

Abstract
The pathophysiology of chronic kidney disease (CKD) is driven by alterations in surviving nephrons to sustain renal function with ongoing nephron loss. Oxygen supply-demand mismatch, due to hemodynamic adaptations, with resultant hypoxia, plays an important role in the pathophysiology in early CKD. We sought to investigate the underlying mechanisms of this mismatch. We utilized the subtotal nephrectomy (STN) model of CKD to investigate the alterations in renal oxygenation linked to sodium (Na) transport and mitochondrial function in the surviving nephrons. Oxygen delivery was significantly reduced in STN kidneys because of lower renal blood flow. Fractional oxygen extraction was significantly higher in STN. Tubular Na reabsorption was significantly lower per mole of oxygen consumed in STN. We hypothesized that decreased mitochondrial bioenergetic capacity may account for this and uncovered significant mitochondrial dysfunction in the early STN kidney: higher oxidative metabolism without an attendant increase in ATP levels, elevated superoxide levels, and alterations in mitochondrial morphology. We further investigated the effect of activation of hypoxia-inducible factor-1α (HIF-1α), a master regulator of cellular hypoxia response. We observed significant improvement in renal blood flow, glomerular filtration rate, and tubular Na reabsorption per mole of oxygen consumed with HIF-1α activation. Importantly, HIF-1α activation significantly lowered mitochondrial oxygen consumption and superoxide production and increased mitochondrial volume density. In conclusion, we report significant impairment of renal oxygenation and mitochondrial function at the early stages of CKD and demonstrate the beneficial role of HIF-1α activation on renal function and metabolism.
Keywords: CKD, hypoxia, metabolism, mitochondria, oxygenation
chronic renal hypoxia has been suggested to be a major contributor to disease progression in chronic kidney disease (CKD) (19, 21, 27, 28, 46). Hence, it is imperative to understand the regulation of kidney oxygenation in CKD. Tissue oxygenation is determined by blood flow as well as oxygen delivery and consumption by cells. There are particular features of renal oxygenation which increase the susceptibility of kidney to hypoxia, and these include inhomogeneous regional renal blood flow (RBF) between cortex and medulla and high renal oxygen consumption (QO2) (46). The high rate of renal QO2 is largely utilized for ATP production to support active sodium transport (TNa). A linear relationship between TNa and QO2 has been repeatedly demonstrated (7, 26, 41). The slope of this relationship has been shown to be altered in experimental (9, 18, 23, 30, 49) and clinical (34, 44, 46) pathophysiological states such as CKD, diabetes, hypertension, and acute kidney injury with high QO2 factored for TNa. In early subtotal nephrectomy (STN) model of CKD, we and others have demonstrated oxygen supply-demand mismatch due to high nephron QO2 per mole of Na reabsorbed or per nephron (8, 9, 18, 30). The underlying mechanisms to account for oxygen supply-demand mismatch include inefficient Na transport (more oxygen utilization per net Na reabsorbed) and/or decreased mitochondrial bioenergetic capacity (more oxygen utilization per net ATP generated). Here, we focused on the latter by investigating in vivo renal oxygenation and mitochondrial function in the STN kidney.
The kidney derives most of its energy by mitochondrial oxidative metabolism (26, 41). Besides ATP generation, mitochondria play a significant role in generation and detoxification of reactive oxygen species (ROS), cell survival/death and other signaling pathways (5). Mitochondrial dysfunction has been strongly implicated in the pathophysiology of highly metabolic organs such as heart, brain, and liver and more recently the kidney. Mitochondrial morphology plays an important role in cellular physiology and pathophysiology (25, 55). There are limited investigations into morphological alterations and associated mitochondrial dysfunction in diseased kidneys, but this is a timely and prolific line of investigation, given the emerging data on changes in mitochondrial metabolism associated with alterations in mitochondrial morphology (15, 25, 36).
Various cellular regulatory factors impact mitochondrial metabolism. One such factor, hypoxia- inducible factor-1α (HIF-1α) regulates cellular adaptation to hypoxia by inducing various genes involved in O2 delivery and consumption (17, 37). HIF-1α has significant effects on mitochondrial metabolism by downregulating mitochondrial oxygen consumption and shifting cells toward glycolytic metabolism during hypoxia adaptation (31, 37, 50). We have previously shown that pharmacological HIF-1α activation lowers oxygen consumption, improves glomerular filtration rate (GFR), and interacts with AMP-activated kinase in cellular hypoxia in STN (8, 24). Here, we examine underlying mechanisms for the salutary effects of HIF-1α activation and its potential to regulate renal oxygenation by targeting mitochondrial function and dynamics in early STN.
METHODS
All experimentation was conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animal experiments were conducted in male Wistar rats with a body weight of 300–350 g (Harlan). All rats received free access to tap water and standard rat chow.
Subtotal nephrectomy.
This procedure was performed with sterile technique as previously described (24, 39). With a right- flank incision, a right unilateral nephrectomy was performed after ligation of renal pedicle and removal of right kidney. Then, a left- flank incision was made, and the left kidney maneuvered to expose the renal artery. Two branches of left renal artery were ligated with 4-0 silk suture. The kidney was replaced back into the body. The fascia was closed with silk suture and skin with steel wound clips. Rats were kept warm with a heating pad until ambulatory and administered a dose of buprenorphine analgesic (0.1 mg/kg ip).
In vivo clearance and renal oxygenation experiments.
Under inactin anesthesia (100 mg/kg intraperitoneally), a tracheostomy tube was placed, and the left internal jugular, left femoral artery, and urinary bladder were cannulated as previously described (8, 40). Mean arterial pressure (MAP) was monitored by connecting the femoral artery catheter to a transducer (DATAQ Instruments, Akron, OH). RBF (ml/min) in the left kidney was monitored continuously with a perivascular ultrasonic transit time flow probe (Transonics T420, Ithaca, NY). After the surgical preparation, the animals were allowed 60 min for stabilization. GFR was measured by clearance of 3H inulin. Blood samples were taken from both the femoral artery and renal vein for measurements of total arterial blood hemoglobin (tHb), O2Hb, Po2, and Pco2 with blood gas analyzer (OPTI CCA and OPTI CCA-TS Blood Gas and Electrolyte Analyzers, Optimedical, Roswell, GA). O2 content (O2ct) was calculated by the formula:
The total left kidney O2 consumption (QO2, ml/min) was calculated from the arteriovenous difference in O2 content multiplied by RBF. TNa is equal to the total amount of sodium filtered (FNa) minus the amount of sodium excreted in the urine (UNaV). Renal oxygen delivery (DO2) was calculated by RBF × arterial O2 content, and fractional O2 extraction (FO2) was calculated by QO2/DO2 × 100.
Reagents and chemicals.
Dimethyloxalyglycine (DMOG) was obtained from Cayman Chemical. For in vivo HIF induction, DMOG (10 mg·kg−1·d−1) was administered subcutaneously from the day of STN surgery until the day of tissue harvesting (8, 24). Kidneys were harvested 7–8 days after STN surgery for molecular analyses.
Isolation of rat kidney mitochondria.
Rat kidneys were harvested for isolation of rat kidney mitochondria as previously described (14). Harvested kidneys were immediately placed in ice-cold mitochondrial isolation buffer A [200 ml: 210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EGTA, 0.5% (weight/volume) fatty acid-free BSA] pH to 7.4 with 1 M KOH. The renal cortex or whole kidneys were minced and homogenized with 5–10 strokes with a prechilled Potter-Elvehjem tissue homogenizer. The homogenate was centrifuged at 800 g for 10 min at 4°C to remove unbroken cells and nuclei. The pellet was discarded, and the supernatant was transferred into two new tubes and centrifuged at 11,000 g at 4°C for 10 min. The resulting pellets were resuspended with buffer A, and the last centrifugation step repeated once more. The final pellet was resuspended in mitochondrial assay solution (MAS) [220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% (weight/volume) fatty acid-free BSA].
Oxygen consumption measurements with Clark electrode assay.
Oxygen chambers with a Clark-type electrode (Hansatech Oxytherm apparatus, PP Systems, Amesbury, MA), calibrated with air-equilibrated water to 228 μmol/l O2 and Na2S2O5-saturated water to zero, were used to measure O2 consumption in rat kidney mitochondria as previously described (14, 35). Mitochondrial protein concentration was determined by using DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). In mitochondria isolated in MAS (0.25 mg/ml), complex I substrates, glutamate-malate (5 mM each), or pyruvate-malate (5 mM each) were added, followed by ADP (400 µM), allowing electron transport to accelerate. This is state 3 respiration and determines the maximal rate of mitochondrial oxygen consumption rates (OCRs) coupled to ATP production. Complex I activity can be bypassed by the complex I inhibitor rotenone (2 µM), while succinate (5 mM) is added to allow oxidation by complex II. State 3 is terminated by adding the ATP synthase inhibitor oligomycin (2.5 µg/ml) to achieve a state 4 rate, where ATP recycling cannot contribute and leads to a reduction in OCR. This is followed by a carefully titrated concentration of a protonophore such as carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP) (200–400 nM), which allows the H+ recycling to continue uncoupled to ATP synthesis to give uncoupled respiration, state 3u. This reflects maximal stimulation of ETC. OCRs were converted from nanomole of O per minute per milliliter to nanomole of O2 per minute per milligram of mitochondrial protein.
ATP assay.
Mitochondria ATP production was measured by a bioluminescence assay, the ATP Determination Kit (Invitrogen, Carlsbad, CA) as previously described (2, 13).
Measurements were performed as described by the manufacturer in fresh isolated mitochondria from the kidneys of the same animals as those for mitochondrial respiration and ROS measurements. Functional mitochondria were normalized to 5 μg/μl, and 10 μl was added to 90 μl of the reaction mix. Samples were loaded into a Tecan Infinite M200 plate reader and luminescence quantified over 10 min.
Superoxide measurements by electron paramagnetic resonance.
Experiments for measuring superoxide generation by electron paramagnetic resonance (EPR) were performed as previously described (2, 13). Immediately after mixing mitochondria (0.1–0.2 mg of protein) with 70 mM 5-(diisopropoxyphosphoryl)-5-ethyl-1-pyrroline-N-oxide (DEPMPO) and appropriate combinations of the substrates, the mixture was loaded into 50-μl glass capillary tubes and introduced into the EPR cavity of a MiniScope MS300 Benchtop Spectrometer. We confirmed that the detected EPR signals are substrate specific, and not due to redox cycling in the studied mixtures, by lack of signals when DEPMPO was mixed with combinations of substrates and inhibitors in the absence of mitochondria. Assignment of the observed signals from mitochondria was confirmed through computer-assisted spectral simulation by using WinSim software (http://epr.niehs.nih.gov/pest.html). The complete removal of these signals upon the inclusion of superoxide dismutase confirmed that a superoxide radical was the exclusive source of the observed EPR-active species. Signals were quantified by measuring the peak amplitudes of the observed spectra and normalized by mitochondrial protein concentrations.
Mitochondrial imaging by electron microscopy at the UC San Diego National Center for Microscopy and Imaging Research.
As per previously described protocols (53), pieces of kidney tissue were fixed in 2% paraformaldehyde plus 2.5% glutaraldehyde (Ted Pella, Redding, CA) in 0.1 M sodium cacodylate (pH 7.4) on ice for 24 h. The samples were washed three times with buffer consisting of 0.1 M of sodium cacodylate plus 3 mM of calcium chloride (pH 7.4) on ice and then postfixed with 1% osmium tetroxide, 0.8% potassium ferrocyanide, 3 mM of calcium chloride in 0.1 M sodium cacodylate (pH 7.4) for 3 h, washed three times with ice-cold distilled water, en bloc stained with 2% uranyl acetate at 4°C for 1 h, dehydrated through graded ethanol solutions, and embedded in Durcupan ACM resin (Fluka, St. Louis, MO). Ultrathin (80 nm) sections were poststained with uranyl acetate and lead salts before imaging with a JEOL 1200FX transmission electron microscope operated at 80 kV. The negatives were digitized at 1,800 dpi with a Nikon CoolScan system, giving an image size of 4,033 × 6,010 pixels and a pixel resolution of 2.35 nm. A stereological analysis to ascertain the ratio of mitochondrial volume to cytoplasmic volume was performed with Adobe Photoshop. dPoint counting was used to determine the mitochondrial volume densities by overlaying a grid on each digitized image. Mitochondria and cytoplasm lying under intercepts were counted. The relative volume of mitochondria was expressed as the ratio of intercepts coinciding with this organelle to the intercepts coinciding with cytoplasm. Cristae abundance was measured as mean cristae surface area normalized to mitochondria outer membrane. Mitochondrial membrane surface areas were measured using ImageJ software (http://www.nih.gov).
Statistical methods.
Data were analyzed by one-way analysis of variance (ANOVA) when more than two groups were compared with Student-Neuman-Keuls tests for post hoc pairwise comparisons using commercial software (SigmaPlot). A P value of <0.05 was considered statistically significant. Unless stated otherwise, results are presented as group means ± SE.
RESULTS
Renal hemodynamics and oxygenation.
We performed in vivo measurements of renal hemodynamics and oxygenation at 1 wk after STN surgery or in control rats. We and others have used DMOG for pharmacological activation of HIF-1α and confirmed HIF-1α induction in the STN kidney with this agent (8, 24, 43, 45, 54). A separate group of STN and control rats treated with DMOG (10 mg·kg−1·d−1 sc) from the day of surgery until the day of experiments were also included. Results are described in Table 1 and Fig. 1. There was a significant increase in MAP in STN compared with control (P < 0.05) group. GFR measured by 3H inulin clearance was significantly lower in STN compared with control (P < 0.01). DMOG treatment improved GFR in STN rats (P < 0.05). Urine flow rates were variable and not significantly different between different groups. Urinary Na excretion was significantly lower in the STN rats compared with controls (P < 0.05).
Table 1.
Renal hemodynamics and oxygenation
| Groups | MAP, mmHg | GFR, ml/min | Urine Flow Rate, μl/min | Urinary Na Excretion, nEq/min | RBF (single kidney), ml/min | DO2, ml/min | FO2, % |
|---|---|---|---|---|---|---|---|
| Con, n = 8 | 102 ± 3 | 2.62 ± 0.15 | 5.7 ± 0.9 | 783 ± 105 | 6.14 ± 0.4 | 1.2 ± 0.1 | 7.8 ± 0.4 |
| STN, n = 11 | 122 ± 6 | 0.54 ± 0.10 | 7.6 ± 1.3 | 375 ± 42 | 3.68 ± 0.3 | 0.77 ± 0.08 | 10.5 ± 0.8 |
| Con+DMOG, n = 7 | 93 ± 6 | 2.58 ± 0.15 | 5.3 ± 1.1 | 797 ± 196 | 6.04 ± 0.3 | 1.2 ± 0.07 | 8.8 ± 0.5 |
| STN+DMOG, n = 14 | 118 ± 4 | 0.85 ± 0.07 | 10.3 ± 1.5 | 581 ± 75 | 4.95 ± 0.3 | 1.1 ± 0.08 | 9.1 ± 0.3 |
| ANOVA analyses | P = 0.002 | P < 0.001 | P = 0.054 | P = 0.018 | P < 0.001 | P = 0.007 | P = 0.02 |
| Post hoc pairwise comparisons | Con vs. STN, P < 0.05 | Con vs. STN, P < 0.01 | Con vs. STN, P < 0.05 | Con vs. STN, P < 0.01 | Con vs. STN, P < 0.01 | Con vs. STN, P = 0.01 | |
| Con vs. Con+D, P = 0.32 | Con vs. Con+D, P = 0.83 | Con vs. Con+D, P = 0.92 | Con vs. Con+D, P = 0.87 | Con vs. Con+D, P = 0.93 | Con vs. Con+D, P = 0.26 | ||
| STN vs. STN+D, P = 0.56 | STN vs. STN+D, P < 0.05 | STN vs. STN+D, P = 0.11 | STN vs. STN+D, P < 0.05 | STN vs. STN+D, P < 0.01 | STN vs. STN+D, P = 0.05 |
Values are means ± SE. MAP, mean arterial pressure; GFR, glomerular filtration rate; RBF, renal blood flow; DO2, O2 delivery; FO2, fractional O2; Con, control; STN, subtotal nephrectomy; D(MOG), dimethyloxalyglycine; Con, control.
Fig. 1.
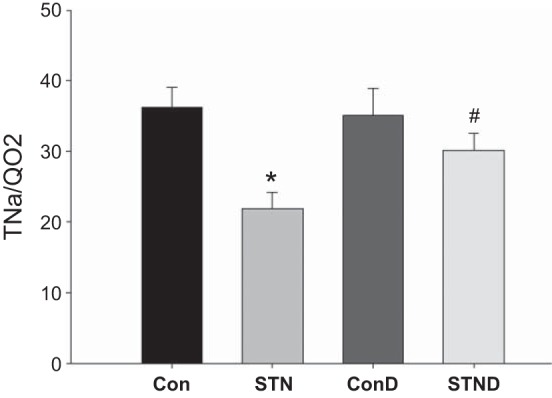
Renal oxygenation and Na transport in subtotal nephrectomy (STN). Reduced active sodium transport (TNa)/renal oxygen consumption (QO2) in STN compared with control significantly improved with dimethyloxalyglycine (DMOG) treatment. ANOVA P = 0.005, post hoc pairwise comparisons: *P < 0.05 STN vs. control (Con), #P < 0.05 STN vs. STN+DMOG (STND). ConD represents control+DMOG; n = 7–14 per group.
We also examined renal oxygenation by measuring RBF, renal oxygen delivery (DO2), renal oxygen consumption (QO2), renal fractional O2 extraction (FO2), and ratio of tubular Na reabsorption (TNa) to QO2. RBF was significantly lower in STN compared with controls (P < 0.01). HIF-1α induction by DMOG treatment increased RBF in STN (P < 0.05). DO2 was significantly reduced in STN kidneys because of a reduction in RBF (P < 0.01). Renal QO2 was similar in STN compared with controls, despite a significant decrease in the number of functioning nephrons. FO2, percentage of O2 consumed per O2 delivered, was significantly higher in STN vs. controls (P = 0.01). DMOG treatment significantly improved DO2 (P < 0.01) and lowered FO2 (P = 0.05). TNa per mole of oxygen consumed was significantly lower in STN compared with controls (P < 0.05) (Fig. 1). DMOG treatment significantly increased TNa/QO2 in STN (P < 0.05), while no significant changes were seen in controls.
Mitochondrial respiration.
To investigate the role of mitochondrial dysfunction in lowering TNa/QO2 in STN kidney, we assessed oxidative metabolism at a mitochondrial level. We measured OCR in fresh isolated mitochondria from kidneys with a Clark-type O2 electrode by using the Hansatech Oxytherm apparatus (PP Systems, Amesbury, MA). Respiratory rates were measured according to established protocols for oxygraphic measurements of oxygen consumption in isolated mitochondria (5, 35) and described in methods.
In paired experiments, OCR in mitochondria from control and STN kidneys were measured. As demonstrated in Fig. 2A, coupled (state 3) rates were elevated in STN mitochondria compared with control in the presence of both complex I substrates pyruvate-malate (P = 0.008) and glutamate-malate (P = 0.007) and complex II (succinate) substrate (P = 0.04). Uncoupled (state 3u) rates (Fig. 2C) were also elevated in STN mitochondria compared with control in the presence of complex I substrates (P < 0.001 for both pyruvate and glutamate) and complex II substrate (succinate, P = 0.05). State 4 rates (Fig. 2B) were also significantly elevated in STN mitochondria in presence of pyruvate (P = 0.015) and glutamate (P = 0.004), while there was a trend for an increase in the presence of succinate (P = 0.08). These results indicate an overall increase in oxidative metabolism in the STN kidney paralleling the in vivo results of tissue oxygen consumption described above. As both state 3 and state 4 rates were elevated, the ratio between the two, the respiratory control ratio, was not significantly different between control and STN. In separate experiments, the mitochondria isolated only from the cortical region did not exhibit significant differences in mitochondrial respiration between STN and control (Fig. 3, A–C), suggesting increased metabolism in the medullary region as driving the increased OCR.
Fig. 2.

Mitochondrial oxygen consumption rates (OCR) in STN A: state 3. Increased mitochondrial OCR in STN with the addition of ADP in the presence of oxidizable complex I substrates (pyruvate and glutamate) and complex II substrate (succinate) after inhibition of complex I by rotenone. *P = 0.008, #P = 0.007, and $P = 0.049 for STN vs. control (Con); n = 8 per group. B: state 4. Increased mitochondrial OCR in STN with the addition of ATP synthase inhibitor oligomycin in the presence of oxidizable complex I substrates (pyruvate and glutamate) and complex II substrate (succinate) after inhibition of complex I by rotenone. *P = 0.015, #P = 0.004, and $P = 0.08 for STN vs. control (Con); n = 8 per group. C: state 3u: Increased mitochondrial OCR in STN with the addition of uncoupler, FCCP, in the presence of oxidizable complex I substrates (pyruvate and glutamate) and complex II substrate (succinate) after inhibition of complex I by rotenone. *P < 0.001, #P < 0.001, and $P = 0.05 for STN vs. control (Con); n = 8 per group.
Fig. 3.

Cortical mitochondrial oxygen consumption rates (OCR). Unchanged cortical mitochondrial OCR for state 3 (A), state 4 (B), and state 3u (C) between STN vs. Con; n = 5 per group.
Separate experiments were conducted in mitochondria isolated from kidneys in STN and controls (Con) treated with DMOG and compared with untreated groups. With HIF-1α induction, in general, mitochondrial respiration was reduced in both controls and STN, but significantly so in the presence of succinate as shown in Fig. 4. Both state 3 and state 3u rates were significantly reduced in DMOG-treated control and STN rats (P < 0.05 Con vs. Con+DMOG and STN vs. STN+DMOG).
Fig. 4.
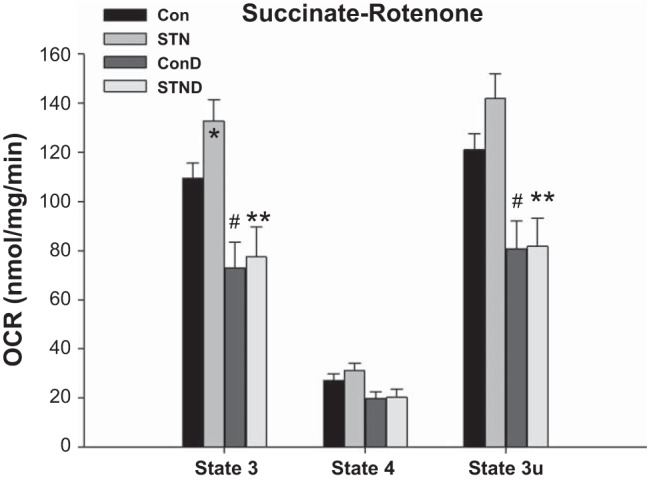
Impact of hypoxia inducible factor-1α (HIF-1α) activation on mitochondrial OCR. Mitochondrial OCR significantly reduced in both controls and STN with HIF-1α activation with DMOG in state 3 and state 3u in the presence of complex II substrate (succinate) after inhibition of complex I by rotenone. For state 3 ANOVA P < 0.001 and state 3u ANOVA P = 0.002, post hoc pairwise comparisons: *P < 0.05 STN vs. Con, #P < 0.05 ConD vs. Con, and **P < 0.05 STN vs. STND; n = 4–8 per group.
Mitochondrial ATP and ROS generation.
ATP production is a fundamental consequence of coupled mitochondrial oxidative phosphorylation and an important parameter to determine the fate of increased oxygen utilization by the mitochondria. Interestingly, despite the increase in mitochondrial respiration, ATP levels were modestly but not significantly higher in STN mitochondria compared with controls. ATP levels were reduced with DMOG treatment in both controls (P = 0.006, Con vs. Con+DMOG) and in STN (P = 0.05, STN vs. STN+DMOG) (Fig. 5A). Mitochondrial superoxide production was measured in fresh isolated mitochondrial with EPR spin trapping spectroscopy. Mitochondrial superoxide production was significantly elevated in STN compared with controls (P < 0.001). HIF-1α induction significantly lowered superoxide production in STN mitochondria (P < 0.001) (Fig. 5B). These results collectively demonstrate significant beneficial effects of HIF-1α induction on mitochondrial function.
Fig. 5.
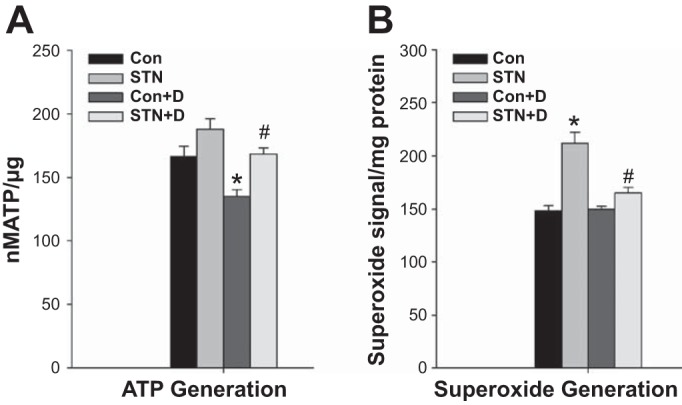
A: ATP content. ATP levels were not significantly different in STN compared with control mitochondria. HIF-1α activation with DMOG significantly lowered ATP levels in both controls and STN. ANOVA P < 0.001, post hoc pairwise comparisons: *P = 0.006 Con vs. Con+D and #P = 0.05 STN vs. STN+D; n = 4 per group. B: superoxide production. Significant increase in superoxide production by mitochondria isolated from STN kidneys. HIF-1α activation with DMOG significantly lowered superoxide levels in STN. ANOVA P < 0.001, post hoc pairwise comparisons: *P < 0.001 STN vs. Con and #P < 0.001 STN vs. STN+D; n = 4 per group.
Mitochondrial morphology.
We performed electron microscopy to correlate functional characteristics with systematic and regional morphological differences in the mitochondria between the STN and control kidneys. Specifically, we assessed mitochondrial length, volume density, number, and cristae abundance.
Significant regional alterations in mitochondrial morphology were found in the STN kidney (Fig. 6). In general, several mitochondria in the STN kidney showed swollen mitochondrial matrix and vesiculated cristae in random samples collected from different sections of the kidney (representative pictures shown in Fig. 6A). Regional differences in morphology are shown in Fig. 6, B–E. Mitochondrial length was similar in cortical and medullary samples from the control kidneys. However, medullary mitochondria in the STN kidneys were significantly shorter than cortical mitochondria (P < 0.01) and also compared with medullary mitochondria in the control kidneys (P < 0.01). Mitochondrial lengths were measured based on two-dimensional electron microscopy images, and mitochondria may be longer if measured in three-dimensional electron microscopy. Medullary mitochondria in the STN kidneys were also more numerous compared with cortical mitochondria in the STN kidneys (P < 0.01) and with medullary mitochondria in the control kidneys (P < 0.01). STN cortical mitochondria demonstrated increased mitochondrial volume density, expressed as the percentage of the cytoplasm occupied by mitochondria, compared with control cortical mitochondria (P < 0.01) and increased cristae abundance, measured as mean cristae surface area normalized to mitochondria outer membrane surface area, compared both to control cortex (P < 0.01) and STN medulla (P < 0.01). To summarize, STN medullary mitochondria were more rounded and increased in number, suggesting increased fragmentation due to mitochondrial fission, whereas STN cortical mitochondria were longer with higher volume density and more cristae indicative of mitochondrial fusion.
Fig. 6.

Mitochondrial morphology by electron microscopy. A: altered morphology of mitochondria in STN (representative pictures). Panel 1: control mitochondria; panel 2: swollen mitochondrial matrix with vesiculated cristae in STN; panel 3: control cortical (left) and medullary (right) mitochondria; panel 4: elongated cortical (left) and rounded medullary (right) mitochondria in STN. The first scale bar on panel 1 applies to both panels 1 and 2 and represents 1,000 nm. The second scale bar on panel 3 applies to both panels 3 and 4 and represents 500 nm. B: mitochondrial length. Mitochondria in STN cortex were significantly longer than control cortex as well as compared with mitochondria in STN medulla. ANOVA P < 0.001, post hoc pairwise comparisons: *P < 0.01 STN cortex vs. Con cortex and #P < 0.01 STN medulla vs. STN cortex and control medulla. No significant differences were seen between control cortical and medullary mitochondria. C: mitochondrial number. Mitochondrial number, counted per square micron, were significantly lower in STN cortex compared with control cortex, whereas the STN medullary mitochondrial were significantly higher than those in STN cortex. ANOVA P < 0.001, post hoc pairwise comparisons: *P < 0.01 STN cortex vs. control cortex and #P < 0.01 STN medulla vs. STN cortex and P = 0.02 STN medulla vs. control medulla. No significant differences were seen between control cortical and medullary mitochondria. D: mitochondrial volume density. In STN kidneys, mitochondrial volume density, expressed as the percentage of the cytoplasm occupied by mitochondria, was significantly higher in STN cortex compared with control cortex. ANOVA P < 0.001, post hoc pairwise comparisons: *P < 0.01 STN cortex vs. control cortex. No significant differences were seen between control cortical and medullary mitochondria. E: mitochondrial cristae abundance. In STN kidneys, cristae abundance, measured as mean cristae surface area normalized to mitochondria outer membrane surface area, were increased in STN cortex compared with control cortex and STN medulla. ANOVA P < 0.001, post hoc pairwise comparisons: *P < 0.01 STN cortex vs. control cortex and #P < 0.01 STN medulla vs. STN cortex. No significant differences were seen between control cortical and medullary mitochondria.
We also examined mitochondrial morphology in DMOG-treated control and STN kidneys. Major differences were seen only in the cortical mitochondria (Fig. 7). There was a significant increase in mitochondrial number with DMOG treatment in STN cortex (P < 0.01) (Fig. 7A). HIF-1α induction also significantly increased mitochondrial volume density in both control and STN cortical mitochondria (P < 0.01) (Fig. 7B). However, no significant effects of DMOG treatment were noted in terms of mitochondrial length or cristae abundance.
Fig. 7.
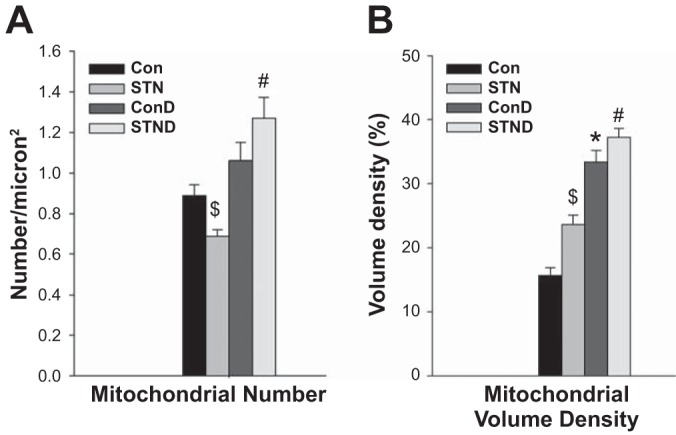
HIF-1α activation and mitochondrial morphology. A: mitochondrial number. Mitochondrial number per square micron were significantly elevated in the STN cortex with DMOG treatment. ANOVA P < 0.001, post hoc pairwise comparisons: #P < 0.01 STN+D vs. STN and $P = 0.02 STN vs. Con. B: mitochondrial volume density. In both control and STN cortical mitochondria, DMOG treatment increased mitochondrial volume density. ANOVA P < 0.001, post hoc pairwise comparisons: *P < 0.01, Con+D vs. Con, #P < 0.01 STN+D vs. STN, and $P < 0.01 STN vs. Con.
DISCUSSION
Pathophysiology of the CKD is independent of specific etiology and is determined by the behavior of the surviving nephrons as they maintain renal function under ongoing stress of rapid and extensive nephron loss. We have utilized the STN model to investigate the hemodynamic and metabolic adaptations that occur in the surviving nephrons following nephron loss (8, 9, 24, 39, 40, 42). At early stages, we observed a significant reduction in TNa per mole of oxygen consumed or high nephron oxygen consumption per surviving nephron. Much of oxygen consumed by the kidney is utilized for ATP production to support Na transport, and the remaining is utilized for other nontransport related activities, referred to as basal metabolism. The ratio of TNa/QO2 has been utilized as a measure of transport efficiency wherein changes in the ratio or slope of the relationship has been interpreted to indicate altered transport efficiency. However, as elegantly discussed recently by Evans et al. (12), this interpretation assumes a fixed basal metabolism, which is unaffected with changes in Na transport. It is important to recognize that maneuvers that change GFR and Na transport can independently change basal O2 consumption, hence TNa/QO2 can change independently of the efficiency of Na transport.
Here, we focus on altered mitochondrial bioenergetic capacity or efficiency (more oxygen utilization per net ATP generated) as a hypothesis for the altered oxygen utilization. We uncovered significant mitochondrial dysfunction in early STN kidney-higher oxygen consumption, mirroring the in vivo results, but without an attendant increase in ATP levels. We also observed elevated superoxide production and alterations in mitochondrial morphology.
The role of mitochondrial dysfunction has recently received significant attention in the pathophysiology of AKI (6, 20, 32, 48, 51, 52) and in diabetic CKD (10, 14, 33, 38), but literature in nondiabetic CKD is limited. Mitochondria respond to changing energy demands in the cells and play a key role in the adaptation to cellular stress (5, 25) such as hypoxia. Mitochondria are also the major source of intracellular ROS, mainly superoxide, which leads to oxidative stress. ROS generation by mitochondria is higher with increased oxidative metabolism when the redox carriers are abundantly charged because of high throughput (1). We observed a significant increase in mitochondrial ROS generation in the STN kidney. The compensatory hyperfiltration in the surviving nephrons likely drives the demand for oxidative metabolism in STN. The decreased oxygen delivery due to reduced RBF and increased oxygen demand can create an oxygen supply-demand mismatch leading to local tissue hypoxia. We and others have demonstrated early tubular hypoxia in STN that precedes any structural changes (27, 39). Interestingly, hypoxia can also lead to increased mitochondrial ROS production and impaired antioxidant mechanisms (1). Excessive ROS production can worsen hypoxia by increasing oxygen utilization, creating a vicious cycle which can result in cellular death.
The morphology of the mitochondrial network is regulated by a balance between fusion and fission events (3, 55). Fission is the division of mitochondria within a cell to form separate mitochondrial compartments (shorter, rounder), while fusion is the merging of two or more mitochondria to form a single compartment (elongated). Changes in the balance of fission and fusion strongly influence mitochondrial metabolism, turnover, bioenergetic capacity, and survival (3, 25, 55). Specifically, attenuated fusion and/or increased fission results in mitochondrial fragmentation and dysfunction (4). While changes in energy demand or supply influence ATP production, morphology of mitochondria also impact bioenergetic capacity and adaptation to metabolic demands of the cell (15, 25, 36). In embryonic fibroblasts, increased ATP synthesis capacity was dependent on decreased fission and increased fusion (15). Decreased fusion was associated with decreased ATP synthesis efficiency and lower expression of ETC proteins in hepatocytes (36). Fragmented mitochondria were found in skeletal muscle from Type 2 diabetic obese subjects associated with decreased ETC activity (22). Fragmented mitochondria show high respiration and reduced ATP synthesis (25). Thus changes in mitochondrial morphology as a mechanism contributing to decreased bioenergetic capacity could explain the dissociation between high respiration without a significant increase in ATP levels in the STN kidney.
We observed significant alterations in mitochondrial morphology. STN medullary mitochondria demonstrated increased fragmentation due to mitochondrial fission, lower mitochondrial volume density, and fewer cristae. Interestingly, STN cortical mitochondria had more fused morphology with greater volume density and cristae abundance compared with the STN medullary mitochondria. These morphological changes are characteristic of mitochondrial adaptation to increased oxidative demand, thus suggesting that cortical mitochondria are better adapted compared with medullary mitochondria in STN. The cortex is well oxygenated, while the blood supply to the medulla is limited to preserve medullary osmotic gradients for urinary concentration but leaves the medullary tissue more prone to hypoxia (11). We believe that the increase in ATP demand due to early hyperfiltration and hyperreabsorption is likely better handled by the adapted mitochondria in the better oxygenated cortical tubules, whereas the medullary mitochondria in a hypoxic environment undergo detrimental changes in mitochondrial dynamics in the early stages, which significantly impacts their function.
HIF-1α activation in acute and chronic kidney injury has been shown to improve GFR and reduce tubular injury (16, 29). We observed several beneficial effects of HIF-1α upregulation on renal hemodynamics in the STN kidney. There was a significant improvement in RBF and GFR. Importantly, HIF-1α activation significantly increased TNa/QO2 ratio, reduced mitochondrial oxygen consumption, and ameliorated other functional abnormalities in STN mitochondria. We observed reduction in mitochondrial respiration with HIF-1α activation, most dramatically in the presence of complex I inhibitor (rotenone) and succinate (complex II substrate). These results suggest that HIF-1α induction may lower mitochondrial respiration in the kidneys via effects on complex II function. HIF-1α activation has been shown to affect mitochondrial metabolism in cells, particularly during hypoxia adaptation. It diminishes NADH supply to the ETC and induces a subunit switch in complex IV of the ETC to optimize its efficiency in hypoxia (50). As a compensatory mechanism in hypoxia, HIF-1α downregulates mitochondrial oxygen consumption and lowers ROS generation (2, 31). HIF-1α induces a glycolytic phenotype by upregulating GLUT transporters to facilitate glucose entry and inducing glycolytic enzymes, which diminish Krebs cycle activity and shifts the cell toward glycolytic metabolism (37). Finally, it induces mitochondrial autophagy as an adaptive metabolic response to prevent increased levels of ROS generation and cell death in hypoxia (56). We also observed significant reduction in ATP levels and superoxide levels with HIF-1α activation in STN. Lastly, we observed beneficial effects on mitochondrial volume density and mitochondrial number with HIF-1α activation.
In conclusion, our findings demonstrate impaired renal oxygenation and mitochondrial dysfunction at the early stages of CKD, which precede tissue injury or proteinuria. These mechanistic findings have significant clinical relevance, given the recent studies demonstrating the association between mitochondrial dysfunction and human kidney disease (38, 47, 51, 52). Our observations also demonstrate that HIF-1α may be a potential therapeutic target to early mitochondria dysfunction and attenuate loss of renal function in CKD.
GRANTS
This work was supported by VA Merit BX002175 (to P. Singh), National Institutes of Health Grants R01 DK-107852 (to P. Singh), R03 DK-101841 (to P. Singh), and UAB-UCSD O’Brien Center (National Institutes of Health Grant P30 DK-079337). Support was also provided by VA Merit BX001963 (to H. Patel) and National Institutes of Health Grants HL-091071 (to H. Patel), HL 107200 (to H. Patel), HL-066941 (to H. Patel), and P41GM103412-28 (supporting National Center for Microscopy and Imaging Research at UC San Diego).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.L.T., H.P., E.H., G.A.P., S.S.A., and P.S. performed experiments; J.L.T., H.P., Y.L., E.H., G.A.P., S.S.A., H.H.P., and P.S. analyzed data; J.L.T., H.P., Y.L., G.A.P., S.S.A., H.H.P., and P.S. interpreted results of experiments; J.L.T., Y.L., G.A.P., S.S.A., H.H.P., and P.S. edited and revised manuscript; H.P., G.A.P., and P.S. prepared figures; P.S. conceived and designed research; P.S. drafted manuscript; P.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Some of the results have been presented as abstracts at American Society of Nephrology and Experimental Biology Annual Meetings.
REFERENCES
- 1.Addabbo F, Montagnani M, Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension 53: 885–892, 2009. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali SS, Hsiao M, Zhao HW, Dugan LL, Haddad GG, Zhou D. Hypoxia-adaptation involves mitochondrial metabolic depression and decreased ROS leakage. PLoS One 7: e36801, 2012. doi: 10.1371/journal.pone.0036801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 369: 2236–2251, 2013. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 4.Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg-Henriksson H, Manco M, Calvani M, Castagneto M, Palacín M, Mingrone G, Zierath JR, Vidal H, Zorzano A. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor α and interleukin-6. Diabetes 54: 2685–2693, 2005. doi: 10.2337/diabetes.54.9.2685. [DOI] [PubMed] [Google Scholar]
- 5.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312, 2011. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119: 1275–1285, 2009. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deetjen P, Kramer K. [The relation of O2 consumption by the kidney to Na re-resorption]. Pflugers Arch Gesamte Physiol Menschen Tiere 273: 636–650, 1961. doi: 10.1007/BF00361632. [DOI] [PubMed] [Google Scholar]
- 8.Deng A, Arndt MA, Satriano J, Singh P, Rieg T, Thomson S, Tang T, Blantz RC. Renal protection in chronic kidney disease: hypoxia-inducible factor activation vs. angiotensin II blockade. Am J Physiol Renal Physiol 299: F1365–F1373, 2010. doi: 10.1152/ajprenal.00153.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng A, Tang T, Singh P, Wang C, Satriano J, Thomson SC, Blantz RC. Regulation of oxygen utilization by angiotensin II in chronic kidney disease. Kidney Int 75: 197–204, 2009. doi: 10.1038/ki.2008.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, Andreyev A, Quach T, Ly S, Shekhtman G, Nguyen W, Chepetan A, Le TP, Wang L, Xu M, Paik KP, Fogo A, Viollet B, Murphy A, Brosius F, Naviaux RK, Sharma K. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 123: 4888–4899, 2013. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epstein FH, Agmon Y, Brezis M. Physiology of renal hypoxia. Ann N Y Acad Sci 718: 72–82, 1994. doi: 10.1111/j.1749-6632.1994.tb55706.x. [DOI] [PubMed] [Google Scholar]
- 12.Evans RG, Harrop GK, Ngo JP, Ow CP, O’Connor PM. Basal renal O2 consumption and the efficiency of O2 utilization for Na+ reabsorption. Am J Physiol Renal Physiol 306: F551–F560, 2014. doi: 10.1152/ajprenal.00473.2013. [DOI] [PubMed] [Google Scholar]
- 13.Fridolfsson HN, Kawaraguchi Y, Ali SS, Panneerselvam M, Niesman IR, Finley JC, Kellerhals SE, Migita MY, Okada H, Moreno AL, Jennings M, Kidd MW, Bonds JA, Balijepalli RC, Ross RS, Patel PM, Miyanohara A, Chen Q, Lesnefsky EJ, Head BP, Roth DM, Insel PA, Patel HH. Mitochondria-localized caveolin in adaptation to cellular stress and injury. FASEB J 26: 4637–4649, 2012. doi: 10.1096/fj.12-215798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friederich-Persson M, Aslam S, Nordquist L, Welch WJ, Wilcox CS, Palm F. Acute knockdown of uncoupling protein-2 increases uncoupling via the adenine nucleotide transporter and decreases oxidative stress in diabetic kidneys. PLoS One 7: e39635, 2012. doi: 10.1371/journal.pone.0039635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunaratnam L, Bonventre JV. HIF in kidney disease and development. J Am Soc Nephrol 20: 1877–1887, 2009. doi: 10.1681/ASN.2008070804. [DOI] [PubMed] [Google Scholar]
- 17.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol 291: F271–F281, 2006. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris DC, Chan L, Schrier RW. Remnant kidney hypermetabolism and progression of chronic renal failure. Am J Physiol 254: F267–F276, 1988. [DOI] [PubMed] [Google Scholar]
- 19.Heyman SN, Khamaisi M, Rosen S, Rosenberger C. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am J Nephrol 28: 998–1006, 2008. doi: 10.1159/000146075. [DOI] [PubMed] [Google Scholar]
- 20.Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, Schnellmann RG. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol 25: 1157–1162, 2014. doi: 10.1681/ASN.2013090952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang DH, Kanellis J, Hugo C, Truong L, Anderson S, Kerjaschki D, Schreiner GF, Johnson RJ. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol 13: 806–816, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 23.Körner A, Eklöf AC, Celsi G, Aperia A. Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes 43: 629–633, 1994. doi: 10.2337/diab.43.5.629. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Satriano J, Thomas JL, Miyamoto S, Sharma K, Pastor-Soler NM, Hallows KR, Singh P. Interactions between HIF-1α and AMPK in the regulation of cellular hypoxia adaptation in chronic kidney disease. Am J Physiol Renal Physiol 309: F414–F428, 2015. doi: 10.1152/ajprenal.00463.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506, 2013. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mandel LJ. Primary active sodium transport, oxygen consumption, and ATP: coupling and regulation. Kidney Int 29: 3–9, 1986. doi: 10.1038/ki.1986.2. [DOI] [PubMed] [Google Scholar]
- 27.Manotham K, Tanaka T, Matsumoto M, Ohse T, Miyata T, Inagi R, Kurokawa K, Fujita T, Nangaku M. Evidence of tubular hypoxia in the early phase in the remnant kidney model. J Am Soc Nephrol 15: 1277–1288, 2004. doi: 10.1097/01.ASN.0000125614.35046.10. [DOI] [PubMed] [Google Scholar]
- 28.Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol 17: 17–25, 2006. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- 29.Nangaku M, Inagi R, Miyata T, Fujita T. Hypoxia and hypoxia-inducible factor in renal disease. Nephron, Exp Nephrol 110: e1–e7, 2008. doi: 10.1159/000148256. [DOI] [PubMed] [Google Scholar]
- 30.Nath KA, Croatt AJ, Hostetter TH. Oxygen consumption and oxidant stress in surviving nephrons. Am J Physiol 258: F1354–F1362, 1990. [DOI] [PubMed] [Google Scholar]
- 31.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197, 2006. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol 306: F734–F743, 2014. doi: 10.1152/ajprenal.00643.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Persson MF, Franzén S, Catrina SB, Dallner G, Hansell P, Brismar K, Palm F. Coenzyme Q10 prevents GDP-sensitive mitochondrial uncoupling, glomerular hyperfiltration and proteinuria in kidneys from db/db mice as a model of type 2 diabetes. Diabetologia 55: 1535–1543, 2012. doi: 10.1007/s00125-012-2469-5. [DOI] [PubMed] [Google Scholar]
- 34.Redfors B, Swärd K, Sellgren J, Ricksten SE. Effects of mannitol alone and mannitol plus furosemide on renal oxygen consumption, blood flow and glomerular filtration after cardiac surgery. Intensive Care Med 35: 115–122, 2009. doi: 10.1007/s00134-008-1206-5. [DOI] [PubMed] [Google Scholar]
- 35.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 6: e21746, 2011. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sebastián D, Hernández-Alvarez MI, Segalés J, Sorianello E, Muñoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Orešič M, Pich S, Burcelin R, Palacín M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528, 2012. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 24: 97–106, 2009. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 38.Sharma K, Karl B, Mathew AV, Gangoiti JA, Wassel CL, Saito R, Pu M, Sharma S, You YH, Wang L, Diamond-Stanic M, Lindenmeyer MT, Forsblom C, Wu W, Ix JH, Ideker T, Kopp JB, Nigam SK, Cohen CD, Groop PH, Barshop BA, Natarajan L, Nyhan WL, Naviaux RK. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol 24: 1901–1912, 2013. doi: 10.1681/ASN.2013020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh P, Blantz RC, Rosenberger C, Gabbai FB, Schoeb TR, Thomson SC. Aberrant tubuloglomerular feedback and HIF-1α confer resistance to ischemia after subtotal nephrectomy. J Am Soc Nephrol 23: 483–493, 2012. doi: 10.1681/ASN.2011020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh P, Deng A, Blantz RC, Thomson SC. Unexpected effect of angiotensin AT1 receptor blockade on tubuloglomerular feedback in early subtotal nephrectomy. Am J Physiol Renal Physiol 296: F1158–F1165, 2009. doi: 10.1152/ajprenal.90722.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh PMA, Thomson SC. Metabolic basis of solute transport. In: Brenner & Rector’s The Kidney, edited by Skorecki K, Chertow GM, Marsden PA, Taal MW, Yu ASL. Philadelphia, PA: Elsevier, 2016, p. 122–143. [Google Scholar]
- 42.Singh P, Thomson SC. Salt sensitivity of tubuloglomerular feedback in the early remnant kidney. Am J Physiol Renal Physiol 306: F172–F180, 2014. doi: 10.1152/ajprenal.00431.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song YR, You SJ, Lee YM, Chin HJ, Chae DW, Oh YK, Joo KW, Han JS, Na KY. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol Dial Transplant 25: 77–85, 2010. doi: 10.1093/ndt/gfp454. [DOI] [PubMed] [Google Scholar]
- 44.Swärd K, Valsson F, Sellgren J, Ricksten SE. Differential effects of human atrial natriuretic peptide and furosemide on glomerular filtration rate and renal oxygen consumption in humans. Intensive Care Med 31: 79–85, 2005. doi: 10.1007/s00134-004-2490-3. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka T, Kojima I, Ohse T, Ingelfinger JR, Adler S, Fujita T, Nangaku M. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest 85: 1292–1307, 2005. doi: 10.1038/labinvest.3700328. [DOI] [PubMed] [Google Scholar]
- 46.Thomson SC, Kashkouli A, Singh P. Glucagon-like peptide-1 receptor stimulation increases GFR and suppresses proximal reabsorption in the rat. Am J Physiol Renal Physiol 304: F137–F144, 2013. doi: 10.1152/ajprenal.00064.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tin A, Grams ME, Ashar FN, Lane JA, Rosenberg AZ, Grove ML, Boerwinkle E, Selvin E, Coresh J, Pankratz N, Arking DE. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the Atherosclerosis Risk in Communities Study. J Am Soc Nephrol 27: 2467–2473, 2016. doi: 10.1681/ASN.2015060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, David S, Burstein D, Karumanchi SA, Stillman IE, Arany Z, Parikh SM. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121: 4003–4014, 2011. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welch WJ, Baumgärtl H, Lübbers D, Wilcox CS. Nephron pO2 and renal oxygen usage in the hypertensive rat kidney. Kidney Int 59: 230–237, 2001. doi: 10.1046/j.1523-1755.2001.00483.x. [DOI] [PubMed] [Google Scholar]
- 50.Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol 300: C385–C393, 2011. doi: 10.1152/ajpcell.00485.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitaker RM, Korrapati MC, Stallons LJ, Jesinkey SR, Arthur JM, Beeson CC, Zhong Z, Schnellmann RG. Urinary ATP synthase subunit β is a novel biomarker of renal mitochondrial dysfunction in acute kidney injury. Toxicol Sci 145: 108–117, 2015. doi: 10.1093/toxsci/kfv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitaker RM, Stallons LJ, Kneff JE, Alge JL, Harmon JL, Rahn JJ, Arthur JM, Beeson CC, Chan SL, Schnellmann RG. Urinary mitochondrial DNA is a biomarker of mitochondrial disruption and renal dysfunction in acute kidney injury. Kidney Int 88: 1336–1344, 2015. doi: 10.1038/ki.2015.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamazaki KG, Andreyev AY, Ortiz-Vilchis P, Petrosyan S, Divakaruni AS, Wiley SE, De La Fuente C, Perkins G, Ceballos G, Villarreal F, Murphy AN. Intravenous (−)-epicatechin reduces myocardial ischemic injury by protecting mitochondrial function. Int J Cardiol 175: 297–306, 2014. doi: 10.1016/j.ijcard.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu X, Fang Y, Ding X, Liu H, Zhu J, Zou J, Xu X, Zhong Y. Transient hypoxia-inducible factor activation in rat renal ablation and reduced fibrosis with L-mimosine. Nephrology (Carlton) 17: 58–67, 2012. doi: 10.1111/j.1440-1797.2011.01498.x. [DOI] [PubMed] [Google Scholar]
- 55.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int 83: 568–581, 2013. doi: 10.1038/ki.2012.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283: 10892–10903, 2008. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]