

Cardiac radiation exposure during radiotherapy increases the risk of heart failure with preserved ejection fraction. In a novel rodent model, cardiac radiation exposure resulted in coronary microvascular rarefaction, oxidative stress, impaired PKG signaling, myocardial fibrosis, mild cardiomyocyte hypertrophy, left ventricular diastolic dysfunction, and elevated left ventricular filling pressures despite preserved ejection fraction.
Keywords: heart failure with preserved ejection fraction, diastolic dysfunction, animal model, radiation, coronary microvasculature
Abstract
Breast cancer radiotherapy increases the risk of heart failure with preserved ejection fraction (HFpEF). Cardiomyocytes are highly radioresistant, but radiation specifically affects coronary microvascular endothelial cells, with subsequent microvascular inflammation and rarefaction. The effects of radiation on left ventricular (LV) diastolic function are poorly characterized. We hypothesized that cardiac radiation exposure may result in diastolic dysfunction without reduced EF. Global cardiac expression of the sodium-iodide symporter (NIS) was induced by cardiotropic gene (adeno-associated virus serotype 9) delivery to 5-wk-old rats. SPECT/CT (125I) measurement of cardiac iodine uptake allowed calculation of the 131I doses needed to deliver 10- or 20-Gy cardiac radiation at 10 wk of age. Radiated (Rad; 10 or 20 Gy) and control rats were studied at 30 wk of age. Body weight, blood pressure, and heart rate were similar in control and Rad rats. Compared with control rats, Rad rats had impaired exercise capacity, increased LV diastolic stiffness, impaired LV relaxation, and elevated filling pressures but similar LV volume, EF, end-systolic elastance, preload recruitable stroke work, and peak +dP/dt. Pathology revealed reduced microvascular density, mild concentric cardiomyocyte hypertrophy, and increased LV fibrosis in Rad rats compared with control rats. In the Rad myocardium, oxidative stress was increased and in vivo PKG activity was decreased. Experimental cardiac radiation exposure resulted in diastolic dysfunction without reduced EF. These data provide insight into the association between cardiac radiation exposure and HFpEF risk and lend further support for the importance of inflammation-related coronary microvascular compromise in HFpEF.
NEW & NOTEWORTHY Cardiac radiation exposure during radiotherapy increases the risk of heart failure with preserved ejection fraction. In a novel rodent model, cardiac radiation exposure resulted in coronary microvascular rarefaction, oxidative stress, impaired PKG signaling, myocardial fibrosis, mild cardiomyocyte hypertrophy, left ventricular diastolic dysfunction, and elevated left ventricular filling pressures despite preserved ejection fraction.
the high doses of thoracic radiation used with radiotherapy for some thoracic tumors and with older breast cancer radiotherapy techniques increase the risk of cardiac disease (9, 50, 51, 54). Advances in radiotherapy planning have substantially reduced cardiac radiation exposure during contemporary thoracic radiotherapy (54). However, cardiac radiation exposure during contemporary breast cancer radiotherapy increases the risk of heart failure (HF) with preserved ejection fraction (HFpEF) in older women and in a dose-dependent manner (42). New patchy perfusion defects develop after thoracic radiotherapy, consistent with microvascular dysfunction, but such perfusion defects are unassociated with reductions in ejection fraction (EF) (31).
In adults, cardiomyocytes do not or rarely divide and, thus, are highly radioresistant (26, 27, 60). Experimental studies have firmly established that cardiac radiation exposure causes coronary microvascular endothelial cell damage and inflammation with subsequent coronary microvascular dysfunction and rarefaction and that myocardial necrosis is a late event (10–12, 60). However, little is known about the effect of radiation on left ventricular (LV) diastolic function.
An emerging HFpEF pathophysiological paradigm (37) postulates that multimorbidity creates a systemic proinflammatory milieu, leading to global coronary microvascular endothelial cell inflammation, myocardial inflammation and fibrosis, and oxidative stress, which limits myocardial nitric oxide (NO)-cGMP-PKG signaling, thus promoting cardiomyocyte hypertrophy and altered phosphorylation of proteins that influence cardiomyocyte relaxation and stiffness. This paradigm has an important correlate, in that an established consequence of coronary microvascular endothelial inflammation is impaired coronary microvascular endothelial function and, ultimately, coronary microvascular rarefaction (15). Indeed, several studies have demonstrated coronary microvascular dysfunction in HFpEF patients, and in an autopsy study of HFpEF, coronary microvascular rarefaction was evident in the LV myocardium and associated with the severity of myocardial fibrosis (23, 34, 35, 49, 57). A study of young patients with HFpEF and no typical risk factors demonstrated microcirculatory dysfunction in association with evidence of parvovirus B19 infection, a virus that specifically infects vascular endothelial (but not myocardial) cells (56).
Recognizing the association between radiation exposure and HFpEF and the potential for microcirculatory compromise in both radiation-induced myocardial damage and typical HFpEF, we hypothesized that global cardiac radiation exposure induces diastolic dysfunction in the absence of reduced EF. To test this hypothesis, we developed a novel model of global cardiac radiation exposure in the normal rat and assessed its effect on cardiac structure and function, myocardial structure, and microvascular density. Furthermore, as inflammation-induced coronary microvascular compromise in human HFpEF is associated with oxidative stress and impaired cGMP-PKG signaling (37), we assessed these pathways in the radiated myocardium.
METHODS
General study design.
External beam radiation causes variable and regional cardiac radiation exposure and may affect the lung and pericardium. In HFpEF, the microvascular inflammation is proposed to be diffuse and affect all cardiac chambers (52, 64). Thus, using adeno-associated virus serotype 9 (AAV9) (39) gene delivery of the rat sodium-iodide symporter (rNIS) gene followed by 131I to deliver cardiac radiation at two different doses [10 and 20 Gray (Gy)], we produced diffuse cardiac radiation exposure in male Sprague-Dawley rats. AAV9-rNIS was delivered via tail vein injection. Cardiac dosimetry was calculated from 125I uptake quantified by SPECT/CT. Animals were observed for 5 mo after 131I administration. Body size, blood pressure, heart rate, and cardiac function (echocardiography) were assessed serially over the study period. At the end of the study, animals underwent treadmill exercise testing, echocardiography, hemodynamic catheterization, and tissue harvest. The study was approved by the Mayo Institutional Animal Care and Use Committee.
Vector preparation.
Recombinant AAV9 vector was manufactured using the Adeno Helper-Free System (Stratagene-Agilent). Polyethylenimine (PEI-Max, Polysciences, Warrington, PA) was used to transfect human embryonic kidney (HEK)-293T cells with pAAV-rNIS encoding the rNIS transgene (courtesy of Dr. Carrasco) (8) under the transcriptional control of the cytomegalovirus promoter pAAV-pRep2Cap9 (courtesy of Dr. Johnston, University of Pennsylvania) and pHelper (AAV Helper-Free System). After 72 h, cells were lysed using freeze-thaw cycles. After treatment with nuclease (Benzonase, 125 U/ml), the lysate was fractionated in iodixanol gradient solution at 400,000 g for 2 h at 4°C. After desalting, the concentration of AAV9-rNIS viral genomes (vg) was determined by quantitative PCR.
AAV9-rNIS dose finding.
Viral dose was determined in preliminary experiments. Sprague-Dawley rats (5 wk old) were injected via tail vein with 2 × 1012, 5 × 1011, 5 × 1010, or 5 × 109 vg of AAV9-rNIS or with PBS (n = 4 for each group). After ≥3 wk, rats were euthanized and NIS expression in different tissues was assessed by immunohistochemistry. With the viral dose of 2 × 1012 or 5 × 1011 vg, NIS expression was observed in both the heart and skeletal muscle (Fig. 1A). With 5 × 1010 vg, immunohistochemistry showed minimal extracardiac NIS expression, whereas cardiac expression was still robust and evenly distributed, suggesting ≈20% cardiomyocyte transduction with robust cardiac 125I uptake on SPECT/CT imaging (Fig. 1B) and rNIS cardiac expression on Western blots (Fig. 1C). With 5 × 109 vg, cardiac expression was minimal/absent on immunohistochemistry, SPECT/CT imaging, and Western blots (Fig. 1, A–C). In the final study, 5 × 1010 vg produced robust cardiac, but not skeletal muscle, expression by Western blot analysis (Fig. 1D).
Fig. 1.

Adeno-associated virus serotype 9 (AAV9)-rat sodium-iodide symporter (rNIS) dose-finding study to produce cardiac-selective NIS expression. A: representative left ventricular [LV; ×5 (top) and ×20 (bottom) magnification] and skeletal muscle (×10 magnification) samples stained for rNIS (brown) for different doses of AAV9-rNIS. Doses of AAV9-rNIS ≥5 × 1011 vg showed saturated rNIS expression in the myocardial cells with marked (2 ×1012 vg) or mild (5 × 1011 vg) skeletal muscle expression. A dose of 5 × 1010 vg achieved heart-specific rNIS expression, while 5 × 109 vg revealed no detectable cardiac rNIS staining. B: SPECT/CT imaging 1 h after 125I administration showed robust 125I uptake in the heart with 5 × 1010 vg, whereas no cardiac signal was seen with 5 × 109 vg. C: cardiac NIS protein was not detectable by Western blot analysis with 5 × 109 vg of AAV9-rNIS, but dose-dependent increases were detected with 5 × 1010 to 2 × 1012 vg. Data are from 2 rats for each dose. D: Western blots showing robust myocardial, but no skeletal muscle, rNIS expression at the end of the study in rats from the experimental groups treated with 5 × 1010 vg of AAV9-rNIS. Data are from 4 rats.
Cardiac dosimetry.
First, cardiac radiation kinetics were defined in AAV9-rNIS-treated rats imaged by SPECT/CT after 125I administration. Micro-SPECT/CT (SPECT reconstruction: MILabs USPECT II, version 3.4f; CT reconstruction: NRecon version 1.6.3.0, SkyScan, Brucer-MicroCT, Kontich, Belgium) images were acquired at 1, 4, 6, 9, and 24 h after 125I injection under anesthesia (2% isoflurane via nose cone) 4 wk after tail vein injection of rNIS-AAV9 (5 × 1010 vg, n = 4) or PBS (n = 1). Cardiac accumulation of radioisotope (Fig. 2A) was analyzed by PMOD Biomedical Image Quantification and Kinetic Modeling Software (PMOD Technologies, Zurich, Switzerland). A volume of interest was drawn around the entire heart using CT scan; the cardiac region was then applied to the fused SPECT scan, and regional activity and volume were calculated. Values are given as ratio of cardiac activity to injected activity over time (Fig. 2B).
Fig. 2.

Cardiac dosimetry. A: representative serial SPECT/CT images after 125I tail vein injection at 9 wk of age (4 wk after AAV9-rNIS injection at 5 × 1010 vg). B: group data for rats injected with 5 × 1010 vg of AAV9-rNIS (n = 4) or PBS (n = 1) demonstrated time-dependent decay of cardiac 125I accumulation in AAV9-rNIS-injected rats but no cardiac accumulation in rats injected with PBS (control). C: calculation of biological cardiac half-life of 125I (see text). D: group data for the 1-h 125I uptake obtained using SPECT/CT in AAV9-rNIS-injected rats used for dosimetry and rats from the study groups to confirmed consistent NIS expression and peak 125I uptake.
The ratio of cardiac to injected activity was plotted as a hemi-logistic graph, and biological cardiac half-life (TbC, time to eliminate half of radioactivity from the heart) was calculated (Fig. 2C) as ln2/0.0706 = 9.8 h. The physical half-life (Tp; time for half of the radioactivity to decay with no other form of elimination of the radioisotope from the body) of 125I is 59.4 days and long enough to be ignored compared with the biological half-life. As the TbC values for 125I and 131I are the same, the effective half-life (Te) for 131I in the heart can be calculated using TbC and Tp of I131 (8 days) as follows: 1/Te = 1/Tp + 1/TbC = 9.32 h. In the four AAV9-rNIS-treated rats, we repeated 125I imaging 2 wk later and confirmed that the cardiac uptake was similar 1 h after 125I at 4 wk (27 ± 7 µCi/mCi) and 6 wk (26 ± 9 µCi/mCi) after AAV9-rNIS injection (P = 0.91). We then calculated the systemic dose (in mCi) of 131I needed to deliver 10 Gy (6.0 ± 0.2 mCi, 14.5 ± 0.9 mCi/kg) and 20 Gy (11.5 ± 0.9 mCi, 29.3 ± 2.2 mCi/kg) to the heart using 131I Te (9.32 h) according to medical internal radiation dosimetry methods (1, 2, 21, 29, 41).
Rats were studied in three groups; in each group, a sample of three to four rats (total: 11 rats) were randomly selected and imaged with SPECT/CT 1 h after 125I injection to validate consistent NIS expression and peak cardiac uptake by SPECT/CT (Fig. 2D), indicating appropriateness of the 131I doses.
131I + barium + l-thyroxine pilot studies.
To establish tolerability and exclude effects from 131I, barium administration (oral gavage) and thyroid supplementation alone, pilot studies were performed in normal rats (n = 10) treated at 10 wk of age with 131I and the same dose of barium and l-thyroxine used in the study rats. Rats were followed until 30 wk of age, as were rats in the other study groups. All rats survived without apparent gastrointestinal effects of 131I or evidence of abnormal thyroid status.
Final study: animal preparation and in vivo experiments.
Four-week-old male Sprague-Dawley rats were purchased from Charles River (Wilmington, MA), fed standard chow and tap water, and housed with a 12:12-h light-dark cycle.
At 5 wk of age, 5 × 1010 vg of AAV9-rNIS or 0.5 ml of PBS was injected via the tail vein. At 10 wk of age (5 wk after AAV9-rNIS injection), the 131I dose to produce a cardiac dose of 10 or 20 Gy was injected via the tail vein after barium administration (oral gavage) to protect the gastric mucosa (53). l-Thyroxine (Sigma-Aldrich) supplementation (5 mg/l in drinking water) was initiated 1 wk before the 131I injection to minimize thyroid uptake (18, 55) and continued until the terminal procedure to prevent hypothyroidism. Evidence of deranged thyroid status was monitored by weekly assessment of general physical condition, body weight, and water intake and monthly assessment of conscious tail cuff blood pressure and heart rate. As no animal displayed clinical evidence of abnormal thyroid hormone status, thyroxine (T4) and free T4 levels were measured 4 mo after 131I administration to exclude subclinical thyroid hormone deficiency. Rats were also monitored weekly for clinical evidence of HF (general condition, edema, respiratory status, and water intake). Under light anesthesia (1.5–2.0% isoflurane via nose cone), venous blood was drawn from the right jugular vein every 4 wk.
Tail-cuff blood pressure was measured every 4 wk using the CODA blood pressure system (Kent Scientific, Torrington, CT). Echocardiography was performed under light anesthesia (1.5–2.0% isoflurane via a nose cone) every 4 wk by a trained rodent cardiac sonographer using a 12-MHz transducer (Vivid 7, GE). Standard short-axis images at the papillary muscle level for anatomic M-mode and two-dimensional and color flow imaging for assessment of pericardial effusion and valvular regurgitation were assessed. End-diastolic and end-systolic LV volumes (EDV and ESV) were calculated using the M-mode cube method. EF was calculated as (EDV − ESV)/EDV.
Treadmill exercise testing was performed 1 wk before the terminal procedure (Panlab Harvard Apparatus, Barcelona, Spain). After 3 days of acclimation sessions, a standardized seven-stage ramped protocol was performed using electric stimuli to maintain effort until exhaustion. Exercise time, distance, and workload [in J, calculated as 9.8 (m/s2) × body weight (om kg) × 0.05 × distance (in m)] were assessed.
Cardiac catheterization.
Under 2% isoflurane anesthesia via nose cone, a 22-gauge venous catheter was inserted in the tail vein. Blood gas, electrolytes (Na+/K+, Ca2+, and ), and blood sugar levels were measured using iStat (Abbott, East Windsor, NJ). Central body temperature was monitored by rectal sensor and regulated at 37.5–38.0°C using an automated heating pad (Physio Suite, Kent Scientific) and a heating lamp if needed. An endotracheal tube was placed for ventilation and isoflurane administration. End-tidal CO2 (LoFlo, Respironics) and arterial O2 saturation were monitored (SomnoSuite, Kent Scientific) (7, 20) and maintained at 35–40 mmHg CO2 and >95% peripheral capillary O2 saturation, respectively, with a tidal volume of 10 ml/kg. After baseline heart rate was recorded, a muscle relaxant (cisatracurium besylate, Nimbex, 2 mg/kg) was injected intravenously every 20 min via a tail vein catheter.
Catheters (ADVantage Admittance Pressure Volume Systems, Transonic Scisense, Ithaca, NY) were immersed in a 1-ml syringe filled with 37.5°C water and calibrated at the level of the posterior axillary line before insertion. Appropriately calibrated 1.9-Fr manometer-tipped pressure (left jugular vein) and pressure admittance (right carotid artery) catheters were placed and advanced to the central vein and LV, respectively, using pressure waveform monitoring (ADVantage Admittance Pressure Volume Systems). Heparin (100 U/kg) was administered, and volume was repleted with normal saline (10 ml/kg body wt). Admittance was converted to volume by Wei’s method (61, 62) with stroke volume calibration from echocardiography. Steady-state pressure, volume, and ECG signals were acquired (Labscribe2, iWorks) at 1,000 Hz using an AV converter (model ETH-256, iWorks). Subsequently, a small subcostal incision was made, and the inferior vena cava (IVC) was temporarily clamped to decrease preload and define the systolic and diastolic pressure-volume relationships (ESPVR and EDPVR, respectively). The LV catheter was withdrawn to the aorta, and KCl was injected to terminate the cardiac cycle in diastole, with continuous monitoring of aortic and central venous pressures to measure mean circulatory filling pressure (MCFP) (16). Upon completion of the procedure, the accuracy of pressure catheter calibration was confirmed.
The ESPVR slope [end-systolic elastance (Ees)], preload recruitable stroke work (PRSW), and maximal rate of rise of LV pressure (dP/dtmax) were assessed as load-insensitive indexes of systolic performance. The slope of the ESPVR in small animals is generally nonlinear, and a single value for ESPVR slope may not represent each rat’s contractile state. Accordingly, statistical analysis of between-group differences used analysis of covariance (ANCOVA), including all end-systolic pressure and volume points during IVC occlusion from all rats, with group as a dummy variable, to compare the slope difference, as recommended by Burkhoff et al. (3) (see Statistical analysis). The time constant of isovolumic relaxation (τ) was calculated using a logistic fit (48). EDPVR was fit as follows: LVEDP = α × eβ × LVEDV, where LVEDP is LV end-diastolic pressure, α is the curve-fitting constant, β is the diastolic stiffness constant, and LVEDV is LV end-diastolic volume. For statistical analysis, the EDPVR was fit to a linear equation using ln(EDP) versus EDV and compared between groups with ANCOVA.
Immunohistochemistry.
Immediately upon completion of the terminal procedure, organs were washed with PBS, fixed in 10% natural buffered formaldehyde, and embedded in paraffin. Blocks were cut to 4 µm thickness and mounted on slides. Before they were stained, slides were incubated at 60°C for 2 h and then rehydrated with 100% xylene and a gradient ethanol bath. After antigen retrieval (10× citrate buffer, pH 6.0, catalog no. ab64214, Abcam) in a steamer for 30 min, innate peroxidase was blocked with 0.3% H2O2 for 20 min. Nonspecific blocking was performed with serum of the secondary antibody species. Slides were incubated with primary antibody (Table 1; CD34 at 1:4,000 dilution and rNIS antibody at 1:8,000 dilution) (36) for 2 h. After three 5-min washes in PBS, slides were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (donkey anti-rabbit HRP, catalog no. ab6802, Abcam) or biotin-conjugated secondary antibody and then revealed with 3,3′-diaminobenzidine (DAB) substrate directly (rNIS: Betazoid DAB chromogen kit, catalog no. BDB-2004H, Biocare Medical) or after signal enhancement with an avidin-biotin kit [CD34: Vectastain Elite ABC HRP kit with rabbit IgG (catalog no. PK-6101) and ImmPACT DAB HRP substrate (catalog no. SK-4105), Vector Laboratories, Burlingame, CA].
Table 1.
Antibodies
| Antibody | Vendor | Catalog No. |
|---|---|---|
| CD34 | Abcam | 81289 |
| Rat NIS | Carrasco Laboratory | |
| Phosphorylated Smad2 | Cell Signaling Technology | 3101 |
| Smad2/3 | Cell Signaling Technology | 8685 |
| Phosphorylated (Ser239) VASP | Cell Signaling Technology | 3114 |
| Phosphorylated (Ser157) VASP | Cell Signaling Technology | 3111 |
| VASP | Cell Signaling technology | 3112 |
| GAPDH | Abcam | ab9482 |
| PKG-1α | Blanton Laboratory |
NIS, sodium-iodide symporter; VASP, vasodilator-stimulated phosphoprotein.
Microvascular density, myocardial fibrosis, and cardiomyocyte size.
LV cross sections stained with CD34 antibody (see Immunohistochemistry) were imaged with whole field digital microscopy (Aperio slide scanner, Leica Biosystems, Buffalo Grove, IL). Sixteen randomly selected segments (8 endocardial and 8 epicardial) per heart were analyzed at ×20 magnification. After exclusion of nontissue area and myocardium with longitudinally sectioned myocytes, the number of microvessels per area (<314 μm2) was measured using an automated ImageJ macro (National Institutes of Health, Bethesda, MD) (32).
To quantify the fibrosis area, heart cross sections were rehydrated as described above and incubated for 2 h with saturated picrosirius solution (picric acid, saturated, aqueous, Electron Microscopy Sciences, Hatfield, PA) with 0.1% sirius red (Direct Red 80, Sigma-Aldrich). Slides were quickly dehydrated with 100% ethanol followed by xylene and mounted using organic medium (Eukitt quick-hardening mounting medium for microscopy, catalog no. 03989, Sigma-Aldrich). The section was scanned by whole-field digital microscopy (as described above) at ×40 magnification, and a ×5-magnified image of total tissue area was reconstructed. Interstitial fibrosis area in total tissue area was measured at ×5 magnification using red color thresholding on the ImageJ software and expressed as percent area of LV myocardial tissue.
Formalin-fixed, paraffin-embedded heart cross sections were stained with FITC-conjugated wheat germ agglutinin and nuclear (4′,6-diamidino-2-phenylindole) staining and used to determine cardiomyocyte size (5). Slides were incubated at 60°C for 2 h and then rehydrated with 100% xylene and a gradient ethanol bath. After antigen retrieval with citrate buffer (pH 6), slides were incubated with FITC-conjugated wheat germ agglutinin (50 ng/ml in PBS with 1 mM CaCl2, catalog no. FL-1021S, Vector Laboratories) for 60 min. After they were washed with PBS, slides were incubated with 4′,6-diamidino-2-phenylindole solution (1 µg/ml, catalog no. D9542, Sigma-Aldrich) for 5 min and then mounted with aqueous medium. Images were acquired using confocal microscopy (model LSM780, Carl Zeiss, Thornwood, NY) and a ×40 aqueous objective. In each of the four LV walls (anterior, lateral, septal, and inferior), 10 appropriately oriented cardiomyocytes for maximal longitudinal dimension and 10 appropriately oriented cardiomyocytes for minimal dimension were measured (6).
Natriuretic peptide mRNA quantification.
Natriuretic peptide mRNA level (normalized to controls) was evaluated by quantitative RT-PCR using SYBR green master mix (Applied Biosystems) and natriuretic peptide primers (Table 2). Total RNA was extracted from snap-frozen tissue using the RNeasy fibrous tissue kit (Qiagen, Germantown, MD), and 1 µg of RNA was subjected to cDNA synthesis (cDNA synthesis kit, Thermo Scientific). mRNA level for intervention groups relative to controls was calculated using the ΔΔCt method, where Ct is threshold cycle.
Table 2.
Primers
| Sequence |
||
|---|---|---|
| Primer | Forward | Reverse |
| ANP | 5′-CTGCGAAGGTCAAGCTGCTTC-3′ | 5′-ATCTTCGGTACCGGAAGCTGTT-3′ |
| BNP | 5′-ACGATGCAGAAGCTGCTGGAG-3′ | 5′-CGCTGTCTTGAGACCTAAGGACT-3′ |
| GAPDH | 5′-ACTCCCATTCTTCCACCTTTG-3′ | 5′-CCCTGTTGCTGTAGCCATATT-3′ |
ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide.
Oxidative stress assessment.
Confocal microscopy imaging of dihydroethidium (DHE)-stained LV cryosections (10 μm) was performed as a semiquantitative assessment of oxidative stress (33). The percent DHE-stained area was quantified by ImageJ. MnSOD activity was quantified in snap-frozen LV tissues using water-soluble tetrazolium salt (WST-1)-based SOD inhibition assay (SOD assay kit, Dojindo Molecular Technologies, Rockville, MD) according to the manufacturer’s instructions (19). The ratio of phosphorylated (p)Smad2 to Smad2/3 protein as a marker of transforming growth factor (TGF)-β activation was assessed by Western blot analysis (see Myocardial cGMP-PKG signaling).
Myocardial cGMP-PKG signaling.
To assess myocardial cGMP concentrations, heart samples were extracted four times in four volumes of ether-water, dried, and reconstituted in cGMP assay buffer. Samples were assayed using a competitive radioimmunoassay cGMP kit (Perkin-Elmer). Samples are corrected for dilution factors and normalized to protein concentration.
In vitro maximal PKG activity was determined by Cyclex colorimetric assay (MBL) according to the manufacturer’s instructions. Absorbance was measured at 450 nm using a plate-reader spectrophotometer, and quantitation was performed against a standard curve generated using full-length cGMP-dependent protein kinase (cGK; MBL). Results are expressed as units per microgram of protein.
In vivo PKG-1α dimer/monomer abundance and in vivo PKG activity as reflected by the Ser239-phosphorylated vasodilator-stimulated phosphoprotein (pVASP)-to-VASP ratio were assessed by Western blot analysis (see Western blot analysis). The Ser157-pVASP-to-VASP ratio as a marker of PKA-mediated VASP phosphorylation was also assessed.
Western blot analysis.
Snap-frozen LV tissue samples were crushed to powder on dry ice, and 20 mg of sample were homogenized with lysis buffer (catalog no. 9803, Cell Signaling Technology) containing 1 mM PMSF and phosphatase inhibitors (1 mM NaF and 2 mM Na3VO4). After centrifugation at 15,000 rpm for 30 min at 4°C, the supernatant was mixed with 4% lithium dodecyl sulfate sample buffer with 5 mM DTT and incubated at 80°C for 5 min; 30 µg of total protein were then separated by SDS-PAGE. Protein was blotted to a 0.44-µm PVDF membrane by wet transfer. For the nonreducing-condition PAGE for PKG-1α dimer/monomer analysis, 20 mg of sample were homogenized with lysis buffer (catalog no. 9803, Cell Signaling Technology) containing 100 mM N-ethylmaleimide to prevent artificial disulfide bond formation. The antibodies used for Western blot analyses are shown in Table 1.
Protein was detected by the chemiluminescense method [Immobilon Western chemiluminescence HRP substrate (Millipore) and ChemiDoc imaging system (Bio-Rad)] and quantified using Image laboratory software (Bio-Rad).
For pVASP and VASP Western blots, pVASP-to-VASP ratios were calculated from the same membrane (antibody stripping, Restore Western blot stripping buffer, Thermo Fisher). To exclude residual pVASP antibody persistence influencing total VASP quantitation, separate membranes were also used to assess pVASP-to-GAPDH and VASP-to-GAPDH ratios, and the pVASP-to-VASP ratio was calculated from the GAPDH-normalized pVASP and VASP. Findings for VASP-to-GAPDH, Ser239-pVASP-to-VASP, and Ser157-pVASP-to-VASP ratios were similar with the two methodologies. For pSmad2 and Smad2 Western blots, signals were calculated from the same membrane after stripping. The pSmad2-to-GAPDH and Smad2-to-GAPDH ratios were calculated. By indexing pSmad2 to Smad2, the factor of GAPDH was mathematically canceled.
Statistical analysis.
Group data are shown as scatterplots with mean and 95% confidence intervals or means ± SD. Comparison between more than two groups was performed using ANOVA with post hoc comparisons with the control group (Dunnett’s test) without correction for multiple comparisons. Given that incremental cardiac doses of radiation were tested, we also performed a post hoc test for trend across groups (control, 10 Gy, and 20 Gy). The relationship between two parameters was analyzed using linear regression except for the analysis of ESPVR and EDPVR slopes, where ANCOVA was used for all ESPVR or EDPVR pressure and volume data points, as recommended by Burkhoff et al. (3). For ANCOVA, between-group comparisons for ESPVR or (linearized) EDPVR slopes (where the overall ANCOVA P < 0.05) used least-squares regression with dummy variables for group. All statistical analysis was performed using JMP version 9 (SAS Institute, Cary, NC) or Prism 7.0 (GraphPad, La Jolla, CA).
RESULTS
Twenty-three male rats were each treated with 5 × 1010 vg of AAV9-rNIS and randomized to receive 131I at doses designed to deliver a cardiac dose of 10 Gy (n = 11) or 20 Gy (n = 12). Twelve age-matched normal male rats served as controls. Three rats (1 rat in the 10-Gy group and 2 rats in the 20-Gy group) died 70-84 days after 131I administration. Analysis used all rats in each group when possible. The number of rats with data for each study variable is shown in Table 3.
Table 3.
Number of animals with data for study procedures
| Control | 10 Gy | 20 Gy | |
|---|---|---|---|
| n | 12 | 11 | 12 |
| Surviving to study end | 12 | 10 | 10 |
| TMET | 10 | 10 | 10 |
| End-study echo | 12 | 10 | 10 |
| Cath ESPVR | 7 | 8 | 7 |
| Cath EDPVR | 7 | 9 | 7 |
| Cath LVEDP | 10 | 10 | 7 |
| Cath CVP | 9 | 10 | 7 |
| Cath MCFP | 11 | 10 | 8 |
| MVD | 12 | 10 | 10 |
| Fibrosis | 12 | 10 | 10 |
| Cardiomyocyte size | 10 | 10 | 9 |
| DHE | 5 | 6 | 5 |
| MnSOD | 10 | 10 | 7 |
| Smad | 8 | 9 | 8 |
| cGMP | 8 | 10 | 8 |
| In vitro PKG activity | 8 | 8 | 8 |
| PKG protein dimer/monomer | 9 | 5 | 5 |
| VASP | 8 | 10 | 8 |
n, Number of animals. TMET, treadmill exercise test; echo, echocardiography; Cath, cardiac catheterization; ESPVR, end-systolic pressure-volume relationship; EDPVR, end-diastolic pressure-volume relationship; LVEDP, left ventricular end-diastolic pressure; CVP, central venous pressure; MCFP, mean circulatory filling pressure; MVD, microvascular density; DHE, dihydroethidium.
Serial assessment of body weight, heart rate, systolic blood pressure, and EF (Fig. 3) and assessment of general condition revealed no evidence of altered thyroid status. At the end of the study, hematocrit was similar across groups (Table 4). T4 and free T4 levels 4 mo after 131I revealed no evidence of hypo- or hyperthyroidism, with levels on l-thyroxine supplementation slightly higher than control but within euthyroid range (Table 4). Serial echocardiography showed no pericardial effusion, significant valvular leakage, or impaired EF. At the end of the study, LV diastolic dimension tended to be smaller in radiated rats, but EF and wall thickness were similar (Table 4) to control rats. While the general condition of the radiated rats (normal fur appearance and no edema) appeared normal at treadmill exercise test, exercise capacity, including exercise duration, distance, and workload, was impaired in radiated rats relative to control rats (Fig. 4, A–C).
Fig. 3.

Serial assessment of body weight, heart rate (in beats/min), systolic blood pressure (BP), and ejection fraction (EF) over the study period. No significant group differences were apparent. BL, baseline.
Table 4.
Characteristics at end study
| Group |
||||
|---|---|---|---|---|
| Control | 10 Gy | 20 Gy | P Value (ANOVA) | |
| n | 12 | 10 | 10 | |
| Body wt, g | 627 ± 75 | 614 ± 63 | 584 ± 56 | 0.31 |
| Conscious heart rate, beats/min | 346 ± 35 | 314 ± 42 | 345 ± 35 | 0.10 |
| Conscious systolic BP, mmHg | 143 ± 17 | 138 ± 18 | 143 ± 17 | 0.75 |
| Hematocrit | 0.55 ± 0.04 | 0.56 ± 0.03 | 0.57 ± 0.02 | 0.34 |
| T4, μg/dl | ||||
| Plasma | 5.4 ± 0.9 | 8.0 ± 2.8* | 7.4 ± 2.1 | 0.014 |
| Plasma free | 2.4 ± 0.4 | 3.3 ± 1.3 | 3.1 ± 0.9 | 0.069 |
| LVEDV, μl | 754 ± 204 | 611 ± 83 | 640 ± 81 | 0.056 |
| Posterior wall thickness, mm | 1.87 ± 0.13 | 1.78 ± 0.24 | 1.98 ± 0.28 | 0.15 |
| Heart rate, beats/min | 324 ± 38 | 311 ± 33 | 306 ± 45 | 0.55 |
| LV systolic pressure, mmHg | 105 ± 17 | 105 ± 20 | 108 ± 20 | 0.93 |
| Arterial elastance, mmHg/ml | 174 ± 69 | 212 ± 60 | 178 ± 40 | 0.38 |
| Heart wt, mg/g | 2.57 ± 0.19 | 2.52 ± 0.36 | 2.69 ± 0.29 | 0.41 |
| Lung wt, mg/g | 3.48 ± 0.50 | 4.08 ± 0.92 | 5.38 ± 3.01 | 0.08 |
| ANP mRNA (normalized to control) | 1.00 ± 1.42 | 2.28 ± 1.36* | 3.76 ± 2.85* | N/A |
| BNP mRNA (normalized to control) | 1.00 ± 0.84 | 6.54 ± 8.88* | 6.66 ± 3.99* | N/A |
Values are means ± SD; n, number of rats. BP, blood pressure; T4, thyroxine; LVEDV, left ventricular end-diastolic volume (echo). Values for heart rate and LV systolic pressure were obtained at cardiac catheterization; N/A, not applicable.
P < 0.05 vs. control.
Fig. 4.

Exercise capacity. A–C: exercise duration, distance, and workload were lower in radiated rats than in control rats at 20 wk after radiation exposure. Data are presented as scatter dot plots, with lines showing mean and 95% confidence interval. *P < 0.05 vs. control. †Post hoc test for linear trend P < 0.003. D–F: severity of exercise intolerance (reduction in workload) was correlated with severity of increases in mean circulatory filling pressure (MCFP), reductions in microvascular density (MVD), and increases in LV fibrosis.
Systolic and diastolic ventricular function.
At catheterization, heart rate, peak ventricular systolic pressure, and effective arterial elastance were similar across groups (Table 4). At the end of the study, EF, Ees, PRSW, and peak +dP/dt (Fig. 5, A–D) were similar in radiated and control rats. The passive stiffness constant (β; Fig. 5E) was greater in both radiated groups than in the control group and greater in 20-Gy rats than in 10-Gy rats (P < 0.001 for all). τ (Fig. 5F) was higher in radiated rats than in control rats, consistent with impaired relaxation. EDP was higher than central venous pressure in all groups, and both were increased in radiated rats compared with control rats (Fig. 5G). MCFP was increased in radiated rats compared with control rats (Fig. 5H). Representative pressure-volume loops during IVC occlusion and all ESPVR and EDPVR data points in the three groups are shown in Fig. 6.
Fig. 5.

Ventricular systolic and diastolic function. At the end of the study, EF (A), slope of the end-systolic pressure-volume relationship (ESPVR; B), preload recruitable stroke work (PRSW; C), and peak rate of increase in LV pressure (+dP/dt; D) did not differ across groups. The passive stiffness constant (β), derived from the end-diastolic pressure-volume relationship (EDPVR; E), varied across groups and was steeper in 10-Gy (P = 0.0008) and 20-Gy (P = 0.0001) rats than in control rats and steeper in 20-Gy rats than in 10-Gy rats (P = 0.0001). The time constant of isovolumic relaxation (τ; F) was higher in radiated rats than in control rats, consistent with impaired relaxation. Filling pressures [central venous pressure (CVP) and LV end-diastolic pressure (LVEDP), G] and MCFP (H) were obtained after volume loading and were higher in radiated rats than in control rats. Data are presented as scatter dot plots, with lines showing mean and 95% confidence interval. *P < 0.05 vs. control. †Post hoc test for linear trend P < 0.007.
Fig. 6.
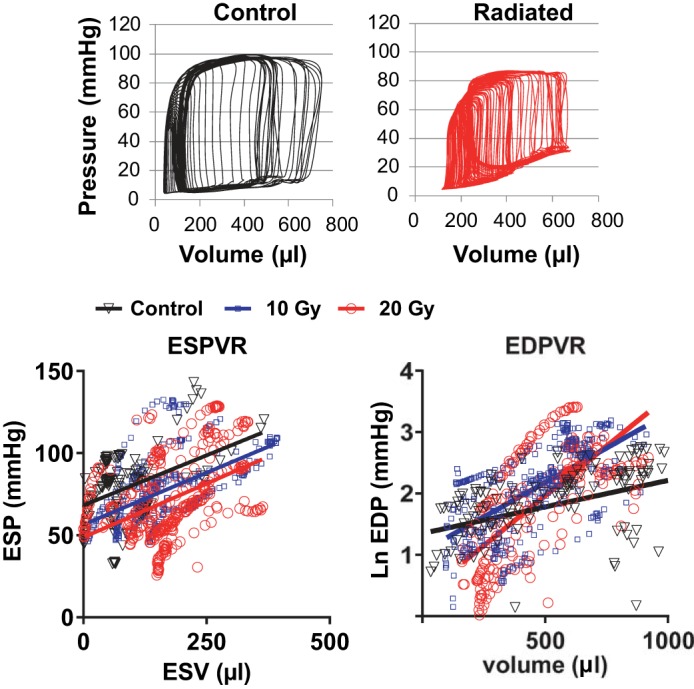
Catheterization data. Top: pressure-volume loops from a control rat and a radiated rat. Bottom: all data points and regression lines for ESPVR (left) and linearized EDPVR (right).
Myocardial histopathology.
At tissue harvest, the pericardium was translucent without adhesions to surrounding tissue or the myocardium. When indexed to body weight, heart weight was not different between groups but lung weight tended to be higher (P = 0.08) in radiated rats (Table 4). However, while cardiomyocyte length (maximum diameter) was not different across groups, cardiomyocyte width (minimum diameter) was increased (Fig. 7 and Fig. 8, A and B), and atrial and brain natriuretic peptide mRNA (Table 4) were increased in radiated rats compared with control rats. Microvascular density was decreased and fibrosis was increased in radiated rats compared with control rats (Fig. 7 and Fig. 8, C and D). Fibrosis was diffuse and not concentrated in the epi- or endocardium (Fig. 7). When adjusted for cardiomyocyte size (minimum dimension), microvascular density was still lower in radiated rats than in control rats (P = 0.002; Table 5). Both fibrosis and β increased as microvascular density decreased (Fig. 8, E and F). While β tended to increase with increasing fibrosis, this relationship was of borderline statistical significance (P = 0.06), and, in a multivariable analysis, when adjusted for fibrosis, microvascular density was still (inversely) associated with β (Table 5). The severity of exercise intolerance was correlated to the severity of elevation in filling pressure, reduction in microvascular density, and increases in LV myocardial fibrosis (Fig. 4, D–F).
Fig. 7.

Myocardial histopathology. Top: representative examples of cardiomyocyte size [wheat germ agglutinin (WGA)-stained LV sections at ×40 magnification, short-axis myocyte orientation) for control, 10-Gy, and 20-Gy groups. Middle: representative examples of MVD (CD34-stained LV sections, ×20 magnification) for control, 10-Gy, and 20-Gy groups. Bottom: representative examples of myocardial fibrosis (picrosirius red-stained heart sections, ×5 magnification) for control, 10-Gy, and 20-Gy groups.
Fig. 8.

Cardiomyocyte size, MVD, and myocardial fibrosis. While maximum (longitudinal) cardiomyocyte dimensions were similar across groups (A), minimum (short-axis) cardiomyocyte dimensions were increased in radiated groups compared with the control group (B). MVD (C) was decreased and fibrosis (D) was increased in radiated groups compared with the control group. Fibrosis increased as MVD decreased (E). LV diastolic stiffness [diastolic stiffness constant (β)] increased as MVD decreased (F). Data are presented as scatter dot plots, with lines showing mean and 95% confidence interval. *P < 0.05 vs. control. †Post hoc test for linear trend P ≤ 0.001.
Table 5.
Multivariable analysis for factors associated with microvascular density and diastolic stiffness constant
| Parameter Estimate | SE of Parameter Estimate | P Value | |
|---|---|---|---|
| MVD (model R2 = 0.62, P < 0.0001) | |||
| Group (radiated vs. control) | −262 | 76 | 0.002 |
| Cardiomyocyte minimum diameter, μm−1 | −25 | 11 | 0.03 |
| Diastolic stiffness constant (model R2 = 0.34, P = 0.045) | |||
| Fibrosis, % | 0.00025 | 0.00016 | 0.13 |
| MVD, 100 μm−2 | −0.005 | 0.00022 | 0.034 |
Oxidative stress.
In the myocardium from radiated rats, there was evidence of oxidative stress with increased DHE staining and decreased MnSOD activity relative to controls (Fig. 9, A and B). The TGF-β downstream target Smad2/3 was activated with increased phosphorylation of Smad2 (Fig. 9C) in the myocardium of radiated rats compared with control rats, consistent with TGF-β activation.
Fig. 9.

Oxidative stress. Left: representative confocal microscopy images (×10 magnification) of dihydroethidium (DHE)-stained LV myocardial sections (top) and Western blots for phosphorylated (p)Smad2 and total Smad2 protein in LV tissue lysates (bottom). Right: group data show increased DHE staining (A) and reduced MnSOD activity (B), consistent with increased oxidative stress, in radiated groups compared with the control group. C: ratio of pSmad2 to total Smad2 protein was increased in radiated rats compared with control rats, consistent with increased transforming growth factor (TGF)-β signaling. Data are presented as scatter dot plots, with lines showing mean and 95% confidence interval. *P < 0.05 vs. control. †Post hoc test for linear trend P < 0.02.
cGMP-PKG signaling.
In myocardial tissue lysates, cGMP concentrations were higher in 20-Gy rats, while in vitro PKG maximal activity and the ratio of the dimer to the monomer PKG form in radiated rats did not differ from those in control rats (Fig. 10). However, the ratio of Ser239-pVASP to total VASP was decreased in radiated rats compared with control rats, suggesting decreased PKG activity in vivo (Fig. 11). Protein levels of total VASP were increased, as previously reported in vivo with myocardial hypertrophy (44). The ratio of Ser157-pVASP to total VASP, a marker of increased PKA activity, in radiated rats did not differ from that in control rats (Fig. 11).
Fig. 10.

Top: myocardial cGMP, maximal in vitro PKG activity, and PKG forms. Total LV myocardial cGMP concentration was increased in radiated rats compared with control rats (A), while in vitro maximal PKG activity (B) and the ratio of dimer to monomer PKG forms (C) did not differ between radiated and control rats. Data are shown as scatter dot plots, with lines showing mean and 95% confidence interval. *P < 0.05 vs. control. †Post hoc test for linear trend P < 0.03. Bottom: representative Western blots for PKG (dimer and monomer forms).
Fig. 11.

In vivo PKG activity assessed by vasodilator-stimulated phosphoprotein (VASP) phosphorylation. Top: representative Western blots for VASP and pVASP. Bottom: the ratio of pVASP (Ser239) to VASP (A) was decreased, while the VASP-to-GAPDH ratio (B) was increased in radiated rats; the ratio of pVASP (Ser157) to VASP (C) was not different between groups. Data are shown as scatter dot plots, with lines showing mean and 95% confidence interval. *P < 0.05 vs. control. †Post hoc test for linear trend P < 0.003.
DISCUSSION
We used a novel model of global cardiac irradiation to show that cardiac radiation exposure can cause diastolic dysfunction (both increased diastolic chamber stiffness and impaired relaxation), elevated filling pressures, and reduced exercise capacity without concomitant reduction of EF. While our data cannot rule out subtle contractile dysfunction or impaired systolic reserve function from our data, radiated rats had normal resting EF, Ees, PRSW, and peak +dP/dt. The diastolic functional abnormalities were associated with decreased microvascular density and increased myocardial fibrosis, with the severity of myocardial fibrosis increasing in proportion to the reduction in microvascular density. Despite normal blood pressure and heart weight, there was evidence of mild concentric cardiomyocyte hypertrophy with increased cardiomyocyte minimum dimension and myocardial atrial natriuretic peptide gene expression in radiated rats. Finally, there was evidence of increased oxidative stress and TGF-β signaling and decreased in vivo PKG activity, as assessed by VASP protein phosphorylation status. The severity of diastolic stiffness correlated more strongly with the extent of microvascular rarefaction than the severity of LV fibrosis, suggesting that, beyond the fibrosis, additional factors may contribute to the LV diastolic dysfunction. These data provide mechanistic insights for clinical studies demonstrating microvascular dysfunction and increased risk of HFpEF after breast cancer radiotherapy. Moreover, these data provide support for the importance of inflammation-driven coronary microvascular compromise in human HFpEF unassociated with radiation exposure.
Coronary microvascular compromise in HFpEF.
The diffuse microvascular endothelial inflammation postulated to contribute to HFpEF pathophysiology (37) can lead to impaired microvascular function related to endothelial dysfunction, as seen in patients with atherosclerotic risk factors (4, 40) and, ultimately, to microvascular rarefaction (15), which can further limit microcirculatory function. In HFpEF, invasive studies have demonstrated evidence of coronary microvascular dysfunction (57), and noninvasive studies have documented reduced coronary flow reserve in HFpEF patients who have normal EF and no epicardial coronary disease (23, 49). Reduced coronary flow reserve is a marker of impaired global microvascular function and cannot discriminate between endothelial dysfunction, impaired myogenic responsiveness, and altered microvascular structure (24). In human HFpEF studies, the severity of microvascular dysfunction correlated with the severity of diastolic dysfunction (57) or markers of HF severity (23, 49). In autopsy specimens from HFpEF patients, coronary microvascular density was decreased in HFpEF relative to sex- and age-appropriate controls (34). Furthermore, the severity of microvascular rarefaction was associated with the severity of myocardial fibrosis, consistent with the proposed relationship between microvascular endothelial inflammation and subsequent myocardial fibrosis and microvascular rarefaction. Ongoing microscopic ischemic foci related to microvascular compromise may also contribute to the fibrosis, diastolic dysfunction, and exercise intolerance observed here and in human HFpEF (40).
While human studies document an association between coronary microvascular dysfunction and the presence of HFpEF, microvascular dysfunction is not specific to HFpEF (4, 40) and associations do not prove causality. It has been suggested that coronary microvascular inflammation due to cardiotropic but endothelial cell-specific viral infections was sufficient to cause microvascular dysfunction and HFpEF in young patients without hypertension or other HFpEF risk factors (56). Here, we provide additional evidence that an insult specific to the coronary microvascular endothelium is sufficient to cause a HFpEF phenotype.
Radiation-induced coronary microvascular compromise.
Experimental studies of radiation-induced cardiac toxicity have used different species, radiation doses, and delivery strategies and examined cardiac toxicity at variable time points. The very high doses of cardiac radiation resulting from thoracic radiotherapy for thoracic tumors and older breast cancer radiotherapy techniques can cause pericardial, valvular, coronary, and myocardial disease (45, 51). As cardiomyocytes have long been known to be resistant to radiation (26), these observations spurred investigation of the mechanism of radiation-induced heart disease. In the 1970s, serial light and electron microscopic studies of rabbits exposed to external beam X-irradiation delivering a cardiac dose of 20 Gy confirmed the absence of cardiomyocyte necrosis and described the presence of marked coronary microvascular endothelial damage with subsequent microvascular rarefaction and myocardial fibrosis (10–12). The pericardium remained normal until the late stages after radiation, and while these early studies suggested that experimental cardiac irradiation led to clinical evidence of HF, cardiac function was not assessed. Subsequent in vivo and in vitro studies confirmed the sensitivity of microvascular endothelial cells to radiation and described subsequent proinflammatory effects with activation of adhesion molecules and proinflammatory cytokines (45). More contemporary rodent studies confirmed the microvascular changes following external beam X-irradiation but found no effects on EF; however, neither hemodynamics nor diastolic function was assessed (13, 26, 47). In humans, breast cancer radiotherapy produces dose-related patchy myocardial perfusion defects in the absence of epicardial coronary disease, consistent with microvascular compromise, but the significance of these findings was questioned, as no effect on EF was observed; however, diastolic function was not assessed (31). A population-based, case-control study found that the odds of HFpEF increased with mean cardiac radiation dose in older women receiving radiotherapy for breast cancer (42). The ongoing study of atomic bomb survivors in Japan has demonstrated that total body radiation exposures of <2.5 Gy lead to marked increases in the incidence of HF (excess risk 22% per Gy) but not ischemic heart disease or myocardial infarction (51); however, information on EF and diastolic function is not available. The effect of radiation-induced microvascular compromise on cardiac function in humans is likely dependent on total and regional cardiac dose variability and the extent of preexisting (comorbidity-driven) microvascular or myocardial dysfunction or remodeling at the time of radiation exposure. The current model provides a time-telescoped model of radiation exposure.
The targeted internal radiation used here was designed to produce global diffuse cardiac exposure and, thus, global microvascular compromise while avoiding the lung and pericardium. The dose effect, time course, and severity of cardiac dysfunction could vary from that produced by external beam radiation. Nonetheless, the myocardial histological findings appear similar to those previously described with external beam radiation.
cGMP-PKG signaling.
In endomyocardial biopsies, myocardial NO and cGMP levels, in vitro maximal PKG activity, and pVASP-to-VASP ratios (assessed by immunohistochemistry) were lower in HFpEF patients than patients with HF with reduced EF or aortic stenosis, suggesting lower in vivo PKG activity (14, 58). The difference in NO-cGMP-PKG signaling between HFpEF patients and nondiseased controls has not been assessed. Here, compared with control rats, radiated rats did not have decreased myocardial cGMP or in vitro maximal PKG activity but did show a decrease in the ratio of Ser239-pVASP to VASP (Western blot analysis), consistent with decreased PKG activity in vivo. The presence of mild cardiomyocyte hypertrophy in the absence of pressure overload is consistent with decreased PKG signaling (37). While PKA can also phosphorylate VASP at Ser239 (30), there was no decrease in the ratio of Ser157-pVASP to VASP to suggest decreased PKA activity. The lack of association between myocardial cGMP levels and reduced in vivo PKG activity may be related to compartmentalization of phosphodiesterases and cGMP pools with differential effects on PKG activity (25, 38) or inability to discriminate myocardial from vascular cGMP in tissue homogenates.
Relation to other experimental HFpEF models.
Ideally, animal models represent a time-telescoped model of the human condition, and the current model provides insights into the mechanism of radiation-induced HFpEF. The inciting event for the microvascular damage, inflammation, and rarefaction used here is different from that in HFpEF unrelated to radiation exposure, although this model shares some features (coronary microvascular inflammation and rarefaction, myocardial fibrosis, diastolic dysfunction, increased oxidative stress and TGF-β, and decreased PKG signaling) of typical human HFpEF (37). However, other large- and small-animal HFpEF models using genetic and/or pharmacological and dietary interventions to recapitulate the clinical risk factors (hypertension, obesity, diabetes, and renal disease) associated with human HFpEF have important differences from our model of radiation-induced diastolic dysfunction (14, 17, 46). The more widely clinically relevant models employing proinflammatory cardiovascular risk factors lack myocardial fibrosis but have LV and cardiomyocyte diastolic dysfunction solely related to deranged NO-cGMP-PKG signaling and altered titin phosphorylation. While microvascular function and structure were not assessed in these models, one would expect coronary microvascular endothelial dysfunction, as occurs in humans with similar vascular risk factors (4, 40), but likely not microvascular rarefaction, which may be a more advanced manifestation of chronic microvascular endothelial inflammation.
Mechanism of diastolic dysfunction with cardiac radiation exposure.
In human HFpEF, increased LV chamber diastolic stiffness may be due to altered myocyte stiffness related to altered titin phosphorylation (58, 59, 65) and/or increased extracellular collagen accumulation (22, 63, 65). Zile et al. (65) demonstrated that both matrix- and titin-based changes contributed to myofiber stiffness in human HFpEF biopsy samples relative to normal and hypertensive controls and emphasized that the contribution of matrix- and titin-based changes vary not just with the relative perturbation in each but also with the operating sarcomere length in vivo. The comorbidity-based HFpEF models (see above) did not show increased fibrosis, and thus myofiber stiffness was solely modified by titin-based changes (14, 17, 46). Here, the decreased PKG activity in vivo and the finding that LV chamber diastolic stiffness correlated better with severity of microvascular rarefaction than fibrosis suggest that both mechanisms are operant at rest. Importantly, with exercise, microvascular compromise with ischemia may contribute to impaired diastolic (or systolic) reserve (40, 57), consistent with the marked reduction in exercise tolerance observed here. Future studies of myofiber passive stiffness and titin phosphorylation in this model, as well as stress data, are needed.
While the invasive measures of systolic function assessed here did not reveal resting systolic dysfunction, over time, microvascular compromise could lead to myocyte death and systolic dysfunction (4, 40). Thus we speculate that the changes described here represent part of a spectrum, with “isolated” resting diastolic dysfunction observed first, but with the future potential for transition to subtle, or even overt, systolic dysfunction. In a study of rats that received radiotherapy (external beam, 20 Gy) alone or radiotherapy + chemotherapy, at 5 mo postexposure, Salata et al. found some apoptotic nuclei TUNEL staining in the radiotherapy group but more striking subjective TUNEL staining in the radiotherapy + chemotherapy group. Only the radiotherapy + chemotherapy group had reduced EF and increased Bax-to-Bcl2 gene expression ratio, suggestive of apoptosis (43). Unfortunately, diastolic function was not assessed in the study of Salata et al. Whether radiation itself or the resulting microvascular compromise induces apoptosis is unclear.
Limitations.
The dose of l-thyroxine supplementation was based on previous rat studies (55) and resulted in slightly higher T4 and free T4 levels than in control rats. However, there was no clinical evidence (body weight and heart rate) of hyperthyroidism, and our pilot studies suggested that 131I and l-thyroxine alone did not alter myocardial properties. Higher T4 levels may induce hypertrophy while inhibiting fibrosis (28) and, thus, result in an underestimation of the severity of fibrosis in our study. However, differences in T4 levels were modest. Furthermore, fibrosis increased with cardiac radiation dose and, when adjusted for dose, did not vary with free T4 levels (P = 0.46). While external beam radiation can cause pericardial disease in the rat, the mode of radiation exposure used here would minimize pericardial damage, and we saw no echocardiographic, gross or microscopic pathological, or hemodynamic evidence of pericardial disease. We did not assess apoptosis, myocyte or myofiber passive stiffness, or titin phosphorylation.
Conclusions.
Using a novel model of experimental global cardiac radiation exposure, we demonstrated that radiation exposure results in diastolic dysfunction without reduced EF and, in association with microvascular rarefaction, myocardial fibrosis, oxidative stress, and decreased in vivo PKG activity. These data provide mechanistic insights for clinical studies demonstrating microvascular dysfunction and increased risk of HFpEF after breast cancer radiotherapy. Moreover, these data provide support for the importance of inflammation-driven coronary microvascular compromise in human HFpEF unassociated with radiation exposure.
GRANTS
H. Saiki was funded by the Mayo Foundation from 2014 to 2016, the Miyata Research Promotion Foundation, Japan, in 2014, and a Research Fellowship from the Uehara Memorial Foundation, Japan, in 2016. M. M. Redfield was funded by the Mayo Foundation and National Heart, Lung, and Blood Institute (NHLBI) Grants P01 HL-76611, U10 HL-110262, and R01 HL-105418. A. J. Guenzel was supported by NHLBI Cardiovasology Training Grant 4T32 HL-007111-39.
DISCLAIMERS
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.S., G.M., K.C., S.J.R., and M.M.R. conceived and designed research; H.S., G.M., A.J.G., W.L., and T.D. performed experiments; H.S., G.M., A.J.G., T.D., and K.C. analyzed data; H.S. and M.M.R. interpreted results of experiments; H.S., A.J.G., and M.M.R. prepared figures; H.S. drafted manuscript; H.S., G.M., A.J.G., W.L., T.D., K.C., L.P., H.H.C., J.C.B.J., S.J.R., and M.M.R. approved final version of manuscript; G.M., S.J.R., and M.M.R. edited and revised manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. Ozgur Ogut for sharing his expertise in aspects of immunoblot assays, Dr. Carrasco for providing the rNIS antibody, and Dr. Blanton for providing the PKG-1α antibody. We thank Bowen Lorna and Jimmy Storlie for meticulous animal care and rodent surgical expertise.
REFERENCES
- 1.Anzai I. Some explanatory notes on the practical use of the MIRD Committee method for internal dose evaluation. Jpn J Radiol Technol 36: 209–225, 1979. [Google Scholar]
- 2.Berman M. Pamphlet No. 12. Kinetic Models for Absorbed Dose Calculations. New York, NY: Society of Nuclear Medicine, 1976. [Google Scholar]
- 3.Burkhoff D, Mirsky I, Suga H. Assessment of systolic and diastolic ventricular properties via pressure-volume analysis: a guide for clinical, translational, and basic researchers. Am J Physiol Heart Circ Physiol 289: H501–H512, 2005. doi: 10.1152/ajpheart.00138.2005. [DOI] [PubMed] [Google Scholar]
- 4.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med 356: 830–840, 2007. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 5.Chazotte B. Labeling membrane glycoproteins or glycolipids with fluorescent wheat germ agglutinin. Cold Spring Harb Protoc 2011: pdb prot5623, 2011. doi: 10.1101/pdb.prot5623. [DOI] [PubMed] [Google Scholar]
- 6.Coelho-Filho OR, Shah RV, Mitchell R, Neilan TG, Moreno H Jr, Simonson B, Kwong R, Rosenzweig A, Das S, Jerosch-Herold M. Quantification of cardiomyocyte hypertrophy by cardiac magnetic resonance: implications for early cardiac remodeling. Circulation 128: 1225–1233, 2013. doi: 10.1161/CIRCULATIONAHA.112.000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crystal GJ. Carbon dioxide and the heart: physiology and clinical implications. Anesth Analg 121: 610–623, 2015. doi: 10.1213/ANE.0000000000000820. [DOI] [PubMed] [Google Scholar]
- 8.Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature 379: 458–460, 1996. doi: 10.1038/379458a0. [DOI] [PubMed] [Google Scholar]
- 9.Darby SC, Ewertz M, McGale P, Bennet AM, Blom-Goldman U, Brønnum D, Correa C, Cutter D, Gagliardi G, Gigante B, Jensen MB, Nisbet A, Peto R, Rahimi K, Taylor C, Hall P. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 368: 987–998, 2013. doi: 10.1056/NEJMoa1209825. [DOI] [PubMed] [Google Scholar]
- 10.Fajardo LF, Stewart JR. Capillary injury preceding radiation-induced myocardial fibrosis. Radiology 101: 429–433, 1971. doi: 10.1148/101.2.429. [DOI] [PubMed] [Google Scholar]
- 11.Fajardo LF, Stewart JR. Experimental radiation-induced heart disease. I. Light microscopic studies. Am J Pathol 59: 299–316, 1970. [PMC free article] [PubMed] [Google Scholar]
- 12.Fajardo LF, Stewart JR. Pathogenesis of radiation-induced myocardial fibrosis. Lab Invest 29: 244–257, 1973. [PubMed] [Google Scholar]
- 13.Franken NA, Camps JA, van Ravels FJ, van der Laarse A, Pauwels EK, Wondergem J. Comparison of in vivo cardiac function with ex vivo cardiac performance of the rat heart after thoracic irradiation. Br J Radiol 70: 1004–1009, 1997. doi: 10.1259/bjr.70.838.9404203. [DOI] [PubMed] [Google Scholar]
- 14.Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschöpe C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail 4: 312–324, 2016. doi: 10.1016/j.jchf.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Goligorsky MS. Microvascular rarefaction: the decline and fall of blood vessels. Organogenesis 6: 1–10, 2010. doi: 10.4161/org.6.1.10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guyton AC. Determination of cardiac output by equating venous return curves with cardiac response curves. Physiol Rev 35: 123–129, 1955. [DOI] [PubMed] [Google Scholar]
- 17.Hamdani N, Franssen C, Lourenço A, Falcão-Pires I, Fontoura D, Leite S, Plettig L, López B, Ottenheijm CA, Becher PM, González A, Tschöpe C, Díez J, Linke WA, Leite-Moreira AF, Paulus WJ. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 6: 1239–1249, 2013. doi: 10.1161/CIRCHEARTFAILURE.113.000539. [DOI] [PubMed] [Google Scholar]
- 18.Harun-Or-Rashid M, Asai M, Sun XY, Hayashi Y, Sakamoto J, Murata Y. Effect of thyroid statuses on sodium/iodide symporter (NIS) gene expression in the extrathyroidal tissues in mice. Thyroid Res 3: 3, 2010. doi: 10.1186/1756-6614-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasegawa T, Okada K, Okita Y, Pinsky DJ. Antioxidant properties of pioglitazone limit nicotinamide adenine dinucleotide phosphate hydrogen oxidase and augment superoxide dismutase activity in cardiac allotransplantation. J Heart Lung Transplant 30: 1186–1196, 2011. doi: 10.1016/j.healun.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Hata K, Goto Y, Kawaguchi O, Takasago T, Saeki A, Nishioka T, Suga H. Hypercapnic acidosis increases oxygen cost of contractility in the dog left ventricle. Am J Physiol Heart Circ Physiol 266: H730–H740, 1994. [DOI] [PubMed] [Google Scholar]
- 21.Howell RW, Wessels BW, Loevinger R, Watson EE, Bolch WE, Brill AB, Charkes ND, Fisher DR, Hays MT, Robertson JS, Siegel JA, Thomas SR. The MIRD perspective 1999. Medical Internal Radiation Dose Committee. J Nucl Med 40: 3S–10S, 1999. [PubMed] [Google Scholar]
- 22.Kasner M, Westermann D, Lopez B, Gaub R, Escher F, Kühl U, Schultheiss HP, Tschöpe C. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol 57: 977–985, 2011. doi: 10.1016/j.jacc.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 23.Kato S, Saito N, Kirigaya H, Gyotoku D, Iinuma N, Kusakawa Y, Iguchi K, Nakachi T, Fukui K, Futaki M, Iwasawa T, Kimura K, Umemura S. Impairment of coronary flow reserve evaluated by phase contrast cine-magnetic resonance imaging in patients with heart failure with preserved ejection fraction. J Am Heart Assoc 5: e002649, 2016. doi: 10.1161/JAHA.115.002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufmann PA, Camici PG. Myocardial blood flow measurement by PET: technical aspects and clinical applications. J Nucl Med 46: 75–88, 2005. [PubMed] [Google Scholar]
- 25.Kokkonen K, Kass DA. Nanodomain regulation of cardiac cyclic nucleotide signaling by phosphodiesterases. Annu Rev Pharmacol Toxicol 57: 455–479, 2017. doi: 10.1146/annurev-pharmtox-010716-104756. [DOI] [PubMed] [Google Scholar]
- 26.Lauk S, Kiszel Z, Buschmann J, Trott KR. Radiation-induced heart disease in rats. Int J Radiat Oncol Biol Phys 11: 801–808, 1985. doi: 10.1016/0360-3016(85)90314-1. [DOI] [PubMed] [Google Scholar]
- 27.Lee CL, Moding EJ, Cuneo KC, Li Y, Sullivan JM, Mao L, Washington I, Jeffords LB, Rodrigues RC, Ma Y, Das S, Kontos CD, Kim Y, Rockman HA, Kirsch DG. p53 functions in endothelial cells to prevent radiation-induced myocardial injury in mice. Sci Signal 5: ra52, 2012. doi: 10.1126/scisignal.2002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HW, Klein LE, Raser J, Eghbali-Webb M. An activator protein-1 (AP-1) response element on pro alpha1(l) collagen gene is necessary for thyroid hormone-induced inhibition of promoter activity in cardiac fibroblasts. J Mol Cell Cardiol 30: 2495–2506, 1998. doi: 10.1006/jmcc.1998.0811. [DOI] [PubMed] [Google Scholar]
- 29.Loeevinger R, Berman M.. A schema for absorbed-dose calculations for biologically-distributed radionuclides. J Nucl Med Suppl 1: 9–14, 1968. [PubMed] [Google Scholar]
- 30.Lohmann SM, Walter U. Tracking functions of cGMP-dependent protein kinases (cGK). Front Biosci 10: 1313–1328, 2005. doi: 10.2741/1621. [DOI] [PubMed] [Google Scholar]
- 31.Marks LB, Yu X, Prosnitz RG, Zhou SM, Hardenbergh PH, Blazing M, Hollis D, Lind P, Tisch A, Wong TZ, Borges-Neto S. The incidence and functional consequences of RT-associated cardiac perfusion defects. Int J Radiat Oncol Biol Phys 63: 214–223, 2005. doi: 10.1016/j.ijrobp.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 32.Mezei T, Horváth E, Turcu M, Gurzu S, Raica M, Jung I. Microvascular density in non-Hodgkin B-cell lymphomas measured using digital morphometry. Rom J Morphol Embryol 53: 67–71, 2012. [PubMed] [Google Scholar]
- 33.Miller FJ Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res 82: 1298–1305, 1998. doi: 10.1161/01.RES.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 34.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 131: 550–559, 2015. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohammed SF, Majure DT, Redfield MM. Zooming in on the microvasculature in heart failure with preserved ejection fraction. Circ Heart Fail 9: e003272, 2016. doi: 10.1161/CIRCHEARTFAILURE.116.003272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N. The Na+/I− symporter mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol 296: C654–C662, 2009. doi: 10.1152/ajpcell.00509.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271, 2013. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 38.Piggott LA, Hassell KA, Berkova Z, Morris AP, Silberbach M, Rich TC. Natriuretic peptides and nitric oxide stimulate cGMP synthesis in different cellular compartments. J Gen Physiol 128: 3–14, 2006. doi: 10.1085/jgp.200509403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther 18: 43–52, 2011. doi: 10.1038/gt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pries AR, Reglin B. Coronary microcirculatory pathophysiology: can we afford it to remain a black box? Eur Heart J 38: 478-488, 2017. doi: 10.1093/eurheartj/ehv760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ricci D, Mennander AA, Miyagi N, Rao VP, Tazelaar HD, Classic K, Byrne GW, Russell SJ, McGregor CG. Prolonged cardiac allograft survival using iodine 131 after human sodium iodide symporter gene transfer in a rat model. Transplant Proc 42: 1888–1894, 2010. doi: 10.1016/j.transproceed.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saiki H, Petersen IA, Scott CG, Bailey KR, Dunlay SM, Finley RR, Ruddy KJ, Yan E, Redfield MM. Risk of heart failure with preserved ejection fraction in older women after contemporary radiotherapy for breast cancer. Circulation 135: 1388–1396, 2017. doi: 10.1161/CIRCULATIONAHA.116.025434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salata C, Ferreira-Machado SC, De Andrade CB, Mencalha AL, Mandarim-De-Lacerda CA, de Almeida CE. Apoptosis induction of cardiomyocytes and subsequent fibrosis after irradiation and neoadjuvant chemotherapy. Int J Radiat Biol 90: 284–290, 2014. doi: 10.3109/09553002.2014.887869. [DOI] [PubMed] [Google Scholar]
- 44.Sartoretto JL, Jin BY, Bauer M, Gertler FB, Liao R, Michel T. Regulation of VASP phosphorylation in cardiac myocytes: differential regulation by cyclic nucleotides and modulation of protein expression in diabetic and hypertrophic heart. Am J Physiol Heart Circ Physiol 297: H1697–H1710, 2009. doi: 10.1152/ajpheart.00595.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultz-Hector S, Trott KR. Radiation-induced cardiovascular diseases: is the epidemiologic evidence compatible with the radiobiologic data? Int J Radiat Oncol Biol Phys 67: 10–18, 2007. doi: 10.1016/j.ijrobp.2006.08.071. [DOI] [PubMed] [Google Scholar]
- 46.Schwarzl M, Hamdani N, Seiler S, Alogna A, Manninger M, Reilly S, Zirngast B, Kirsch A, Steendijk P, Verderber J, Zweiker D, Eller P, Höfler G, Schauer S, Eller K, Maechler H, Pieske BM, Linke WA, Casadei B, Post H. A porcine model of hypertensive cardiomyopathy: implications for heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 309: H1407–H1418, 2015. doi: 10.1152/ajpheart.00542.2015. [DOI] [PubMed] [Google Scholar]
- 47.Seemann I, Gabriels K, Visser NL, Hoving S, te Poele JA, Pol JF, Gijbels MJ, Janssen BJ, van Leeuwen FW, Daemen MJ, Heeneman S, Stewart FA. Irradiation induced modest changes in murine cardiac function despite progressive structural damage to the myocardium and microvasculature. Radiother Oncol 103: 143–150, 2012. doi: 10.1016/j.radonc.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 48.Senzaki H, Kass DA. Analysis of isovolumic relaxation in failing hearts by monoexponential time constants overestimates lusitropic change and load dependence: mechanisms and advantages of alternative logistic fit. Circ Heart Fail 3: 268–276, 2010. doi: 10.1161/CIRCHEARTFAILURE.109.865592. [DOI] [PubMed] [Google Scholar]
- 49.Srivaratharajah K, Coutinho T, deKemp R, Liu P, Haddad H, Stadnick E, Davies RA, Chih S, Dwivedi G, Guo A, Wells GA, Bernick J, Beanlands R, Mielniczuk LM. Reduced myocardial flow in heart failure patients with preserved ejection fraction. Circ Heart Fail 9: e002562, 2016. doi: 10.1161/CIRCHEARTFAILURE.115.002562. [DOI] [PubMed] [Google Scholar]
- 50.Stewart FA. Mechanisms and dose-response relationships for radiation-induced cardiovascular disease. Ann ICRP 41: 72–79, 2012. doi: 10.1016/j.icrp.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 51.Stewart FA, Seemann I, Hoving S, Russell NS. Understanding radiation-induced cardiovascular damage and strategies for intervention. Clin Oncol (R Coll Radiol) 25: 617–624, 2013. doi: 10.1016/j.clon.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Su MY, Lin LY, Tseng YH, Chang CC, Wu CK, Lin JL, Tseng WY. CMR-verified diffuse myocardial fibrosis is associated with diastolic dysfunction in HFpEF. JACC Cardiovasc Imaging 7: 991–997, 2014. doi: 10.1016/j.jcmg.2014.04.022. [DOI] [PubMed] [Google Scholar]
- 53.Suksanpaisan L, Pham L, McIvor S, Russell SJ, Peng KW. Oral contrast enhances the resolution of in-life NIS reporter gene imaging. Cancer Gene Ther 20: 638–641, 2013. doi: 10.1038/cgt.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor CW, Wang Z, Macaulay E, Jagsi R, Duane F, Darby SC. Exposure of the heart in breast cancer radiation therapy: a systematic review of heart doses published during 2003 to 2013. Int J Radiat Oncol Biol Phys 93: 845–853, 2015. doi: 10.1016/j.ijrobp.2015.07.2292. [DOI] [PubMed] [Google Scholar]
- 55.Trujillo MA, Oneal MJ, McDonough SJ, Morris JC. Viral dose, radioiodide uptake, and delayed efflux in adenovirus-mediated NIS radiovirotherapy correlates with treatment efficacy. Gene Ther 20: 567–574, 2013. doi: 10.1038/gt.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tschöpe C, Bock CT, Kasner M, Noutsias M, Westermann D, Schwimmbeck PL, Pauschinger M, Poller WC, Kühl U, Kandolf R, Schultheiss HP. High prevalence of cardiac parvovirus B19 infection in patients with isolated left ventricular diastolic dysfunction. Circulation 111: 879–886, 2005. doi: 10.1161/01.CIR.0000155615.68924.B3. [DOI] [PubMed] [Google Scholar]
- 57.van Empel VP, Mariani J, Borlaug BA, Kaye DM. Impaired myocardial oxygen availability contributes to abnormal exercise hemodynamics in heart failure with preserved ejection fraction. J Am Heart Assoc 3: e001293, 2014. doi: 10.1161/JAHA.114.001293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126: 830–839, 2012. doi: 10.1161/CIRCULATIONAHA.111.076075. [DOI] [PubMed] [Google Scholar]
- 59.van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, Ijsselmuiden AJ, Schalkwijk CG, Bronzwaer JG, Diamant M, Borbély A, van der Velden J, Stienen GJ, Laarman GJ, Niessen HW, Paulus WJ. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 117: 43–51, 2008. doi: 10.1161/CIRCULATIONAHA.107.728550. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Boerma M, Zhou D. Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res 186: 153–161, 2016. doi: 10.1667/RR14445.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei CL, Valvano JW, Feldman MD, Nahrendorf M, Peshock R, Pearce JA. Volume catheter parallel conductance varies between end-systole and end-diastole. IEEE Trans Biomed Eng 54: 1480–1489, 2007. doi: 10.1109/TBME.2007.890732. [DOI] [PubMed] [Google Scholar]
- 62.Wei CL, Valvano JW, Feldman MD, Pearce JA. Nonlinear conductance-volume relationship for murine conductance catheter measurement system. IEEE Trans Biomed Eng 52: 1654–1661, 2005. doi: 10.1109/TBME.2005.856029. [DOI] [PubMed] [Google Scholar]
- 63.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschöpe C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4: 44–52, 2011. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 64.Zakeri R, Moulay G, Chai Q, Ogut O, Hussain S, Takahama H, Lu T, Wang XL, Linke WA, Lee HC, Redfield MM. Left atrial remodeling and atrioventricular coupling in a canine model of early heart failure with preserved ejection fraction. Circ Heart Fail 9: e003238, 2016. doi: 10.1161/CIRCHEARTFAILURE.115.003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 131: 1247–1259, 2015. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]