

The mechanism by which EET acts on obesity-induced cardiomyopathy is unknown. Here, we describe a previously unrecognized function of EET infusion that inhibits nephroblastoma overexpressed (NOV) levels and activates Wnt1, hence identifying NOV inhibition and enhanced Wnt1 expression as novel pharmacological targets for the prevention and treatment of cardiomyopathy and heart failure.
Keywords: cardiomyopathy, metabolic syndrome, oxidative stress, hypertension, myocardial biology, nephroblastoma overexpressed, heme oxygenase-1
Abstract
We have previously reported that epoxyeicosatrienoic acid (EET) has multiple beneficial effects on vascular function; in addition to its antiapoptotic action, it increases insulin sensitivity and inhibits inflammation. To uncover the signaling mechanisms by which EET reduces cardiomyopathy, we hypothesized that EET infusion might ameliorate obesity-induced cardiomyopathy by improving heme oxygenase (HO)-1, Wnt1, thermogenic gene levels, and mitochondrial integrity in cardiac tissues and improved pericardial fat phenotype. EET reduced levels of fasting blood glucose and proinflammatory adipokines, including nephroblastoma overexpressed (NOV) signaling, while increasing echocardiographic fractional shortening and O2 consumption. Of interest, we also noted a marked improvement in mitochondrial integrity, thermogenic genes, and Wnt 1 and HO-1 signaling mechanisms. Knockout of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) in EET-treated mice resulted in a reversal of these beneficial effects including a decrease in myocardial Wnt1 and HO-1 expression and an increase in NOV. To further elucidate the effects of EET on pericardial adipose tissues, we observed EET treatment increases in adiponectin, PGC-1α, phospho-AMP-activated protein kinase, insulin receptor phosphorylation, and thermogenic genes, resulting in a “browning” pericardial adipose phenotype under high-fat diets. Collectively, these experiments demonstrate that an EET agonist increased Wnt1 and HO-1 signaling while decreasing NOV pathways and the progression of cardiomyopathy. Furthermore, this report presents a portal into potential therapeutic approaches for the treatment of heart failure and metabolic syndrome.
NEW & NOTEWORTHY The mechanism by which EET acts on obesity-induced cardiomyopathy is unknown. Here, we describe a previously unrecognized function of EET infusion that inhibits nephroblastoma overexpressed (NOV) levels and activates Wnt1, hence identifying NOV inhibition and enhanced Wnt1 expression as novel pharmacological targets for the prevention and treatment of cardiomyopathy and heart failure.
Listen to this article's corresponding podcast at http://ajpheart.physiology.org/content/early/2017/05/31/ajpheart.00093.2017.
excess body fat and impaired fat metabolism negatively influence the health of an individual and are associated with the development of various downstream diseases, including type 2 diabetes and heart disease.
Epoxyeicosatrienoic acid (EET) has pleiotropic effects that are either produced and act locally or circulate in blood and improve coronary and pulmonary vascular function (14, 28, 42). The sustained levels of EET by soluble epoxide hydrolase (sEH) inhibitors in cardiovascular disease (9, 21, 26, 56), heart failure (33), and blood pressure (53) are of intense interest. Specific overexpression of cytochrome P-450 (CYP)2J2 and increases in circulating EET attenuate diabetic cardiomyopathy in male animals and hypertension (3, 31).
EETs affect multiple signaling pathways of phosphatidlyinositol 3-kinase (PI3K) and ATP-sensitive K+ channels and other signaling molecules, including peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), as well as mitochondrial (42) channels (for a review, see Ref. 5); however, the effect of EETs on the wingless-related integration site (Wnt) family and NOV signaling pathways has not been studied. The Wnt family of proteins controls diverse biological processes, whereas nephroblastoma overexpressed CCN3 (NOV/CCN3) is involved in many pathophysiological processes and insulin resistance (32). Induction of NOV results in increased adipose tissue deposition and enhanced cholesterol and plasma triglyceride formation in human cardiometabolic patients (38).
Increased ROS production causes cell death and the dissemination of new, highly reactive free radicals that induce adverse cardiac remodeling and regulate cardiac function via mitochondrially derived ROS (15, 23, 40). The important function of cardiac mitochondria in energy metabolism and cardiovascular disease has been extensively studied, mainly involving the role of mitochondrial oxidative stress in cardiac pathophysiology (22, 51).
Heme oxygenase (HO)-1 and EET are synergistically linked in cellular protection. An increase in HO activity increases the antioxidants CO and bilirubin, which decreases oxidative stress and reduces pathological remodeling of the heart, renal function, and obesity (for reviews, see Refs. 2 and 19). Dyslipidemia reduces HO-1 expression, resulting in degeneration of respiratory capacity (1), a decline in energy expenditure, and mitochondrial dysfunction (6, 23, 35). Ectopic fat accumulation and inflammation adjacent to the epicardium and pericardium result in a local decrease in HO-1 and increases in ROS with deleterious effects on cardiac structure and function, further contributing to cardiomyopathy (30, 39, 41). Moreover, oxidative mutations of mitochondrial fusion and fission proteins disable the ability of the mitochondria to maintain mtDNA integrity, further enhancing ROS-mediated oxidative stress (10, 43). Furthermore, increased HO-1 expression decreases fat deposition, adipocyte terminal differentiation, and hypertrophy with a concurrent reduction in inflammatory TNF-α, IL-6, and monocyte chemoattractant protein (MCP)-1 levels (7, 49, 59). Low levels of circulating EET have also been associated with obesity (61).
Another signaling protein affected by EET is PGC-1α. PGC-1α is a master regulator of mitochondrial biogenesis that regulates several key components of the adaptive thermogenesis program and β-fatty acid oxidation (12, 55, 60). PGC-1α expression levels in cardiomyocytes are regulated in mitochondrial function and polarity, cardiac energy supply, and O2 consumption (V̇o2) (44, 60). In mice, constitutive overexpression of PGC-1α in cardiac tissue activates mitochondrial biogenesis and proliferation (44). Furthermore, mice that lack PGC-1α in adipose tissue and are fed a high-fat diet (HFD) develop insulin resistance and increased circulating lipids (27), as PGC-1α controls the energy state (4). Since the underlying mechanism of how EET affects obesity-induced cardiomyopathy and ameliorates heart failure is not well assessed, we explored the effectiveness of an EET agonist on the regulation of NOV, WNT1, and β-catenin signaling pathways. Our results uncover a previously unknown function of EET as a negative regulator of NOV signaling.
MATERIALS AND METHODS
Animal protocols.
db/db mice were divided into three treatment groups after a 16-wk acclimatization period: 1) control, 2) EET agonist (EET-A; 1.5 mg/100 g body wt, 8-wk treatment), and 3) EET-A-Ln-PGC-1α-shRNA. All mice were fed a HFD that contained 58% fat (from lard), 25.6% carbohydrate, and 16.4% protein with total calories of 23.4 kJ/g (Teklad Laboratory Animal Diets, Harlan). Mice (n = 6 animals/group) were treated as follows: 1) control, 2) injected intraperitoneally with EET-A twice per week for 8 wk at a dose of 1.5 mg/100 g body wt, and 3) EET-A injected twice per week for 8 wk intraperitoneally at a dose of 1.5 mg/100 g body wt. Two bolus injections of PGC-1α (sh) lentivirus (40–70 × 106 TU/mouse at 80- to 100-μl volume) were administered (Dharmacon, Lafayette, CO) into the retroorbital at the 4th and 6th weeks of EET administration. EET-A is a single enantiomer (>98% ee) and >95% pure as determined by 1H/13C NMR. It is an inhibitor of sEH (IC50 = 392 n M) and not metabolized by this enzyme (16). At the end of the study, mice were anesthetized with pentobarbital sodium (65 mg/kg ip). Body weight and fat content were measured at euthanasia. All animal experiments followed a PLA Hospital (Beijing, China) and New York Medical College Institional Animal Care and Use Committee institutionally approved protocol in accordance with National Institutes of Health guidelines.
Fasting blood glucose, glucose tolerance test, and blood pressure measurements.
Fasting blood glucose levels were measured after a 12-h fast. For the glucose tolerance test, mice were injected intraperitoneally with glucose (2.0 g/kg body wt). Measurements of blood glucose levels were evaluated through blood samples obtained every 30 min for a total of 120 min. Blood pressure was measured by the tail-cuff method using the CODA tail-cuff system (Kent Scientific, Torrington, CT) at the end of the experiment and compared with each of the three groups. All measurements were determined at the same time of day, and the systolic blood pressure was recorded (in mmHg).
Echocardiogram measurement of fractional shortening.
Echocardiography was performed using a 12-MHz probe on ketamine-xylazine-anesthetized mice as previously described (24). Left ventricular (LV) end-diastolic and end-systolic proportions, fractional shortening (FS), and wall thickness quantification measurements were derived from M-mode tracings acquired from a two-dimensional echocardiograph midventricular short-axis view.
Determination of V̇o2 and respiratory quotient.
db/db mice groups were allowed to acclimatize in the V̇o2 chambers for a total of 3 wk. Adaptation periods for the 3-wk duration were executed in 2-h increments three times per week. The Oxylet gas analyzer and air flow unit (Oxylet, Panlab-Bioseb, Vitrolles, France) were used to determine mouse V̇o2. Each mouse was placed individually in the machine, and V̇o2, CO2 production (V̇co2), and respiratory quotient (RQ) (as V̇co2/V̇o2) were calculated (49, 50).
Cell culture of the 3T3-L1 mouse embryonic fibroblast cell line.
3T3-L1 murine embryonic fibroblast preadipocytes were purchased from the American Type Culture Collection (ATCC, Manassas, VA). After being thawed, 3T3-L1 cells were cultured at 37°C in a 5% CO2 incubator in α-minimal essential medium (α-MEM; Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated FBS (Invitrogen) and 1% antibiotic/antimycotic solution (Invitrogen). The medium was changed after 48 h and every 3–4 days thereafter as previously described (10). For adipogenesis experiments, the medium was replaced with adipogenic medium [DMEM with high glucose (Invitrogen) supplemented with 10% (vol/vol) FBS, 10 μg/ml insulin (Sigma-Aldrich, St. Louis, MO), 0.5 mM dexamethasone (Sigma-Aldrich), and 0.1 mM indomethacin (Sigma-Aldrich)], and cells were cultured for an additional 8 days. Cells were cultured in the absence and presence of EET-A in a dose of 10 μM. At the experimental end points, cells were collected by trypsinization, washed once with PBS, and then lysed for RNA extraction.
Real-time quantitative PCR and Western blot analysis.
Total RNA was extracted from heart and pericardial adipose tissue with TRIzol (Ambion, Austin, TX). A Biotek plate reader and the Take3 plate (Biotek Winooski, VT) were used to determine RNA at an absorbance of 260 nm (A260). RNA measurements were subsequently evaluated by the A260-to-A280 ratio. A High-Capacity cDNA Reverse Transcription Kit (Applied BioSystems) was used to synthesize cDNA from total RNA (Applied Bio Systems). TaqMan Fast Universal Master Mix (2×) on a 7500 HT Fast Real-Time PCR System (Applied BioSystems) was used to perform real-time PCR. Specific TaqMan Gene Expression Assay probes for mouse HO-1, PGC-1α, NOV, β-catenin, Wnt10b, cytochrome c oxidase subunit I (COX-I), mitochondrial fusion protein mitofusion 2 (Mfn2), TNF-α, IL-6, and GAPDH were used as previously described (7, 18, 58). Hearts were lysed in RIPA lysis buffer supplemented with protease and phosphatase inhibitors (CompleteMini and PhosSTOP, Roche Diagnostics, Indianapolis, IN) for Western blot analysis. Frozen pooled pericardial mouse adipose tissue was ground under liquid nitrogen and suspended in homogenization buffer [containing (in mmol/l) 10 phosphate buffer, 250 sucrose, 1.0 EDTA, 0.1 PMSF, and 0.1% (vol/vol) tergitol, pH 7.5]. Immunoblot analysis for Wnt1, Wnt5b, β-catenin, NOV, PGC-1α, HO-1, sirtuin (SIRT)1, uncoupling protein-1 (UCP1), IL-6, matrix metalloproteinase (MMP)-2, phospho-AKT (pAKT), AKT, phospho-AMP-activated protein kinase (pAMPK), AMPK, phospho-insulin receptor (Tyr972) (pIR972), and β-actin was performed as previously described (7, 18, 58).
Measurement of EETs.
Fat tissue was homogenized in 66% methanol containing a 500-pg mixture of internal standards [PGE2-d4, 8(9)-EET-d11, and 11(12)-EET-d8]. EETs were extracted using solid-phase C18-ODS AccuBond II 500-mg cartridges (Agilent Technologies, Santa Clara, CA). In brief, each sample was centrifuged for 15 min at 4°C. The EET concentration was calculated as previously described (52).
Statistical analysis.
Data values are expressed as means ± SE. One-way ANOVA was performed with individual group means compared by Student’s t-test using Bonferroni’s correction for multiple comparisons. The null hypothesis was rejected at P < 0.05.
RESULTS
Effect of EET-A on weight gain, blood glucose levels, blood pressure, heart weight, EET levels, and V̇o2 in db/db mice and its relation to HO-1 and PGC-1α.
db/db control mice exhibited an average weight gain of 0.8 g/wk with an average final weight of 61.8 ± 2.2 g. EET-A decreased weight gain (P < 0.05) compared with db/db control mice. EET-A-Ln-PGC-1α (sh) reversed the EET-A-mediated reduction of weight gain (Fig. 1A). The final weights after EET-A Ln-PGC-1α (sh) administration were 43.3 ± 1.65 and 56.8 ± 1.33 g, respectively (P < 0.05). db/db mice gained weight at a faster absolute rate after Ln-PGC-1α (sh) injection compared with the initial rate of weight loss prompted by EET before PGC-1α lentiviral injection in the same experimental group (Fig. 1A).
Fig. 1.

Effect of EET agonist (EET-A) treatment on body weight, fasting blood glucose, glucose tolerance, and heart weight in db/db mice. A: body weight. B: fasting blood glucose. C: glucose tolerance. D: blood pressure. E and F: effect of EET-A treatment on heart weight (E) and EET levels in cardiac tissues (F). G−I: respiratory quotation (RQ; G), mouse respiratory O2 consumption (V̇o2; H), and release of CO2 (V̇co2; I) in db/db control (Con) mice, db/db mice treated with EET-A, and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α)-deficient db/db mice treated with EET-A. Results are means ± SE; n = 6. *P < 0.05 vs. db/db control mice; #P < 0.05 vs. db/db mice treated with EET-A.
Blood glucose levels in control db/db mice, EET-treated db/db mice, and EET-A with PGC-1α (sh) db/db mice were 416.7 ± 61.5, 162.85 ± 16.4, and 275.6 ± 28.02 mg/dl, respectively (Fig. 1B). PGC-1α lentivirus reversed the effects of EET-A treatment. Blood glucose concentrations in EET-A-PGC-1α (sh) mice were 112 ± 12.3 mg/dl higher than in EET-A-treated mice (Fig. 1B). EET-A treatment increased the tolerance to glucose challenge (P < 0.05) compared with db/db control mice (Fig. 1C). The EET-A-mediated effect was reversed (P < 0.05) by Ln- PGC-1α (sh) (Fig. 1C).
Systolic blood pressure was increased in control db/db mice. Furthermore, EET-A decreased blood pressure levels (P < 0.01) in mice compared with control, whereas treatment with EET-A in PGC-1α-deficient mice showed no beneficial effect on blood pressure (Fig. 1D).
db/db control mice exhibited an average heart weight of 0.16 g, and EET-A significantly decreased heart weight (P < 0.05) compared with db/db control mice. EET-A-Ln-PGC-1α (sh) reversed the EET-A-mediated reduction of heart weight (Fig. 1E). Total EET levels were measured in cardiac tissue of mice as shown in Fig. 1F. EET levels were significantly (P < 0.05) higher in EET-A treated mice compared with db/db control mice. The inhibition of PGC-1α in EET-A-treated mice significantly (P < 0.05) prevented the induction of EET levels and reduced them to the levels observed in db/db control mouse cardiac tissue (Fig. 1F).
We examined the effect of EET-A on both V̇o2 and the ratio (RQ) of CO2/O2 in db/db mice. We found that EET-A treatment exhibited a significant (P < 0.05) increase in V̇o2 with a concomitant lowering of V̇co2/V̇o2 compared with control db/db mice. PGC-1α-deficient animals treated with EET-A displayed a significant (P < 0.05) decrease in V̇o2 and an elevated V̇co2/V̇o2 (Fig. 1, G−I).
Effect of EET-A administration on FS.
The effect of EET-A treatment on FS is shown in Fig. 2. FS was reduced in untreated db/db control mice (Fig. 2, A and D), whereas administration of EET-A restored myocyte function (P < 0.05) as demonstrated by an increase in FS (Fig. 2, B and D). EET-A-PGC-1α (sh) reversed the effects of EET-A treatment by reducing FS to levels more profoundly depressed than those seen in control animals (Fig. 2, C and D). PGC-1α inhibition significantly (P < 0.05) diminished FS in relation to EET-treated mice alone.
Fig. 2.

Effect of EET-A on left ventricular (LV) dilation and fractional shortening (FS) in db/db mice. Sample M-mode short-axis echocardiographic images show LV dilation and FS in db/db control mice (A), db/db mice treated with EET-A (B), and PGC-1α-deficient db/db mice treated with EET-A (C). D: graph showing LV FS in each cohort. n = 9. *P = 0.05 vs. control; #P < 0.02 vs. EET-A.
Effect of EET-A administration on Wnt1, Wnt5b, and β-catenin in the heart and adipose tissue and NOV expression in the heart.
EET-A induction significantly (P < 0.05) upregulated the expression of Wnt1 protein expression levels in cardiac and adipose tissues compared with control mice. Basal levels of Wnt1 were higher in adipose tissue compared with heart after the induction of EET (Fig. 3, A, B, E, and F). In contrast, EET-A decreased the expression of Wnt5b in heart and adipose tissues (P < 0.05) compared with control mice (Fig. 3, A, C, E, and H). Furthermore, expression of β-catenin was significantly (P < 0.05) greater in heart and adipose tissues of mice after treatment with EET-A (Fig. 3, A, D, E, and I). Importantly, knockdown of PGC-1α prevented the EET-A-mediated effect on Wnt1, Wnt5b, and β-catenin protein expression compared with EET-A-treated mice (Fig. 3, E−I). We further analyzed the effect of EET-A on the inflammatory adipokine NOV/CCN3 in heart tissue of db/db mice. Western blot densitometry analysis revealed that EET-A treatment significantly (P < 0.05) decreased NOV protein expression in heart tissue compared with control mice. The EET-A-mediated decrease in NOV expression was moderately impaired in PGC1-deficient mice (Fig. 3, F and J).
Fig. 3.

Effect of EET-A treatment on Wnt1, Wnt5b, and β-catenin expression in cardiac and adipose tissue of db/db mice. A−D: representative Western blots (A) and densitometry analysis of Wnt1 (B), Wnt5b (C), and β-catenin (D) in cardiac and adipose tissues of db/db control mice and db/db mice treated with EET-A. Results are means ± SE; n = 5. *P < 0.05 vs. db/db control mice in cardiac tissue, #P < 0.05 vs. db/db control mice in adipose tissue. E: EET suppression of nephroblastoma overexpressed (NOV) mRNA. F−J: representative Western blots (F) and densitometry analysis of Wnt1 (G), Wnt5b (H), β-catenin (I), and NOV/CCN3 expression (J) in cardiac tissues of db/db control mice, db/db mice treated with EET-A, and PGC-1α-deficient db/db mice treated with EET-A. Results are means ± SE; n = 3. *P < 0.05 vs. db/db control mice; #P < 0.05 vs. db/db mice treated with EET-A.
Effect of EET-A administration on PGC-1α, HO-1, SIRT1, SIRT3, and pIR972 protein expression levels in heart tissues in control and PGC-1α-deficient mice.
Western blot analysis of heart tissues was performed in all groups of db/db mice. Heart tissues of control db/db mice exhibited lower protein expression of PGC-1α, HO-1, SIRT1, SIRT3, and pIR972. EET-A produced a significant (P < 0.05) increase in the cardiac expression of PGC-1α, HO-1, SIRT1, SIRT3, and pIR972 protein (Fig. 4, A–F). Notably, knockdown of PGC-1α significantly (P < 0.05) prevented the EET-A-mediated beneficial effect on PGC-1α, HO-1, SIRT1, and pIR972 protein expression (Fig. 4, A−D and F). Moreover, knockdown of PGC-1α reduced the EET-A mediated effect on SIRT3 protein expression compared with the EET-A-treated group (Fig. 4E).
Fig. 4.

Effect of EET-A treatment on PGC-1α, heme oxygenase (HO)-1, sirtuin (SIRT)1, SIRT3, and phospho-insulin receptor (Tyr972) (pIR972) in cardiac tissue of db/db mice. A−F: representative Western blots (A) and densitometry analysis of PGC-1α (B), HO-1 (C), SIRT1 (D), SIRT3 (E), and pIR972 (F). Effects of EET-A treatment on TNF-α, IL-6, and matrix metalloproteinase-2 (MMP-2) in cardiac tissues of db/db mice are shown. G−I: mRNA expression of TNF-α (G) and IL-6 (H) as well as representative Western blots and densitometry analysis of MMP-2 (I) in cardiac tissues of db/db control mice, db/db mice treated with EET-A, and PGC-1α-deficient db/db mice treated with EET-A. Results are mean ± SE; n = 4. *P < 0.05 vs. db/db control mice; #P < 0.05 vs. db/db mice treated with EET-A.
EET-A administration decreases expression of inflammatory and cardiac remodeling mice.
We performed RT-PCR to measure the mRNA expression of inflammatory TNF-α and IL-6 and Western blot analysis to demonstrate MMP-2 levels. As shown in Fig. 4, G and H, the mRNA levels of TNF-α and IL-6 in EET-treated db/db mice were reduced compared with control db/db mice. This EET-A effect was prevented in PGC-1α-deficient mice (Fig. 4, G and H). Fibrotic protein expression in db/db mouse cardiac tissue, as measured by the expression of MMP-2 protein, was reduced in EET-A-treated mice compared with control mice (P < 0.05), an effect that was prevented by Ln-PGC-1α (sh) (Fig. 4I).
Effect of EET-A administration on Wnt1, Wnt5b, and β-catenin in adipose tissue of mice.
The expression of NOV in adipose tissue was significantly (P < 0.05) higher compared with that in heart tissue in control db/db mice (Fig. 5A). To corroborate our heart tissue analysis, we measured Wnt1, Wnt5b, and β-catenin expression in adipose tissues of mice. EET-A significantly (P < 0.05) increased the expression of Wnt1 protein expression in adipose tissue compared with control mice (Fig. 5, B and C). In contrast, EET-A treatment decreased the expression of Wnt5b in adipose tissue (P < 0.05) compared with control mice (Fig. 5, B and D). Furthermore, the expression of β-catenin was significantly (P < 0.05) higher in adipose tissue after treatment with EET-A (Fig. 5, B and E). Knockdown of PGC-1α moderately prevented the EET-A-mediated effect on Wnt1, Wnt5b, and β-catenin protein expression compared with the EET-A-treated group of mice (Fig. 5, B–E).
Fig. 5.

Effect of EET-A treatment on Wnt1, Wnt5b, and β-catenin expression in adipose tissue of db/db mice. A: mRNA expression of NOV/CCN3 in adipose tissue versus the heart. B−E: representative Western blots (B) and densitometry analysis of Wnt1 (C), Wnt5b (D), and β-catenin (E) in adipose tissues of db/db control mice, db/db mice treated with EET-A, and PGC-1α-deficient db/db mice treated with EET-A. Effects of EET-A treatment on PGC-1α, HO-1, SIRT1, pIR972, and adiponectin in adipose tissue of db/db mice are shown. F−K: representative Western blots (F) and densitometry analysis of PGC-1α (G), HO-1 (H), SIRT1 (I), pIR972 (J), and adiponectin (K) in cardiac tissues of db/db control mice, db/db mice treated with EET-A, and PGC-1α-deficient db/db mice treated with EET-A. Results are means ± SE; n = 4. *P < 0.05 vs. db/db control mice; #P < 0.05 vs. db/db mice treated with EET-A.
Effect of EET-A administration on PGC-1α, HO-1, SIRT1, insulin receptor phosphorylation, and adiponectin in adipose tissue.
EET-A led to a twofold upregulation of PGC-1α compared with db/db control mice (Fig. 5, F and G). The EET-A-mediated increase in PGC-1α levels was completely prevented in Ln-PGC-1α (sh) mice (Fig. 5, F and G). EET-A increased HO-1 levels compared with db/db control mice (P < 0.05; Fig. 5, F and H). The inhibition of PGC-1α in EET-treated mice completely prevented the EET-A-mediated induction of HO-1 protein expression and reduced it to the level observed in db/db control mice (Fig. 5, F and H). EET-A-treated mice expressed higher SIRT1 levels than control db/db mice (P < 0.05), whereas lentiviral-mediated suppression of PGC-1α reversed the effect of EET-A treatment (Fig. 5, F and I). Furthermore, EET-A upregulated phosphorylation of IR972 (P < 0.05) compared with control db/db mice (Fig. 5, F and J). PGC-1α (sh) induction in EET-A-treated animals prevented EET-mediated phosphorylation of IR972, reducing it to that of untreated db/db mice (Fig. 5, F and J). Moreover, adiponectin protein expression was significantly higher (P < 0.05) in adipose tissue of EET-A-treated db/db mice compared with db/db control mice, an effect that was significantly (P < 0.05) abrogated in PGC-1α-deficient db/db mice treated with EET-A (Fig. 5K).
Effect of EET-A administration on Mfn2, MnSOD, pAKT, pAMPK, and UCP1 in adipose tissue of db/db mice.
EET-A increased Mfn2 and MnSOD protein expression levels severalfold compared with db/db control mice (P < 0.05; Fig. 6, A−C). The inhibition of PGC-1α in EET-treated mice through Ln-PGC-1α (sh) completely prevented the EET-A-mediated induction of MnSOD protein expression (Fig. 6, A−C). Untreated db/db mice and EET-A-PGC-1α db/db mice exhibited decreases in both pAKT and pAMPK in heart tissues (P < 0.05) compared with EET-treated db/db mice (Fig. 6, D and E). EET-A-treated mice expressed higher UCP1 protein expression compared with control db/db mice (P < 0.05), whereas lentiviral-mediated suppression of PGC-1α reversed the effect of EET-A treatment (Fig. 6F).
Fig. 6.

Effect of EET-A treatment on mitofusion 2 (Mfn2), MnSOD, phospho-AKT (pAKT), phospho-AMP-activated protein kinase (pAMPK), and uncoupling protein-1 (UCP1) protein expression in adipose tissue of db/db mice. A−F: representative Western blots (A) and densitometry analysis of Mfn2 (B), MnSOD (C), pAKT (D), pAMPK (E), and UCP1 (F) in adipose tissues of db/db control mice, db/db mice treated with EET-A, and PGC-1α-deficient db/db mice treated with EET-A. Results are means ± SE; n = 3. *P < 0.05 vs. db/db control mice; #P < 0.05 vs. db/db mice treated with EET-A.
EET-A administration decreases expression of adipogenic and inflammatory mediators in adipose tissue of db/db mice.
NOV protein expression in adipose tissue of EET-A-treated mice was decreased (P < 0.05) compared with control db/db mice adipose tissue (Fig. 7, A and B). The effect of EET-A was reversed by lentiviral-mediated suppression of PGC-1α (Fig. 7, A and B). Mice treated with EET-A had significantly (P < 0.01) decreased protein expression of adipogenic fatty acid synthase (FAS), mesoderm-specific transcript (MEST), and adipocyte protein 2 (aP2) in adipose tissue compared with control db/db mice (Fig. 7, A and C−E). However, the levels of these adipogenic markers were not impacted by lentiviral-mediated suppression of PGC-1α (Fig. 7, A and C−E).
Fig. 7.

Effect of EET-A on protein expression of adipogenic markers. A: representative Western blots of NOV, fatty acid synthase (FAS), mesoderm-specific transcript (MEST), and adipocyte protein 2 proteins. B−E: densitometry analyses of NOV (B), FAS (C), MEST proteins (D), and aP2 (E) in adipose tissues of db/db control mice, db/db mice treated with EET-A, and PGC-1α-deficient db/db mice treated with EET-A. Results are means ± SE; n = 3. *P < 0.05 vs. db/db control mice; #P < 0.05 vs. db/db mice treated with EET-A.
Effect of EET-A administration on HO-1, PGC-1α, NOV, β-catenin, Wnt10b, and mitochondrial fusion in the 3T3-L1 mouse embryonic fibroblast cell line.
To corroborate our in vivo results, we performed in vitro experiments on the 3T3-L1 mouse embryonic fibroblast cell line. mRNA expression levels of HO-1, PGC-1α, β-catenin, and Wnt10b were significantly (P < 0.05) increased with the EET treatment (Fig. 8, A−D), whereas expression of NOV mRNA was significantly (P < 0.05) decreased in EET-A-treated 3T3-L1 cells (Fig. 8E). mRNA expression levels of mitochondrial COX-I was significantly (P < 0.05) increased in EET-A-treated cells compared with wild-type cells (Fig. 8F). Furthermore, Mfn2 levels were significantly elevated in wild-type cells treated with EET-A compared with WT control cells (Fig. 8G).
Fig. 8.

Effect of EET-A treatment on wild-type (WT) 3T3-L1 adipocyte cells on gene expression of HO-1, PGC-1α, NOV, β-catenin, Wnt10b, and genes involved in mitochondrial biogenesis and dynamics. A−G: mRNA expression of HO-1(A), PGC-1α (B), NOV (C), β-catenin (D), Wnt10b (E), cytochrome c oxidase subunit I (COX-I; F), and Mfn2 (G) in 3T3-L1-derived adipocyte cells in the absence and presence of EET-A (10 μM) treatment. Results are means ± SE; n = 3–4. *P < 0.05 vs. WT cells.
DISCUSSION
In the present report, we describe a previously unrecognized function of EET as a regulator of NOV, which itself acts as a negative regulator of cardiac function and a potentiator of pericardial fat dysfunction. EET also acts as a positive regulator of Wnt1-β-catenin and improves FS. These actions are associated with increased levels of HO-1 and PGC-1α and attenuation of cardiomyopathy in obese mice. EET agonists reduce oxidative stress and inflammation and increase mitochondrial function and insulin sensitivity in cardiac as well as in adipose tissue. More importantly, EET increases Wnt1-β-catenin signaling and reduces levels of NOV, which consequently increases PGC-1α expression and inhibits pericardial and visceral fat deposition, all of which prevents cardiac remodeling in diabetic cardiomyopathy. The beneficial effects of EET-A on glucose metabolic parameters are abolished in PGC-1α-deficient db/db mice (27). These improvements in the global metabolic parameters and cardiovascular function are associated with increased Wnt1, β-catenin, Sirt1, Sirt3, and HO-1 protein levels and decreased NOV protein levels in both adipose and heart tissue.
Importantly, the effect of EET-A on Wnt1, β-catenin, and Wnt5b protein expression was, to a certain degree, mitigated in both heart and adipose tissues of PGC-1α-deficient db/db mice, hence confirming its role in adipose tissue homeostasis. Wnt1 and Wnt10b maintain preadipocytes in an undifferentiated state through inhibition of the adipogenic markers MEST, aP2, C/EBP, and Wnt5b proteins. Furthermore, both EET and HO-1 attenuate adiposity and vascular dysfunction in obese mice and improve insulin sensitivity and adiponectin expression (58) via an increase of antioxidants (34, 45, 54). Several reports have demonstrated an association between adipogenesis and Wnt signaling in the regulation of adult tissue remodeling and homeostasis. Enhanced levels of EET in mice subjected to myocardial infarction leads to an increase in the Wnt1/β-catenin canonical signaling cascade, which reduces infarct size and ameliorates cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway (8). siRNA-mediated downregulation and inhibition of Wnt1 prevents the EET-HO-1 axis from increasing β-catenin and Wnt1 responsive genes and abrogates these beneficial effects (7, 8). Although Wnt/β-catenin signaling has vital functions during myocardial infarction, pressure-volume overload, inflammatory diseases, cardiomyopathy, chronic hypertension, or congenital heart disease, Wnt/β-catenin levels may vary depending on the injury model or stage of heart remodeling (37).
EET augmentation significantly decreases NOV protein expression in cardiac and adipose tissue, which concurrently upregulates PGC-1α-mediated downstream signaling, enhances mitochondrial function and energy metabolism, and prevents the development of cardiac remodeling in cardiomyopathy. Furthermore, EET reduces insulin resistance via increases in Wnt1 and PGC-1α levels in the heart and adipose tissue of db/db mice. These result in improved metabolic parameters in db/db mice, highlighting the observation that NOV, PGC-1α, and HO-1 are interrelated and have a significant role to play in the development of obesity, diabetes, and cardiomyopathy in the db/db mouse. The observed reduction in NOV expression and upregulation of Wnt1-β-catenin, PGC-1α, and HO-1 in cardiac tissue does not occur in PGC-1α-deficient cells, indicating that PGC-1α is located downstream of NOV in the signaling pathway. Enhanced NOV expression is linked with increases in several inflammatory cytokines that might deleteriously affect insulin signaling, resulting in obesity, adipose tissue deposition, and insulin resistance in human cardiometabolic patients (38). Moreover, downregulation of NOV is associated with a reduction in adipose tissue deposition and inflammatory cytokines as well as enhanced insulin sensitivity in HFD-fed mice coupled with upregulation of PGC-1α in adipose tissue (32).
More importantly, we found that EET-induced Wnt1 and PGC-1α expression and the resulting increase in adipose tissue adiponectin is abolished in PGC-1α-deficient db/db mice, highlighting that the EET-Wnt1-PGC-1α-adiponectin axis plays a significant role in adiposity, diabetes, and cardiac remodeling. Some investigators have reported that an increase in adiponectin expression leads to decreased oxidative stress and inflammation under conditions of obesity induced by a HFD animal model. Similarly, an increase in adiponectin levels decreases diabetes-induced endothelial dysfunction and protects against oxidative stress-induced cardiac myocyte remodeling through the induction of many signaling proteins (13, 15, 29, 48).
This work also highlights the importance of mitochondrial function in the genesis of obesity-induced cardiomyopathy. Enhanced EET activity increases mitochondrial fusion-related Mfn2 protein expression in adipose tissue of mice, an effect that is reversed in PGC-1α-deficient mice. HO-1 decreases heme levels resulting in decreased levels of ROS and oxidative stress (1), both known causes of mitochondrial fragmentation and cardiomyopathy (25, 35). HO-1 expression is increased by EET-A (46, 47), which further enhances mitochondrial biogenesis and viability (1). The EET-A-mediated increases in Mfn2 levels confirm that HO activity is mediated by PGC-1α, a finding that is consistent with prior observations that increased levels of HO-1 and CO-releasing molecules exert cardioprotective, antifibrotic, and antiapoptotic effects in both ischemic and nonischemic cardiomyopathy (reviewed in 36).
Mice treated with EET-A demonstrate a significant improvement in FS, a finding that is reversed in PGC-1α-deficient db/db mice. This is consistent with prior observations that PGC-1α deficiency is associated with LV dilation and poor cardiac contractility (4). The data presented here suggest that it is the EET-induced reduction in NOV production that leads to an improvement in Wnt1-PGC-1α-HO-1 signaling that, in turn, improves cardiac mitochondrial function, energy metabolism, and FS in mice.
Ectopic fat deposition around the heart (pericardial fat) is known to be involved in cardiac remodeling because it produces oxidative stress and inflammatory cytokines that cause cardiovascular disease. Hence, the observed decrease in pericardial fat deposition in the EET-A-treated db/db mouse may, itself, be a prominent cause of improved FS. Ectopic fat deposition around the heart in humans is linked to cardiac remodeling and cardiomyopathy, which itself increases the likelihood of further obesity, insulin resistance, and even more LV dysfunction (17, 20, 30, 39, 41).
In addition to NOV, EET augmentation also reduces levels of TNF-α and IL-6 while increasing MnSOD, UCP1, and SIRT1 in adipose tissue. Normalization of mitochondrial function and reduction in inflammatory cytokine levels has the ability to reprogram adipocytes and reverse their phenotype from inflammatory to a healthy, functional beige adipose tissue phenotype that expresses normal mitochondrial quality and thermogenic genes responsible for the attenuation of cardiomyopathy. UCP1 is associated with increased energy expenditure in, and “browning” of, adipose tissue, leading to synthesis of adipokines and the reduction of obesity (11). EET augmentation also decreases MMP-2 expression in cardiac tissue, which provides a mechanism for the prior observation that EET decreases cardiac remodeling. Induction of HO-1 reduces MMP-2 and cardiac remodeling (for a review, see Ref. 1). The decrease in MMP-2 and improvement in FS are also accompanied by increased levels of AKT and AMPK in adipose tissue, a result of either EET or EET-mediated increases in PGC-1α-HO-1. Similarly, in metabolic organs such as adipose tissue, an increase in AMPK activates catabolic function and brown adipocyte-like cells (57), which may increase the conversion of white fat to brown fat-like cells and may improve cardiac function.
Finally, in addition to a reduction in NOV, TNF-α, and IL-6 levels, an increase in Wnt1 and β-catenin levels, and an increase in PGC-1α-HO-1 gene expression, we observed a significant reduction in levels of the adipogenic markers FAS, aP2, and MEST in the adipose tissue of mice. The reduction of adipogenic and inflammatory markers in mice treated with EET-A is coupled with increased levels of adiponectin and HO-1, both key to the maintenance of adipocyte maturation in favor of early-stage adipocyte differentiation, that is, healthy adipocytes (7, 49, 50, 59).
In summary, EET decreases NOV and increases Wnt1b in both cardiac and pericardial fat that results in a decrease in inflammatory cytokines and an increase in adiponectin, PGC-1α, HO-1, FS, and mitochondrial integrity. This study describes how EET acts through NOV and Wnt1-β-catenin to attenuate obesity-induced cardiomyopathy. As shown in Fig. 9, EET-A reduces body fat mass, maintains blood glucose homeostasis, reduces pericardial fat deposition, and enhances mitochondrial function. Most importantly, EET treatments restore thermogenic genes, characteristic of a brown fat cell-like phenotype and myocardial systolic function through an increase in Wnt1-β-catenin, PGC-1α, and mitochondrial fusion. Therefore, EET leads to the reprogramming of the pericardial fat phenotype to a brown fat cell phenotype to express functional PGC-1α, thermogenic genes (as evidenced by a decrease in FAS), and an increase in insulin receptor phosphorylation, which contribute to the improvement in cardiac function. The findings that EET activates Wnt-β-catenin and inhibits NOV levels that control PGC-1α-HO-1 in visceral adipose and cardiac tissues identifies both NOV inhibition and increases in Wnt1 as novel pharmacological targets to attenuate and perhaps reverse cardiac remodeling in obesity-induced cardiomyopathies. Hence, EET offers a multifactorial clinical approach to the treatment of cardiomyopathy and concomitant metabolic disorders.
Fig. 9.
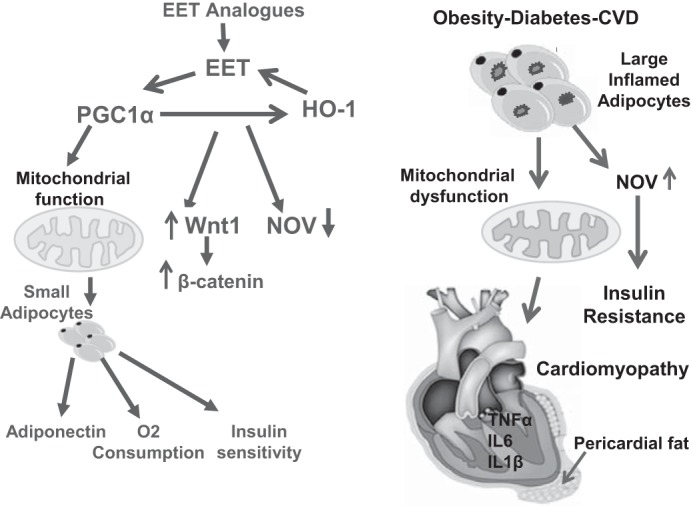
Schematic description of EET-mediated perturbations in the Wnt1-NOV-PGC-1α-HO-1 pathway in cardiac and adipose tissue of db/db mice. Obesity-mediated decrease in EET, HO-1, and PGC-1α promotes mitochondrial dysfunction and thermogenic genes that contribute to the development of cardiomyopathy. EET induction leads to a decrease in adipogenic FAS, MEST, and aP2 expression in adipose tissue. The EET-A-mediated stimulation of Wnt signaling causes a reduction in NOV and subsequent increases in PGC-1α and HO-1 that lead to increased V̇o2, increased mitochondrial integrity, thermogenic protein expression levels, and insulin sensitivity that, in concert, results in an improvement in cardiac function.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-34300 to N. G. Abraham and HL-109015 to J. I. Shapiro, and by The Renfield Foundation, The Rockefeller University.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.G.A. conceived and designed research; J.C., S.P.S., G.J., L.V., I.B., and H.J. performed experiments; S.P.S. prepared figures; J.M., L.V., and N.G.A. analyzed data; J.M., performed ECHO; J.R.F., EET agonist; M.A., J.I.S., and N.G.A. edited and revised manuscript; S.P.S. and N.G.A. interpreted results of experiments; J.I.S. and N.G.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Jennifer Brown for editorial assistance in preparing the manuscript.
REFERENCES
- 1.Abraham NG, Junge JM, Drummond GS. Translational significance of heme oxygenase in obesity and metabolic syndrome. Trends Pharmacol Sci 37: 17–36, 2016. doi: 10.1016/j.tips.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham NG, Rezzani R, Rodella L, Kruger A, Taller D, Li Volti G, Goodman AI, Kappas A. Overexpression of human heme oxygenase-1 attenuates endothelial cell sloughing in experimental diabetes. Am J Physiol Heart Circ Physiol 287: H2468–H2477, 2004. doi: 10.1152/ajpheart.01187.2003. [DOI] [PubMed] [Google Scholar]
- 3.Abraham NG, Sodhi K, Silvis AM, Vanella L, Favero G, Rezzani R, Lee C, Zeldin DC, Schwartzman ML. CYP2J2 targeting to endothelial cells attenuates adiposity and vascular dysfunction in mice fed a high-fat diet by reprogramming adipocyte phenotype. Hypertension 64: 1352–1361, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 1: 259–271, 2005. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Bodiga S, Zhang R, Jacobs DE, Larsen BT, Tampo A, Manthati VL, Kwok WM, Zeldin DC, Falck JR, Gutterman DD, Jacobs ER, Medhora MM. Protective actions of epoxyeicosatrienoic acid: dual targeting of cardiovascular PI3K and KATP channels. J Mol Cell Cardiol 46: 978–988, 2009. doi: 10.1016/j.yjmcc.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudina S, Bugger H, Sena S, O’Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, Theobald H, Khalimonchuk O, Wayment B, Sheng X, Rodnick KJ, Centini R, Chen D, Litwin SE, Weimer BE, Abel ED. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119: 1272–1283, 2009. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao J, Peterson SJ, Sodhi K, Vanella L, Barbagallo I, Rodella LF, Schwartzman ML, Abraham NG, Kappas A. Heme oxygenase gene targeting to adipocytes attenuates adiposity and vascular dysfunction in mice fed a high-fat diet. Hypertension 60: 467–475, 2012. doi: 10.1161/HYPERTENSIONAHA.112.193805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao J, Tsenovoy PL, Thompson EA, Falck JR, Touchon R, Sodhi K, Rezzani R, Shapiro JI, Abraham NG. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat 116-117: 76–86, 2015. doi: 10.1016/j.prostaglandins.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhary KR, Abukhashim M, Hwang SH, Hammock BD, Seubert JM. Inhibition of soluble epoxide hydrolase by trans-4- [4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is protective against ischemia-reperfusion injury. J Cardiovasc Pharmacol 55: 67–73, 2010. doi: 10.1097/FJC.0b013e3181c37d69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289, 2010. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, Wu J, Gunawardana SC, Banks AS, Camporez JP, Jurczak MJ, Kajimura S, Piston DW, Mathis D, Cinti S, Shulman GI, Seale P, Spiegelman BM. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156: 304–316, 2014. doi: 10.1016/j.cell.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, Vetto AP, Petrosyan S, Marsala M, Murphy AN, Williams DS, Spiegelman BM, Cleveland DW. Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab 15: 778–786, 2012. doi: 10.1016/j.cmet.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeMarco VG, Dellsperger KC. Adipocyte-derived adiponectin is cardioprotective: fat cells can be our friends. Am J Physiol Heart Circ Physiol 291: H2588–H2589, 2006. doi: 10.1152/ajpheart.00862.2006. [DOI] [PubMed] [Google Scholar]
- 14.Dhanasekaran A, Al-Saghir R, Lopez B, Zhu D, Gutterman DD, Jacobs ER, Medhora M. Protective effects of epoxyeicosatrienoic acids on human endothelial cells from the pulmonary and coronary vasculature. Am J Physiol Heart Circ Physiol 291: H517–H531, 2006. doi: 10.1152/ajpheart.00953.2005. [DOI] [PubMed] [Google Scholar]
- 15.Essick EE, Ouchi N, Wilson RM, Ohashi K, Ghobrial J, Shibata R, Pimentel DR, Sam F. Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling. Am J Physiol Heart Circ Physiol 301: H984–H993, 2011. doi: 10.1152/ajpheart.00428.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falck JR, Koduru SR, Mohapatra S, Manne R, Atcha R, Manthati VL, Capdevila JH, Christian S, Imig JD, Campbell WB. 14,15-Epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates: carboxylate modifications. J Med Chem 57: 6965–6972, 2014. doi: 10.1021/jm500262m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res 118: 1786–1807, 2016. doi: 10.1161/CIRCRESAHA.115.306885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaiani S, Avogaro A, Bombonato GC, Bolognesi M, Amor F, Vigili de Kreutzenberg S, Guarneri G, Sacerdoti D. Nonalcoholic fatty liver disease (NAFLD) in nonobese patients with diabetes: prevalence and relationships with hemodynamic alterations detected with Doppler sonography. J Ultrasound 12: 1–5, 2009. doi: 10.1016/j.jus.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodman AI, Chander PN, Rezzani R, Schwartzman ML, Regan RF, Rodella L, Turkseven S, Lianos EA, Dennery PA, Abraham NG. Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J Am Soc Nephrol 17: 1073–1081, 2006. doi: 10.1681/ASN.2004121082. [DOI] [PubMed] [Google Scholar]
- 20.Granér M, Pentikäinen MO, Nyman K, Siren R, Lundbom J, Hakkarainen A, Lauerma K, Lundbom N, Nieminen MS, Petzold M, Taskinen MR. Cardiac steatosis in patients with dilated cardiomyopathy. Heart 100: 1107–1112, 2014. doi: 10.1136/heartjnl-2013-304961. [DOI] [PubMed] [Google Scholar]
- 21.Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol 42: 687–691, 2007. doi: 10.1016/j.yjmcc.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guarini G, Kiyooka T, Ohanyan V, Pung YF, Marzilli M, Chen YR, Chen CL, Kang PT, Hardwick JP, Kolz CL, Yin L, Wilson GL, Shokolenko I, Dobson JG Jr, Fenton R, Chilian WM. Impaired coronary metabolic dilation in the metabolic syndrome is linked to mitochondrial dysfunction and mitochondrial DNA damage. Basic Res Cardiol 111: 29, 2016. doi: 10.1007/s00395-016-0547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gutterman DD. Mitochondria and reactive oxygen species: an evolution in function. Circ Res 97: 302–304, 2005. doi: 10.1161/01.RES.0000179773.18195.12. [DOI] [PubMed] [Google Scholar]
- 24.Hong SJ, Park CG, Seo HS, Oh DJ, Ro YM. Associations among plasma adiponectin, hypertension, left ventricular diastolic function and left ventricular mass index. Blood Press 13: 236–242, 2004. doi: 10.1080/08037050410021397. [DOI] [PubMed] [Google Scholar]
- 25.Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B, Joseph R, Prabhu SD, Suliman HB, Piantadosi CA, Agarwal A, George JF. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 1: e85817, 2016. doi: 10.1172/jci.insight.85817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev 92: 101–130, 2012. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleiner S, Mepani RJ, Laznik D, Ye L, Jurczak MJ, Jornayvaz FR, Estall JL, Chatterjee Bhowmick D, Shulman GI, Spiegelman BM. Development of insulin resistance in mice lacking PGC-1α in adipose tissues. Proc Natl Acad Sci USA 109: 9635–9640, 2012. doi: 10.1073/pnas.1207287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsen BT, Campbell WB, Gutterman DD. Beyond vasodilatation: non-vasomotor roles of epoxyeicosatrienoic acids in the cardiovascular system. Trends Pharmacol Sci 28: 32–38, 2007. doi: 10.1016/j.tips.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Zhang H, Chen J, Dellsperger KC, Hill MA, Zhang C. Adiponectin abates diabetes-induced endothelial dysfunction by suppressing oxidative stress, adhesion molecules, and inflammation in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 303: H106–H115, 2012. doi: 10.1152/ajpheart.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Fox CS, Hickson DA, May WL, Ding J, Carr JJ, Taylor HA. Pericardial fat and echocardiographic measures of cardiac abnormalities: the Jackson Heart Study. Diabetes Care 34: 341–346, 2011. doi: 10.2337/dc10-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma B, Xiong X, Chen C, Li H, Xu X, Li X, Li R, Chen G, Dackor RT, Zeldin DC, Wang DW. Cardiac-specific overexpression of CYP2J2 attenuates diabetic cardiomyopathy in male streptozotocin-induced diabetic mice. Endocrinology 154: 2843–2856, 2013. doi: 10.1210/en.2012-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinerie C, Garcia M, Do TT, Antoine B, Moldes M, Dorothee G, Kazazian C, Auclair M, Buyse M, Ledent T, Marchal PO, Fesatidou M, Beisseiche A, Koseki H, Hiraoka S, Chadjichristos CE, Blondeau B, Denis RG, Luquet S, Fève B. NOV/CCN3: a new adipocytokine involved in obesity-associated insulin resistance. Diabetes 65: 2502–2515, 2016. doi: 10.2337/db15-0617. [DOI] [PubMed] [Google Scholar]
- 33.Merabet N, Bellien J, Glevarec E, Nicol L, Lucas D, Remy-Jouet I, Bounoure F, Dreano Y, Wecker D, Thuillez C, Mulder P. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J Mol Cell Cardiol 52: 660–666, 2012. doi: 10.1016/j.yjmcc.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 34.Mügge A, Elwell JH, Peterson TE, Harrison DG. Release of intact endothelium-derived relaxing factor depends on endothelial superoxide dismutase activity. Am J Physiol Cell Physiol 260: C219–C225, 1991. [DOI] [PubMed] [Google Scholar]
- 35.Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW II, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D, Youle RJ; American Heart Association Council on Basic Cardiovascular Sciences, Council on Clinical Cardiology, and Council on Functional Genomics and Translational Biology . Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ Res 118: 1960–1991, 2016. doi: 10.1161/RES.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otterbein LE, Foresti R, Motterlini R. Heme oxygenase-1 and carbon monoxide in the heart: the balancing act between danger signaling and pro-survival. Circ Res 118: 1940–1959, 2016. doi: 10.1161/CIRCRESAHA.116.306588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozhan G, Weidinger G. Wnt/β-catenin signaling in heart regeneration. Cell Regen (Lond) 4: 3, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pakradouni J, Le Goff W, Calmel C, Antoine B, Villard E, Frisdal E, Abifadel M, Tordjman J, Poitou C, Bonnefont-Rousselot D, Bittar R, Bruckert E, Clément K, Fève B, Martinerie C, Guérin M. Plasma NOV/CCN3 levels are closely associated with obesity in patients with metabolic disorders. PLoS One 8: e66788, 2013. doi: 10.1371/journal.pone.0066788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pucci G, Battista F, de Vuono S, Boni M, Scavizzi M, Ricci MA, Lupattelli G, Schillaci G. Pericardial fat, insulin resistance, and left ventricular structure and function in morbid obesity. Nutr Metab Cardiovasc Dis 24: 440–446, 2014. doi: 10.1016/j.numecd.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 40.Pung YF, Sam WJ, Hardwick JP, Yin L, Ohanyan V, Logan S, Di Vincenzo L, Chilian WM. The role of mitochondrial bioenergetics and reactive oxygen species in coronary collateral growth. Am J Physiol Heart Circ Physiol 305: H1275–H1280, 2013. doi: 10.1152/ajpheart.00077.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riehle C, Abel ED. Insulin signaling and heart failure. Circ Res 118: 1151–1169, 2016. doi: 10.1161/CIRCRESAHA.116.306206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romashko M, Schragenheim J, Abraham NG, McClung JA. Epoxyeicosatrienoic acid as therapy for diabetic and ischemic cardiomyopathy. Trends Pharmacol Sci 37: 945–962, 2016. doi: 10.1016/j.tips.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, Ricquier D. The biology of mitochondrial uncoupling proteins. Diabetes 53, Suppl 1: S130–S135, 2004. doi: 10.2337/diabetes.53.2007.S130. [DOI] [PubMed] [Google Scholar]
- 44.Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res 107: 825–838, 2010. doi: 10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sacerdoti D, Bolognesi M, Di Pascoli M, Gatta A, McGiff JC, Schwartzman ML, Abraham NG. Rat mesenteric arterial dilator response to 11,12-epoxyeicosatrienoic acid is mediated by activating heme oxygenase. Am J Physiol Heart Circ Physiol 291: H1999–H2002, 2006. doi: 10.1152/ajpheart.00082.2006. [DOI] [PubMed] [Google Scholar]
- 46.Sacerdoti D, Colombrita C, Di Pascoli M, Schwartzman ML, Bolognesi M, Falck JR, Gatta A, Abraham NG. 11,12-Epoxyeicosatrienoic acid stimulates heme-oxygenase-1 in endothelial cells. Prostaglandins Other Lipid Mediat 82: 155–161, 2007. doi: 10.1016/j.prostaglandins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Sacerdoti D, Pesce P, Di Pascoli M, Bolognesi M. EETs and HO-1 cross-talk. Prostaglandins Other Lipid Mediat 125: 65–79, 2016. doi: 10.1016/j.prostaglandins.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Shibata R, Ouchi N, Murohara T. Adiponectin and cardiovascular disease. Circ J 73: 608–614, 2009. doi: 10.1253/circj.CJ-09-0057. [DOI] [PubMed] [Google Scholar]
- 49.Singh SP, Bellner L, Vanella L, Cao J, Falck JR, Kappas A, Abraham NG. Downregulation of PGC-1α prevents the beneficial effect of EET-heme oxygenase-1 on mitochondrial integrity and associated metabolic function in obese mice. J Nutr Metab 2016: 9039754, 2016. doi: 10.1155/2016/9039754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh SP, Schragenheim J, Cao J, Falck JR, Abraham NG, Bellner L. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat 125: 8–18, 2016. doi: 10.1016/j.prostaglandins.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal 12: 537–577, 2010. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther 331: 906–916, 2009. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun D, Cuevas AJ, Gotlinger K, Hwang SH, Hammock BD, Schwartzman ML, Huang A. Soluble epoxide hydrolase-dependent regulation of myogenic response and blood pressure. Am J Physiol Heart Circ Physiol 306: H1146–H1153, 2014. doi: 10.1152/ajpheart.00920.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turkseven S, Kruger A, Mingone CJ, Kaminski P, Inaba M, Rodella LF, Ikehara S, Wolin MS, Abraham NG. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am J Physiol Heart Circ Physiol 289: H701–H707, 2005. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- 55.Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab 3: 333–341, 2006. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 56.Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, Fiehn O, Hammock BD, Weiss RH. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol 52: 314–323, 2008. doi: 10.1097/FJC.0b013e318185fa3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Dam AD, Kooijman S, Schilperoort M, Rensen PC, Boon MR. Regulation of brown fat by AMP-activated protein kinase. Trends Mol Med 21: 571–579, 2015. doi: 10.1016/j.molmed.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 58.Vanella L, Sodhi K, Kim DH, Puri N, Maheshwari M, Hinds TD Jr, Bellner L, Goldstein D, Peterson SJ, Shapiro JI, Abraham NG. Increased heme-oxygenase 1 expression in mesenchymal stem cell-derived adipocytes decreases differentiation and lipid accumulation via upregulation of the canonical Wnt signaling cascade. Stem Cell Res Ther 4: 28, 2013. doi: 10.1186/scrt176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waldman M, Bellner L, Vanella L, Schragenheim J, Sodhi K, Singh SP, Lin D, Lakhkar A, Li J, Hochhauser E, Arad M, Darzynkiewicz Z, Kappas A, Abraham NG. Epoxyeicosatrienoic acids regulate adipocyte differentiation of mouse 3T3 cells, via PGC-1α activation, which is required for HO-1 expression and increased mitochondrial function. Stem Cells Dev 25: 1084–1094, 2016. doi: 10.1089/scd.2016.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 61.Zha W, Edin ML, Vendrov KC, Schuck RN, Lih FB, Jat JL, Bradbury JA, DeGraff LM, Hua K, Tomer KB, Falck JR, Zeldin DC, Lee CR. Functional characterization of cytochrome P450-derived epoxyeicosatrienoic acids in adipogenesis and obesity. J Lipid Res 55: 2124–2136, 2014. doi: 10.1194/jlr.M053199. [DOI] [PMC free article] [PubMed] [Google Scholar]