiabetic controls. The presence of diabetes significantly reduced glomerular renin protein levels in KO Akita mice.
DISCUSSION
Experiments using selective COX2 inhibitors in rodent models of diabetic kidney disease have generally had beneficial actions on the disease process, including reducing albuminuria (5, 36) and mesangial sclerosis (5), as well as decreasing GBM thickness (33). In contrast, podocyte-specific COX2 deletion in diabetic Akita mice significantly enhanced albuminuria and did not improve the histologic features of diabetic kidney disease at either the light microscopic or ultrastructural level. Thus, the role of eicosanoids in diabetic kidney disease is complex. In this regard, the product of COX2 enzymatic activity (PGH2) is converted to prostaglandins and/or thromboxanes by cellular synthases, which then act on the diverse family of G protein-coupled eicosanoid receptors (14). Given the rapid degradation of eicosanoids in biologic systems, the biologic actions of eicosanoids on a given cell type are, therefore, dependent on the profile of locally generated and locally acting eicosanoids and the receptor subtypes present on the cell surface (14). Thus, the effects of COX2 deletion specifically in podocytes are likely restricted to autocrine effects and/or paracrine effects on immediately adjacent cells, such as mesangial cells and glomerular endothelial cells. In diabetes, the loss of locally acting eicosanoids generated by podocytes adversely affects glomerular filtration barrier function and does not improve the histopathologic features of the disease. In contrast, the net effect of broadly inhibiting COX2 enzymatic activity in diabetes favorably alters the profile of eicosanoids generated by the multiple cell types that compose the kidney, which apparently counterbalances the loss of podocyte eicosanoid generation during COX2 inhibition and ameliorates the disease process.
The autocrine effects of podocyte eicosanoid generation are also important in modulating podocyte apoptosis and promoting cellular attachment (3, 44). For example, we found that generation of PGE2 by cultured podocytes promotes apoptosis by activating the EP1 receptor (44), and EP1 receptor blockade has beneficial effects on diabetic kidney disease in STZ-treated rats (28). Similarly, a thromboxane receptor antagonist attenuates apoptosis induced by overexpression of COX2 in podocytes (3). The effects of COX2 metabolites on podocyte biology are, however, complex. In this regard, podocytes lacking COX2 exhibit both increased apoptosis and reduced cellular attachment in vitro (3). Although these changes in apoptosis and cellular attachment would tend to enhance podocyte loss in vivo, podocyte number was similar in both WT and KO diabetic and nondiabetic mice (Table 3), although podocyte density was decreased in both groups of diabetic mice due to the increase in glomerular volume in diabetic animals (Table 3). Nevertheless, podocyte eicosanoid generation does appear to have important biological actions in the diabetic milieu, because KO of podocyte COX2 decreased the integrity of the glomerular filtration barrier, enhancing albuminuria in Akita mice. This loss of glomerular filtration barrier integrity may reflect the complex actions of locally generated eicosanoids on the podocyte cytoskeleton. Indeed, cultured podocytes from mice lacking COX2 demonstrate cytoskeletal disorganization and prominent stress fibers (3), suggesting an important influence of podocyte eicosanoid generation on cytoskeletal dynamics. Given the important role of the actin cytoskeleton in glomerular filtration barrier function (10), these observations are consistent with the notion that eicosanoid-induced alternations in the podocyte cytoskeleton and, in turn, a loss of glomerular filtration barrier integrity may have contributed to enhanced albuminuria in KO Akita mice.
High glucose levels are reported to increase renin activity in cultured podocytes, and this increase is prevented by pharmacologic inhibition of COX2 (4). Therefore, we determined the effect of COX2 KO on glomerular expression of renin mRNA and protein. As shown in Fig. 6C, glomerular renin mRNA levels were decreased in both groups of Akita mice compared with controls. The decrease in glomerular renin activity was unexpected given that the intrarenal renin-angiotensin system is suggested to be a disease accelerant in rodent models of diabetic kidney disease (6, 20). Several studies have suggested, however, that renin mRNA and protein levels are decreased in kidneys from diabetic rodents with established diabetic kidney disease (22, 23). In contrast, renin mRNA is increased in proximal tubules of kidneys from diabetic rats 2 wk after induction of diabetes with streptozotocin (53). While further studies will be required to better define mechanisms, it may be that renin expression is either compartmentalized within the kidney or differentially regulated in early vs. later stages of the disease process.
BP is an important variable that affects glomerular filtration barrier integrity. We think this potential confounder is unlikely to contribute to enhanced albuminuria in KO Akita mice because: 1) BP measurements were similar in WT and KO Akita mice (Table 2), and 2) COX2 KO was restricted to podocytes, so the systemic effects of deleting COX2 were limited. We do acknowledge, however, the limitations of tail cuff plethysmography (52) used in the present study, including 1) the mice may continue to experience some degree of anxiety, which could affect systemic BP, 2) measurements are operator dependent, and 3) tail cuff BP responses may not reflect central arterial pressure. These limitations should be considered when interpreting the BP results in the present study.
We were also concerned that podocyte-specific KO of COX2 might have adverse effects on kidney development given that systemic KO of COX2 causes renal developmental abnormalities in the postnatal period characterized by smaller kidneys, tubular dilation, cyst formation, glomerular hypoplasia, periglomerular fibrosis, variable glomerulosclerosis, and diffuse interstitial fibrosis (35). However, none of these abnormalities were observed in the present study (Fig. 2). Moreover, no differences in podocyte ultrastructure were observed by TEM in nondiabetic mice (Fig. 3). There are several possible explanations for the lack of kidney development effects. First, podocyte COX2 expression may not be required for the kidney developmental program. Second, systemic KO deletes COX2 from all cells throughout kidney development, which appears to be important for postnatal development of the kidney (35). In contrast, KO of podocyte COX2 in the present study was dependent on activation of the podocin promoter, which occurs as podocytes differentiate into mature cells (38). If expression of COX2 is required before this differentiation process to cause postnatal abnormalities, then deletion of COX2 in podocytes would occur after the critical period of nephrogenesis, requiring COX2 expression in the mouse kidney. Third, KO of COX2 is dependent on the ability of the podocin promoter to promote Cre-mediated recombination, which is likely incomplete (11). In this scenario, some residual podocyte COX2 generation may be sufficient for normal development.
Lastly, while expression of COX2 in podocytes was reduced in KO Akita mice (Fig. 1), expression of COX2 in mesangial areas was not affected in diabetic mice lacking COX2 in podocytes. As a result, we were surprised at the marked reduction in COX2 mRNA expression in glomerular preparations (Fig. 1C), suggesting dissociation between COX mRNA and COX2 protein levels. In this regard, regulation of COX2 expression is complex and includes both transcriptional and post-transcriptional mechanisms (15). These complex regulatory mechanisms may have contributed to the apparent dissociation between COX2 mRNA and COX2 protein in the present studies.
In summary, podocyte-specific KO of COX2 enhanced albuminuria and did not attenuate the histologic features of diabetic kidney disease. These findings are in contrast to the generally beneficial effects of COX2 inhibitors in rodent models of diabetic kidney disease. Taken together with published studies (3), these data suggest that 1) some basal level of COX2 expression in podocytes is required to attenuate the adverse effects of diabetes mellitus on glomerular filtration barrier integrity, and 2) KO of COX2 specifically in podocytes does not ameliorate diabetic kidney disease in Akita mice.
GRANTS
These studies were supported by grants R01DK-087707 (R. F. Spurney) from the National Institutes of Health, 1I01BX-002984 (R. F. Spurney) from the Veterans Administration (VA) Merit Review Program and the VA Career Development Award 2 (CDA-2) IK2BX-002240 (M. A. Sparks). Drs. Spurney and Wang received salary support from a grant funded by the Juvenile Diabetes Research Foundation (no. 201302131). Portions of the research reported by this publication were supported by the National Institute of Diabetes, Digestive and Kidney Diseases of the National Institutes of Health under award number P30DK-096493.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.W., Y.S., J.B., W.E., M.A.S., and R.F.S. performed experiments; L.W., Y.S., M.A.S., A.F.B., and R.F.S. analyzed data; L.W., A.F.B., and R.F.S. interpreted results of experiments; R.F.S. conceived and designed research; R.F.S. prepared figures; R.F.S. drafted manuscript; R.F.S. edited and revised manuscript; R.F.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The results presented in this paper have not been published previously, in whole or in part, except in abstract format.
REFERENCES
- 1.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S; RENAAL Study Investigators . Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 2.Chang JH, Paik SY, Mao L, Eisner W, Flannery PJ, Wang L, Tang Y, Mattocks N, Hadjadj S, Goujon JM, Ruiz P, Gurley SB, Spurney RF. Diabetic kidney disease in FVB/NJ Akita mice: temporal pattern of kidney injury and urinary nephrin excretion. PLoS One 7: e33942, 2012. doi: 10.1371/journal.pone.0033942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng H, Fan X, Guan Y, Moeckel GW, Zent R, Harris RC. Distinct roles for basal and induced COX-2 in podocyte injury. J Am Soc Nephrol 20: 1953–1962, 2009. doi: 10.1681/ASN.2009010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng H, Fan X, Moeckel GW, Harris RC. Podocyte COX-2 exacerbates diabetic nephropathy by increasing podocyte (pro)renin receptor expression. J Am Soc Nephrol 22: 1240–1251, 2011. doi: 10.1681/ASN.2010111149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng HF, Wang CJ, Moeckel GW, Zhang MZ, McKanna JA, Harris RC. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of diabetes and hypertension. Kidney Int 62: 929–939, 2002. doi: 10.1046/j.1523-1755.2002.00520.x. [DOI] [PubMed] [Google Scholar]
- 6.Conway BR, Rennie J, Bailey MA, Dunbar DR, Manning JR, Bellamy CO, Hughes J, Mullins JJ. Hyperglycemia and renin-dependent hypertension synergize to model diabetic nephropathy. J Am Soc Nephrol 23: 405–411, 2012. doi: 10.1681/ASN.2011060577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalla Vestra M, Masiero A, Roiter AM, Saller A, Crepaldi G, Fioretto P. Is podocyte injury relevant in diabetic nephropathy? Studies in patients with type 2 diabetes. Diabetes 52: 1031–1035, 2003. doi: 10.2337/diabetes.52.4.1031. [DOI] [PubMed] [Google Scholar]
- 8.Das AK, Pickett TM, Tungekar MF. Glomerular basement membrane thickness —a comparison of two methods of measurement in patients with unexplained haematuria. Nephrol Dial Transplant 11: 1256–1260, 1996. doi: 10.1093/ndt/11.7.1256. [DOI] [PubMed] [Google Scholar]
- 9.Ding G, Reddy K, Kapasi AA, Franki N, Gibbons N, Kasinath BS, Singhal PC. Angiotensin II induces apoptosis in rat glomerular epithelial cells. Am J Physiol Renal Physiol 283: F173–F180, 2002. doi: 10.1152/ajprenal.00240.2001. [DOI] [PubMed] [Google Scholar]
- 10.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 17: 428–437, 2007. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP, Hartleben G, Wiech T, Fornoni A, Nelson RG, Kretzler M, Wanke R, Pavenstädt H, Kerjaschki D, Cohen CD, Hall MN, Rüegg MA, Inoki K, Walz G, Huber TB. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121: 2197–2209, 2011. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol 290: F214–F222, 2006. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- 13.Gurley SB, Mach CL, Stegbauer J, Yang J, Snow KP, Hu A, Meyer TW, Coffman TM. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am J Physiol Renal Physiol 298: F788–F795, 2010. doi: 10.1152/ajprenal.90515.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao CM, Breyer MD. Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int 71: 1105–1115, 2007. doi: 10.1038/sj.ki.5002192. [DOI] [PubMed] [Google Scholar]
- 15.Harper KA, Tyson-Capper AJ. Complexity of COX-2 gene regulation. Biochem Soc Trans 36: 543–545, 2008. doi: 10.1042/BST0360543. [DOI] [PubMed] [Google Scholar]
- 16.Hirose K, Osterby R, Nozawa M, Gundersen HJ. Development of glomerular lesions in experimental long-term diabetes in the rat. Kidney Int 21: 689–695, 1982. doi: 10.1038/ki.1982.82. [DOI] [PubMed] [Google Scholar]
- 17.Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int 74: 22–36, 2008. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- 18.Jensen EB, Gundersen HJ, Osterby R. Determination of membrane thickness distribution from orthogonal intercepts. J Microsc 115: 19–33, 1979. doi: 10.1111/j.1365-2818.1979.tb00149.x. [DOI] [PubMed] [Google Scholar]
- 19.Jia J, Ding G, Zhu J, Chen C, Liang W, Franki N, Singhal PC. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol 28: 500–507, 2008. doi: 10.1159/000113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly DJ, Wilkinson-Berka JL, Allen TJ, Cooper ME, Skinner SL. A new model of diabetic nephropathy with progressive renal impairment in the transgenic (mRen-2)27 rat (TGR). Kidney Int 54: 343–352, 1998. doi: 10.1046/j.1523-1755.1998.00019.x. [DOI] [PubMed] [Google Scholar]
- 21.Komers R, Lindsley JN, Oyama TT, Schutzer WE, Reed JF, Mader SL, Anderson S. Immunohistochemical and functional correlations of renal cyclooxygenase-2 in experimental diabetes. J Clin Invest 107: 889–898, 2001. doi: 10.1172/JCI10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komers R, Xu B, Fu Y, McClelland A, Kantharidis P, Mittal A, Cohen HT, Cohen DM. Transcriptome-based analysis of kidney gene expression changes associated with diabetes in OVE26 mice, in the presence and absence of losartan treatment. PLoS One 9: e96987, 2014. doi: 10.1371/journal.pone.0096987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosugi T, Heinig M, Nakayama T, Matsuo S, Nakagawa T. eNOS knockout mice with advanced diabetic nephropathy have less benefit from renin-angiotensin blockade than from aldosterone receptor antagonists. Am J Pathol 176: 619–629, 2010. doi: 10.2353/ajpath.2010.090578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 54: 687–697, 1998. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 25.Lemley KV, Bertram JF, Nicholas SB, White K. Estimation of glomerular podocyte number: a selection of valid methods. J Am Soc Nephrol 24: 1193–1202, 2013. doi: 10.1681/ASN.2012111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group . Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345: 851–860, 2001. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 27.Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am 39: 481–497, 2010. doi: 10.1016/j.ecl.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makino H, Tanaka I, Mukoyama M, Sugawara A, Mori K, Muro S, Suganami T, Yahata K, Ishibashi R, Ohuchida S, Maruyama T, Narumiya S, Nakao K. Prevention of diabetic nephropathy in rats by prostaglandin E receptor EP1-selective antagonist. J Am Soc Nephrol 13: 1757–1765, 2002. doi: 10.1097/01.ASN.0000019782.37851.BF. [DOI] [PubMed] [Google Scholar]
- 29.Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 42: 1341–1344, 1999. doi: 10.1007/s001250051447. [DOI] [PubMed] [Google Scholar]
- 30.Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35: 39–42, 2003. doi: 10.1002/gene.10164. [DOI] [PubMed] [Google Scholar]
- 31.Molitch ME, DeFronzo RA, Franz MJ, Keane WF, Mogensen CE, Parving HH, Steffes MW; American Diabetes Association . Nephropathy in diabetes. Diabetes Care 27, Suppl 1: S79–S83, 2004. doi: 10.2337/diacare.27.2007.S79. [DOI] [PubMed] [Google Scholar]
- 32.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 45: 593–605, 2007. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 33.Nasrallah R, Robertson SJ, Karsh J, Hébert RL. Celecoxib modifies glomerular basement membrane, mesangium and podocytes in OVE26 mice, but ibuprofen is more detrimental. Clin Sci (Lond) 124: 685–694, 2013. doi: 10.1042/CS20120543. [DOI] [PubMed] [Google Scholar]
- 34.National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals., Institute for Laboratory Animal Research (U.S.), and National Academies Press (U.S.) Guide for the Care and Use of Laboratory Animals. Washington, D.C.: National Academies Press, 2011, p. xxv. [Google Scholar]
- 35.Norwood VF, Morham SG, Smithies O. Postnatal development and progression of renal dysplasia in cyclooxygenase-2 null mice. Kidney Int 58: 2291–2300, 2000. doi: 10.1046/j.1523-1755.2000.00413.x. [DOI] [PubMed] [Google Scholar]
- 36.Quilley J, Santos M, Pedraza P. Renal protective effect of chronic inhibition of COX-2 with SC-58236 in streptozotocin-diabetic rats. Am J Physiol Heart Circ Physiol 300: H2316–H2322, 2011. doi: 10.1152/ajpheart.01259.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramage IJ, Howatson AG, McColl JH, Maxwell H, Murphy AV, Beattie TJ, Ramage IJ. Glomerular basement membrane thickness in children: a stereologic assessment. Kidney Int 62: 895–900, 2002. doi: 10.1046/j.1523-1755.2002.00527.x. [DOI] [PubMed] [Google Scholar]
- 38.Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attié T, Gubler MC, Antignac C. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol 160: 131–139, 2002. doi: 10.1016/S0002-9440(10)64357-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spurney RF, Coffman TM. Stressed-out podocytes in diabetes? J Am Soc Nephrol 19: 2035–2037, 2008. doi: 10.1681/ASN.2008090955. [DOI] [PubMed] [Google Scholar]
- 40.Teiken JM, Audettey JL, Laturnus DI, Zheng S, Epstein PN, Carlson EC. Podocyte loss in aging OVE26 diabetic mice. Anat Rec (Hoboken) 291: 114–121, 2008. doi: 10.1002/ar.20625. [DOI] [PubMed] [Google Scholar]
- 41.Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, Chowdhury M, Hodgin J, Wiggins PA, Wiggins RC. Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol 25: 1118–1129, 2014. doi: 10.1681/ASN.2013080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vogt L, de Zeeuw D, Woittiez AJ, Navis G. Selective cyclooxygenase-2 (COX-2) inhibition reduces proteinuria in renal patients. Nephrol Dial Transplant 24: 1182–1189, 2009. doi: 10.1093/ndt/gfn644. [DOI] [PubMed] [Google Scholar]
- 43.Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, Zhang H, Hui Y, Cheng Y, Petrenko N, Yu Y, FitzGerald GA. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci USA 106: 7548–7552, 2009. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L, Chang JH, Paik SY, Tang Y, Eisner W, Spurney RF. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol Endocrinol 25: 1376–1386, 2011. doi: 10.1210/me.2011-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, Ellis MJ, Gomez JA, Eisner W, Fennell W, Howell DN, Ruiz P, Fields TA, Spurney RF. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int 81: 1075–1085, 2012. doi: 10.1038/ki.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Flannery PJ, Rosenberg PB, Fields TA, Spurney RF. Gq-dependent signaling upregulates COX2 in glomerular podocytes. J Am Soc Nephrol 19: 2108–2118, 2008. doi: 10.1681/ASN.2008010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L, Jirka G, Rosenberg PB, Buckley AF, Gomez JA, Fields TA, Winn MP, Spurney RF. Gq signaling causes glomerular injury by activating TRPC6. J Clin Invest 125: 1913–1926, 2015. doi: 10.1172/JCI76767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White KE, Bilous RW, Marshall SM, El Nahas M, Remuzzi G, Piras G, De Cosmo S, Viberti G. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 51: 3083–3089, 2002. doi: 10.2337/diabetes.51.10.3083. [DOI] [PubMed] [Google Scholar]
- 49.Whitesall SE, Hoff JB, Vollmer AP, D’Alecy LG. Comparison of simultaneous measurement of mouse systolic arterial blood pressure by radiotelemetry and tail-cuff methods. Am J Physiol Heart Circ Physiol 286: H2408–H2415, 2004. doi: 10.1152/ajpheart.01089.2003. [DOI] [PubMed] [Google Scholar]
- 50.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71: 1205–1214, 2007. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 51.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 54: 1626–1634, 2005. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 52.Zhao X, Ho D, Gao S, Hong C, Vatner DE, Vatner SF. Arterial pressure monitoring in mice. Curr Protoc Mouse Biol 1: 105–122, 2011. doi: 10.1002/9780470942390.mo100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zimpelmann J, Kumar D, Levine DZ, Wehbi G, Imig JD, Navar LG, Burns KD. Early diabetes mellitus stimulates proximal tubule renin mRNA expression in the rat. Kidney Int 58: 2320–2330, 2000. doi: 10.1046/j.1523-1755.2000.00416.x. [DOI] [PubMed] [Google Scholar]