

In a porcine model of ventricular ischemia, spinal cord stimulation decreased sympathetic nerve activation regionally in ischemic myocardium with no effect on normal myocardium, demonstrating that the antiarrhythmic effects of spinal cord stimulation are likely due to attenuation of local sympathoexcitation in the ischemic myocardium and not changes in global myocardial electrophysiology.
Keywords: spinal cord stimulation, sympathetic nervous system, ventricular excitability, cardiac arrhythmias
Abstract
Myocardial ischemia creates autonomic nervous system imbalance and can trigger cardiac arrhythmias. We hypothesized that neuromodulation by spinal cord stimulation (SCS) will attenuate local cardiac sympathoexcitation from ischemia-induced increases in afferent signaling, reduce ventricular arrhythmias, and improve myocardial function during acute ischemia. Yorkshire pigs (n = 20) were randomized to SCS (50 Hz at 200-μs duration, current 90% motor threshold) or sham operation (sham) for 30 min before ischemia. A four-pole SCS lead was placed percutaneously in the epidural space (T1–T4), and a 56-electrode mesh was placed over the heart for high-resolution electrophysiological recordings, including activation recovery intervals (ARIs), activation time, repolarization time, and dispersion of repolarization. Electrophysiological and hemodynamic measures were recorded at baseline, after SCS/sham, during acute ischemia (300-s coronary artery ligation), and throughout reperfusion. SCS 1) reduced sympathoexcitation-induced ARI and repolarization time shortening in the ischemic myocardium; 2) attenuated increases in the dispersion of repolarization; 3) reduced ventricular tachyarrythmias [nonsustained ventricular tachycardias: 24 events (3 sham animals) vs. 1 event (1 SCS animal), P < 0.001]; and 4) improved myocardial function (dP/dt from baseline to ischemia: 1,814 ± 213 to 1,596 ± 282 mmHg/s in sham vs. 1,422 ± 299 to 1,380 ± 299 mmHg/s in SCS, P < 0.01). There was no change in ventricular electrophysiology during baseline conditions without myocardial stress or in the nonischemic myocardium. In conclusion, in a porcine model of acute ventricular ischemia, SCS reduced regional myocardial sympathoexcitation, decreased ventricular arrhythmias, and improved myocardial function. SCS decreased sympathetic nerve activation locally in the ischemic myocardium with no effect observed in the normal myocardium, thus providing mechanistic insights into the antiarrhythmic and myocardial protective effects of SCS.
NEW & NOTEWORTHY In a porcine model of ventricular ischemia, spinal cord stimulation decreased sympathetic nerve activation regionally in ischemic myocardium with no effect on normal myocardium, demonstrating that the antiarrhythmic effects of spinal cord stimulation are likely due to attenuation of local sympathoexcitation in the ischemic myocardium and not changes in global myocardial electrophysiology.
spinal cord stimulation (SCS) of the thoracic dorsal column has traditionally been used to treat refractory angina and has now been suggested to have myocardial protective and antiarrhythmic effects in the heart during myocardial ischemia (19, 21, 23, 33). Direct neural recordings have demonstrated that coronary artery occlusion activates local afferent sympathetic nerve endings in the ischemic myocardium (12, 25). The cardiac afferent nerve fibers travel to the dorsal column of the thoracic spinal cord and synapse within a complex neural circuit to result in reflex efferent sympathoexcitation (12, 25). Increased spinal sympathoexcitation leads to acute physiological changes, including tachycardia and hypertension as well as long-term neuronal remodeling of the intrathoracic, extracardiac ganglia and the intrinsic cardiac nervous system (3, 31, 37). In addition, increased sympathetic stimulation can impact myocardial electrophysiology and trigger ventricular arrhythmias and sudden cardiac death (32, 39).
Reduction of sympathoexcitation through surgical sympathectomy or spinal neuraxial modulation via thoracic epidural anesthesia or SCS has been demonstrated to reduce malignant ventricular arrhythmias (1, 10, 36). SCS may inhibit spinothalamic tract neurons in the spinal cord and influence the intrathoracic cardiac nervous system, thus reducing reflex sympathoexcitation during ventricular ischemia (4, 6, 11, 13). Current research and clinical evidence suggest that, during myocardial ischemia, SCS decreases myocardial O2 demand, improves lactate metabolism, and attenuates autonomic nervous system imbalance (19, 21, 23, 24, 36). In animal studies, SCS reduced ischemia-induced ventricular arrhythmias by 37% and was more effective than pharmacological treatment with β-blockers in preventing spontaneous ventricular arrhythmias (17, 23).
While the beneficial effects of SCS have been suggested in both clinical and animal studies, the precise mechanisms of SCS’ antiarrhythmic effects during ischemia have not been fully described (6, 17, 23, 26, 36). It is unknown if the effects of spinal neural modulation are due to changes in global myocardial electrophysiology or instead due to attenuation of local sympathoexcitation in the affected ischemic myocardium. Therefore, the aim of this study was to investigate the effects of SCS on cardiac ventricular electrophysiology at rest and during focal acute myocardial ischemia with increased afferent signaling and reflex efferent sympathetic stimulation. We hypothesized that neuromodulation by SCS will 1) reduce cardiac sympathoexcitation from ischemia-induced increases in afferent signaling in affected myocardium, 2) reduce ventricular arrhythmias, and 3) improve myocardial function during acute ischemia.
METHODS
All animal experiments were devised in accordance with guidelines set by the University of California Institutional Animal Care and Use Committee and the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of California-Los Angeles Animal Research Committee. Yorkshire pigs, n = 20 (male or female), weighing 45 ± 4 kg, were sedated with intramuscular telazol (4–6 mg/kg), intubated, and mechanically ventilated. General anesthesia was maintained with inhaled isoflurane (1.5–2.5%) and intravenous boluses of fentanyl (total: 10–30 μg/kg) during surgical preparation. In the prone position, using the loss of resistance technique, an 18-gauge Touhy needle was placed in the epidural space followed by fluoroscopy-guided insertion of a four-pole spinal cord stimulating lead (Octrode, Advanced Neuromodulation Systems, Plano, TX). In the supine position, animals then underwent median sternotomy to expose the heart. Heart rate (HR) and surface 12-lead electrocardiogram (ECG) were monitored using the ECG. The femoral artery was catheterized to monitor arterial blood pressure, and the femoral vein was catheterized for intravenous saline infusion (10 ml/kg). Hourly arterial blood gas was tested, and adjustments via ventilation or infusion of sodium bicarbonate were performed as necessary to maintain acid-base homeostasis. General anesthesia was transitioned to intravenous α-chloralose (50 mg/kg initial bolus followed by a 20 mg·kg−1·h−1 continuous infusion) after surgical preparation was completed. Use of intravenous α-chloralose as an anesthetic has been previously shown to be least disruptive of autonomic nervous system activity and has been used extensively in investigational studies (31).
SCS
A four-pole spinal cord stimulating lead was placed in the epidural space using fluoroscopy guidance. The lead was inserted at the T1–T4 spinal cord level, with the most cranial pole of the lead at T1 (Fig. 1A). Functional position was verified via electrical current from both rostral and caudal poles. A stimulator (model S88 stimulator, Grass Instruments), led through a constant-current isolation unit, produced the stimuli. Motor threshold (MT) was obtained by increasing stimulus intensity until muscle contractions were induced in the shoulder. Animals were rotated to the supine position, and MT was reestablished. SCS [or sham operation (sham)] was performed for 30 min preischemia. In the SCS group, SCS was delivered for 30 min at 50 Hz, 400-μs duration. The current had an intensity of 90% MT, a range of 0.12–1.60 mA, and a mean of 0.76 mA.
Fig. 1.

A: fluoroscopy guidance was used during insertion of a four-pole spinal cord stimulation (SCS) electrode at the T1-T4 level. B: 20 Yorkshire pigs were randomized to 2 treatment protocols with or without spinal cord stimulation. Both groups underwent coronary artery ligation to produce acute ventricular ischemia for 300 s (5 min) followed by 60 min of reperfusion.
Experimental Protocols
All animals were randomized to SCS or sham groups. In the SCS group (n = 10), after placement of the electrode, SCS was delivered for 30 min before ischemia and then stopped. In the sham group (n = 10), the spinal cord stimulating electrode was placed but not turned on during a 30-min period before the coronary artery occlusion. After the 30 min SCS or sham period, coronary artery occlusion was performed for 300 s (5 min) in both experimental groups, and reperfusion was then followed for 60 min. Electrophysiological and hemodynamic measurements were recorded at baseline, throughout ischemia, and for 1 h of reperfusion after the coronary artery occlusion (Fig. 1B).
Coronary artery occlusion.
A 4-0 prolene suture was placed around the second diagonal branch of the left anterior descending coronary artery (LAD). The suture was led through a short polyethylene tubing segment, which was then used to ligate the coronary artery to induce ischemia for 300 s.
Hemodynamic assessment and surface ECG recordings.
Left ventricular (LV) end-systolic pressure (LVESP), LV end-diastolic pressure (LVEDP), HR, and maximum and minimum rate rise of LV pressure (dP/dtmax and dP/dtmin) were measured using a 12-pole conductance, high-fidelity pressure monitoring pigtail catheter (5-Fr) inserted into the LV via the left carotid artery and connected to a MPVS Ultra Pressure Volume Loop System (Millar Instruments, Houston, TX). ECG data (12-lead) were continuously recorded via a Prucka CardioLab system (GE Healthcare, Fairfield, CT). Precordial lead electrodes (V1−V6) were positioned posteriorly in a manner that reflects standard anterior precordial lead electrode placement and records the horizontal plane. ECGs were manually analyzed.
Electrophysiological recordings and analysis.
A 56-electrode nylon mesh was placed around the heart and unipolar electrograms were measured using a Prucka CardioLab electrophysiology mapping system (GE Healthcare, Fairfield, CT) (Fig. 2A). All electrophysiological measures were recorded at baseline, after 30 min of SCS or sham, during acute ischemia (at 0, 45, 90, and 300 s from the ligation), and throughout 60 min of reperfusion.
Fig. 2.

A: 56-electrode nylon mesh placed around the heart for measuring biventricular surface electrograms. B: activation recovery interval (ARI) was measured from unipolar epicardial electrograms. ARI is the difference between repolarization time (RT) and activation time (AT).
Whole heart activation recovery interval (ARI), which has been shown to be a surrogate for action potential duration, was measured and analyzed using iScalDyn software (University of Utah, Salt Lake City, UT) (2, 34). ARI is measured from the most negative deflection of change in voltage over time (dV/dT) of the activation wave and the most positive deflection of dV/dT of the repolarization wave. Activation time (AT) was measured as the minimum dV/dT in the QRS complex, repolarization time (RT) was measured as the maximum dV/dT in the T wave, and ARI was calculated by subtracting AT from RT (Fig. 2B). Specifically, ARI was analyzed in electrodes showing ST elevation or depression, as previously described (15). Ischemia can complicate measurement of ARI when the maximum derivative of the T wave and the minimum derivative of the action potential downstroke become less distinct. To ensure accuracy of ARI measurement, each electrogram with ST segment changes was both measured by semiautomated accepted software and then checked by hand following the guidelines described by Haws and Lux (15) for ARI measurements in ischemia and carefully measured across four to five beats.
Epicardial electrograms were measured across the whole heart and then analyzed in two regions: ischemic myocardium and remote myocardium. The ischemic myocardium was defined by surface unipolar electrograms showing ST elevation (0.1 mV above baseline) or ST depression (0.1 mV below baseline), and the remote myocardium was defined as the distant myocardium not in distribution of coronary artery ligation and by electrodes without ST changes (9). Dispersion of AT, RT, and ARI were measured as variance in AT, RT, and ARI in the whole heart as well as in the two regions described above. Tpeak-Tend was measured via the clearest T wave recording using surface 12-lead ECG leads. T wave analysis was able to be completed on n = 8 animals in both SCS and sham groups. The highest amplitude of T wave deflection was used to define the peak of the T wave (Tpeak), and the point where the tangent on the descending limb of the T wave intersects the isoelectric line was used to define the end of the T wave (Tend) (29). Three-dimensional sock AT, RT, and ARI were projected onto two-dimensional polar maps using publicly available Map3D software (University of Utah; http://www.sci.utah.edu/cibc/software/107-map3d.html).
A continuous 12-lead ECG was used for manual classification of nonsustained ventricular tachycardias (NSVTs) and premature ventricular contractions (PVCs) during 5-min acute ischemia and 60-min reperfusion. NSVTs were classified as three or more beats of ventricular tachycardia (VT) at a rate of >120 beats/min and lasting <30 s. PVCs were defined as abnormal heart beats containing an irregularity in QRS complex morphology, a decrease in R wave-to-R wave time interval, and an absence of a P wave. There were no episodes of sustained VTs or ventricular fibrillation.
Statistical Analysis
Electrophysiological and hemodynamic data at each time point are reported as means ± SD. Paired t-tests were used to compare two electrophysiological or hemodynamic measures, such as before and after SCS. Two-way repeated measures ANOVA, followed by Tukey’s post hoc test for multiple comparisons, was used to compare changes in electrophysiological and hemodynamic measures from baseline throughout multiple ischemia and reperfusion time points in SCS and sham groups. Differences in magnitude of change from baseline to peak ischemia (5 min) and end reperfusion (60 min) between SCS and sham groups were compared using linear mixed-effects models with fixed terms for group (SCS vs. sham), time, the group × time interaction, and a random pig effect. We also included the size of the ischemic myocardium during coronary artery ligation in the models. Pairwise contrasts of interest between groups were estimated and formally tested with the model. Mean differences from baseline to peak ischemia and end reperfusion are presented as conditional means and 95% confidence intervals. Results were considered statistically significant with two-tailed P values < 0.05. Analysis was performed using Sigma Plot (version 12.5) except for the linear mixed-effects models, which were performed using SAS version 9.4 (Cary, NC).
RESULTS
Effect of SCS on Basal Cardiac Electrophysiology and Hemodynamics
One animal in the sham group was unstable before initiation of the protocol, and, therefore, data were analyzed and reported for n = 19 sham (n = 9) and SCS (n = 10) animals. Electrophysiological indexes were measured before and after 30 min of SCS or sham to assess changes in baseline electrophysiology associated with SCS. There were no significant changes in global myocardial excitability as measured by ARI, AT, RT, and dispersion of repolarization after SCS or sham time control (Figs. 3 and 4). There was also no significant change in T wave peak to end interval after SCS or sham (SCS: 44 ± 11.8 ms at baseline to 41 ± 9.7 ms at 30 min SCS, P = 0.09; sham: 45 ± 8.0 at baseline to 48 ± 10.9 ms at 30 min SCS, P = 0.08).
Fig. 3.

Representative polar maps of ARI (top), AT (middle), and RT (bottom) during sinus rhythm after 30 min of sham or SCS. Left: sham. Right: SCS treatment. There was no change in ARI, AT, or RT in sham or SCS groups.
Fig. 4.

There was no change from before to 30 min after SCS or sham in global ARI (all P > 0.89; A), global RT (all P > 0.93; B), global AT (all P > 0.23; C), or global ventricular dispersion of repolarization (DOR; all P > 0.79; D). Sham: n = 9; SCS: n = 10.
Hemodynamics remained largely stable during SCS, with an SCS-associated change in only mean arterial pressure (MAP), which slightly decreased from baseline after 30 min of SCS but was unchanged in the sham group (Table 1). Hemodynamic measures of HR, LVESP, LVEDP, dP/dtmax, and dP/dtmin showed no significant change after SCS (Table 1).
Table 1.
Hemodynamic changes associated with SCS
| Sham (n = 9) |
SCS (n = 10) |
|||
|---|---|---|---|---|
| Baseline | 30 min | Baseline | 30 min | |
| HR, beats/min | 71 ± 15 | 70 ± 15 | 71 ± 12.0 | 72 ± 11 |
| LVESP, mmHg | 109 ± 8 | 107 ± 11 | 108 ± 19 | 106 ± 18 |
| LVEDP, mmHg | 11 ± 4 | 9 ± 4 | 9 ± 3 | 9 ± 3 |
| MAP, mmHg | 109 ± 7 | 109 ± 8 | 105 ± 20 | 100 ± 18* |
| dP/dtmax, mmHg/s | 1,802 ± 261 | 1,807 ± 256 | 1,511 ± 285 | 1,480 ± 279 |
| dP/dtmin, mmHg/s | −2,435 ± 601 | −2,379 ± 670 | −2,335 ± 682 | −2,332 ± 648 |
Values are means ± SE; n, no. of animals. Sham, sham operation; SCS, spinal cord stimulation; HR, heart rate; LVESP, left ventricular end-systolic pressure; LVEDP, left ventricular end-diastolic pressure; MAP, mean arterial pressure; dP/dtmax and dP/dtmin, maximum and minimum rate rise of LV pressure, respectively.
P = 0.039 vs. SCS at baseline. All other P values were >0.17.
Effect of SCS on Cardiac Electrophysiological and Hemodynamic Measures During Acute Ischemia
Electrophysiological measures.
Electrophysiological measures were recorded across the whole heart and analyzed regionally in ischemic and nonischemic zones of the myocardium. In the ischemic regions, SCS was found to attenuate cardiac sympathetic excitation during acute ischemia, as measured by ARI, AT, and RT as well as the dispersion of ARI and dispersion of repolarization, with no change in nonischemic regions (Fig. 5). In addition, ARI data were analyzed both with and without electrograms showing severe ST segment changes to ensure that those leads were not skewing the data. The results were unchanged, and the correlation coefficients between the measures with and without ST segment changes were 1–0.98.
Fig. 5.

Representative polar maps of ARI (top), AT (middle), and RT (bottom) during sinus rhythm with and without SCS at baseline and during myocardial ischemia induced by left anterior descending artery (LAD) ligation. Left: sham. Right: SCS treatment. The black dashed line indicates the ischemic zone. LAD ligation caused a reduction in ARI and RT and an increase in AT. However, SCS pretreatment attenuated the reduction in ARI and RT and prolonged the increase in AT.
In the ischemic myocardium, there was an expected increase in sympathoexcitation and shortening of ARI duration throughout ischemia. However, ARI shortening was attenuated with SCS treatment compared with sham (Fig. 6A). SCS was also associated with a greater increase in AT (Fig. 6B) and an attenuation in RT reduction from baseline to 300-s LAD ligation (Fig. 6C). SCS treatment was not associated with any significant changes in electrophysiological parameters in the remote, nonischemic myocardium during acute ischemia. There were no changes from baseline to 300-s LAD ligation in ARI (Fig. 6D), AT (Fig. 6E), or RT (Fig. 6F) in the remote mycardium.
Fig. 6.

Change in electrophysiological parameters in the ischemic and remote regions from baseline to 45-, 90-, and 300-s ischemia. Data are presented as mean differences from baseline at each time point in the sham versus SCS group. In the ischemic myocardium, SCS attenuated sympathetic excitation associated ARI reduction (*P = 0.035 vs. sham; A), was associated with a greater increase in AT (*P = 0.001 vs. sham; B), and was associated with a greater reduction in RT (*P = 0.008 vs. sham; C). In the remote unaffected myocardium, there was no change in ARI (D), AT (E), or RT (F) in the SCS or sham groups (all P > 0.28). Sham: n = 9; SCS: n = 10.
SCS reduced ventricular arrhythmia risk as shown by the attenuation of the increase in dispersion of ARI and repolarization in the ischemic myocardium (Fig. 7, A and B). There was no change in the dispersion of ARI or RT in the remote myocardium (Fig. 7, C and D).
Fig. 7.

Change in dispersion in the ischemic and remote regions from baseline to 45-, 90-, and 300-s ischemia. Data are presented as mean differences from baseline at each time point in the sham versus SCS group. In the ischemic myocardium, SCS attenuated the increase in the dispersion of ARI (*P = 0.006 vs. sham; A) and dispersion of RT (*P = 0.002 vs. sham; B). In the remote unaffected myocardium, there was no change in the dispersion of ARI (C) or dispersion of RT (D) (all P > 0.16 in SCS or sham groups). Sham: n = 9; SCS: n = 10.
Global electrophysiological measures.
Increased Tpeak-Tend interval is associated with a greater risk for ventricular arrhythmia. After 300 s of ischemia, Tpeak-Tend interval increased significantly from baseline in the sham group; however, there was no increase in Tpeak-Tend during ischemia in the SCS-treated group (Fig. 8). No significant change was observed in global ARI measured from baseline immediately before LAD ligation to 300 s into LAD ligation (SCS: 361.0 ± 36.0 to 337.0 ± 35.0 ms vs. sham: 356.0 ± 66.0 to 325.0 ± 66.0 ms, P = 0.43). However, SCS was associated with a significant increase in global AT from baseline to peak ischemia at 300 s into LAD ligation compared with sham treatment (SCS: 26.0 ± 3.0 to 28.0 ± 3.0 ms vs. sham: 26.0 ± 5.0 to 26.0 ± 5.0 ms, P = 0.004). No significant changes between SCS and sham treatment from baseline to peak ischemia were observed in global RT (SCS: 388.0 ± 38.0 to 365.0 ± 35.0 ms vs. sham: 382.0 ± 69.0 to 352.0 ± 70.0 ms, P = 0.39) or dispersion of repolarization (SCS: 575.0 ± 164.0 to 1,465.0 ± 1,079.0 ms2 vs. sham: 572.0 ± 254.0 to 1,445.0 ± 1,578.0 ms2, P = 0.68).
Fig. 8.
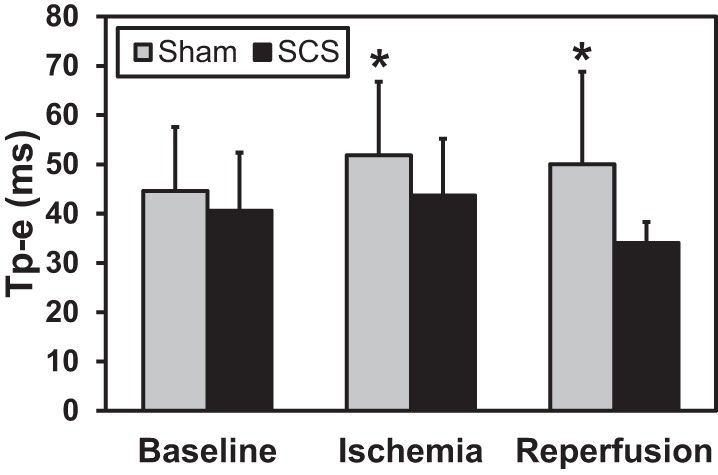
During ischemia (300 s) and after reperfusion (60 min), the T wave peak to end interval (Tp-e) was prolonged in the sham group (all *P < 0.009 vs. sham baseline), whereas Tp-e was unchanged in the SCS group (all P > 0.193 vs. SCS baseline). Baseline measurement was taken immediately before LAD ligation. Sham: n = 8; SCS: n = 8.
Hemodynamic measures.
SCS treatment attenuated the decrease in LV function during acute ischemia, as measured by LV dP/dtmax (Table 2). SCS also attenuated the increase in LVEDP from baseline to 300 s of ischemia. HR, LVESP, MAP, and dP/dtmin were not significantly changed by SCS from baseline to the end of ischemia (Table 2).
Table 2.
Hemodynamic changes associated with SCS during acute ischemia and reperfusion
| Sham (n = 9) |
SCS (n = 10) |
|||||
|---|---|---|---|---|---|---|
| Baseline | Ischemia | Reperfusion | Baseline | Ischemia | Reperfusion | |
| HR, beats/min | 71 ± 14 | 76 ± 14 | 77 ± 14 | 71 ± 15 | 73.0 ± 15 | 77 ± 15 |
| LVESP, mmHg | 108 ± 17 | 102 ± 17 | 103 ± 17 | 109 ± 17 | 108 ± 17 | 103 ± 17 |
| LVEDP, mmHg | 8 ± 4 | 10 ± 4 | 9 ± 4 | 9 ± 4 | 10 ± 4‡ | 8 ± 4† |
| MAP, mmHg | 110 ± 7 | 111 ± 19 | 109 ± 14 | 103 ± 17 | 100 ± 17 | 96 ± 19 |
| dP/dtmax, mmHg/s | 1,814 ± 282 | 1,596 ± 282* | 1,559 ± 282* | 1,442 ± 299 | 1,380 ± 299† | 1,390 ± 300† |
| dP/dtmin, mmHg/s | −2,212 ± 704 | −1,894 ± 704 | −2,222 ± 706 | −2,269 ± 746 | −2,068 ± 746 | −2,203 ± 748 |
Values are means ± SE; n, no. of animals. Baseline was measured before left anterior descending coronary artery ligation; ischemia was measured at 300 s of ischemia; reperfusion was measured at 60 min of reperfusion.
P = 0.001, change from baseline.
P ≤ 0.009, magnitude of mean difference from baseline in sham vs. SCS.
P ≤ 0.037, magnitude of mean difference from baseline in sham vs. SCS. All other P values were >0.11.
Effect of SCS on Ventricular Arrhythmias During Ischemia
SCS treatment was associated with fewer ventricular arrhythmias during acute ischemia compared with sham, demonstrating a functional and beneficial effect of SCS on arrhythmogenesis (Table 3). Ventricular arrhythmias were measured in episodes of 1) NSVTs [1 episode was recorded in 1 SCS animal vs. 25 episodes in 3 sham animals (P < 0.001, Table 3)] and 2) PVCs [17 PVCs recorded in 8 animals in the SCS group (range = 0–17 PVCs) vs. 105 PVCs in 9 animals in the sham group (range = 0–105 PVCs, P < 0.001)].
Table 3.
Ventricular arrhythmias associated with SCS
| NSVT |
PVC |
|||
|---|---|---|---|---|
| Sham | SCS | Sham | SCS | |
| Acute ischemia (5 min) | 24 (3) | 1 (1)* | 105 (9) | 17 (8)* |
| Reperfusion (60 min) | 1 (1) | 0 (0) | 14 (8) | 10 (8) |
Values are no. of arrhythmic events (with no. of animals in parentheses). NSVT, nonsustained ventricular tachycardia; PVC, premature ventricular contraction.
P = 0.001 vs. sham. All other P values were >0.29.
Effect of SCS on Cardiac Electrophysiological and Hemodynamic Measures During Reperfusion
Electrophysiological measures.
Electrophysiological measures in the ischemic myocardium were not significantly affected by SCS during reperfusion. From baseline to 60-min reperfusion, there were no significant changes in ARI (SCS: 368 ± 37 to 355 ± 39 ms vs. sham: 364 ± 66 to 353 ± 74 ms, P = 0.31), AT (SCS: 26 ± 3 to 26 ± 3 ms vs. sham: 24 ± 6 to 23 ± 5 ms, P = 0.63), RT (SCS: 394 ± 38 to 381 ± 39 ms vs. sham: 388 ± 70 to 375 ± 75 ms, P = 0.28), or dispersion of repolarization (SCS: 493 ± 337 to 491 ± 348 ms2 vs. sham: 511 ± 323 to 434 ± 375 ms2, P = 0.91).
Electrophysiological parameters in the nonischemic myocardium were similarly not significantly altered by SCS from baseline to 60-min reperfusion: ARI (SCS: 576 ± 271 to 636 ± 460 ms vs. sham: 561 ± 331 to 612 ± 453 ms, P = 0.26), AT (SCS: 27 ± 3 to 27 ± 3 ms vs. sham: 26 ± 5 to 26 ± 5 ms, P = 0.25), RT (SCS: 385 ± 34 to 377 ± 37 ms vs. sham: 379 ± 68 to 366 ± 74 ms, P = 0.23), and dispersion of repolarization (SCS: 576 ± 271 to 636 ± 460 ms2 vs. sham: 561 ± 331 to 612 ± 453 ms2, P = 0.53).
Global electrophysiological measures.
Myocardial electrophysiology and hemodynamic parameters were measured for 60 min following acute ischemia to assess SCS-mediated effects on overall cardiac function during reperfusion, a postischemic phase of cardiac stress. During reperfusion, Tpeak-Tend interval continued to be prolonged in the sham group and did not return to baseline after 60-min reperfusion (Fig. 8). However, in the SCS treatment group, Tpeak-Tend interval was unchanged from baseline throughout reperfusion, suggesting an SCS-mediated reduction of ventricular arrhythmogenic potential. SCS treatment was not associated with any significant changes from baseline (measured immediately before LAD ligation) to 60 min of reperfusion in global myocardial electrophysiological measures of ARI (SCS: 361 ± 36 to 352 ± 38 ms vs. sham: 356 ± 66 to 344 ± 72 ms, P = 0.24), AT (SCS: 26 ± 3 to 26 ± 3 ms vs. sham: 26 ± 5 to 25 ± 5 ms, P = 0.25), RT (SCS: 388 ± 38 to 378 ± 38 ms vs. sham: 382 ± 69 to 369 ± 74 ms, P = 0.23), or dispersion of repolarization (SCS: 575 ± 164 to 577 ± 324 ms2 vs. sham: 572 ± 254 to 590 ± 362 ms2, P = 0.72).
Hemodynamic measures.
LV function improved in the SCS group throughout reperfusion, as shown by the attenuation in dP/dtmax reduction (Table 2). LVEDP was reduced from baseline to 60 min of reperfusion in the SCS group, whereas it increased in the sham group (Table 2). HR, LVESP, MAP, and dP/dtmin were not significantly changed by SCS from baseline to the end of reperfusion.
Effect of SCS on Ventricular Arrhythmias During Reperfusion
Both SCS and sham groups had fewer ventricular arrhythmias during reperfusion than during ischemia, with no significant effect of SCS on the number of PVCs (Table 3).
DISCUSSION
In a porcine model of acute myocardial ischemia and reperfusion, thoracic SCS attenuated regional myocardial sympathetic excitation, decreased ventricular arrhythmias, and improved myocardial function. The major findings of this study are that SCS 1) reduced sympathoexcitation-induced ARI and RT shortening in the ischemic myocardium, 2) had no effect on ventricular electrophysiology in the nonischemic myocardium or during baseline conditions without myocardial stress, 3) attenuated increases in dispersion of repolarization, 4) reduced VTs, and 5) improved myocardial function during acute ischemia.
Effects of SCS on Myocardial Electrophysiology During Acute Ischemia
Our porcine model of acute ischemia with high-resolution cardiac electrophysiological mapping provides new insights into the mechanism of SCS’ antiarrhythmic effects during myocardial ischemia. The results show that SCS attenuated regional myocardial sympathetic excitatability (ARI and dispersion of repolarization) only in the ischemic ventricular myocardium. SCS was not found to have any significant effect on ventricular electrophysiology in resting conditions before ischemia or in the remote, nonischemic myocardium. ARI, which is a measure of action potential duration, is impacted by both AT and RT. AT is reflective of the rate of phase 0 action potential depolarization and is influenced by myocardial resting membrane potential and conduction velocity through myocardial gap junctions (5). Reflex sympathoexcitation during acute ischemia increases conduction velocity and reduces epicardial AT (5). RT, on the other hand, is reflective of phase 3 of the action potential and is affected by changes in K+ channel kinetics during sympathoexcitation (34). Therefore, altered ventricular electrophysiology during myocardial ischemia and SCS may be reflective of changes in cellular ionic currents and local sympathetic nerve activity.
Coronary artery occlusion has been shown to activate myocardial sensory nerves, increase afferent neural input to the thoracic spinal cord, and cause a reflex efferent sympathoexcitatory response within the local intrinsic cardiac nervous system (12, 22, 25, 31). Our results are supported by Odenstedt et al. (27) who described, using vectorcardiographic analysis in pigs, that SCS reduced repolarization alterations during ischemia-reperfusion. However, in canines, Wang et al. (36) reported that SCS mitigated the decrease in monophasic action potential duration in select ischemic regions. Importantly, they also demonstrated that SCS treatment reduced efferent neural activity in the stellate ganglia during ischemia. The exact mechanism of action through which SCS attenuates myocardial sympathoexcitation is not known. However, SCS has been shown to inhibit spinothalamic tract neurons in the spinal cord and influence the intrathoracic cardiac nervous system during ventricular ischemia (4, 6, 11, 13). Thus, the findings from previous studies are expanded on by our results, and together they suggest that SCS modulates the cardiac neural afferent activity and reduces the reflex efferent activation of regional intrinsic cardiac neurons in the ischemic myocardium.
Myocardial ischemia creates a substrate for VTs. VTs are most commonly caused by reentry circuits and involve a complex interaction between activation and repolarization of electrical wave fronts (8, 14, 28, 30). Increased heterogeneity (dispersion) of ARIs or repolarization, as seen in myocardial ischemia, is a major precursor to reentrant VTs, and previous studies have shown that PVCs and VTs originate in the ischemic regions of the myocardium (7, 18, 20). Here, we show that SCS attenuated the increase in dispersion of ARI and repolarization in the ischemic myocardium, reduced Tpeak-Tend, and was associated with fewer ventricular arrhythmias.
To our knowledge, this is the first study to use comprehensive electrophysiological mapping to demonstrate that the antiarrhythmic effect of SCS is likely due to the reduction in regional sympathoexcitation, which decreased the heterogeneity of myocardial repolarization in ischemic areas, thus stabilizing myocardial excitability and reducing the risk of reentrant VTs and sudden cardiac death (7, 18, 20, 34). Increase in dispersion of repolarization is affected more by direct sympathetic nerve stimulation than by circulating catecholamines (38). The localized reduction of sympathoexcitation during focal ischemia is an important new finding in understanding the therapeutic benefits of SCS. There are regional differences in sympathetic nerve innervation across the LV and right ventricle (34, 35). Furthermore, after myocardial infarction, there is regional remodeling of the intrinsic cardiac nervous system, thus altering afferent neural signals and neural processing. Cardiac injury is associated with changes in sympathetic innervation and a heightened sympathetic state. These changes most frequently occur at the ischemic border zone and dynamically amplify the risk for ventricular arrhythmias (7, 18, 20). These results, suggesting that SCS’ therapeutic effect may be due to regional attenuation of sympathoexcitation in the areas most affected, as opposed to global sympatholysis, may provide insights into the mechanisms behind the therapeutic benefit of SCS in mitigating ventricular arrhythmias during acute ischemia and after myocardial infarction in heart failure as well (31).
SCS Improves Myocardial Function During Acute Ischemia
In addition to reducing sympathoexcitation and decreasing ventricular arrhythmias, SCS was also found to improve myocardial function during acute ischemia. The improvement in myocardial function, despite a reduction in myocardial sympathoexcitation, suggests that SCS is providing myocardial protection through additional local mechanisms. In previous studies of chronic ischemic heart failure, SCS improved cardiac contractile function through reduced myocardial O2 consumption and improved lactate metabolism (19, 21, 23, 24). In an elegant study, Lopshire and colleagues (23), demonstrated in a canine model of postinfarction heart failure that SCS improved ventricular function better than medical management with β-blockers. Liu et al. (21) demonstrated in a porcine model of heart failure that even 15 min of SCS is enough to improve ventricular function, as measured by dP/dt, speckle tracking strain imaging echocardiography, and ejection fraction. The improvement in regional and global ventricular function persisted even after SCS had been discontinued. While in our study mechanisms underlying beneficial effects of SCS on myocardial function during acute ischemia were not characterized, studies in chronic ischemic disease support our findings and provide some insights.
Effects of SCS During Reperfusion
In this study, SCS was not found to have an effect on local ventricular electrophysiology during reperfusion. Acute coronary artery occlusion for 5 min causes activation of cardiac sympathetic afferent nerve fibers without causing infarction or permanent myocardial damage (16, 25). Therefore, this acute duration of ischemia likely did not induce a significant reperfusion injury in the ischemic myocardium. Once the coronary artery occlusion was released, the cardiac afferent stimulation was no longer present. The electrophysiological parameters returned to near baseline, and the effects of SCS on reflex sympathoexcitation were no longer observed.
In addition to the local electrophysiological measures, Tpeak-Tend and ventricular function returned to baseline after ischemia with SCS treatment. However, in the sham group, the risk of ventricular arrhythmias, as measured by Tpeak-Tend and ventricular function, did not return to baseline levels, even after 60 min of reperfusion. Increase in Tpeak-Tend is a strong predictor of the risk of ventricular arrhythmias and sudden cardiac death (34, 38). SCS attenuated the Tpeak-Tend increase throughout ischemia and reperfusion in this study. This is supported by previous studies involving longer periods of ischemia, which found that SCS reduced Tpeak-Tend during ischemia and was associated with fewer ventricular arrhythmias; however, the effects throughout reperfusion were not reported (17, 27).
Limitations
While this study provides mechanistic insights into the therapeutic benefits of SCS, it does have limitations. First, all of our experiments were performed in healthy animals with acute ischemia to see the effect of SCS on normal ventricular electrophysiology. However, the findings of this study may not be applicable to hearts with infarction or cardiomyopathy. Second, all measures were taken in an anesthetized animal (inhaled isoflurane and then intravenous α-chloralose). Therefore, the resulting sympathoexcitation associated with ischemia may be blunted in an awake animal. In addition, the effect of ischemia may be lessened due to the possible protection of isoflurane to myocardium during ischemia. However, this was constant in both SCS and sham groups, and, therefore, the differences seen here between the two groups should not be affected. Finally, in this study, we investigated the effects of preemptive, traditional, thoracic SCS on acute ischemia, and, therefore, these results may not be applicable to other SCS treatment modalities, such as reactive, high-frequency, or burst stimulation.
Conclusions
In a porcine model of acute ventricular ischemia with increased cardiac afferent signaling, SCS reduced local efferent sympathoexcitation, decreased ventricular arrhythmias, and improved myocardial function. SCS decreased sympathetic nerve activation regionally in the ischemic myocardium with no effect observed on the normal myocardium. These findings provide important mechanistic insights into the antiarrhythmic and myocardial protective effects of thoracic SCS.
GRANTS
K. Howard-Quijano is supported by the Foundation for Education and Research Mentored Research Training Grant. A. Mahajan is supported by National Heart, Lung, and Blood Institute Research Project Grant R01-HL-084261.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.H.-Q., T.T., and A.M. conceived and designed research; K.H.-Q., T.T., E.A.D., Y.K., and A.A. performed experiments; K.H.-Q., T.T., E.A.D., J.K., Y.K., T.G., A.A., K.S., and A.M. analyzed data; K.H.-Q., T.T., E.A.D., J.K., Y.K., T.G., A.A., K.S., and A.M. interpreted results of experiments; K.H.-Q., T.T., E.A.D., J.K., Y.K., A.A., and A.M. prepared figures; K.H.-Q., E.A.D., J.K., A.A., K.S., and A.M. drafted manuscript; K.H.-Q., T.T., E.A.D., J.K., Y.K., T.G., A.A., K.S., and A.M. edited and revised manuscript; K.H.-Q., T.T., E.A.D., J.K., Y.K., T.G., A.A., K.S., and A.M. approved final version of manuscript.
REFERENCES
- 1.Ajijola OA, Lellouche N, Bourke T, Tung R, Ahn S, Mahajan A, Shivkumar K. Bilateral cardiac sympathetic denervation for the management of electrical storm. J Am Coll Cardiol 59: 91–92, 2012. doi: 10.1016/j.jacc.2011.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ajijola OA, Vaseghi M, Zhou W, Yamakawa K, Benharash P, Hadaya J, Lux RL, Mahajan A, Shivkumar K. Functional differences between junctional and extrajunctional adrenergic receptor activation in mammalian ventricle. Am J Physiol Heart Circ Physiol 304: H579–H588, 2013. doi: 10.1152/ajpheart.00754.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajijola OA, Yagishita D, Patel KJ, Vaseghi M, Zhou W, Yamakawa K, So E, Lux RL, Mahajan A, Shivkumar K. Focal myocardial infarction induces global remodeling of cardiac sympathetic innervation: neural remodeling in a spatial context. Am J Physiol Heart Circ Physiol 305: H1031–H1040, 2013. doi: 10.1152/ajpheart.00434.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ardell JL, Cardinal R, Vermeulen M, Armour JA. Dorsal spinal cord stimulation obtunds the capacity of intrathoracic extracardiac neurons to transduce myocardial ischemia. Am J Physiol Regul Integr Comp Physiol 297: R470–R477, 2009. doi: 10.1152/ajpregu.90821.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arenal A, Villemaire C, Nattel S. Mechanism of selective epicardial activation delay during acute myocardial ischemia in dogs. Circulation 88: 2381–2388, 1993. doi: 10.1161/01.CIR.88.5.2381. [DOI] [PubMed] [Google Scholar]
- 6.Armour JA, Linderoth B, Arora RC, DeJongste MJ, Ardell JL, Kingma JG Jr, Hill M, Foreman RD. Long-term modulation of the intrinsic cardiac nervous system by spinal cord neurons in normal and ischaemic hearts. Auton Neurosci 95: 71–79, 2002. doi: 10.1016/S1566-0702(01)00377-0. [DOI] [PubMed] [Google Scholar]
- 7.Behrens S, Li C, Franz MR. Effects of myocardial ischemia on ventricular fibrillation inducibility and defibrillation efficacy. J Am Coll Cardiol 29: 817–824, 1997. doi: 10.1016/S0735-1097(96)00571-2. [DOI] [PubMed] [Google Scholar]
- 8.Ben-David J, Zipes DP. Differential response to right and left ansae subclaviae stimulation of early afterdepolarizations and ventricular tachycardia induced by cesium in dogs. Circulation 78: 1241–1250, 1988. doi: 10.1161/01.CIR.78.5.1241. [DOI] [PubMed] [Google Scholar]
- 9.Boon D, van Goudoever J, Piek JJ, van Montfrans GA. ST segment depression criteria and the prevalence of silent cardiac ischemia in hypertensives. Hypertension 41: 476–481, 2003. doi: 10.1161/01.HYP.0000054980.69529.14. [DOI] [PubMed] [Google Scholar]
- 10.Bourke T, Vaseghi M, Michowitz Y, Sankhla V, Shah M, Swapna N, Boyle NG, Mahajan A, Narasimhan C, Lokhandwala Y, Shivkumar K. Neuraxial modulation for refractory ventricular arrhythmias: value of thoracic epidural anesthesia and surgical left cardiac sympathetic denervation. Circulation 121: 2255–2262, 2010. doi: 10.1161/CIRCULATIONAHA.109.929703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandler MJ, Brennan TJ, Garrison DW, Kim KS, Schwartz PJ, Foreman RD. A mechanism of cardiac pain suppression by spinal cord stimulation: implications for patients with angina pectoris. Eur Heart J 14: 96–105, 1993. doi: 10.1093/eurheartj/14.1.96. [DOI] [PubMed] [Google Scholar]
- 12.Felder RB, Thames MD. The cardiocardiac sympathetic reflex during coronary occlusion in anesthetized dogs. Circ Res 48: 685–692, 1981. doi: 10.1161/01.RES.48.5.685. [DOI] [PubMed] [Google Scholar]
- 13.Foreman RD, Linderoth B, Ardell JL, Barron KW, Chandler MJ, Hull SS Jr, TerHorst GJ, DeJongste MJ, Armour JA. Modulation of intrinsic cardiac neurons by spinal cord stimulation: implications for its therapeutic use in angina pectoris. Cardiovasc Res 47: 367–375, 2000. doi: 10.1016/S0008-6363(00)00095-X. [DOI] [PubMed] [Google Scholar]
- 14.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res 96: 459–466, 2005. doi: 10.1161/01.RES.0000156891.66893.83. [DOI] [PubMed] [Google Scholar]
- 15.Haws CW, Lux RL. Correlation between in vivo transmembrane action potential durations and activation-recovery intervals from electrograms. Effects of interventions that alter repolarization time. Circulation 81: 281–288, 1990. doi: 10.1161/01.CIR.81.1.281. [DOI] [PubMed] [Google Scholar]
- 16.Holland RP, Brooks H. The QRS complex during myocardial ischemia. An experimental analysis in the porcine heart. J Clin Invest 57: 541–550, 1976. doi: 10.1172/JCI108309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Issa ZF, Zhou X, Ujhelyi MR, Rosenberger J, Bhakta D, Groh WJ, Miller JM, Zipes DP. Thoracic spinal cord stimulation reduces the risk of ischemic ventricular arrhythmias in a postinfarction heart failure canine model. Circulation 111: 3217–3220, 2005. doi: 10.1161/CIRCULATIONAHA.104.507897. [DOI] [PubMed] [Google Scholar]
- 18.Kimura S, Bassett AL, Kohya T, Kozlovskis PL, Myerburg RJ. Simultaneous recording of action potentials from endocardium and epicardium during ischemia in the isolated cat ventricle: relation of temporal electrophysiologic heterogeneities to arrhythmias. Circulation 74: 401–409, 1986. doi: 10.1161/01.CIR.74.2.401. [DOI] [PubMed] [Google Scholar]
- 19.Kingma JG Jr, Linderoth B, Ardell JL, Armour JA, DeJongste MJ, Foreman RD. Neuromodulation therapy does not influence blood flow distribution or left-ventricular dynamics during acute myocardial ischemia. Auton Neurosci 91: 47–54, 2001. doi: 10.1016/S1566-0702(01)00285-5. [DOI] [PubMed] [Google Scholar]
- 20.Kurz RW, Xiao-Lin R, Franz MR. Increased dispersion of ventricular repolarization and ventricular tachyarrhythmias in the globally ischaemic rabbit heart. Eur Heart J 14: 1561–1571, 1993. doi: 10.1093/eurheartj/14.11.1561. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Yue WS, Liao SY, Zhang Y, Au KW, Shuto C, Hata C, Park E, Chen P, Siu CW, Tse HF. Thoracic spinal cord stimulation improves cardiac contractile function and myocardial oxygen consumption in a porcine model of ischemic heart failure. J Cardiovasc Electrophysiol 23: 534–540, 2012. doi: 10.1111/j.1540-8167.2011.02230.x. [DOI] [PubMed] [Google Scholar]
- 22.Longhurst JC, Tjen-A-Looi SC, Fu LW. Cardiac sympathetic afferent activation provoked by myocardial ischemia and reperfusion. Mechanisms and reflexes. Ann N Y Acad Sci 940: 74–95, 2001. doi: 10.1111/j.1749-6632.2001.tb03668.x. [DOI] [PubMed] [Google Scholar]
- 23.Lopshire JC, Zhou X, Dusa C, Ueyama T, Rosenberger J, Courtney N, Ujhelyi M, Mullen T, Das M, Zipes DP. Spinal cord stimulation improves ventricular function and reduces ventricular arrhythmias in a canine postinfarction heart failure model. Circulation 120: 286–294, 2009. doi: 10.1161/CIRCULATIONAHA.108.812412. [DOI] [PubMed] [Google Scholar]
- 24.Mannheimer C, Camici P, Chester MR, Collins A, DeJongste M, Eliasson T, Follath F, Hellemans I, Herlitz J, Lüscher T, Pasic M, Thelle D. The problem of chronic refractory angina; report from the ESC Joint Study Group on the Treatment of Refractory Angina. Eur Heart J 23: 355–370, 2002. doi: 10.1053/euhj.2001.2706. [DOI] [PubMed] [Google Scholar]
- 25.Minisi AJ, Thames MD. Activation of cardiac sympathetic afferents during coronary occlusion. Evidence for reflex activation of sympathetic nervous system during transmural myocardial ischemia in the dog. Circulation 84: 357–367, 1991. doi: 10.1161/01.CIR.84.1.357. [DOI] [PubMed] [Google Scholar]
- 26.Murray S, Carson KG, Ewings PD, Collins PD, James MA. Spinal cord stimulation significantly decreases the need for acute hospital admission for chest pain in patients with refractory angina pectoris. Heart 82: 89–92, 1999. doi: 10.1136/hrt.82.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Odenstedt J, Linderoth B, Bergfeldt L, Ekre O, Grip L, Mannheimer C, Andrell P. Spinal cord stimulation effects on myocardial ischemia, infarct size, ventricular arrhythmia, and noninvasive electrophysiology in a porcine ischemia-reperfusion model. Heart Rhythm 8: 892–898, 2011. doi: 10.1016/j.hrthm.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Opthof T, Coronel R, Vermeulen JT, Verberne HJ, van Capelle FJ, Janse MJ. Dispersion of refractoriness in normal and ischaemic canine ventricle: effects of sympathetic stimulation. Cardiovasc Res 27: 1954–1960, 1993. doi: 10.1093/cvr/27.11.1954. [DOI] [PubMed] [Google Scholar]
- 29.Perkiömäki JS, Koistinen MJ, Yli-Mäyry S, Huikuri HV. Dispersion of QT interval in patients with and without susceptibility to ventricular tachyarrhythmias after previous myocardial infarction. J Am Coll Cardiol 26: 174–179, 1995. doi: 10.1016/0735-1097(95)00122-G. [DOI] [PubMed] [Google Scholar]
- 30.Qu Z, Weiss JN, Garfinkel A. Cardiac electrical restitution properties and stability of reentrant spiral waves: a simulation study. Am J Physiol Heart Circ Physiol 276: H269–H283, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Rajendran PS, Nakamura K, Ajijola OA, Vaseghi M, Armour JA, Ardell JL, Shivkumar K. Myocardial infarction induces structural and functional remodelling of the intrinsic cardiac nervous system. J Physiol 594: 321–341, 2016. doi: 10.1113/JP271165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saffitz JE. Sympathetic neural activity and the pathogenesis of sudden cardiac death. Heart Rhythm 5: 140–141, 2008. doi: 10.1016/j.hrthm.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 33.Southerland EM, Milhorn DM, Foreman RD, Linderoth B, DeJongste MJ, Armour JA, Subramanian V, Singh M, Singh K, Ardell JL. Preemptive, but not reactive, spinal cord stimulation mitigates transient ischemia-induced myocardial infarction via cardiac adrenergic neurons. Am J Physiol Heart Circ Physiol 292: H311–H317, 2007. doi: 10.1152/ajpheart.00087.2006. [DOI] [PubMed] [Google Scholar]
- 34.Vaseghi M, Yamakawa K, Sinha A, So EL, Zhou W, Ajijola OA, Lux RL, Laks M, Shivkumar K, Mahajan A. Modulation of regional dispersion of repolarization and T-peak to T-end interval by the right and left stellate ganglia. Am J Physiol Heart Circ Physiol 305: H1020–H1030, 2013. doi: 10.1152/ajpheart.00056.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaseghi M, Zhou W, Shi J, Ajijola OA, Hadaya J, Shivkumar K, Mahajan A. Sympathetic innervation of the anterior left ventricular wall by the right and left stellate ganglia. Heart Rhythm 9: 1303–1309, 2012. doi: 10.1016/j.hrthm.2012.03.052. [DOI] [PubMed] [Google Scholar]
- 36.Wang S, Zhou X, Huang B, Wang Z, Liao K, Saren G, Lu Z, Chen M, Yu L, Jiang H. Spinal cord stimulation protects against ventricular arrhythmias by suppressing left stellate ganglion neural activity in an acute myocardial infarction canine model. Heart Rhythm 12: 1628–1635, 2015. doi: 10.1016/j.hrthm.2015.03.023. [DOI] [PubMed] [Google Scholar]
- 37.Webb SW, Adgey AA, Pantridge JF. Autonomic disturbance at onset of acute myocardial infarction. BMJ 3: 89–92, 1972. doi: 10.1136/bmj.3.5818.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yagishita D, Chui RW, Yamakawa K, Rajendran PS, Ajijola OA, Nakamura K, So EL, Mahajan A, Shivkumar K, Vaseghi M. Sympathetic nerve stimulation, not circulating norepinephrine, modulates T-peak to T-end interval by increasing global dispersion of repolarization. Circ Arrhythm Electrophysiol 8: 174–185, 2015. doi: 10.1161/CIRCEP.114.002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou S, Jung BC, Tan AY, Trang VQ, Gholmieh G, Han SW, Lin SF, Fishbein MC, Chen PS, Chen LS. Spontaneous stellate ganglion nerve activity and ventricular arrhythmia in a canine model of sudden death. Heart Rhythm 5: 131–139, 2008. doi: 10.1016/j.hrthm.2007.09.007. [DOI] [PubMed] [Google Scholar]