

Absence of lysyl oxidase (Lox) causes thoracic aortic aneurysms. The aortic mechanical behavior of Lox−/− mice is consistent with reduced elastin and collagen cross-linking but demonstrates vascular location-specific differences. Lox−/− aortas show upregulation of matrix remodeling genes and location-specific differential expression of other matrix and smooth muscle cell gene sets.
Keywords: matrisome, elastin, collagen, mechanics, gene array, aneurysm, dissection, tortuosity
Abstract
Mutations in lysyl oxidase (LOX) are associated with thoracic aortic aneurysm and dissection (TAAD). Mice that do not express Lox (Lox−/−) die soon after birth and have 60% and 40% reductions in elastin- and collagen-specific cross-links, respectively. LOX inactivation could also change the expression of secreted factors, the structural matrix, and matrix-associated proteins that constitute the aortic matrisome. We hypothesized that absence of Lox will change the mechanical behavior of the aortic wall because of reduced elastin and collagen cross-linking and alter the expression levels of matrisome and smooth muscle cell (SMC) genes in a vascular location-specific manner. Using fluorescence microscopy, pressure myography, and gene set enrichment analysis, we visualized the microarchitecture, quantified the mechanical behavior, and examined matrisome and SMC gene expression from ascending aortas (AAs) and descending aortas (DAs) from newborn Lox+/+ and Lox−/− mice. Even though Lox−/− AAs and DAs have fragmented elastic laminae and disorganized SMCs, the unloaded outer diameter and wall thickness were similar to Lox+/+ AAs and DAs. Lox−/− AAs and DAs have altered opening angles, circumferential stresses, and circumferential stretch ratios; however, only Lox−/− AAs have increased pressurized diameters and tangent moduli. Gene set enrichment analysis showed upregulation of the extracellular matrix (ECM) regulator gene set in Lox−/− AAs and DAs as well as differential expression of secreted factors, collagens, ECM-affiliated proteins, ECM glycoproteins, and SMC cell cycle gene sets that depend on the Lox genotype and vascular location. These results provide insights into the local chemomechanical changes induced by Lox inactivation that may be important for TAAD pathogenesis.
NEW & NOTEWORTHY Absence of lysyl oxidase (Lox) causes thoracic aortic aneurysms. The aortic mechanical behavior of Lox−/− mice is consistent with reduced elastin and collagen cross-linking but demonstrates vascular location-specific differences. Lox−/− aortas show upregulation of matrix remodeling genes and location-specific differential expression of other matrix and smooth muscle cell gene sets.
lysyl oxidase (LOX) is a copper-dependent enzyme that cross-links elastin and collagen in the extracellular matrix (ECM). LOX is part of an enzyme family with four additional members: LOX-like 1–4 [LOXL1–4 (38)]. These enzymes share a highly conserved carboxyl-terminal catalytic domain that includes the copper-binding site and a cytokine receptor-like domain. In addition to their cross-linking activity, LOX and LOXL enzymes can influence cell proliferation, chemotactic responses, and epithelial-to-mesenchymal transition (29). Mice that do not express lysyl oxidase (Lox−/−) have fragmented elastic fibers in the arteries, lungs, and skin and die within a few hours of birth with a ruptured diaphragm, impaired airway development, and thoracic aortic aneurysm and dissection (TAAD) (19, 31, 32). Recently, point mutations in LOX have been linked to TAAD in humans (16, 27).
A hallmark of TAAD is medial degeneration, characterized by loss or fragmentation of elastic fibers in the aortic wall (14). Medial degeneration may be caused by accelerated degradation of elastic fibers or by impaired elastic fiber assembly. Elastic fiber assembly is a multistep process that incorporates tropoelastin (soluble elastin) into ever more complex structures leading to mature elastic fibers. This process is coordinated by LOX and a number of other ECM proteins, including fibrillins and fibulins (44). Mutations in some of these critical elastic fiber proteins, including LOX, fibrillin-1, and fibulin-4, are associated with TAAD, whereas mutations in others, including elastin and fibulin-5, are not (28). Early models of TAAD pathogenesis focused on mechanical failure of the aortic wall due to medial degeneration (28). Mechanical strength and stiffness of the wall are critical for withstanding physiological loads and for maintaining a normal smooth muscle cell (SMC) phenotype (39). However, in addition to their structural role, ECM proteins regulate the availability of signaling molecules in the extracellular environment. Contemporary models of TAAD pathogenesis consider the ECM, associated SMCs, and signaling molecules as an integrated chemomechanical system (22).
The local chemomechanical environment of the vascular wall can be defined by expression levels and amounts of ECM-associated genes and proteins. The “matrisome” is a collection of proteins that includes ECM core components (i.e., elastin and other glycoproteins, collagens, and proteoglycans) and ECM-associated components (i.e., cross-linking enzymes, growth factors, and proteases). Quantification of matrisome proteins has been used to compare and contrast ECM composition in normal and malignant tissues (35) but has not been applied to cardiovascular disease. Characterization of the local mechanical properties and matrisome signature of the aortic wall in healthy and diseased states may uncover important mechanisms of vascular pathologies, including TAAD. We hypothesized that absence of LOX will change the mechanical behavior of the aortic wall because of the lack of elastin and collagen cross-linking and alter the expression levels of matrisome and SMC phenotype genes because of modulation of cell signaling in a vascular location-specific manner. We examined the morphology, wall structure, mechanical behavior, and gene expression of ascending and descending segments of the thoracic aorta in newborn Lox+/+ and Lox−/− mice to determine whether genotype- and vascular location-specific changes in the chemomechanical environment may be linked to TAAD pathogenesis.
MATERIALS AND METHODS
Animals and tissue collection.
All animal protocols were approved by the Institutional Animal Care and Use Committee. Lox+/− mice were bred to produce Lox+/+ and Lox−/− pups (19). Pups were used within 24 h of birth and euthanized by CO2 inhalation. The thoracic aorta was cut into two segments: 1) the ascending aorta (AA) from the aortic valve to the left common carotid artery and 2) the descending aorta (DA) from the ductus arteriosus to the diaphragm, to analyze differences between vascular locations. The aortic segments were used for morphology (n = 5–8 per group), fluorescence microscopy (n = 3 per group), blood pressure measurement (n = 3–20 per group), mechanical testing (n = 5–8 per group), gene array (n = 2 per group of 8 pooled samples), and quantitative PCR (qPCR; n = 3 per group of 8 pooled samples).
Morphology and fluorescence microscopy.
To visualize thoracic aortic morphology, yellow latex (Ward’s Natural Science) was injected into the vascular tree, as previously described (13). Images were taken with a digital camera mounted on a dissecting microscope. To visualize aortic wall structure, frozen section preparation and fluorescence staining were performed as previously described (26). Alexa fluor 633 hydrazide (0.6 µM, Life Technologies) was used for elastin staining (8, 40). CNA35 (kindly provided by Magnus Hook, Texas A&M University) labeled with Oregon green 488 (Life Technologies) was used for collagen staining (5 µM) (24). Hoechst 34580 (5 µM, Life Technologies) was used for nuclear staining. Imaging was performed using a Zeiss ×40 oil-immersion lens (numerical aperture: 1.3) on a Zeiss LSM 710 confocal microscope. Image postprocessing and quantification were performed using ImageJ (National Institutes of Health) and Zen (Zeiss). To quantify elastic fiber integrity, the average circumferential length of the continuous elastic lamina within a constant image area was measured as previously described (9). To quantify cellular organization, the average nuclear aspect ratio of all cells within a constant image area was calculated by dividing the long-axis length by the short-axis length.
Blood pressure measurement.
Left ventricular (LV) blood pressure was measured as previously described (43). Pups were anesthetized with 1.5% isoflurane and placed on a heating pad with additional radiant heat to keep warm. A 30-gauge needle and tubing filled with 0.2% heparin sodium salt in PBS were connected to a fluid-filled pressure transducer (Uniflow; Baxter). Under ultrasound guidance (Vevo 2100, VisualSonics), the needle was advanced through apical puncture into the LV chamber. After stable pressure data were recorded, the needle was flushed, and the pressure data were recorded again to confirm that there were no clots in the needle tip. Mean LV pressure and heart rate were averaged over the stable range of pressure data. Assuming that the LV diastolic pressure is zero, the reported systolic LV pressure is double the measured mean pressure.
Mechanical testing and data analysis.
Mechanical testing and data analysis were performed as previously described (23). The in vivo stretch ratio (IVSR) in the longitudinal direction was measured from images of the aortic segments taken before and after dissection. The aortic segments were stored in physiological saline solution (PSS) for up to 3 days before mechanical testing (2). Mechanical tests were performed on the aortic segments using a Myograph 110P (Danish Myotechnology). The AA or DA was mounted in the myograph bath on custom stainless steel cannulae, stretched longitudinally to its approximate IVSR, and pressurized with PSS from 5 to 60 mmHg in 5-mmHg increments. After three preconditioning cycles, the lumen pressure, outer diameter, and longitudinal force were recorded at 1 Hz for three additional loading cycles. After mechanical testing, two to three cross-sectional rings of 150–250 µm in length were cut and imaged to determine the unloaded thickness and diameter. To determine residual strain, as measured by the opening angle, a single radial cut was made in each ring, and images were acquired after a 5-min equilibration period (7).
Pressure-outer diameter curves were fitted to the following equation:
| (1)) |
where do is the outer diameter, P is the lumen pressure, and ai are constants determined by regression in MATLAB (MathWorks) (12). The compliance () at each applied pressure was calculated as follows:
| (2)) |
Average circumferential stretch (λθ) was calculated as follows:
| (3)) |
where ri and ro are the loaded inner and outer radii, respectively, and Ri and Ro are the unloaded inner and outer radii, respectively. Average circumferential stress (σθ) was calculated as follows:
| (4)) |
Stress-stretch curves were fit to the following equation:
| (5)) |
where bi are constants determined by regression in MATLAB. The tangent modulus () at each applied pressure was calculated as follows:
| (6)) |
Gene expression analyses.
Aortic segments were processed for gene array analysis as previously described (23). Segments were flash frozen in liquid nitrogen and stored at −80°C. Eight aortic segments from each genotype were pooled together. RNA was isolated using the RNAeasy Plus Mini Kit (Qiagen), and RNA purity was confirmed by a measured ratio of absorbance at 260 and 280 nm between 1.8 and 2.0. For two pooled samples per group, the isolated RNA was further processed by the Genome Technology Access Center (GTAC) at Washington University for use with the Affymetrix Mouse Gene 2.0 array. Expression Console (Affymetrix) was used by GTAC for quality control and to generate gene expression values with robust multiarray analysis background correction, median polish summarization, and quantile normalization. The array-control probe sets and unknown genes were removed before statistical analysis. All gene array data are accessible through series record GSE89227 at the National Center for Biotechnology Information Gene Expression Omnibus.
To determine whether other genes from the Lox family are upregulated to compensate for the absence of Lox, the changes in gene array expression of Lox and Loxl1–4 for each vascular location and genotype were calculated relative to the expression level of Lox in Lox+/+ AAs or DAs. Results for Lox and Loxl1–4 gene expression were confirmed by qPCR. For qPCR, isolated RNA from three pooled samples per group was processed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosciences). qPCR was performed on a QuantStudio 12K machine (Applied Biosystems) with TaqMan Fast Advanced Master Mix (Applied Biosystems). TaqMan Gene Expression Assays (Life Technologies) for Lox and Loxl1–4 were used for primers, and all experiments were run in triplicate. The threshold cycle (Ct) from each triplicate was averaged and normalized to the average expression level of β2-microglobulin (B2m), hypoxanthine phosphoribosyl transferase 1 (Hprt), and cyclin-dependent kinase inhibitor 1B (Cdkn1b).
To investigate the effects of Lox absence on the expression of many genes that may act in concert, we performed gene set enrichment analysis (GSEA) according to Subramanian et al. (41). Normalized expressions for genes in the array were arranged into a list ranked according to differential expression based on genotype (Lox−/− vs. Lox+/+, regardless of vascular location) or by genotype and vascular location (Lox−/− AA vs. Lox+/+ AA and Lox−/− DA vs. Lox+/+ DA). The locations within the ranked list of genes making up subsets that encode ECM core proteins, ECM-associated proteins (34), or proteins specific to SMC phenotypes (25) were then determined. The fraction of genes in a subset that match genes located at the top or bottom of the ranked list, weighted by the fraction of genes not in the subset and normalized by the size of subset, was used to calculate the normalized enrichment score (NES). The NES value is high and positive when genes in the subset are concentrated at the top of the ranked list, high and negative when genes in the subset are concentrated at the bottom of the ranked list, and close to zero when genes in the subset are randomly distributed in the ranked list. High, positive NES indicates increased expression of a specific subset in Lox−/− compared with Lox+/+, whereas high, negative NES indicates decreased expression of a specific subset in Lox−/− compared with Lox+/+. The running enrichment score (ES) for each subset was calculated up to a given position in the ranked list. The leading-edge genes in a subset are those that appear before ES is maximal (for positive NES) or after ES reaches a minimum (for negative NES). The leading-edge genes contribute the most to the overall NES.
Statistical analyses.
Prism (GraphPad) or SPSS (IBM) was used to perform comparisons between groups using ordinary two-way ANOVA with Tukey’s post hoc analysis for multiple comparisons or unpaired, two-tailed Student’s t-test for comparison between two groups. The sources of variation are reported as genotype, vascular location, or combination of genotype × vascular location. Data are shown as means ± SE, and P < 0.05 was considered significant. For GSEA, enrichment of a specific subset was considered significant at a false discovery rate q value of <0.25 and nominal P value of <0.05.
RESULTS
Aortic abnormalities in Lox−/− mice.
The gross morphology of the thoracic aorta was evaluated by injecting a latex-based dye into the arterial tree. All Lox−/− mice exhibited tortuosity in the DA, whereas 22% had patent ductus arteriosus (Fig. 1). In 28% of Lox−/− mice, aneurysms were visible in the AA and aortic arch. In 36% of Lox−/− mice, aneurysms or pseudoaneurysms were visible in the DA just distal to the left subclavian artery (Fig. 1, E and F). Aortic tortuosity in Lox−/− mice is likely due to lengthening of the aorta in vivo (10, 11). Although we did not measure the entire aortic length in this study, the in vivo length of the AA from the root to the innominate artery is 1,470 ± 70 μm in Lox−/− mice and 1,130 ± 60 μm in Lox+/+ mice (P = 0.003).
Fig. 1.
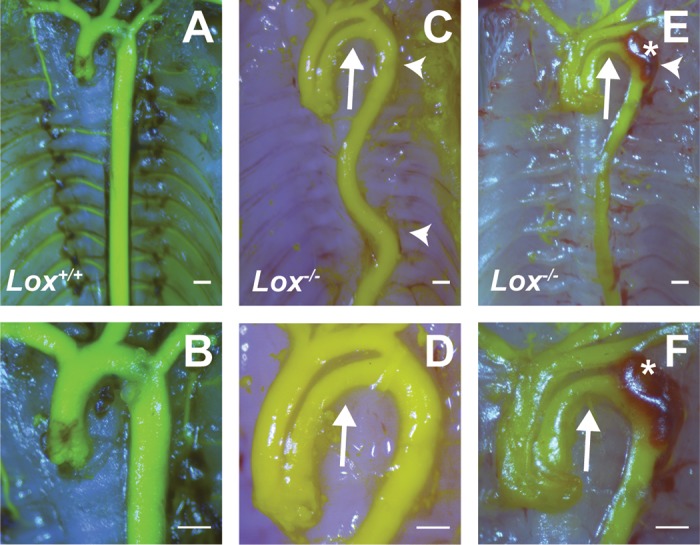
Gross aortic morphology in newborn lysyl oxidase (Lox) mice. A and B: representative images of the Lox+/+ ascending aorta (AA) and descending aorta (DA). Ligamentum arteriosum, left over after the full closure of the ductus arteriosus, was removed during dissection. C and D: representative images of the Lox−/− AA and DA. The Lox−/− AA was longer than the Lox+/+ AA. The Lox−/− DA was tortuous (arrowheads). The arrows indicate patent ductus arteriosus, a common occurrence in Lox−/− mice. E and F: additional images of the Lox−/− thoracic aorta with tortuosity of the proximal DA (arrowhead), patent ductus arteriosus (arrows), and pseudoaneurysm (*), which may indicate dissection of the proximal DA wall. B, D, and F show higher-magnification images of A, C, and E, respectively. Scale bars = 500 µm. n = 5–8 per group.
A more detailed evaluation of the aortic wall structure was performed using fluorescence microscopy. Example images of the AA are shown in Fig. 2. DA images were similar (not shown). Aortas from Lox−/− mice exhibited areas of low elastin staining compared with Lox+/+ mice, interrupting the continuity of the elastic laminae (Fig. 2, A and B). As a result, the uninterrupted elastic laminae length in the Lox−/− AA and DA was two to three times shorter than those in Lox+/+ mice, with genotype accounting for 77% of the variance between groups (Fig. 2I). We did not quantify collagen organization, as the collagen staining was highly variable between samples (Fig. 2, C and D). The cell nuclei were more rounded (aspect ratio closer to 1) in Lox−/− aortic segments compared with Lox+/+ aortic segments, indicating more disorganized SMCs, with genotype accounting for 92% of the variance between groups (Fig. 2, E, F, and J).
Fig. 2.

Aortic wall structure in newborn Lox mice. The elastic laminae structure in Lox+/+ (A) and Lox−/− (B) AAs showed fragmented laminae in Lox−/− compared with Lox+/+ mice. Collagen staining in Lox+/+ (C) and Lox−/− (D) AAs showed abundant adventitial collagen in both genotypes. Nuclear staining in Lox+/+ (E) and Lox−/− (F) AAs showed more rounded nuclei in the Lox−/− AA compared with the Lox+/+ AA. G and H: composite images of Lox+/+ (G) and Lox−/− (H) AAs. L indicates the location of the vascular lumen. Scale bars = 10 µm. I: quantification of the uninterrupted elastic laminae length confirmed elastic fiber fragmentation in the Lox−/− AA and DA. J: quantification of the nuclear aspect ratio confirmed altered cellular organization in the Lox−/− AA and DA. n = 3 images/group, with at least 8 elastic laminae and 20 nuclei per image analyzed. &Genotype accounted for significant variance between groups (P < 0.05). *Groups were significantly different from each other (P < 0.05).
Our results indicate that the absence of Lox results in gross vascular abnormalities with variable penetrance of specific defects (i.e., tortuosity, aneurysms, and patent ductus arteriosus). The morphological abnormalities are accompanied by microstructural changes in the elastin architecture and nuclear shape changes in the cells within the wall of the AA and DA.
Blood pressure.
LV blood pressure and heart rate were measured using ultrasound guidance of a small-gauge needle inserted through the chest wall. The systolic LV pressure of newborn mice was not significantly different between genotypes and averaged 23 mmHg (Fig. 3A). The average heart rate was 20% lower in Lox−/− mice compared with Lox+/+ mice (Fig. 3B). The heart rate may be slower in Lox−/− mice because of cardiovascular and pulmonary abnormalities that lead to death within a few hours of birth. The sample size for the Lox−/− group was low (n = 3) because of difficulties in obtaining Lox−/− mice that were not having difficulty breathing immediately after birth. The results indicate that differences in aortic structure, mechanical behavior, or matrisome gene expression between Lox+/+ and Lox−/− mice are not due to differences in the systolic blood pressure applied to the aortic wall.
Fig. 3.

Left ventricular (LV) blood pressure in newborn Lox mice. A: there was no significant difference in systolic blood pressure between Lox−/− and Lox+/+ mice. B: heart rate [in beats/min (bpm)] of Lox−/− mice was significantly lower than Lox+/+ mice. *Groups were significantly different from each other (P < 0.05). n = 3 for Lox−/− and n = 20 for Lox+/+ mice. Blood pressure was measured in newborn mice before genotyping, and only mice that appeared to be breathing normally were used.
Structural and material properties of Lox−/− aortas.
The residual strains and unloaded dimensions of the aortas were measured to investigate the effects of the absence of Lox on vascular remodeling. Longitudinal residual strain, as measured by IVSR, was not significantly different between vascular locations (AA or DA; Fig. 4A). However, genotype accounted for 13% of the variation in IVSR between groups. Circumferential residual strain, as measured by opening angle, was 40% smaller in Lox−/− aortic segments compared with Lox+/+ aortic segments, with genotype accounting for 55% of the variation between groups (Fig. 4B). Opening angle did not vary with vascular location. Unloaded aortic wall thickness was not significantly different between groups or vascular locations (Fig. 4C). The unloaded aortic outer diameter was not significantly affected by genotype, but vascular location accounted for 40% of the variation between groups (Fig. 4D). The absence of Lox did not cause significant remodeling of the unloaded dimensions of the AA or DA wall but did cause changes in residual strains that may affect the mechanical stimuli experienced by SMCs.
Fig. 4.

Residual strain and unloaded dimensions of the AA and DA from newborn Lox mice. A: genotype accounts for significant variance between groups for the in vivo stretch ratio (IVSR) (&P = 0.045); however, there was no significant difference between Lox−/− and Lox+/+ aortic segments at either vascular location. B: genotype accounts for significant variance in opening angle (&P = 0.0001). Lox−/− opening angle was significantly smaller than Lox+/+ in both the AA (*P = 0.001) and DA (*P = 0.01). C: unloaded wall thickness is not different between Lox−/− and Lox+/+ genotypes or between AA and DA locations. D: vascular location accounted for significant variance in unloaded outer diameter (#P = 0.0007), and there was a significant difference in Lox−/− mice between AA and DA locations (*P = 0.0027). There was no difference between genotypes at either vascular location. n = 5–8 per group.
Mechanical tests were performed to determine the structural properties of the AA and DA wall. Structural properties depend on the material and geometry of the vascular wall. For all groups, the aortic outer diameter increased nonlinearly with pressure (Fig. 5A), as expected (23, 43). The aortic outer diameter varied significantly with genotype, vascular location, and combination of genotype × vascular location at all measured pressures. The Lox−/− AA had 18–26% larger outer diameters than the Lox+/+ AA and was also significantly larger than the Lox−/− DA at all pressures. The outer diameters of Lox−/− and Lox+/+ DAs were not significantly different. The source of variation in the compliance, or slope of the pressure-diameter curve, changed with applied pressure (Fig. 5B). The compliance of the Lox−/− AA was 105% larger than the Lox+/+ AA at 5 mmHg and 44–58% smaller at pressures between 30 and 50 mmHg, just beyond the measured systolic pressure. The compliance of the Lox−/− DA was not significantly different from the Lox+/+ DA. Despite similar changes in morphology and microstructure for Lox−/− AAs and DAs compared with Lox+/+ AAs and DAs, the structural properties were altered only in the Lox−/− AA.
Fig. 5.

Structural properties of the AA and DA from newborn Lox mice. A: genotype, vascular location, and genotype × vascular location had a significant effect on the variance of the mean outer diameter of the pressurized aorta. At all pressures, Lox−/− AA outer diameter was significantly larger than the Lox+/+ AA and Lox−/− DA (*P < 0.05). B: effects of genotype, vascular location, and genotype × vascular location on the aortic compliance varied with applied pressure. Lox−/− AA compliance was significantly lower than the Lox+/+ AA between 30 and 50 mmHg (*P < 0.05). Genotype (&), vascular location (#), or genotype × vascular location (%) accounted for significant variance between groups (P < 0.05). n = 5–8 per group.
The unloaded dimensions and mechanical test data were used to determine the circumferential material properties of the aortic wall. Material properties are independent of geometry. Genotype was the major source of variation in the circumferential stretch ratio (Fig. 6A). The circumferential stretch ratio of the Lox−/− AA and DA was 17–32% higher than the Lox+/+ AA and DA at all pressures. Genotype was also the major source of variation in circumferential stress (Fig. 6B). Circumferential stress was higher in the Lox−/− AA and DA compared with the Lox+/+ AA and DA at pressures between 15 and 60 mmHg. As a result of the changes in circumferential stretch and stress at each pressure, the stretch-stress curves for the Lox−/− AA and DA were shifted to the right of the Lox+/+ AA and DA (Fig. 6C). The tangent modulus, or slope of the stretch-stress curves, was significantly different because of genotype between 15 and 35 mmHg and because of the interaction between genotype and vascular location at pressures between 30 and 60 mmHg (Fig. 6D). The tangent modulus of the Lox−/− AA was 83–162% higher than the Lox+/+ AA at pressures between 15 and 60 mmHg. The tangent modulus was not significantly different between Lox−/− and Lox+/+ DAs. In Lox−/− aortas, circumferential material properties were altered in a manner consistent with changes in the microstructural organization and content of the vascular wall.
Fig. 6.

Material properties of the AA and DA from newborn Lox mice. A: mean circumferential stretch ratio was significantly larger in the Lox−/− AA and DA compared with the Lox+/+ AA and DA (*P < 0.05). B: mean circumferential stress was significantly larger in the Lox−/− AA compared with the Lox+/+ AA at pressures between 15 and 60 mmHg (*P < 0.05). C: circumferential stress-stretch plots for the Lox−/− AA and DA were shifted to the right of the Lox+/+ AA and DA. D: the tangent modulus depended on genotype alone from 15 to 35 mmHg and on genotype × vascular location between 30 and 60 mmHg. The tangent modulus was significantly larger in the Lox−/− AA compared with the Lox+/+ AA from 15 to 60 mmHg (*P < 0.05). Genotype (&) and genotype × vascular location (%) accounted for significant variance between groups (P < 0.05). n = 5–8 per group.
Gene expression analyses of Lox−/− aortas.
Gene arrays and qPCR were used to analyze Lox family gene expression levels for each genotype and vascular location. The normalized expression of Lox in the Lox−/− AA and DA was 15% of Lox+/+ AA and DA values from the gene array (Fig. 7A) and 4–6% of Lox+/+ AA and DA values from the qPCR experiments (Fig. 7B). In the AA, there were no significant changes in Loxl1–4 in Lox−/− mice compared with Lox+/+ mice by gene array or qPCR. In the DA, normalized expression levels of Loxl1 in Lox−/− mice were increased in the gene array from 56% to 70% and in qPCR from 40% to 89%. Loxl2 and Loxl4 expressions were not changed in the Lox−/− DA compared with the Lox+/+ DA. Loxl3 expression in the Lox−/− DA was upregulated in the gene array, but these findings were not confirmed in the qPCR analysis. Note that expression levels of Loxl2–4 were low compared with Lox and near the minimum detection limit.
Fig. 7.

Normalized gene expression profiles of Lox and Loxl1–4 in newborn Lox aortas. Normalized gene expression levels of Lox from the Lox+/+ AA and DA from the microarray results (A) or quantitative (q)PCR (B) were set to 100%, and expression of all other Lox family genes was scaled to these values. *P < 0.05. n = 2 per group of 8 pooled samples for gene array, and n = 3 per group of 8 pooled samples for qPCR.
Normalized gene expression, grouped by genotype regardless of vascular location or grouped by genotype at each vascular location, was used with GSEA software to determine differential expression levels of matrisome and SMC phenotype genes. For the matrisome, six gene subsets (ECM glycoproteins, proteoglycans, collagens, ECM regulators, secreted factors, and ECM-affiliated proteins) were downloaded from http://matrisomeproject.mit.edu (34). For the SMC phenotype, one gene subset called “Kegg VSMC Contraction” (where VSMC is vascular SMC) (SMC contractility) was downloaded from http://software.broadinstitute.org/. Two additional SMC phenotype subsets were defined from Larsson et al. (25) to describe gene expression patterns associated with SMCs in a synthetic phase and producing ECM proteins (SMC matrix) or in a proliferative phase and actively multiplying (SMC cell cycle). Lox deletion, regardless of vascular location, results in increased expression levels of genes related to ECM maintenance and remodeling (ECM regulators), secreted factors with modulatory activity (secreted factors), ECM structural components (ECM glycoproteins and collagens), and cell proliferation (SMC cell cycle; Table 1). The leading-edge genes contributing to the significant enrichment of these subsets are listed in Supplemental Tables S1–S5 in the Supplemental Material (Supplemental Material for this article is available at the American Journal of Physiology-Heart and Circulatory Physiology website).
Table 1.
Summary of gene set enrichment analysis for Lox−/− aortas compared with Lox+/+ aortas, regardless of vascular location and separately for the AA and DA
|
Lox−/− vs. Lox+/+ |
Lox−/− vs. Lox+/+ AA |
Lox−/− vs. Lox+/+ DA |
|||||
|---|---|---|---|---|---|---|---|
| Gene Subset | Size | NES | P value | NES | P value | NES | P value |
| ECM regulators | 299 | 2.16 | <0.0001* | 1.87 | <0.0001* | 1.85 | <0.0001* |
| Secreted factors | 348 | 1.69 | <0.0001* | 2.01 | <0.0001* | 1.06 | 0.294 |
| ECM glycoproteins | 193 | 1.61 | <0.0001* | 1.22 | 0.063 | 1.49 | 0.002* |
| SMC cell cycle | 15 | 1.71 | 0.012* | 1.22 | 0.202 | 1.66 | 0.017* |
| Collagens | 43 | 1.46 | 0.041* | 1.59 | 0.011* | 1.07 | 0.332 |
| SMC matrix | 36 | 1.16 | 0.24 | 1.25 | 0.137 | 0.89 | 0.701 |
| ECM-affiliated proteins | 160 | 1.11 | 0.210 | 1.36 | 0.017* | −1.35 | 0.03* |
| Proteoglycans | 36 | 1.08 | 0.342 | 1.02 | 0.401 | 0.93 | 0.555 |
| SMC contractility | 102 | −0.77 | 0.936 | −0.61 | 0.996 | −0.65 | 0.993 |
The name of the gene subset, number of genes in the subset (Size), normalized enrichment score (NES), and nominal P values are shown. The gene subsets are ordered from highest to lowest NES values, based on the lysyl oxidase (Lox)−/− versus Lox+/+ results regardless of vascular location. Positive NES indicates upregulation of the gene set, whereas negative NES indicates downregulation of the gene set in Lox−/− compared with Lox+/+ mice. Extracellular matrix (ECM) regulators were significantly upregulated at both locations. Secreted factors, collagens, and ECM-affiliated proteins genes were significantly upregulated only in the Lox−/− ascending aorta (AA) compared with the Lox+/+ AA. ECM glycoproteins and smooth muscle cell (SMC) cell cycle were significantly upregulated, and ECM-affiliated proteins were significantly downregulated only in the Lox−/− descending aorta (DA) compared with the Lox+/+ DA. Leading-edge genes for the significantly enriched subsets are listed in Supplemental Tables S1−S13.
P < 0.05.
Separating the array data by vascular location, to compare Lox−/− AAs vs. Lox+/+ AAs or Lox−/− DAs vs. Lox+/+ DAs, revealed the existence of locational differences in gene expression (Table 1). The absence of Lox in AAs led to a significant upregulation of the gene subsets of ECM regulators, secreted factors, collagens, and ECM-affiliated proteins. The absence of Lox in DAs resulted in an upregulation of the gene subsets of ECM regulators, ECM glycoproteins, and SMC cell cycle and downregulation of ECM-affiliated proteins. The leading-edge genes contributing to the significant enrichment of these subsets are listed in Supplemental Tables S6–S13.
The only gene subset that was consistently upregulated in Lox−/− mice in the combined comparison and in the AA and DA comparisons was ECM regulators. All other gene subsets were differentially regulated by the absence of Lox in a vascular location-specific manner. Changes in opposite directions for the AA and DA for the ECM-affiliated proteins subset led to a cancellation of their effects in the combined analysis. Our results show that the aortic chemomechanical environment is altered through differential expression of matrisome and SMC genes in response to the absence of Lox, combined with location-specific changes associated with the AA or DA.
DISCUSSION
Elastin and collagen fibers in the aortic wall provide elasticity and strength as well as a suitable microenvironment for SMCs and a repository for growth factors (45). Modifications to ECM proteins affect mechanical properties of the wall, SMC phenotype, and growth factor signaling. Genetic mutations that alter this chemomechanical environment are linked to TAAD in humans (22). Mouse models of TAAD have been instrumental in identifying mechanisms for possible therapeutic intervention. We used newborn Lox−/− mice as a model of TAAD to investigate the morphological, structural, mechanical, and genetic changes associated with severe reductions in elastin and collagen cross-linking in the ascending and descending thoracic aorta. Consistent with previous results (19, 31), we found that Lox−/− mice have aortic aneurysms, aortic tortuosity, and fragmented elastic fibers. Adding to previous morphological and structural results, we show that Lox−/− mice have patent ductus arteriosus, aneurysms at consistent locations along the thoracic aorta, increased aortic length, and disorganized cells in the aortic wall.
Lox−/− mice have increased AA length and DA tortuosity. Newborn fibulin-4 (Fbln4)−/− (23) and elastin knockout (Eln−/−) mice (43) as well as adult mice lacking fibulin-5 (Fbln5−/−) and with SMC-specific deletion of Fbln4 (Fbln4SMKO) (20) also have aortic tortuosity, indicating that mechanically functional elastic fibers are necessary for longitudinal aortic stability. A computational model of stress-mediated aortic growth during development suggests that if elastin is never produced, the aorta will lengthen without bounds (1). Cells in the Lox−/− aorta are more disorganized than those in the Lox+/+ aorta, as indicated by their nuclei, which are not preferentially aligned circumferentially between the elastic laminae. This change in nuclear alignment may be due to growth and proliferation of Lox−/− SMCs in the longitudinal direction.
In this study, we analyzed the mechanical behavior of the Lox−/− aorta in vitro. Quantification of the circumferential structural and material properties of the Lox−/− aorta is critical for understanding the contribution of elastin and collagen cross-links to cardiovascular function and to the mechanical environment of SMCs in the aortic wall. The structural properties of the Lox−/− aorta change in a vascular location-specific manner. The Lox−/− AA has larger outer diameters and reduced compliance values compared with the Lox+/+ AA when pressurized, whereas the Lox−/− DA and Lox+/+ DA have similar diameter and compliance values. The protection of the Lox−/− DA from structural property changes may be related to the increased expression of Loxl1, SMC cell cycle, and ECM glycoproteins observed in this vascular segment. Because of the structural property changes, the Lox−/− AA will be ~25% larger and less compliant than the Lox+/+ AA in vivo at a systolic blood pressure of 23 mmHg in newborn mice (21, 43). The changes in size and compliance of the AA will affect the local chemomechanical environment of SMCs in the wall as well as global cardiac output, pulse propagation, and wave reflection, leading to immediate and long-term changes in cardiovascular mechanics.
In contrast to the unique structural properties of the Lox−/− AA, both the Lox−/− AA and DA showed altered material properties compared with the Lox+/+ AA and DA, as evidenced by the circumferential stretch-stress behavior. The shift in material properties is consistent with previous results in newborn Fbln4−/− (23) and Eln−/− (43) aortas and with the loss of elastin contribution at low stretch (45). The residual circumferential strain, as measured by the opening angle, is decreased in Lox−/− compared with Lox+/+ aortic segments. We also found decreased opening angles in newborn Fbln4−/− (23) and Eln−/− (43) aortas. Residual strain normalizes the strain and stress distribution across the aortic wall, so that SMCs at the inner and outer boundaries are under approximately the same loaded conditions (7). Reduced opening angle would lead to higher strains at the inner wall compared with the outer wall and may contribute to the disorganization of cell nuclei observed in the Lox−/− aorta. Our results show that cross-linked elastic fibers are important for the development and maintenance of the material properties and residual stresses that contribute to mechanical homeostasis of the aortic wall (22).
Lox−/− mice have phenotypic similarities to Fbln4−/− mice. FBLN4 binds to the propeptide of LOX, and the direct interaction of these two proteins is important for elastic fiber assembly (18). Loss of FBLN4 decreases LOX activity and leads to alterations in collagen localization, organization, and cross-linking, suggesting that FBLN4 and LOX interactions are also important for collagen fiber maturation (37). In comparing the mechanical changes in newborn Fbln4−/− (23) and Lox−/− aortic segments, we found key similarities and differences. The Fbln4−/− AA is larger than the wild-type AA in the unloaded state, whereas the Lox−/− AA is not. Fbln4−/− mice have a 94% reduction in desmosine elastin cross-links in the aorta (33) compared with a 60% reduction in desmosine for the Lox−/− aorta (19); hence, the AA dilation in the unloaded state may be related to the almost complete absence of elastin cross-linking in the Fbln4−/− aorta. Despite the large differences in elastin cross-linking, we found similar shifts in the structural and material properties for Fbln4−/− and Lox−/− AAs and DAs compared with their respective wild-type controls, indicating that FBLN4 and LOX may work together in elastin and collagen fiber assembly and cross-linking.
Comparing expression profiles of matrisome genes in Lox−/− and Lox+/+ aortas, we identified a matrix signature induced by Lox deletion and the reduction of elastin and collagen cross-linking. On the basis of leading-edge analyses, we found that 27% of the ECM regulators gene subset were enriched in Lox−/− compared with Lox+/+ aortas, regardless of vascular location. In the list of leading-edge genes, we found matrix metallopeptidases (i.e., Mmps, Adamts, and Adams) and their regulators (i.e., tissue inhibitors of metalloproteinases and serpins), suggesting that a positive feedback loop is initiated by the absence of Lox that encourages ECM remodeling (Supplemental Table S1). Mmp8, a neutrophil collagenase that degrades collagen types I, II, and III, showed the highest upregulation of all Mmp family members and is also upregulated in the Fbln4−/− aorta (23). The ECM regulators gene set includes members of the Lox family, but only Loxl3 appeared in the leading-edge gene list. Examination of expression values for individual family members by gene array and qPCR confirmed that Lox had the highest expression in the newborn aorta, followed by Loxl1 at ~50% of wild-type Lox levels, with little comparative expression of Loxl2–4. Loxl1 was unchanged in the Lox−/− AA but was upregulated in the Lox−/− DA, which may explain the lack of structural mechanical changes in the Lox−/− DA. Similar expression patterns in the newborn aorta for Lox family members and a lack of compensation for inactive Lox were also reported by Lee et al. (27).
Besides ECM regulators, all other matrisome and SMC gene sets had changes in expression that were specific to vascular location. Among other characteristics, the AA and DA have different geometries, flow profiles (4), and SMC embryonic origins (neural crest for the AA and somatic mesoderm for the DA) (30) that may be responsible for differential responses to the absence of Lox. In the AA, 25% of the secreted factors and 16% of the collagens gene subsets were enriched in Lox−/− compared with Lox+/+ mice. The leading-edge genes in the secreted factors subset were proinflammatory factors such as Il6 and chemokine (C-C motif) ligand 2 (Ccl2), which have been linked to aortic dissection in mice (Supplemental Table S7) (42). The leading-edge genes in the collagen subset for the Lox−/− AA versus Lox+/+ AA did not encode the major collagens in the aortic wall (collagen types I or III) but nonfibrillar collagens (Col8a1 and Col8a2) and minor fibrillar collagens (Col11a1; Supplemental Table S8). Col8a1 is also upregulated in the Fbln4−/− aorta (23). The results demonstrate important roles for inflammation and nontraditional collagens in AA remodeling and SMC response to an altered local chemomechanical environment.
In the DA, 17% of ECM glycoproteins and 80% of SMC cell cycle gene subsets were enriched in Lox−/− compared with Lox+/+ mice. Elastin is not one of the genes in the leading edge that contributed to differential regulation of the glycoprotein subset, implying that elastin cross-linking is not linked to expression. Genes in the leading edge of the glycoprotein subset for the Lox−/− DA versus Lox+/+ DA included those related to thrombus formation (i.e., Fgg and Fgb), which are likely caused by the observed aortic dissections, and matricellular proteins [i.e., thrombospondin (Thbs)1 and Thbs4], which play important roles in tissue repair (Supplemental Table S11). Genes in the leading edge of the SMC cell cycle subset included Cdc25a, hyaluronan mediated motility receptor (Hmmr), and hepatoma-derived growth factor (Hdgf), which have been implicated in SMC proliferation and migration (Supplemental Table S12) (6, 15, 36). The increases in glycoprotein and SMC cell cycle gene expression may help protect the Lox−/− DA from the structural mechanical changes observed in the Lox−/− AA.
The ECM-affiliated proteins subset was upregulated in the Lox−/− AA compared with the Lox+/+ AA and downregulated in the Lox−/− DA compared with the Lox+/+ DA. Genes that contribute to differential expression of the ECM-affiliated proteins subset include lectins (i.e., Clec1b and Clec4b1), which may modulate hemostasis (3), and mucins (i.e., Muc19) and surfactant-associated proteins (i.e., Sftpa1), which may be related to the pulmonary defects and diaphragmatic hernias observed in Lox−/− mice (Supplemental Tables S9 and S13) (19). Expression of syndecans (i.e., Sdc3 and Sdc1), which play a role in cell adhesion and ECM assembly (47), also contributed to the overexpression of ECM-affiliated proteins in the Lox−/− AA compared with the Lox+/+ AA. The GSEA results highlight the importance of locational differences in gene expression resulting from the interplay between the absence of Lox and specific AA or DA characteristics.
As SMC contractile genes have been implicated in TAAD (17, 46, 48) and we observed changes in cellular morphology in the Lox−/− aorta, we expected to see changes in genes related to the SMC phenotype in the Lox−/− aorta compared with the Lox+/+ aorta. However, our results only showed upregulation of the SMC cell cycle subset in the Lox−/− DA compared with the Lox+/+ DA, emphasizing the role of vascular location in determining the SMC phenotype. Genes in subsets defining SMC matrix phenotype (25) or SMC contractility were not upregulated because of the absence of Lox. These results indicate that it is important to look at several different SMC phenotype markers.
In conclusion, we investigated the mechanical alterations and changes in matrisome and SMC phenotype gene expression in the thoracic aorta of newborn Lox−/− mice. The absence of Lox results in TAAD characterized by fragmented elastic fibers and disorganized cells. Although the aneurysm phenotype in the Lox−/− aorta was of variable penetrance by gross examination, the Lox−/− AA consistently showed larger diameters than the Lox+/+ AA when pressurized, demonstrating the need to evaluate cardiovascular phenotypes under physiological loads. The Lox−/− AA and DA have altered circumferential stress and stretch ratios compared with the Lox+/+ AA and DA, confirming the role of elastin and collagen cross-linking in determining the material behavior of the aortic wall. Our gene expression analysis indicates that TAAD in the Lox−/− aorta arises from an altered chemomechanical environment characterized by changes in signaling of ECM regulators and not from changes in the expression of major ECM structural components. The GSEA results also highlight the locational dependence of the gene expression changes. We summarized the morphological, mechanical, and gene expression changes of the Lox−/− AA and DA in Fig. 8.
Fig. 8.

Summary of morphological, mechanical, and gene expression data in Lox−/− AAs and DAs. All changes are described with respect to the Lox+/+ AA and DA. Changes that were common to both the AA and DA are shown in the overlapping region. circ, Circumferential.
Limitations and future directions.
For this study, we quantified only the circumferential mechanical behavior of the aortic segments, although the longitudinal mechanical behavior and the interaction between the two directions are also important (5). The small size of the newborn mouse aorta and incomplete cleaning of surrounding tissue may affect the structural behavior and calculations of the material behavior. We pooled multiple samples for each group for the microarray analysis, thus reducing the statistical power of our results. Microarray data are only indicative of the expression level of the genes, and additional studies are necessary to determine how altered gene expression is translated into changes in proteins in the aortic wall. GSEA is sensitive to the size and comprehensive nature of the included gene subsets. The matrisome gene subsets have been confirmed by proteomics studies. The gene subsets defining SMC phenotypes were chosen from published literature and likely represent polarized states of the synthetic, proliferative, or contractile phenotype. The genetic signatures identified in this study are associated with the pathogenic and mechanical changes encountered in TAAD. However, our study cannot determine whether the enriched genes are necessary and/or sufficient for TAAD. Future studies are needed that concentrate on pharmacological or genetic modulation of pathways related to the enriched gene subsets.
GRANTS
This work was partially funded by National Institutes of Health Grants HL-115560 (to J. E. Wagenseil), HL-053325 (to R. P. Mecham), HL-105314 (to R. P. Mecham and J. E. Wagenseil), and S10-RR-026949 (to the Center for Cardiovascular Research) and Washington University Institute of Clinical and Translational Sciences Grant UL1-TR-000448.
DISCLAIMERS
This publication is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.E.W. conceived and designed research; M.C.S. and J.K. performed experiments; M.C.S. and J.K. analyzed data; M.C.S., J.K., R.P.M., and J.E.W. interpreted results of experiments; M.C.S. and J.E.W. prepared figures; M.C.S., J.K., and J.E.W. drafted manuscript; M.C.S., J.K., R.P.M., and J.E.W. edited and revised manuscript; M.C.S., J.K., R.P.M., and J.E.W. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Genome Technology Access Center at the Washington University School of Medicine for help with the genomic analysis. We thank Carla Weinheimer and Attila Kovacs of the Center for Cardiovascular Research at the Washington University School of Medicine for help with the blood pressure experiments.
REFERENCES
- 1.Alford PW, Taber LA. Computational study of growth and remodelling in the aortic arch. Comput Methods Biomech Biomed Engin 11: 525–538, 2008. doi: 10.1080/10255840801930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amin M, Kunkel AG, Le VP, Wagenseil JE. Effect of storage duration on the mechanical behavior of mouse carotid artery. J Biomech Eng 133: 071007, 2011. doi: 10.1115/1.4004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, Vögtle T, Remer K, Braun A, Bösl M, Watson SP, Nieswandt B. Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol 33: 926–934, 2013. doi: 10.1161/ATVBAHA.112.300672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benim AC, Nahavandi A, Assmann A, Schubert D, Feindt P, Suh SH. Simulation of blood flow in human aorta with emphasis on outlet boundary conditions. Appl Math Model 35: 3175–3188, 2011. doi: 10.1016/j.apm.2010.12.022. [DOI] [Google Scholar]
- 5.Cardamone L, Valentín A, Eberth JF, Humphrey JD. Origin of axial prestretch and residual stress in arteries. Biomech Model Mechanobiol 8: 431–446, 2009. doi: 10.1007/s10237-008-0146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen S, Law CS, Grigsby CL, Olsen K, Gardner DG. A role for the cell cycle phosphatase Cdc25a in vitamin D-dependent inhibition of adult rat vascular smooth muscle cell proliferation. J Steroid Biochem Mol Biol 122: 326–332, 2010. doi: 10.1016/j.jsbmb.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chuong CJ, Fung YC. On residual stresses in arteries. J Biomech Eng 108: 189–192, 1986. doi: 10.1115/1.3138600. [DOI] [PubMed] [Google Scholar]
- 8.Clifford PS, Ella SR, Stupica AJ, Nourian Z, Li M, Martinez-Lemus LA, Dora KA, Yang Y, Davis MJ, Pohl U, Meininger GA, Hill MA. Spatial distribution and mechanical function of elastin in resistance arteries: a role in bearing longitudinal stress. Arterioscler Thromb Vasc Biol 31: 2889–2896, 2011. doi: 10.1161/ATVBAHA.111.236570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui JZ, Tehrani AY, Jett KA, Bernatchez P, van Breemen C, Esfandiarei M. Quantification of aortic and cutaneous elastin and collagen morphology in Marfan syndrome by multiphoton microscopy. J Struct Biol 187: 242–253, 2014. doi: 10.1016/j.jsb.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Dobrin PB, Schwarcz TH, Baker WH. Mechanisms of arterial and aneurysmal tortuosity. Surgery 104: 568–571, 1988. [PubMed] [Google Scholar]
- 11.Ertugrul A. Diffuse tortuosity and lengthening of the arteries. Circulation 36: 400–407, 1967. doi: 10.1161/01.CIR.36.3.400. [DOI] [PubMed] [Google Scholar]
- 12.Fonck E, Prod’hom G, Roy S, Augsburger L, Rüfenacht DA, Stergiopulos N. Effect of elastin degradation on carotid wall mechanics as assessed by a constituent-based biomechanical model. Am J Physiol Heart Circ Physiol 292: H2754–H2763, 2007. doi: 10.1152/ajpheart.01108.2006. [DOI] [PubMed] [Google Scholar]
- 13.Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, Judge DP, Cooke SK, Lindsay ME, Rouf R, Myers L, ap Rhys CM, Kent KC, Norris RA, Huso DL, Dietz HC. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest 124: 448–460, 2014. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-β signaling and vascular smooth muscle cell contractility. Circ Res 113: 327–340, 2013. doi: 10.1161/CIRCRESAHA.113.300675. [DOI] [PubMed] [Google Scholar]
- 15.Gouëffic Y, Guilluy C, Guérin P, Patra P, Pacaud P, Loirand G. Hyaluronan induces vascular smooth muscle cell migration through RHAMM-mediated PI3K-dependent Rac activation. Cardiovasc Res 72: 339–348, 2006. doi: 10.1016/j.cardiores.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 16.Guo DC, Regalado ES, Gong L, Duan X, Santos-Cortez RLP, Arnaud P, Ren Z, Cai B, Hostetler EM, Moran R, Liang D, Estrera A, Safi HJ, Leal SM, Bamshad MJ, Shendure J, Nickerson DA, Jondeau G, Boileau C, Milewicz DM; University of Washington Center for Mendelian Genomics . LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ Res 118: 928–934, 2016. doi: 10.1161/CIRCRESAHA.115.307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39: 1488–1493, 2007. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 18.Horiguchi M, Inoue T, Ohbayashi T, Hirai M, Noda K, Marmorstein LY, Yabe D, Takagi K, Akama TO, Kita T, Kimura T, Nakamura T. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci USA 106: 19029–19034, 2009. doi: 10.1073/pnas.0908268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hornstra IK, Birge S, Starcher B, Bailey AJ, Mecham RP, Shapiro SD. Lysyl oxidase is required for vascular and diaphragmatic development in mice. J Biol Chem 278: 14387–14393, 2003. doi: 10.1074/jbc.M210144200. [DOI] [PubMed] [Google Scholar]
- 20.Huang J, Davis EC, Chapman SL, Budatha M, Marmorstein LY, Word RA, Yanagisawa H. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ Res 106: 583–592, 2010. doi: 10.1161/CIRCRESAHA.109.207852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, Guo X, Kassab GS. Axial nonuniformity of geometric and mechanical properties of mouse aorta is increased during postnatal growth. Am J Physiol Heart Circ Physiol 290: H657–H664, 2006. doi: 10.1152/ajpheart.00803.2005. [DOI] [PubMed] [Google Scholar]
- 22.Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science 344: 477–479, 2014. doi: 10.1126/science.1253026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Procknow JD, Yanagisawa H, Wagenseil JE. Differences in genetic signaling, and not mechanical properties of the wall, are linked to ascending aortic aneurysms in fibulin-4 knockout mice. Am J Physiol Heart Circ Physiol 309: H103–H113, 2015. doi: 10.1152/ajpheart.00178.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krahn KN, Bouten CV, van Tuijl S, van Zandvoort MA, Merkx M. Fluorescently labeled collagen binding proteins allow specific visualization of collagen in tissues and live cell culture. Anal Biochem 350: 177–185, 2006. doi: 10.1016/j.ab.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Larsson E, McLean SE, Mecham RP, Lindahl P, Nelander S. Do two mutually exclusive gene modules define the phenotypic diversity of mammalian smooth muscle? Mol Genet Genomics 280: 127–137, 2008. doi: 10.1007/s00438-008-0349-y. [DOI] [PubMed] [Google Scholar]
- 26.Le VP, Cheng JK, Kim J, Staiculescu MC, Ficker SW, Sheth SC, Bhayani SA, Mecham RP, Yanagisawa H, Wagenseil JE. Mechanical factors direct mouse aortic remodelling during early maturation. J R Soc Interface 12: 20141350, 2015. doi: 10.1098/rsif.2014.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee VS, Halabi CM, Hoffman EP, Carmichael N, Leshchiner I, Lian CG, Bierhals AJ, Vuzman D, Mecham RP, Frank NY, Stitziel NO; Brigham Genomic Medicine . Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci USA 113: 8759–8764, 2016. doi: 10.1073/pnas.1601442113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindsay ME, Dietz HC. The genetic basis of aortic aneurysm. Cold Spring Harb Perspect Med 4: a015909, 2014. doi: 10.1101/cshperspect.a015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci 63: 2304–2316, 2006. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol 27: 1248–1258, 2007. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 31.Mäki JM, Räsänen J, Tikkanen H, Sormunen R, Mäkikallio K, Kivirikko KI, Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation 106: 2503–2509, 2002. doi: 10.1161/01.CIR.0000038109.84500.1E. [DOI] [PubMed] [Google Scholar]
- 32.Mäki JM, Sormunen R, Lippo S, Kaarteenaho-Wiik R, Soininen R, Myllyharju J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am J Pathol 167: 927–936, 2005. doi: 10.1016/S0002-9440(10)61183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaughlin PJ, Chen Q, Horiguchi M, Starcher BC, Stanton JB, Broekelmann TJ, Marmorstein AD, McKay B, Mecham R, Nakamura T, Marmorstein LY. Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol Cell Biol 26: 1700–1709, 2006. doi: 10.1128/MCB.26.5.1700-1709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol 49: 10–24, 2016. doi: 10.1016/j.matbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 3: e01308, 2014. doi: 10.7554/eLife.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narron JV, Stoops TD, Barringhaus K, Matsumura M, Everett AD. Hepatoma-derived growth factor is expressed after vascular injury in the rat and stimulates smooth muscle cell migration. Pediatr Res 59: 778–783, 2006. doi: 10.1203/01.pdr.0000219299.24435.4f. [DOI] [PubMed] [Google Scholar]
- 37.Papke CL, Tsunezumi J, Ringuette LJ, Nagaoka H, Terajima M, Yamashiro Y, Urquhart G, Yamauchi M, Davis EC, Yanagisawa H. Loss of fibulin-4 disrupts collagen synthesis and maturation: implications for pathology resulting from EFEMP2 mutations. Hum Mol Genet 24: 5867–5879, 2015. doi: 10.1093/hmg/ddv308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodríguez C, Martínez-González J, Raposo B, Alcudia JF, Guadall A, Badimon L. Regulation of lysyl oxidase in vascular cells: lysyl oxidase as a new player in cardiovascular diseases. Cardiovasc Res 79: 7–13, 2008. doi: 10.1093/cvr/cvn102. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol 2: a005066, 2010. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen Z, Lu Z, Chhatbar PY, O’Herron P, Kara P. An artery-specific fluorescent dye for studying neurovascular coupling. Nat Methods 9: 273–276, 2012. doi: 10.1038/nmeth.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A III, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest 119: 3637–3651, 2009. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. Reduced vessel elasticity alters cardiovascular structure and function in newborn mice. Circ Res 104: 1217–1224, 2009. doi: 10.1161/CIRCRESAHA.108.192054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today 81: 229–240, 2007. doi: 10.1002/bdrc.20111. [DOI] [PubMed] [Google Scholar]
- 45.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev 89: 957–989, 2009. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 87: 701–707, 2010. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xian X, Gopal S, Couchman JR. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res 339: 31–46, 2010. doi: 10.1007/s00441-009-0829-3. [DOI] [PubMed] [Google Scholar]
- 48.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 38: 343–349, 2006. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.