

This study establishes that pharmacological modification of hemoglobin O2 affinity can be a promising and novel therapeutic strategy for the treatment of hypoxic hypoxia and paves the way for the clinical development of molecules that prevent hypoxemia.
Keywords: oxygen, hemoglobin, hypoxia, microcirculation, tissue partial pressure of oxygen, oxygen delivery, partial pressure of oxygen at which hemoglobin is 50% saturated with oxygen
Abstract
Adaptation to hypoxia requires compensatory mechanisms that affect O2 transport and utilization. Decreased hemoglobin (Hb) O2 affinity is considered part of the physiological adaptive process to chronic hypoxia. However, this study explores the hypothesis that increased Hb O2 affinity can complement acute physiological responses to hypoxia by increasing O2 uptake and delivery compared with normal Hb O2 affinity during acute severe hypoxia. To test this hypothesis, Hb O2 affinity in mice was increased by oral administration of 2-hydroxy-6-{[(2S)-1-(pyridine-3-carbonyl)piperidin-2yl] methoxy}benzaldehyde (GBT1118; 70 or 140 mg/kg). Systemic and microcirculatory hemodynamics and oxygenation parameters were studied during hypoxia in awake-instrumented mice. GBT1118 increased Hb O2 affinity and decreased the Po2 at which 50% of Hb is saturated with O2 (P50) from 43 ± 1.1 to 18.3 ± 0.9 mmHg (70 mg/kg) and 7.7 ± 0.2 mmHg (140 mg/kg). In a dose-dependent fashion, GBT1118 increased arterial O2 saturation by 16% (70 mg/kg) and 40% (140 mg/kg) relative to the control group during 5% O2 hypoxia. In addition, a GBT1118-induced increase in Hb O2 affinity reduced hypoxia-induced hypotension compared with the control group. Moreover, microvascular blood flow was higher during hypoxia in GBT1118-treated groups than the control group. The increased O2 saturation and improved blood flow in GBT1118-treated groups preserved higher interstitial tissue Po2 than in the control group during 5% O2 hypoxia. In conclusion, increased Hb O2 affinity enhanced physiological tolerance to hypoxia, as evidenced by improved hemodynamics and tissue oxygenation. Therefore, pharmacologically induced increases in Hb O2 affinity become a potential therapeutic approach to improve tissue oxygenation in pulmonary diseases characterized by severe hypoxemia.
NEW & NOTEWORTHY This study establishes that pharmacological modification of hemoglobin O2 affinity can be a promising and novel therapeutic strategy for the treatment of hypoxic hypoxia and paves the way for the clinical development of molecules that prevent hypoxemia.
birds and mammals that live at high altitudes adapt to environmental hypoxia, in part by genetic changes that increase hemoglobin (Hb) affinity for O2, creating “high-affinity Hb.” This high-affinity Hb increases O2 uptake by Hb in the lungs and thus increases arterial O2 saturation () during environmental hypoxia (2, 31). In humans, adaptation to hypoxia includes a decrease in Hb O2 affinity, which is typically mediated by an increase in red blood cell (RBC) 2,3-diphosphoglycerate concentration, a decrease in the pH of blood (Bohr effect), and an increase in CO2 (Haldane effect) (21, 23). Under normoxic conditions, where O2 uptake in the lungs is not limited, decreased Hb O2 affinity can be beneficial, as it increases O2 off loading to tissues (7). However, during hypoxic hypoxia, a decrease in Hb O2 affinity may be maladaptive, since it can lead to decreased O2 uptake in the lungs, decreased , and hypoxemia. The result is impaired exercise performance, acute mountain sickness, high-altitude cerebral edema, high-altitude pulmonary edema, or death (9). Nearly 25% of people who travel above an elevation of 2,590 m (8,500 feet) develop manifestations of high-altitude illness, as their acute physiological adaptive response to hypoxia fails to maintain O2 delivery and extraction (24).
In mammals and other vertebrates, O2 transport is optimized by tight regulation of ventilation and Hb mass. Early responses to hypoxia include neuronal stimulation of respiration by the carotid bodies and rate-limiting processes in the synthesis of the responsible neurotransmitter (19), whereas late responses to hypoxia stimulate erythropoiesis (34). Hypoxia elicits equally important physiological responses at the tissue level. Hypoxemic lung diseases, resulting from ventilation-perfusion mismatch or O2 diffusion limitation, decrease O2 uptake and delivery to tissue (11, 25). With decreased O2 delivery to tissue, tissue metabolism may be anaerobic, leading to reduced cellular function, lactic acidosis, and, subsequently, cell death (25). When O2 loading of Hb is compromised in the lungs, an increase in Hb O2 affinity may be beneficial by improving the loading of Hb with O2 and, consequently, increasing arterial O2 content and, potentially, O2 delivery to tissues. Aromatic aldehydes, such as agents being developed for treatment of sickle cell disease, such as 5-hydroxymethylfurfural (5-HMF) (1) and GBT440, are natural allosteric modulators of Hb O2 affinity. While 5-HMF and GBT440 increase Hb O2 affinity and form a stable imine intermediate with intracellular Hb, GBT440 has been shown to improve pharmacokinetic (PK) and pharmacodynamic (PD) properties compared with 5-HMF (1, 28).
This study investigated the hypothesis that increasing Hb O2 affinity during acute severe hypoxia in mice by oral administration of 2-hydroxy-6-{[(2S)-1-(pyridine-3-carbonyl)piperidin-2yl]methoxy} benzaldehyde (GBT1118; a structural analog of GBT440) is protective, in that this agent preserves O2 delivery to tissues. GBT1118 binds covalently and reversibly via an imine intermediate to the NH2-terminal valine of the Hb α-chain and allosterically increases intracellular Hb affinity for O2. To test our hypothesis, GBT1118 was administered orally, and mice were subjected to sequential hypoxic challenges: mild hypoxia (normobaric 15% inspired O2), moderate hypoxia (normobaric 10% inspired O2), and severe hypoxia (normobaric 5% inspired O2). Systemic and microvascular hemodynamics, blood gases, and interstitial tissue Po2 levels were studied during hypoxia.
MATERIALS AND METHODS
PK and PD Experiments with GBT1118
All PK and PD experiments were conducted by Portola Pharmaceuticals (South San Francisco, CA). Protocols were approved by the Portola Pharmaceuticals Institutional Animal Care and Use Committee, an Association for Assessment and Accreditation of Laboratory Animal Care-accredited institution, and conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). GBT1118 was synthesized by Global Blood Therapeutics. GBT1118 concentration in plasma and RBCs and Hb O2 affinity were used to study PK and PD, respectively. Briefly, precannulated (with an indwelling jugular vein cannula) 8- to 12-wk-old male mice (C57BL/6) were obtained from Charles River Laboratories (Hollister, CA) and acclimated for ≥1 day before GBT1118 administration.
Oral administration.
GBT1118 was formulated in dimethylacetamide-polyethylene glycol (PEG)-400 and 40% Cavitron at a ratio of 1:5:4 and administered via oral gavage (70, 100, or 140 mg/kg po) as a single dose of 5 ml/kg. PK and PD were studied at 0, 2, 4, 6, 8, and 24 h after oral administration.
Intravenous administration.
GBT1118 was formulated in sterilized saline at 10 mg/kg and administered as a single dose of 1 ml/kg. PK and PD were studied at 0, 10, and 30 min and 1, 2, 4, 6, and 8 h after intravenous administration.
PK and PD samples.
For each time point, a subgroup of mice was used. They were anesthetized with ketamine, xylazine, and acepromazine (40, 2.5, and 0.75 mg/kg, respectively) intraperitoneally at 0.1 ml/20 g body wt, exsanguinated via cardiac puncture, and euthanized by cervical dislocation. Three mice (n = 3) were sampled per time point.
PK Processing and Data Analysis
GBT1118 concentration in blood and plasma samples was analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Briefly, blood and plasma samples, standard samples, and quality control samples were prepared for LC-MS/MS. GBT1118 was separated on a Hypersil GOLD column (Thermo Fisher Scientific, Waltham, MA). The peak area of the m/z 341 → 203 product ion (GBT1118) was measured in positive ion mode. Blood and plasma concentrations of GBT1118 were analyzed by noncompartmental analysis using Phoenix WinNonlin software (Centara, Princeton, NJ) to obtain PK parameters. Hb occupancy, which represents the GBT1118-to-Hb concentration ratio, was calculated by dividing the GBT1118 concentration in blood by the Hb concentration in blood.
Blood O2 Equilibrium Curves
Blood O2 equilibrium curves (OECs) were measured using a HEMOX analyzer (TCS Scientific). The hematocrit (Hct) for each individual sample was determined before the OEC was measured. To measure changes in the O2 affinity, blood samples were diluted 100-fold in HEMOX buffer. In the HEMOX analyzer, samples were first saturated with compressed air (21% O2) and then deoxygenated with pure N2. Absorbance at isosbestic points for Hb (570 nm) and deoxy Hb (560 nm) were recorded as a function of sample Po2. During deoxygenation, Po2 and Hb O2 saturation (So2) were recorded to obtain an OEC. The Po2 at which Hb is 50% saturated with O2 (P50) was calculated using regression analysis with the HEMOX analyzer software. The percent change in P50 (%ΔP50) was calculated as follows: %ΔP50 = 100 × [(P50 of vehicle − P50 of dose)/P50 of dose].
Animal Preparation for Hypoxia Experiments
All protocols were approved by the Institutional Animal Care and Use Committee of the University of California-San Diego and conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). Male 8- to 10-wk-old C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were implanted with dorsal windows. The window chamber model is widely used for microvascular studies in the unanesthetized state; the complete surgical technique has been described in detail elsewhere (4). The dorsal window chamber model includes muscle and connective tissue and has stable O2 consumption. As such, changes in O2 supply determine the tissue O2 state (3). Briefly, animals were anesthetized using pentobarbital sodium (50 mg/kg ip). After hair removal, sutures were used to lift the dorsal skin away from the animal, and one frame of the chamber was positioned on the animal’s back. A chamber consisted of two identical titanium frames with a 12-mm-diameter circular window. The intact skin of the other side was exposed to the ambient environment. Animals were allowed 2 days for recovery. Finally, animals were anesthetized again, and an arterial catheter (PE-50) was implanted in the carotid artery. Catheters were tunneled under the skin, exteriorized at the dorsal side of the neck, and securely attached to the window frame.
Inclusion Criteria
Animals were suitable for the experiments if 1) mean arterial blood pressure (MAP) was >80 mmHg at baseline, 2) heart rate (HR) was >400 beats/min at baseline, and 3) systemic Hct was >45%.
Preparation of GBT1118
GBT1118 was provided by Global Blood Therapeutics and formulated in dimethylacetamide, PEG-400, and 40% Cavitron at a ratio of 1:5:4.
Acute Hypoxia Protocol
GBT1118 (70 or 140 mg/kg) or vehicle was administered orally, and animals were assigned to the following groups: GBT1118 (70 mg/kg), GBT1118 (140 mg/kg), and control (vehicle only). Animals were returned to their cages for 2 h. Conscious animals were placed in a restraining tube and secured to the microscope stage for observation. First, animals were ventilated with compressed room air [21% O2-79% N2 (Airgas, San Diego, CA)] and allowed 20 min to adjust to the experimental environment before measurements were taken. Animals were then ventilated with 15% O2-85% N2 (Airgas) to induce normobaric 15% O2 hypoxia and allowed 15 min to adjust to the new gas environment before measurements were taken. Subsequently, animals were subjected to moderate hypoxia by ventilation with 10% O2-90% N2 (Airgas) into the restraining tube and allowed 15 min to adjust to the new gas environment before measurements were taken. Finally, mice were subjected to severe hypoxia by ventilation with 5% O2-95% N2 into the restraining tube and allowed 15 min to adjust to the new gas environment before measurements were taken. At each time point, systemic parameters, blood gases, and microvascular hemodynamics were studied. Blood P50 was measured at the end of the experiment. Tolerance to hypoxia was determined in animals maintained for an additional 1.5 h at 5% O2 hypoxia and defined as the ability to maintain MAP at >60 mmHg. An animal was considered to fail tolerance to hypoxia if MAP decreased to <60 mmHg for a period of 5 min, and hypoxia was stopped, according to the tolerance-to-hypoxia criteria, to prevent animal stress or discomfort. All gases were warmed and humidified before they were introduced into the restraining tube. The experimental protocol is shown in Fig. 1A.
Fig. 1.
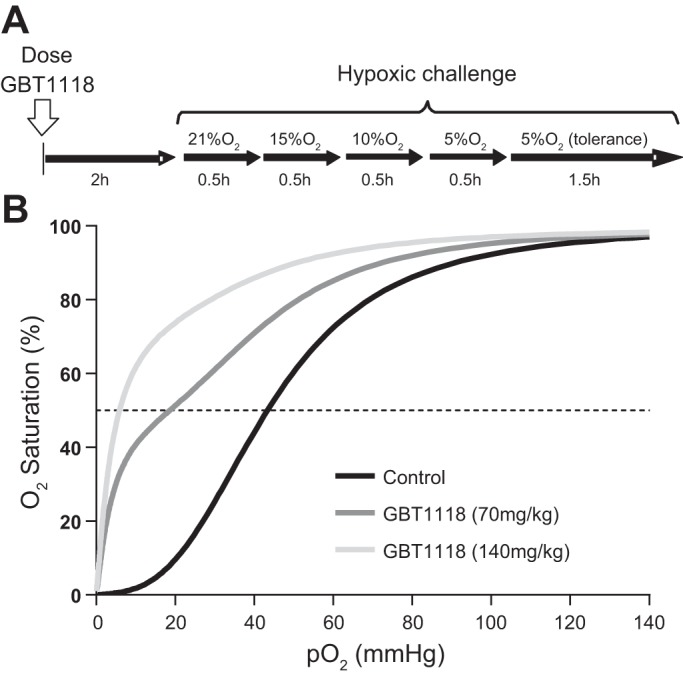
A: hypoxic challenge design. Mice were dosed orally with GBT1118 (70 or 140 mg/kg) or vehicle only. At 1 h after GBT1118 or vehicle administration, mice were exposed to hypoxia by stepwise decreases in O2 concentration to 15%, 10%, and 5%. Animals were kept at each hypoxic level for 0.5 h. To assess tolerance to hypoxia, mice were kept at 5% O2 hypoxia for an additional 1.5 h. B: changes in O2 affinity after GBT1118 administration, shown as representative O2 equilibrium curves (OECs) of blood (40% hematocrit) from vehicle- and GBT1118-treated mice (n = 6 animals/group). GBT1118 at 70 or 140 mg/kg decreased the Po2 at which Hb is 50% saturated with O2 (P50) of blood from 43 ± 1.1 to 18.3 ± 0.9 and 7.7 ± 0.2 mmHg, respectively. A leftward shift in the OEC, representing a decrease in P50, indicates a GBT1118 dose-dependent increase in Hb O2 affinity relative to vehicle-only control.
Hypoxia Experimental Groups
Animals were randomly assigned to three experimental groups: 1) GBT1118 (140 mg/kg), which received GBT1118 at 140 mg/kg po, 2) GBT1118 (70 mg/kg), which received GBT1118 at 70 mg/kg po, and 3) control, which received vehicle alone, orally. Changes in blood OECs with oral administration of GBT1118 are shown in Fig. 1B.
Systemic Parameters
MAP and HR were recorded continuously on a data acquisition and analysis system (model MP 150, Biopac Systems, Santa Barbara, CA). Respiratory rate was continuously acquired via the barometric method (4). Briefly, respiratory rate was calculated from the differential pressure (model TSD160A, Biopac Systems) based on inspiratory and expiratory changes in pressure inside the restraining tube relative to the atmospheric pressure. Respiratory rate was expressed as the number of breaths per minute, obtained from a 20-s pressure trace via a fast Fourier transform to obtain the power spectral density. Hct was measured from centrifuged arterial blood samples taken in heparinized capillary tubes. Hb content was determined spectrophotometrically (B-Hemoglobin, Hemocue, Stockholm, Sweden). Arterial blood gases [arterial Po2() and arterial Pco2 ()] and pH were measured using a blood gas analyzer (RAPIDLab 248, Bayer, Norwood, MA). Arterial Hb saturations were measured on a CO-oximeter system (model IL 482, Instrumentation Laboratory, Lexington, MA).
Microvascular Experimental Setup
The animal in the restraining tube with the protruding window chamber was fixed to the microscopic stage for transillumination with an intravital microscope (model BX51WI, Olympus, New Hyde Park, NY). The tissue image was projected onto a charge-coupled device camera (model 4815, Cohu Industries) connected to a videocassette recorder and viewed on a monitor. Measurements were carried out using a ×40 (LUMPFL-WIR, numerical aperture 0.8, Olympus) water-immersion objective. The same sites of study were followed throughout the experiment to enable direct comparisons with baseline levels.
Microhemodynamics
The video image-shearing method was used to measure vessel diameter (D) (13). Changes in arteriolar and venular diameter from baseline were used as indicators of a change in vascular tone. Arteriolar and venular centerline velocities were measured using the photodiode cross-correlation method (photo diode/velocity tracker, model 102B, Vista Electronics, San Diego, CA). The measured centerline velocity was corrected according to vessel size to obtain the mean RBC velocity (V) (12). Microvascular blood flow (Q̇), which corresponds to volumetric blood flow, was calculated from the measured values as follows: Q̇ = π × V(D/2)2. This calculation assumes a parabolic velocity profile and has been found to be applicable to 15- to 80-μm-internal diameter tubes and 6−60% Hct (12, 22). The same sites of study were followed throughout the experiment to enable direct comparisons with baseline levels.
Microvascular Po2
High-resolution noninvasive microvascular Po2 measurements were made using phosphorescence quenching microscopy (PQM) (15). PQM is based on the relationship between the decay rate of excited palladium-mesotetra-(4-carboxyphenyl)porphyrin (Frontier Scientific Porphyrin Products, Logan, UT) bound to albumin and O2 concentration, which is determined using the Stern-Volmer equation (32). This method has been used in microcirculatory studies to determine Po2 levels in different tissues (15). PQM measurements of Po2 were obtained for both groups as follows: 1) the probe with the phosphorescence complex (15 mg/kg iv at 10 mg/ml) was injected 10 min before O2 measurements, 2) the tissue was illuminated with a pulse of 420-nm-wavelength light to excite the probe to its triplet state, 3) the emitted phosphorescence (680-nm wavelength) was collected and analyzed to yield the phosphorescence lifetime, and 4) the phosphorescence lifetime was converted to O2 concentration (Po2). The phosphorescence lifetimes are concentration independent, which permits extravascular fluid Po2 measurements, although the amount of dye-albumin complex that extravasates is very small. Interstitial tissue Po2, which corresponds to extravascular fluid Po2, was measured in regions between functional capillaries.
Vital Organ Hypoxic Areas
Immunohistochemical staining of pimonidazole bound to hypoxic zones in vital tissues during hypoxia was accomplished by intraperitoneal injection of the hypoxic marker Hypoxyprobe-1 (40 mg/kg pimonidazole) and Hoechst 33342 (5 mg/kg, Invitrogen, Carlsbad, CA) diluted in PBS (total volume 100 μl). For mice exposed to 10% hypoxia, in vivo hypoxic staining was followed by a second intraperitoneal injection of Hypoxyprobe-1 and Hoechst 33342 during 5% hypoxia (33). After euthanasia, vital organs were removed and split sagittally and then transversely. Tissues were fixed by immersion in formalin for 24 h at room temperature before transfer to 70% (vol/vol) ethanol and then cut into 100-μm-thick sections.
Hypoxyprobe-1 Immunohistochemistry
Monoclonal antibody directed against pimonidazole (included in the Hypoxyprobe-1 green kit) was used for immunohistochemical staining of the tissue sections. Fluorescence microscopy was performed using a fixed-stage upright microscope (model BX51WI, Olympus) equipped with a digital camera (ORCA 285, Hamamatsu, Hamamatsu City, Japan) and the appropriate fluorescence filters (XF100-2 and XF02-2, Omega Optical, Brattleboro, VT). Images of areas positive for pimonidazole and Hoechst staining were collected using Wasabi imaging software (Hamamatsu). The ratio of pixels stained for pimonidazole in each region to the total cellular area of the image was calculated. Ten images were analyzed, by sections, and the results were pooled to determine the mean and SD. For visualization of pimonidazole and Hoechst colocalization, both images were superimposed (33).
Statistical Analysis
Values are means ± SD. Data are presented as absolute values and relative to baseline. A ratio of 1.0 signifies no change from baseline, whereas lower or higher ratios are indicative of changes proportionally lower or higher than baseline. Grubbs’ method was used to assess closeness for all measured parameters at baseline. Statistically significant changes between solutions and time points were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons test for post hoc analysis when appropriate. All statistics were calculated using Prism 6 (GraphPad, San Diego, CA). Results were considered statistically significant if P < 0.05.
Animal Experimentation Statement
Research was conducted in compliance with the United States Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and with principles stated in the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 2011).
RESULTS
GBT1118 PK and PD Characteristics in Mice
GBT1118 showed a high partition into RBCs, with RBC-to-plasma ratios of 51:1 and 34:1 for intravenous (10 mg/kg) and oral (100 mg/kg) administration, respectively (Fig. 2A and Table 1). The volume of distribution was small in blood (0.072 l/kg) but much larger in plasma (2.95 l/kg), suggesting that GBT1118 is preferentially distributed into RBCs relative to plasma. GBT1118 showed low clearance in both blood (0.057 ml·min−1·kg−1) and plasma (3.21 ml·min−1·kg−1), indicating that GBT1118 was mostly bound to Hb (Fig. 2A and Table 1). The terminal half-life of GBT1118 was 13.9 and 11.3 h in blood and plasma, respectively. GBT1118 was well absorbed, and absolute bioavailability after 100 mg/kg po in whole blood and plasma was 45.8% and 33.5%, respectively (Fig. 2A and Table 2). Oral and intravenous administration of GBT1118 decreased P50 relative to the control group (Fig. 2B). GBT1118 blood concentrations and the change in Hb O2 affinity (Fig. 2C and Table 2) showed a strong correlation (r2 = 0.89).
Fig. 2.

A: GBT1118 pharmacokinetics in male mice after intravenous (IV) dosing at 10 mg/kg and oral (PO) dosing at 100 mg/kg (n = 3 mice/time point). Blood samples were collected up to 8 h (intravenous) and 24 h (oral) after administration of GBT1118. Mean blood and plasma concentration-time profiles are shown. B: GBT1118 pharmacodynamics, shown as the effect of GBT1118 on OECs after intravenous and oral administration. Blood samples were collected for hemoximetry at different time points after single intravenous (10 mg/kg) and oral (100 mg/kg) GBT1118 administration. Both doses/routes of administration elicited a leftward shift in the blood OEC and a subsequent decrease in P50 relative to vehicle, indicating an increase in Hb O2 affinity (P50 = 38, 31.6, and 13.6 mmHg for vehicle control, intravenous GBT1118, and oral GBT1118, respectively). C: GBT1118 pharmacokinetics and pharmacodynamics, shown as pharmacokinetics-pharmacodynamics correlation after single intravenous and oral doses of GBT1118. Blood samples were collected at maximum concentration after a single intravenous (10 mg/kg) or oral (70, 100 or 140 mg/kg) dose of GBT1118. Blood samples were analyzed for GBT1118 blood concentrations as well as for effects on Hb O2 affinity using OECs (n = 18 total mice for the various indicated doses).
Table 1.
Blood and plasma pharmacokinetic parameters in male mice after intravenous and oral administration of GBT1118
| 10 mg/kg iv GBT1118 |
100 mg/kg po GBT1118 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T½, h | AUC(0-∞), μg·h·ml−1 | Vss, l/kg | CL, ml·min−1·kg−1 | Blood-to-plasma ratio | Tmax, h | Cmax, μg/ml | AUC(0-∞), μg·h·ml−1 | F, % | Blood-to-plasma ratio | |
| Blood | 13.9 | 2,929 | 0.07 | 0.06 | 51.4 | 2 | 318 | 13,428 | 45.8 | 34.1 |
| Plasma | 11.3 | 60 | 2.95 | 3.21 | 8 | 12 | 224 | 33.5 | ||
Blood samples were collected at maximum concentration in plasma (Cmax) after a single intravenous (10 mg/kg) or oral (100 mg/kg) dose of GBT1118. T1/2, terminal half-life; AUC, area under the curve; Vss, volume of distribution; CL, clearance; Tmax, time to reach Cmax; F, bioavailability.
Table 2.
Pharmacokinetics/pharmacodynamics after a single intravenous or oral dose of GBT1118 to mice
| Dose, mg/kg | n | Route | Tmax, h | Blood Cmax, µM | ΔP50, % | %Hb Occupancy |
|---|---|---|---|---|---|---|
| 10 | 24 | iv | 0.17 | 408 ± 70 | 17 ± 2 | 19 ± 3 |
| 70 | 9 | po | 4–6 | 682 ± 68 | 54 ± 4 | 32 ± 3 |
| 100 | 18 | po | 4–6 | 836 ± 93 | 64 ± 4 | 39 ± 4 |
| 140 | 9 | po | 4–6 | 1,134 ± 190 | 77 ± 6 | 54 ± 9 |
Values are means ± SD; n, number of mice. Blood samples were also analyzed for effects on Hb O2 affinity using O2 equilibrium curves. Percent occupancy was calculated by dividing GBT1118 blood concentration obtained at Cmax by Hb concentration (~2.15 mM) in blood, as defined by percent hematocrit (43% hematocrit). An increase in GBT1118 concentration in blood was accompanied by an increase in Hb O2 affinity, as evidenced by increase in ∆P50.
Hypoxic Challenge
Eighteen animals were included in the hypoxic challenge study: six animals were treated with GBT1118 (70 mg/kg), six animals were treated with GBT1118 (140 mg/kg), and six animals were treated with vehicle (control). All animals tolerated the experimental protocol without signs of stress or discomfort. Animals passed the Grubbs’ test, ensuring that all parameters at baseline were similar within a population (P > 0.3). After oral administration of GBT1118, animals were exposed to hypoxia by stepwise decreases in %O2 from 21% to 15%, then to 10%, and later to 5%. Each 5% step in O2 was maintained for ∼30 min; 5% O2 was maintained for 2 h to assess tolerance to hypoxia (Fig. 1A).
Changes in Blood O2 Affinity
A single oral dose of GBT1118 at 70 or 140 mg/kg decreased P50 from 43 ± 1.1 to 18.3 ± 0.9 mmHg and then to 7.7 ± 0.2 mmHg, respectively (Fig. 1B). GBT1118-induced changes in P50 were dose dependent. The GBT1118-induced decrease in P50 was maintained throughout the protocol. According to the PK/PD results, the expected Hb occupancies achieved by oral administration of GBT1118 at 70 and 140 mg/kg in mice are ~32% and 54%, respectively.
, Blood Gases, and Metabolic Acidosis During Hypoxia
decreased with decreasing percent O2 (Fig. 3A) and was similar in all groups. GBT1118 dose-dependently increased relative to the control group during hypoxia (Fig. 3B). The GBT1118-induced increase in Hb O2 affinity increased pulmonary O2 loading. The control group showed a decrease in arterial pH (Fig. 4A) and an increase in lactate (Fig. 4B) compared with GBT118-treated groups during hypoxia. Differences in arterial pH and lactate increased with the reduction in percent O2. The control group showed an increased respiratory rate during 10% and 5% O2 hypoxia compared with GBT118-treated groups (Fig. 4C). Acidosis during 10% and 5% O2 hypoxia in the control group was accompanied by a decrease in both arterial blood CO2 (Fig. 4D) and arterial blood (Fig. 4E) compared with GBT118-treated groups. Although GBT118-treated groups increased their respiratory rate (Fig. 4C), the hyperpnea did not elicit a drastic decrease in (Fig. 4D). In contrast to the control group, no difference in arterial blood pH at 10% O2 hypoxia was observed in either GBT1118-treated group. The increase in O2 affinity protection during hypoxia due to GBT1118 was noteworthy at 5% O2 hypoxia, as the decrease in arterial blood pH was only a fraction of that in the control group.
Fig. 3.

A: arterial Po2 () decreased during hypoxia. B: GBT1118 increased arterial O2 saturation () during hypoxia. Values are means ± SD of 6 animals/group. *P < 0.05 vs. control at each percent O2; ξP < 0.05 vs. GBT1118 (70 mg/kg); †P < 0.05 vs. baseline (BL).
Fig. 4.

Blood gases, respiratory rates, and interstitial tissue Po2. GBT1118 prevented acidosis and improved O2 delivery to tissues during hypoxia. A–F: changes in arterial blood pH, lactate concentration, respiration rate (in breaths/min), arterial blood Pco2 (), arterial blood , and interstitial tissue Po2 during hypoxia. Values are means ± SD of 6 animals/group. *P < 0.05 vs. control at each percent O2; ξP < 0.05 vs. GBT1118 (70 mg/kg); †P < 0.05 vs. 21% O2. Dashed line, BL values (pH 7.3, lactate = 1.17 mmol/l, respiration rate = 173.5 breaths/min, = 38.4 mmHg, and = 20 meq/l).
Interstitial Tissue Po2 During 5% O2 Hypoxia
Interstitial tissue Po2 during 5% O2 hypoxia was higher, in a dose-dependent manner, in GBT1118-treated groups than in the control group (Fig. 4F). In addition, the increased O2 affinity protection against acidosis and lactic acidosis after GBT1118 administration during hypoxia could be explained by improved tissue oxygenation. Therefore, the GBT1118-induced increase in Hb O2 affinity increased not only but also O2 delivery compared with the control group. The increased interstitial tissue Po2 during 5% O2 hypoxia confirms that the GBT1118-induced increase in O2 affinity does not prevent off loading of O2 to tissue and that the O2 bound to GBT1118-modified Hb was available for tissue O2 metabolism, despite the GBT1118-induced increase in Hb O2 affinity.
Blood Pressure and HR During Hypoxia
Consistent with the increased O2 affinity protection against acidosis and lactic acidosis and the preservation of interstitial Po2 during hypoxia, the increased O2 affinity reduced hypoxia-induced hypotension and bradycardia. Cardiovascular changes were blunted in GBT1118-treated groups compared with the control group during progressive hypoxia. MAP and HR decreased in the control group, whereas MAP (Fig. 5A) and HR (Fig. 5B) were significantly higher in GBT1118-treated groups than in the control group in a dose-dependent fashion.
Fig. 5.

Systemic response during hypoxia. A and B: change in mean arterial pressure and heart rate (in beats/min) during hypoxia. Values are means ± SD of 6 animals/group. *P < 0.05 vs. control at each percent O2; ξP < 0.05 vs. GBT1118 (70 mg/kg); †P < 0.05 vs. BL.
Microvascular Diameter and Blood Flow During Hypoxia
Hypoxia produced vasodilation in all groups (Fig. 6A). Although in the groups with increased Hb O2 affinity the hypoxia-induced vasodilation relative to normoxia occurred at 15% and 10% O2 hypoxia, in the control group the hypoxia-induced vasodilation only occurred at 15% O2 hypoxia. The GBT1118 (140 mg/kg)-treated group showed significant vasodilation compared with the GBT1118 (70 mg/kg)-treated group and control group at 10% and 5% O2 hypoxia, respectively. Microvascular blood flows were similar at baseline in both GBT1118-treated groups compared with the control group. Microvascular blood flow increased with 15% O2 hypoxia in all groups (Fig. 6B). However, microvascular blood flow decreased drastically in the GBT1118 (70 mg/kg)-treated group and control group at 10% and 5% O2 hypoxia compared with the GBT1118 (140 mg/kg)-treated group. Microvascular blood flow was significantly increased in the GBT1118 (140 mg/kg)-treated group compared with the GBT1118 (70 mg/kg)-treated group and control group at 10% and 5% O2 hypoxia, respectively.
Fig. 6.

GBT1118 preserves microvascular blood flow during hypoxia. A and B: changes in arteriolar diameter and blood flow during hypoxia. Values are means ± SD of 6 animals/group. †P < 0.05 vs. 21% O2.
Vital Organ Hypoxia and 5% O2 Hypoxia Tolerance
The GBT1118 (70 and 140 mg/kg)-induced increase in O2 affinity reduced pimonidazole-positive hypoxic staining in the intestine and liver compared with the control group (Fig. 7A). In addition, the GBT1118 (140 mg/kg)-induced increase in O2 affinity reduced pimonidazole-positive hypoxic staining in the brain, heart, and kidney compared with the control group (Fig. 7A). All animals tolerated the initial 30 min of 5% O2 hypoxia; however, the control group failed to tolerate 5% O2 hypoxia over 90 min of exposure. Tolerance to 2 h of 5% O2 hypoxia was 17% and 83% in the GBT1118 (70 mg/kg)- and GBT1118 (140 mg/kg)-treated groups, respectively. In summary, the GBT1118-induced increase in O2 affinity reduced tissue hypoxia and improved tolerance to severe hypoxia.
Fig. 7.

GBT1118 reduces tissue hypoxia and improves tolerance to hypoxia in mice. A: hypoxic tissues (brain, heart, kidney, intestine, and liver) positively stained by pimonidazole relative to control during extreme hypoxia. B: tolerance to hypoxia in mice. Values are means ± SD of 6 animals/group.
DISCUSSION
The principal finding of the present study is that the GBT1118-induced increase in Hb O2 affinity increased , prevented metabolic acidosis, preserved cardiovascular function, and reduced vital organ hypoxia during progressive hypoxia. Moreover, the GBT1118-induced increase in Hb O2 affinity improved tolerance to 5% O2 hypoxia. Therefore, the GBT1118-induced increase in Hb O2 affinity increased O2 delivery during hypoxia, thus preserving MAP, HR, blood flow, and aerobic metabolism. The GBT1118-induced increase in Hb O2 affinity increased blood O2 loading in the lungs and allowed for increased O2 delivery during hypoxia. The increase in O2 delivery during hypoxia preserved MAP and HR and interstitial tissue Po2 compared with the control group during hypoxia. Consequently, the GBT1118 (70 and 140 mg/kg)-induced increase in Hb O2 affinity decreased hypoxia in vital tissues and lowered lactate levels.
O2 transport in mammals and other vertebrates is accomplished by the Hb inside RBCs, which captures O2 in the lungs and, driven by the heart, delivers O2 to tissues. Under conditions of severe environmental hypoxic hypoxia, high altitude, or limited respiratory O2 loading (such as lung diseases), hypoxemia causes insufficient O2 delivery to tissues, resulting in altitude sickness, acute respiratory failure, and even death (18). Reduced inspired O2 limits the amount of O2 bound to Hb in the pulmonary circulation and provides a good model to evaluate the effects of Hb O2 affinity on O2 delivery. The GBT1118-induced increase in Hb O2 affinity in healthy mice subjected to reduced inspired O2 prevented the hemodynamic disturbance produced by hypoxia and maintained microvascular blood flow and tissue oxygenation compared with animals with normal Hb O2 affinity. Hypoxia led to alterations in arterial blood gases in the control group compared with GBT1118-treated groups. Arterial blood Pco2 and were higher in GBT1118-treated groups than in the control group during hypoxia. Analysis of arterial blood gases indicates that the GBT1118-induced increase in Hb O2 affinity during reduced inspired O2 prevented metabolic acidosis and accumulation of lactate, which confirms the pimonidazole-positive hypoxic staining in vital organs in the GBT1118-treated groups.
As multiple signaling pathways are activated by hypoxic stimuli (such as O2, CO2, and lactate), they are integrated in areas including respiratory centers to modulate the response to hypoxia. The reduced lactate levels from the GBT1118-induced increase in Hb O2 affinity during hypoxia appear to reduce severe hypoxia-induced mixed blood acid-base disorder in the control group. Protective effects due to the GBT1118-induced increase in Hb O2 affinity are more pronounced at 5% O2 hypoxia because of prevention of acute hyperventilation, hypocapnia, and lactate acidosis relative to the control group. This interpretation could be strengthened by calculation or estimation of the strong ion difference and total weak acid concentration using the principles of Stewart for acid-base analysis (29). The mechanisms responsible for protection from mixed acid-base disorder are not fully understood, but they are likely related to improved O2 delivery and reduced vital organ hypoxia. Recently, new important transduction mechanisms of carotid body response have been complemented with the lactate-mediated olfactory activation, which, in addition to the carotid body chemosensory reflex resulting from low during hypoxia, mediates an integrated ventilatory and cardiovascular response to hypoxia (5). This mechanism is compatible with mitochondrial signaling responses to hypoxia sensing, as NADH accumulates and serves as a cosubstrate to produce lactate in carotid body glomus cells (8). The measured partially blunted respiratory rate response to hypoxia in GBT1118-treated groups is part of the increase in O2 protection to hypoxia via a decrease in the compensatory responses to severe hypoxia by the carotid body. Changes in these arterial parameters of O2, CO2, and pH depolarize the membrane of glomus cells, increasing cytosolic Ca2+, which results in the release of excitatory neurotransmitters, increasing the activity of the carotid sinus nerve. Thus moderate gas disturbances during hypoxia affect the activity of the carotid body and the firing activity of the central neural system. Future studies of modified Hb O2 affinity during hypoxia should aim to investigate the chemosensory drive of the carotid body chemoreceptors.
O2 availability is a critical metabolic regulator of microvascular blood flow. Several mechanisms, including responses to intraluminal pressure (myogenic), shear stress on the endothelial lining of vessels (shear dependent), metabolite concentrations in vessels and/or tissue (metabolic), and neural stimuli, contribute to changes in vascular tone (14, 20). Vascular responses to hypoxia are the result of chemoreflex control, mediated primarily through sympathetic and local vascular regulation (10). The GBT1118-induced increase in Hb O2 affinity did not affect arteriolar diameter or blood flow (Fig. 6) at baseline relative to the control group. However, all groups experienced hypoxic vasodilation and increased blood flow at 15% O2 hypoxia. As the severity of hypoxia increased, arteriolar diameter and blood flow decreased in the control group, which could be explained by systemic hypotension triggering a myogenic peripheral response to vasoconstriction to preserve the circular wall stress (which is proportional to pressure ÷ radius). Consequently, the vasoconstriction and the hypotension in the control group drastically reduced microvascular blood flow with the degree of the hypoxia. The GBT1118-induced increase in Hb O2 affinity extended the hypoxic vasodilation to 10% O2 hypoxia, in part by preserving blood pressure and O2 delivery at lower percent O2 than in the control group. Even though blood flow was significantly lower than baseline during 5% inspired O2 in the GBT1118 (70 mg/kg)-treated group, a hypoxic protective activity was observed, as indicated by a minimal decrease in pH and lower blood lactate concentration and respiratory rate than in the control group at 5% O2 hypoxia. A previous study (6) has established that the nitrite reductase activity of RBCs and Hb is a function of Hb O2 affinity. This study indicated that the O2 affinity of Hb in the RBCs regulates the production of nitric oxide by Hb nitrite reductase activity and mediates nitric oxide-dependent vasodilation. At ∼40-60% So2, the nitrite reductase activity of Hb can generate sufficient nitric oxide to stimulate relaxation of the smooth muscle and produce vasodilation. Analysis of the changes in diameter at baseline and during hypoxia in our study suggests that changes in diameter during hypoxia as a function of microvascular So2 follow a quadratic function. Furthermore, maximal vasodilation during hypoxia for the control and GBT1118-treated groups was detected at ∼50% microvascular So2. Although maximal hypoxic vasodilation is not a direct indicator of nitrite reductase activity of Hb in the RBCs, it reflects the contribution of all hypoxia-mediated vasodilation, including vasodilation due to nitrite reductase.
Oral administration of GBT1118 at 70 and 140 mg/kg achieved Hb occupancies of ~32% and 54%, respectively. OECs showed a dose-dependent increase in Hb O2 affinity and decrease in P50 in GBT1118-treated groups relative to the control group. As the hypoxic challenge used in this study was controlled based on the inspired Po2, blood gas analysis showed no difference in terms of between the GBT1118-treated groups and the control group; however, the GBT1118-induced increase in Hb O2 affinity resulted in a dose-dependent increase in . Given that O2 content, rather than Po2, is the critical determinant of O2 delivery to tissues (33), the GBT1118-mediated increase in translated into an improved O2 delivery to tissues. The arterial blood pH and lactate levels in the GBT1118-treated groups suggest an improvement in O2 delivery to tissues, metabolic activity, and tissue oxygenation compared with the control group. High blood lactate levels indicate an increased production and accumulation of anaerobic metabolites, limited O2 delivery to tissues, metabolic acidosis, and tissue hypoxia (17, 26). Administration of GBT1118 before severe hypoxia maintained normal arterial blood pH of ~7.35. In contrast, arterial blood pH of the control group markedly decreased to ~7 during hypoxia (5% O2), suggesting insufficient O2 delivery. In support of the latter observation, blood lactate levels were significantly higher in the control group (5 mM) than in GBT1118-treated groups (<3.5 mM), and a severe compensation by hyperventilation and hypocapnia was observed in the control group but not in GBT1118-treated groups. Moreover, higher microvascular tissue Po2 levels, together with the GBT1118-mediated increase in , indicate an improvement in O2 delivery to tissues in GBT1118-treated groups compared with the control group.
GBT1118-mediated improvement in O2 delivery to tissues is also substantiated by changes in systemic and microvascular hemodynamic parameters as well as changes in respiration during hypoxia. Compensatory changes in response to hypoxia in terms of MAP, HR, and respiration rate were less pronounced in GBT1118-treated groups than in the control group. MAP and HR decreased to 50 mmHg and 375 beats/min in the control group, whereas a dose-dependent increase in MAP and HR and an improved tolerance to 5% O2 hypoxia were noted in GBT1118-treated groups. In addition, microvascular blood flow and tissue oxygenation were preserved and, consequently, hypoxic areas in tissues were reduced in GBT1118-treated groups compared with the control group during hypoxia. The increase in microvascular blood flow in GBT1118-treated animals exposed to more severe hypoxia suggests that GBT1118 significantly changes cardiac output and total peripheral vascular resistance during severe hypoxia compared with control animals. The decreased cardiac output during severe hypoxia has been well documented and found to be 0.45 of baseline in conscious mice at 5% O2 (30). In our present study, the cardiac output was not measured, but it is expected to decrease to a similar extent in the control animals. The effect of GBT1118 on cardiac output during severe hypoxia will be further examined in future studies.
Since direct O2 measurements in vital organs are not applicable, bioreductive drugs, such as pimonidazole, provide an alternative approach for assessing the extent of hypoxia (27). Pimonidazole is reduced to a reactive intermediate that binds covalently to molecules containing thiol groups at <10 mmHg Po2 (27), which results in pimonidazole-protein complexes that can be detected by simple immunohistochemistry. The GBT1118-induced increase in Hb O2 affinity reduced hypoxia in tissues positively stained by pimonidazole relative to the control group. Furthermore, in the GBT1118 (70 mg/kg)-treated group, the liver and intestine were protected from hypoxia; in the GBT1118 (140 mg/kg)-treated group, all organs were protected from hypoxia. In addition, the control group did not tolerate exposure to 5% O2 hypoxia for >90 min, whereas 16% of animals in the GBT1118 (70 mg/kg)-treated group and 83% of animals in the GBT1118 (140 mg/kg)-treated group tolerated exposure to 5% O2 hypoxia for >120 min. OEC measurements of blood samples require dilution in a solution (HEMOX) that is strongly buffered (pH 7.3) and oxygenated and deoxygenated in the absence of CO2. As a result, the OECs and P50 values measured using the HEMOX analyzer do not reflect the expected decrease in Hb O2 affinity, which is a function of the acid-base status of the sample (Bohr and Haldane effects). In our previous studies, we used pH and Pco2 changes induced by 10% and 5% O2 hypoxia in mice to replicate the conditions used for the OEC measurements obtained using the HEMOX analyzer. The results indicate that the P50 values, independent of blood Pco2 and pH, were lowered by 3.2% at 10% O2 hypoxia and by 4.4% at 10% O2 hypoxia. Our previous results agree with the effects of Pco2 and pH on O2 dissociation curves for whole blood reported by Zwart et al. (35). Hypoxia-induced hypometabolism has been shown to occur only in small species, as allometric variation (mass-specific metabolic rate and O2 composition-to-body mass relationship) is higher in smaller-sized species than in larger-sized species (16). Therefore, direct extrapolation of the extent to which increases in Hb O2 affinity provide protection from hypoxia still needs to be evaluated in larger animals and humans. Reduced and its effect on cerebral blood flow are an important problem during hypoxia. This study, however, did not include cerebral blood flow and carbonic anhydrase inhibition as an additional control group; these factors act as an adjuvant to the protection from hypoxia achieved with increased Hb O2 affinity. Further investigation of the protection from hypoxia as a result of increased Hb O2 affinity should include cerebral blood flow measurements and carbonic anhydrase inhibitors. Finally, our results relative to tolerance to 5% O2 hypoxia reflect the previous stepwise increase in hypoxia to achieve 5% O2 and are expected to be different from results in animals directly exposed to 5% O2 hypoxia.
The benefits of modifying Hb O2 affinity to improve hypoxemia and prevent dyspnea can potentially reduce associated comorbidities of hypoxic hypoxia. Our results suggest that increased Hb O2 affinity complements acute adaptation to hypoxia via an increase in at low . In conclusion, our in vivo hypoxia design induced physiologically significant decreases in and O2 delivery in tissues and served as a good experimental model for environmental hypoxia as well as lung diseases resulting in hypoxemia. GBT1118-treated animals maintained higher and demonstrated less cardiac decompensation and hypoxic injury to various organ tissue beds than untreated hypoxic control animals. While a GBT1118-mediated increase in Hb O2 affinity could theoretically decrease O2 release from Hb, in vitro data show that GBT1118-modified Hb remains sensitive to the Bohr effect, with intact unloading of O2 under low pH conditions (data not shown). Moreover, the results from this study demonstrate that increased affinity of Hb for O2 from GBT1118 dosing did not limit efficient O2 extraction at the tissue level, as evidenced by decreased lactic acidosis, improved cardiovascular function, and, ultimately, tolerance to 5% O2 hypoxia. Together, these findings demonstrate the therapeutic potential of pharmacologically increasing Hb O2 affinity in people who reside at higher altitudes or patients with lung diseases, in whom hypoxia plays a key role in morbidity and mortality.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R56 HL-123015 and R01 HL-126945.
DISCLOSURES
K. Dufu, A. Hutchaleelala, Q. Xu, Z. Li, N. Vlahakis, D. Oksenberg, and J. Lehrer-Graiwer are employees and shareholders of Global Blood Therapeutics Incorporated. All other authors declare no competing financial interests by the results presented in this report. Financial support was received from Global Blood Therapeutics Incorporated for the completion of the study. Global Blood Therapeutics Incorporated did not participate in the implementation of the experimental study.
AUTHOR CONTRIBUTIONS
K.D., O.Y., A.H., Q.X., Z.L., D.O., J.L.-G., and P.C. conceived and designed research; K.D., O.Y., and P.C. prepared figures; K.D., O.Y., N.V., D.O., J.L.-G., and P.C. drafted manuscript; K.D., O.Y., E.S.A.-i., A.H., Q.X., Z.L., N.V., D.O., J.L.-G., and P.C. edited and revised manuscript; K.D., O.Y., E.S.A.-i., A.H., Q.X., Z.L., N.V., D.O., J.L.-G., and P.C. approved final version of manuscript; O.Y., E.S.A.-i., and P.C. performed experiments; O.Y. and P.C. analyzed data; O.Y., N.V., and P.C. interpreted results of experiments.
REFERENCES
- 1.Abdulmalik O, Safo MK, Chen Q, Yang J, Brugnara C, Ohene-Frempong K, Abraham DJ, Asakura T. 5-Hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br J Haematol 128: 552–561, 2005. doi: 10.1111/j.1365-2141.2004.05332.x. [DOI] [PubMed] [Google Scholar]
- 2.Black CP, Tenney SM. Oxygen transport during progressive hypoxia in high-altitude and sea-level waterfowl. Respir Physiol 39: 217–239, 1980. doi: 10.1016/0034-5687(80)90046-8. [DOI] [PubMed] [Google Scholar]
- 3.Cabrales P, Meng F, Acharya SA. Tissue oxidative metabolism after extreme hemodilution with PEG-conjugated hemoglobin. J Appl Physiol 109: 1852–1859, 2010. doi: 10.1152/japplphysiol.00344.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabrales P, Tsai AG, Frangos JA, Intaglietta M. Role of endothelial nitric oxide in microvascular oxygen delivery and consumption. Free Radic Biol Med 39: 1229–1237, 2005. doi: 10.1016/j.freeradbiomed.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Chang AJ, Ortega FE, Riegler J, Madison DV, Krasnow MA. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 527: 240–244, 2015. doi: 10.1038/nature15721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, Darley-Usmar VM, Kerby JD, Lang JD Jr, Kraus D, Ho C, Gladwin MT, Patel RP. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 107: 566–574, 2006. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douglas SW, Adamson JW. The anemia of chronic disorders: studies of marrow regulation and iron metabolism. Blood 45: 55–65, 1975. [PubMed] [Google Scholar]
- 8.Fernández-Agüera MC, Gao L, González-Rodríguez P, Pintado CO, Arias-Mayenco I, García-Flores P, García-Pergañeda A, Pascual A, Ortega-Sáenz P, López-Barneo J. Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab 22: 825–837, 2015. doi: 10.1016/j.cmet.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Gallagher SA, Hackett PH. High-altitude illness. Emerg Med Clin North Am 22: 329–355, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Heistad DD, Abboud FM. Dickinson W. Richards Lecture: Circulatory adjustments to hypoxia. Circulation 61: 463–470, 1980. doi: 10.1161/01.CIR.61.3.463. [DOI] [PubMed] [Google Scholar]
- 11.Henig NR, Pierson DJ. Mechanisms of hypoxemia. Respir Care Clin N Am 6: 501–521, 2000. doi: 10.1016/S1078-5337(05)70087-3. [DOI] [PubMed] [Google Scholar]
- 12.Intaglietta M, Tompkins WR. Capillary video red blood cell velocimetry by cross-correlation and spatial filtering. Microvasc Res 34: 108–115, 1987. doi: 10.1016/0026-2862(87)90083-5. [DOI] [PubMed] [Google Scholar]
- 13.Intaglietta M, Tompkins WR. Microvascular measurements by video image shearing and splitting. Microvasc Res 5: 309–312, 1973. doi: 10.1016/0026-2862(73)90042-3. [DOI] [PubMed] [Google Scholar]
- 14.Johnson PC, Intaglietta M. Contributions of pressure and flow sensitivity to autoregulation in mesenteric arterioles. Am J Physiol 231: 1686–1698, 1976. [DOI] [PubMed] [Google Scholar]
- 15.Kerger H, Groth G, Kalenka A, Vajkoczy P, Tsai AG, Intaglietta M. Po2 measurements by phosphorescence quenching: characteristics and applications of an automated system. Microvasc Res 65: 32–38, 2003. doi: 10.1016/S0026-2862(02)00027-4. [DOI] [PubMed] [Google Scholar]
- 16.Kleiber M. The Fire of Life: an Introduction to Animal Energetics. New York: Wiley, 1961. [Google Scholar]
- 17.Kraut JA, Madias NE. Metabolic acidosis: pathophysiology, diagnosis and management. Nat Rev Nephrol 6: 274–285, 2010. doi: 10.1038/nrneph.2010.33. [DOI] [PubMed] [Google Scholar]
- 18.Kulkarni AC, Kuppusamy P, Parinandi N. Oxygen, the lead actor in the pathophysiologic drama: enactment of the trinity of normoxia, hypoxia, and hyperoxia in disease and therapy. Antioxid Redox Signal 9: 1717–1730, 2007. doi: 10.1089/ars.2007.1724. [DOI] [PubMed] [Google Scholar]
- 19.Kumar GK. Hypoxia. 3. Hypoxia and neurotransmitter synthesis. Am J Physiol Cell Physiol 300: C743–C751, 2011. doi: 10.1152/ajpcell.00019.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laughlin MH, Davis MJ, Secher NH, van Lieshout JJ, Arce-Esquivel AA, Simmons GH, Bender SB, Padilla J, Bache RJ, Merkus D, Duncker DJ. Peripheral circulation. Compr Physiol 2: 321–447, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Lenfant C, Ways P, Aucutt C, Cruz J. Effect of chronic hypoxic hypoxia on the O2-Hb dissociation curve and respiratory gas transport in man. Respir Physiol 7: 7–29, 1969. doi: 10.1016/0034-5687(69)90066-8. [DOI] [PubMed] [Google Scholar]
- 22.Lipowsky HH, Zweifach BW. Application of the “two-slit” photometric technique to the measurement of microvascular volumetric flow rates. Microvasc Res 15: 93–101, 1978. doi: 10.1016/0026-2862(78)90009-2. [DOI] [PubMed] [Google Scholar]
- 23.Mairbäurl H, Weber RE. Oxygen transport by hemoglobin. Compr Physiol 2: 1463–1489, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Mason F. High altitude illness: avoiding the perils of the peaks. AMAA J 18: 13–15, 2005. [Google Scholar]
- 25.McLellan S, Walsh T. Oxygen delivery and haemoglobin. Contin Educ Anaesth Crit Care Pain 4: 123–126, 2004. doi: 10.1093/bjaceaccp/mkh033. [DOI] [Google Scholar]
- 26.Mizock BA, Falk JL. Lactic acidosis in critical illness. Crit Care Med 20: 80–93, 1992. doi: 10.1097/00003246-199201000-00020. [DOI] [PubMed] [Google Scholar]
- 27.Nordsmark M, Loncaster J, Aquino-Parsons C, Chou S-C, Ladekarl M, Havsteen H, Lindegaard JC, Davidson SE, Varia M, West C, Hunter R, Overgaard J, Raleigh JA. Measurements of hypoxia using pimonidazole and polarographic oxygen-sensitive electrodes in human cervix carcinomas. Radiother Oncol 67: 35–44, 2003. doi: 10.1016/S0167-8140(03)00010-0. [DOI] [PubMed] [Google Scholar]
- 28.Oksenberg D, Dufu K, Patel MP, Chuang C, Li Z, Xu Q, Silva-Garcia A, Zhou C, Hutchaleelaha A, Patskovska L, Patskovsky Y, Almo SC, Sinha U, Metcalf BW, Archer DR. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br J Haematol 175: 141–153, 2016. doi: 10.1111/bjh.14214. [DOI] [PubMed] [Google Scholar]
- 29.Stewart PA. Modern quantitative acid-base chemistry. Can J Physiol Pharmacol 61: 1444–1461, 1983. doi: 10.1139/y83-207. [DOI] [PubMed] [Google Scholar]
- 30.Stobdan T, Zhou D, Ao-Ieong E, Ortiz D, Ronen R, Hartley I, Gan Z, McCulloch AD, Bafna V, Cabrales P, Haddad GG. Endothelin receptor B, a candidate gene from human studies at high altitude, improves cardiac tolerance to hypoxia in genetically engineered heterozygote mice. Proc Natl Acad Sci USA 112: 10425–10430, 2015. doi: 10.1073/pnas.1507486112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Storz JF. Hemoglobin function and physiological adaptation to hypoxia in high-altitude mammals. J Mammal 88: 24–31, 2007. doi: 10.1644/06-MAMM-S-199R1.1. [DOI] [Google Scholar]
- 32.Vanderkooi JM, Maniara G, Green TJ, Wilson DF. An optical method for measurement of dioxygen concentration based upon quenching of phosphorescence. J Biol Chem 262: 5476–5482, 1987. [PubMed] [Google Scholar]
- 33.Yalcin O, Cabrales P. Increased hemoglobin O2 affinity protects during acute hypoxia. Am J Physiol Heart Circ Physiol 303: H271–H281, 2012. doi: 10.1152/ajpheart.00078.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon D, Ponka P, Prchal JT. Hypoxia. 5. Hypoxia and hematopoiesis. Am J Physiol Cell Physiol 300: C1215–C1222, 2011. doi: 10.1152/ajpcell.00044.2011. [DOI] [PubMed] [Google Scholar]
- 35.Zwart A, Kwant G, Oeseburg B, Zijlstra WG. Oxygen dissociation curves for whole blood, recorded with an instrument that continuously measures Po2 and So2 independently at constant t, Pco2, and pH. Clin Chem 28: 1287–1292, 1982. [PubMed] [Google Scholar]