

Abstract
Tuberculosis (TB) is one of the oldest known human diseases and is transmitted by the bacteria Mycobacterium tuberculosis (Mtb). TB has a rich history with evidence of TB infections dating back to 5,800 b.c. TB is unique in its ability to remain latent in an individual for decades, with the possibility of later reactivation, causing widespread systemic symptoms. Currently, it is estimated that more than one-third of the world’s population (~2 billion people) are infected with Mtb. Prolonged periods of therapy and complexity of treatment regimens, especially in active infection, have led to poor compliance in patients being treated for TB. Therefore, it is vitally important to have a thorough knowledge of the pathophysiology of Mtb to understand the disease progression, as well as to develop novel diagnostic tests and treatments. Alveolar macrophages represent both the primary host cell and the first line of defense against the Mtb infection. Apoptosis and autophagy of macrophages play a vital role in the pathogenesis and also in the host defense against Mtb. This review will outline the role of these two cellular processes in defense against Mtb with particular emphasis on innate immunity and explore developing therapies aimed at altering host responses to the disease.
Keywords: tuberculosis, apoptosis, endoplasmic reticulum stress, autophagy
tuberculosis (TB), transmitted by the bacteria Mycobacterium tuberculosis (Mtb), is a global epidemic and 1 of the top 10 causes of mortality worldwide. In 2015, the disease accounted for 1.4 million deaths, surpassing the mortality of other epidemic diseases, such as human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS) and malaria (144). The disease burden of TB extends beyond mortality, also contributing to high levels of disability-adjusted life years (DALYs) in many regions (38). In fact, estimates show that Mtb surpasses malaria for DALYS due to illness (65 million DALYs for TB vs. 50 million DALYs for malaria; 144). Commonly regarded as a disease of the poor and disenfranchised, TB is more common in developing countries, with up to 40% of some populations being carriers of the bacteria (101). In 2015, there were 10.4 million new cases of TB, with 6 countries accounting for 60% of these cases: India, Indonesia, China, Nigeria, Pakistan, and South Africa (144).
Individuals infected with Mtb commonly present with pulmonary or generalized complaints, including cough, chest pain, fatigue, and unintentional weight loss (65). Though the lungs are the most common organs affected, the bacteria can disseminate intravascularly and seed multiple organs and structures within the body. Immunocompromised individuals are especially susceptible to disseminated disease and have higher rates of mortality associated with infection (85). Furthermore, disseminated disease can present as a diagnostic challenge, as disease manifestation can vary among different individuals (104, 110, 135).
The prolonged incubation of the bacteria, along with their ability to remain latent in an individual for extended periods of time, makes disease eradication almost impossible. In fact, it is estimated that >90% of TB infections are latent (152). Because of this, advances in clinical diagnosis are of paramount importance in controlling the spread of disease. Latent infections are commonly tested by the purified protein derivative test or the IFN-γ release assay (3). Both assays test the cell-mediated immune response and have comparable sensitivities (86). In active infection, diagnostic recommendations are chest radiograph followed by three sputum specimens for both acid-fast staining and culture (23). In addition, the Centers for Disease Control and Prevention now recommends nucleic acid amplification of respiratory specimens for all individuals in which the diagnosis of Mtb is considered. However, if clinical suspicion for infection is high, empiric therapy may be initiated even before the presence of a positive test.
Although the presence of TB dates back thousands of years (20, 151), efficacious treatments have only been present for the last half-century. Whereas initial treatments were crude and involved isolating patients in sanatoriums (42), management now consists of long-term antibiotics. Streptomycin, the first effective treatment for Mtb, was developed in 1946, and many other antibiotics were developed shortly thereafter (42). Isoniazid and pyrazinamide, discovered in the 1950s, are still in use today. Isoniazid therapy for 9 mo is the current preferred therapy for latent Mtb infection (83). For uncomplicated active infections, the treatment of choice consists of using four drugs: rifampin, isoniazid, pyrazinamide, and ethambutol (RIPE therapy) for 2 mo, followed by rifampicin and isoniazid for another 4 mo (144).
Prolonged periods of therapy and complicated treatment regimens have led to poor compliance in patients being treated for TB. The World Health Organization estimates that ~4% of new cases of Mtb are already multidrug resistant and only half of patients who receive treatment for Mtb are considered cured on follow-up (144). Poor compliance, along with inappropriate use of antimicrobials, has effectively paved the way for the development of drug-resistant, multidrug-resistant (MDR-TB), and extensively drug-resistant tuberculosis (10). Treatments for these cases vary depending on patterns of drug resistance and, in some cases, may require surgical resection (29). With these limitations in diagnosis and treatment, a fundamental understanding of the pathophysiology of TB is necessary to develop both novel diagnostic tools as well as therapeutics.
General Pathogenesis of Tuberculosis
M. tuberculosis (Mtb) is a nonmotile, facultative intracellular bacteria of macrophages. Entry into resident alveolar macrophages following exposure is an absolute prerequisite for Mtb to cause lung infection in a susceptible host. Experiments have demonstrated that the depletion of resident alveolar macrophages in mice using liposome-encapsulated, dichloromethylene diphosphonate protects mice from Mtb infection (68) but increases the susceptibility of mice to other infections, such as Streptococcus pneumoniae (60). These results suggest that although alveolar macrophages protect the host against typical extracellular bacterial pathogens, they facilitate the establishment of Mtb at least during the initial stages of infection. Once Mtb comes in contact with the alveolar macrophages, they are readily engulfed by the granulocytes using a variety of phagocytic receptors. The complement receptors (C receptors), mannose receptors, surfactant protein-A (SP-A), cluster of differentiation 14 (CD14) receptor, and scavenger receptors are some of the important receptors involved in the adhesion and entry of the Mtb in the macrophages (27, 106, 139). C receptors are important for bacterial opsonization (a process by which a pathogen is marked for ingestion and eliminated by phagocytes; 117). Mannose receptors assist the macrophages in phagocytosing unopsonized bacteria (117). SP-A acts as an opsonin and may also modulate the activity of other receptors to enhance macrophage binding and uptake of Mtb (32). This initial uptake of bacteria and subsequent intracellular growth inside alveolar macrophages establish the infection within the host. However, late in the infection, Mtb can exist as an extracellular microbe in the necrotic cavities of lung parenchyma that connect to airways, which provide an oxygen-rich environment and facilitate aerosol transmission between hosts. Although resident alveolar macrophages are the primary cells involved in the initial uptake of Mtb, during the later stages of infection, dendritic cells and monocyte-derived macrophages also participate in the phagocytic process (41, 131). The interaction between Mtb and these antigen-presenting cells ultimately activates the macrophages and stimulates the production of cytokines and chemokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1B, IL-6, IL-12, IL-15, IL-18, and interferon (IFN)-γ (132). These cytokines further stimulate macrophages and play a crucial role in the inflammatory response and the outcome of mycobacterial infection.
The pathogenesis of TB is dependent on the delicate interplay between the mechanisms involved in host defense and the various survival strategies employed by Mtb (115). Alveolar macrophages can facilitate both innate and adaptive defense mechanisms to combat Mtb infection. The mechanisms involved in the development of adaptive immunity to Mtb require a close interaction between CD4+ T cells and dendritic cells. First, the major histocompatibility complex (MHC) class II molecules on dendritic cells present processed Mtb antigens to CD4+ T cells. Subsequent macrophage activation by type 1 T helper (Th1) cells involves the production of IFN-γ by Th1 cells and the interaction of its T-cell receptor and CD4 molecule with Mtb peptide-MHC class II complex on macrophages in the presence of costimulation by CD40 ligand (CD40L) on the Th1 cell and CD40 on the macrophage. The dendritic cells, which are able to uptake the infected apoptotic macrophages and extracellular Mtb, stimulate CD8+ T cells by cross-presenting Mtb antigen via MHC class I molecules. Then, the MHC class I molecules, expressed on all nucleated cells, present mycobacterial proteins to activated, antigen-specific CD8+ T cells and are eliminated. The stimulation of T cells is facilitated by the costimulatory signals B7.1 (CD80) and B7.2 (CD86), which are expressed on macrophages and dendritic cells and bind to CD28 on naive T cells. In addition, several cytokines produced by the macrophages and dendritic cells, such as IL-2, IL-12, IL-18, and IL-23, help in the stimulation of T lymphocytes (98). The strategies employed by Mtb to circumvent adaptive immunity include 1) increasing the production of anti-inflammatory cytokines or decreasing the expression of proinflammatory cytokines, leading to the dampening of T-cell stimulation and reducing the expression of antigen-presenting molecules in macrophages (33, 103); and 2) attenuating the expression of the costimulatory molecule, CD80 (111), and impairing adequate costimulatory signaling leading to T-cell anergy and apoptosis (43, 44). These considerations, taken together, undoubtedly demonstrate that the interactions between Mtb and host macrophages play a central role in the pathogenesis of TB.
Macrophages can also destroy the microbes directly, without requiring the activation of the adaptive T-cell response. In this regard, the active metabolite of vitamin D, 1,25-dihydroxyvitamin D, has been shown to assist macrophages in attenuating the growth of Mtb (22, 107). Interestingly, vitamin D deficiency has been reported in some patients with TB (21) and in susceptible populations at higher risk of being infected with TB (141). Inside the activated macrophages, the putative mechanisms involved in the killing of Mtb include the generation of reactive oxygen and nitrogen species. In an experimental model of Mtb infection, it was determined that mice that lack the ability to produce superoxide suffered from increased early outgrowth of mycobacteria (19). However, the increased susceptibility to TB is not seen in patients with chronic granulomatous disease, a disease associated with the defective production of reactive oxygen species (143). The precise role of reactive nitrogen species in TB is also debatable and remains to be elucidated, although the production of nitric oxide by the macrophages in response to mycobacterial infection has been shown to be bactericidal (108). In addition, natural resistance-associated macrophage protein (Nramp1), an integral membrane protein expressed exclusively in macrophage/monocytes and polymorphonuclear leukocytes, influences the rate of intracellular replication of Mtb in macrophages (9). Nramp1 belongs to a family of transporters that facilitate the cellular entry of metal ions such as Fe2+, which ultimately induce macrophages to produce antimicrobial toxic radicals (153). Experiments have shown that the addition of small quantities of iron to resident alveolar macrophages stimulates their antimicrobial activity and that this effect is abrogated by the addition of antioxidants, catalase, or mannitol, suggesting that the Fe(II)-mediated inhibition of mycobacterial growth is mediated by the Fenton/Haber-Weiss reaction and free hydroxyl radicals (153). Interestingly, studies from West Africa have shown that a functional polymorphism in the promoter region of Nramp1 is associated with reduced gene expression and increased susceptibility to TB (8, 11). However, further mechanistic studies in animal models and humans are required to clearly delineate the role of this protein in the pathogenesis of TB. Apoptosis and autophagy are other effector mechanisms that the infected host uses to limit the outgrowth of Mtb and will be discussed in detail below.
Apoptosis and Endoplasmic Reticulum Stress in Tuberculosis
The endoplasmic reticulum (ER) participates in the modification and proper folding of newly synthesized proteins. ER is also the major storage site of intracellular Ca2+ and synthesis of lipids. Cellular infection or stress induces a loss of Ca2+ from the ER and an increase in the intracellular redox state, resulting in impaired folding and subsequent accumulation of misfolded proteins in the ER. The aggregation of misfolded proteins ultimately results in ER stress (ERS; 15). This pathological event triggers an essential cellular response called the unfolded protein response (UPR; 94), which halts early protein synthesis to attenuate the accumulation of unfolded or misfolded proteins in the ER, restoring normal cellular function. However, prolonged or uncontrolled ERS activates downstream signaling pathways that push the cell toward apoptosis. There are three major ER-localized signaling pathways involved in ERS: inositol-requiring-1α (IRE1α), double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6; 148). The pathway for ERS-induced apoptosis also includes CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP); glucose-regulated protein-78 (GRP78/BiP), a major UPR target protein that targets misfolded proteins for degradation (100); and phosphorylated alpha subunit of eukaryotic initiation factor 2 (eIF2α), which inhibits protein synthesis. The role of ERS in promoting the apoptosis (90, 129) of macrophages infected with Mtb, and thereby limiting the spread of infection, is discussed below.
Necrosis and apoptosis are the two known mechanisms by which cells die. Necrosis is caused by accidental and irreversible damage to the plasma membrane, which destroys the integrity of the cell, whereas apoptosis is a programmed cell death associated with changes in nuclear organization and chromatin fragmentation. In apoptosis, the cytoplasm and other cellular components never leave the dying cell, and hence apoptosis is immunologically silent (125). Although apoptosis in microbial infections causes tissue damage, the initiation of apoptosis may be beneficial to the host, as it promotes the removal of the microorganisms (125). Apoptosis plays a vital role in host defenses against intracellular pathogens, including Mtb, by preventing the release of intracellular pathogens and the spread of mycobacterial infection. The apoptosis of macrophages can limit mycobacterial infection by activating both innate and adaptive immune response (79, 102). Apoptotic bodies containing cell cytoplasm, other cellular organelles, and microorganisms are taken up by macrophages and dendritic cells via receptor-mediated phagocytosis and then undergo degradation and are presented via MHC class II complexes. This process is defined as efferocytosis and may promote both innate and adaptive immunity. There is currently intense investigation and debate over the mechanisms and functional consequences of macrophage apoptosis in TB. Recent studies have shown that alveolar macrophages undergo apoptosis as an innate defense response against Mtb (7). In an experiment conducted to show the role of apoptosis and necrosis in the viability of mycobacteria, researchers exposed peripheral blood monocytes infected with replicating and viable M. bovis bacillus Calmette-Guérin (BCG) to the cellular inducers of either necrosis (hydrogen peroxide) or apoptosis (adenosine triphosphate). The treatment killed the infected monocytes. However, the necrosis of monocytes had no effect on the microbial viability, but the apoptosis of monocytes reduced the mycobacterial viability (88). Therefore, it is evident that macrophage death by either apoptosis or necrosis appears to have drastically different outcomes for the course of infection. Apoptotic cell death of Mtb-infected macrophages is associated with mycobacterial killing (64, 77, 88, 95) and stimulation of T-cell responses via antigen presentation (116, 142). In contrast, necrotic cell death of macrophages aggravates infection by allowing the release of viable mycobacteria for subsequent reinfection (67, 88, 126). Although some recent work has demonstrated that necrosis, too, can follow a programmed series of events, a beneficial role of necrosis in Mtb infection has yet to be found (13). In TB, necrosis may also be the primary pathological event in granuloma formation, which causes liquefaction, cavitation, and tissue damage, resulting in massive expansion of bacillary numbers (78).
Almost all mycobacteria can induce apoptosis. Mildly virulent mycobacterial strains, such as BCG, and the avirulent strain, Mtb H37Ra, are better inducers of macrophage apoptosis compared with the more virulent mycobacteria, such as Mtb H37Rv (31, 55, 56, 63, 105). In fact, studies have shown that virulent Mtb may not elicit macrophage apoptosis, but rather induce macrophage necrosis to avoid bacterial killing (66). These virulent mycobacteria have effective mechanisms to evade apoptosis and survive. To escape apoptosis, virulent Mtb manipulate the ultrastructural architecture of the macrophage ER. Macrophages infected with the virulent strain, H37Rv, have predominantly rough ER (RER) compared with macrophages infected with the avirulent strain, H37Ra, which have a smooth ER phenotype. The functional consequences of this phenotype change result in increased cytosolic Ca2+ levels and the simultaneous induction of phosphatidyl choline/phosphatidyl ethanolamine (PC/PE) expression in the macrophages infected with the H37Ra strain, which facilitates apoptosis. However, in macrophages infected with the H37Rv strain, cholesterol homeostasis is disturbed, resulting in the inhibition of apoptosis and sustained infection (114).
Furthermore, the secretion of TNF-α, a proinflammatory cytokine, in an autocrine/paracrine manner has been shown to play a significant role in the apoptosis of macrophages (87). Interestingly, TNF-α levels are significantly higher at the site of disease in patients with TB (5). TNF-knockout mice, when infected with Mtb, display increased lung infection, which can be ameliorated by the administration of recombinant TNF-α (52). TNF-α also plays an important role in the containment of latent infection in granulomata (87). The administration of mice with MP6-XT22, a monoclonal antibody that neutralizes TNF-α, results in the fatal reactivation of TB, increased tissue bacillary burden, squamous metaplasia, and fluid accumulation in the alveolar spaces (87). TNF-α mediates its proapoptotic activity through its receptors 1 and 2 (TNFR1 and TNFR2). TNFR1 (55), the FAS receptor (FasR), also known as apoptosis antigen 1 (APO-1 or APT) or tumor necrosis factor receptor superfamily member 6 (TNFRSF6), and the FAS ligand (FasL) play important roles in the induction of apoptosis. The binding of FasL to FasR results in signal transduction, leading to the apoptosis (92) of human macrophages infected with Mtb (95). Strangely, the level of TNF-α production induced by the Mtb avirulent strain, H37Ra, is comparable to that induced by the virulent strain, H37Rv, even though apoptosis is higher in H37Ra (4, 55). It was, however, observed that virulent Mtb employs tactics to combat TNF-α-induced apoptosis. First, H37Rv induces the release of the soluble TNF-α receptor (TNFR2), which forms inactive TNF-α-TNFR2 complexes that account for the decrease in TNF-α bioactivity (4). The release of soluble TNFR2 by H37Rv-infected macrophages is dependent on IL-10 production (4). Thus the pathogenic strains of Mtb may selectively induce IL-10, leading to decreased TNF-α activity and subdued macrophage apoptosis. Second, virulently infected macrophages also show reduced susceptibility to FasL-induced apoptosis, correlating with a reduced level of FasR expression (95). Finally, the virulent Mtb strains may interfere with TNF-α signaling by upregulating the expression of the antiapoptotic MCL1 gene, a member of the B-cell lymphoma (BCL2) gene family (123).
ER stress response plays a significant role in limiting the survival of Mtb by inducing apoptosis (62). Mtb induces ER stress in macrophages by disrupting Ca2+ homeostasis and affecting macrophage polarization. Depending on the stimuli, macrophages can acquire a distinct phenotype during the process of polarization. M1 and M2 are the well-described phenotypes, also referred to as classically or alternatively activated macrophages, respectively. Classical activation is stimulated by microbial products and results in M1 macrophages characterized by high antigen presentation and high production of IL-1, IL-6, IL-12, IL-23, TNF-α, nitric oxide, and reactive oxygen intermediates (133). These M1 macrophages increase inflammatory response and mediate resistance against intracellular microbes (75). In contrast, alternative/M2 activation restrains inflammation and upregulates surface molecules, such as dectin-1, mannose receptor, and scavenger receptors A and B1 (35, 81). M2 macrophages also produce high levels of IL-10 (89). Interestingly, macrophages infected with attenuated Mtb strain, H37Ra, display a M1 phenotype, whereas the M2 phenotype is dominant in macrophages infected with the virulent Mtb, H37Rv (74). The mannose receptor and scavenger receptors on M2 macrophages further facilitate H37Rv entry, and IL-10 inhibits macrophage apoptosis, as mentioned earlier. ER stress stimulates M1 polarization of macrophages and facilitates Mtb removal, suggesting that ER stress may be an important component of the host immune response to Mtb (74).
Mycobacteria are composed of several virulence factors, such as the 38-kDa antigen (Ag; Rv0934) (113), 19-kDa Ag (p19; Rv3763) (17), early secreted antigenic target of 6 kDa (ESAT-6; Rv3875) (14), heparin-binding hemagglutinin Ag (HBHA; Rv0475) (15), and PE_PGRS33 (Rv1818c) (6). All of these factors have been shown to cause ER stress and affect major apoptogenic factors on host cells. The 19-kDa Mtb glycolipoprotein, p19, shown to be both cell wall associated and secreted, is capable of inducing apoptosis in macrophages in both a dose- and time-dependent manner (77). This effect of p19 is mediated by Toll-like receptor 2 (TLR2), as the incubation of cells with anti-TLR2 monoclonal antibody inhibited p-19-induced apoptosis. López et al. also established that the viability of Mtb was significantly reduced in cells undergoing apoptosis induced by p19 (77). Additionally, the early secreted mycobacterial antigen, ESAT-6, increases intracellular Ca2+ concentration, reactive species accumulation, and ER stress-induced apoptosis in macrophages (14). Furthermore, the 38-kDa Ag has been demonstrated to increase the expression of ER molecular chaperones, including CHOP and BiP, and phosphorylated eIF2α in bone marrow-derived macrophages and to induce apoptosis via the activation of caspase-12, caspase-9, and caspase-3 (73). Recently, Sohn et al. demonstrated that the Mtb HBHA, a virulence factor and diagnostic antigen involved in extrapulmonary dissemination of Mtb, effectively induces apoptosis in macrophages. HBHA increased reactive oxygen species production, DNA fragmentation, nuclear condensation, caspase activation, and poly (ADP-ribose) polymerase cleavage in apoptotic macrophages (124). Furthermore, Choi et al. demonstrated that HBHA stimulation induced the ER stress sensor molecule, CHOP, in a caspase-dependent manner. In addition, enhanced reactive oxygen species production and elevated cytosolic Ca2+ levels are essential for HBHA-induced ER stress responses (15). In fact, Lim et al. showed that infection with M. kansasii also induced ER stress-mediated apoptosis of macrophages. They found that the apoptosis was associated with calpain activation, as the inhibition of calpain prevented the induction of CHOP and BiP in infected macrophages (71). The exogenous silencing of CHOP expression enhanced the survival of Mtb, whereas increasing ER stress reduced the rate of Mtb survival (72). In fact, calpain activation has also been associated with the development of pleural fibrosis during Mtb infection (145). It is, however, important to note that although initially Mtb increases ERS, the bacteria gradually decrease ERS over time in an attempt to survive within macrophages (72), suggesting that ERS plays an important role in limiting Mtb infection (Fig. 1).
Fig. 1.

Role of apoptosis in mycobacterial tuberculosis (Mtb) infection. The major pathophysiological events that lead to either induction or inhibition of apoptosis in macrophages by the avirulent Mtb strain (H37Ra) and virulent Mtb strain (H37Rv) are summarized. Macrophages infected with H37Ra strain attain M1 phenotype, have predominantly smooth endoplasmic reticulum (ER), and have high levels of ER stress, resulting in increased apoptosis and Mtb death. However, macrophages infected with H37Rv strain attain M2 phenotype, have predominantly rough ER, and have comparatively lower levels of ER stress, resulting in the attenuation of apoptosis and improved Mtb survival.
The role of ERS and apoptosis is particularly evident at the site of a pulmonary tubercle or granuloma formation in Mtb infection. In immunocompetent hosts, the control of TB infection occurs during granuloma formation. Mtb granulomata are highly cellular and are characterized by differentiated myeloid cells surrounded by lymphocytes (13). These macrophage-rich areas of the lung that are infected with virulent Mtb show increased levels of ERS markers, such as CHOP, ATF3, and phosphorylated Ire1α and eIF2α (119). Tubercles, characteristic of active Mtb disease, are distinguished from other granulomata by the extent of necrosis, which leads to liquefaction and cavitation at the necrotic center. This necrotic center causes the hallmark symptomatology of tissue destruction and bloody sputum in pulmonary TB. Interestingly, ERS markers and apoptotic cells are more abundant in the granulomatous tissue surrounding the centralized areas of caseation (119). Therefore, ERS-induced macrophage apoptosis is beneficial in protecting the host from Mtb infection and may limit the dissemination of the bacteria in advanced granulomata (140).
Summary of apoptosis and endoplasmic reticulum stress in tuberculosis.
First, macrophage apoptosis is beneficial to the host as it clears the intracellular bacteria and activates the host innate and adaptive immune response; however, macrophage necrosis is detrimental to the host as it allows the expansion of Mtb numbers and subsequent tissue damage. Second, macrophages can differentiate into M1 and M2 phenotypes: M1 macrophages promote inflammatory responses and increase apoptosis, whereas M2 macrophages attenuate inflammation and decrease apoptosis. Third, ER stress pushes macrophages to the M1 subtype and apoptosis and thereby facilitates Mtb clearance. Fourth, virulent Mtb selectively induce the M2 phenotype in macrophages to suppress apoptosis and killing by the host. Fifth, strategies employed by virulent strains of Mtb to evade macrophage apoptosis include 1) abundance of RER and impaired cholesterol homeostasis, 2) induction of IL-10 and subsequent decrease in TNF-α-mediated apoptotosis, 3) reduced FasR expression and susceptibility to FasL-induced apoptosis, and 4) upregulation of the antiapoptotic MCL1 gene. Finally, several virulence factors of Mtb (38-kDa antigen, p19, ESAT-6, HBHA, and PE_PGRS33) are known to induce macrophage apoptosis in an ER stress-dependent manner.
Autophagy in Tuberculosis
Autophagy is upregulated in several lung pathologies (16, 45, 70, 149, 150). The major difference between apoptosis and autophagy is that during apoptosis the characteristic cell changes (blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation) finally lead to cell death. However, during autophagy, cells degrade dysfunctional and unnecessary cellular components, a process that may or may not lead to cell death. Oxidative stress and other environmental stressors commonly induce autophagy (2, 50). Autophagy prolongs cell survival by turning over cellular constituents and preserving cellular homeostasis (50). During autophagy, these cellular constituents, such as proteins, lipids, and organelles, are sequestered into double-membrane vesicles called autophagosomes that are transported to endosomes or lysosomes to become autophagolysosomes. Within the autophagolysosomes, hydrolase enzymes digest the autophagic components to their basic elements (i.e., amino acids and fatty acids) to be reused for cell building and energy generation. Autophagy maintains organelle quality control by disposing of dysfunctional or damaged cellular organelles. Autophagy of ribosomes, peroxisomes, and mitochondria is termed “ribophagy,” “pexophagy,” and “mitophagy,” respectively.
Autophagy is upregulated by the activation of adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and by the inhibition of the mechanistic target of rapamycin (mTOR) pathway (109). Two ubiquitin-like conjugation systems, microtubule-associated protein-1 light chain 3 (LC3) and autophagy protein 5 (ATG5)-ATG12, are required for the formation of the double-membrane autophagosomes (28, 51, 128). The autophagosome formation can be detected by measuring the conversion of LC3-I (unconjugated cytosolic form) to LC3-II (autophagosomal membrane-associated phosphatidylethanolamine-conjugated form; 28, 51, 128). Additionally, the SQSTM1/p62 protein, also known as sequestosome-1, recognizes cellular components marked for degradation and targets them for autophagy by the virtue of its ubiquitin association domain (UBA) and a LC3-interacting region (LIR; 46). Impairment of autophagy results in the buildup of p62 and cellular dysfunction (47, 61). Autophagy is also upregulated by the association of an autophagy-related protein, beclin 1, and VPS34, a class III phosphatidylinositol-3-kinase (PI3KC3; 40, 53). Beclin 1 can also influence apoptosis and necrosis of cells through its interactions with the B-cell lymphoma-2 (BCL2) family of proteins, BCL2-associated X protein (BAX) and BCL2-antagonistic/killer (BAK; 53, 54, 138).
Xenophagy, the process in which autophagy eliminates bacteria by degradation, was first reported in Mtb (39). It was shown that the physiological stimulation of autophagy in macrophages caused mycobacterial phagosomes to mature into phagolysosomes and led to the suppression of intracellular survival of Mtb (39). During Mtb infection, the increased production of IFN-γ activates macrophages to induce autophagy and traffic Mtb to lysosomes for degradation (25). Induction of autophagy by IFN-γ is associated with protective immunity against TB (25). In addition, studies have shown the role of several autophagy factors, such as ATG5, ATG12, ATG16L1, p62, nuclear dot protein 52 (NDP52), beclin 1 (BECN1), and LC3 (25, 48, 112, 120, 136), in the pathogenesis of infection with Mtb and other infections, where stimulation of autophagy has been shown to increase bacterial killing (39, 112, 134, 146), whereas the suppression of autophagy increases Mtb survival (25, 39, 112, 136).
Transcriptional upregulation of autophagy by Mtb is a complex phenomenon requiring the involvement of numerous genes in multiple steps of autophagy, including the biogenesis of the phagophore, autophagosome and lysosome fusion, and the targeting/degradation of substrate (121). Transcription factor EB (TFEB) is a critical regulator of genes involved in autophagy activation, assembly, and target degradation (121). Recently, Kim et al. identified the role of the nuclear receptor peroxisome proliferator-activated receptor-α (PPARα) in host defense against Mtb and BCG infections via the upregulation of TFEB (59). PPARα binds to the PPAR response elements in the promoter region of numerous target genes (80) and regulates key cellular activities, such as mitochondrial and peroxisomal function and energy metabolism (57, 118). The authors demonstrated that PPARα is essential for host antimycobacterial responses, as the deficiency of PPARα increased the bacterial load and exaggerated the inflammatory responses to mycobacterial infection (59). Interestingly, this immune modulator role of PPARα was mediated by the upregulation of Lamp2, Rab7, and Tfeb, genes involved in autophagy and lysosomal biogenesis (59). These results suggest that further studies are required to explore the possibility of using PPARα agonists in anti-TB management.
The process of elimination of intracellular bacteria by autophagy can involve LC3-associated phagocytosis, the sequestration of phagosomes within autophagosomes, but is largely dependent on targeting cytosolic bacteria for ubiquitination. Mtb evades its destruction in phagolysosomes and can translocate from the phagosome into the cytosol of macrophages and dendritic cells, where it is susceptible to ubiquitination. During autophagy, ubiquitination of intracellular bacteria recruits several key proteins that mediate bacterial delivery to autophagosomes for degradation. Although the process of Mtb ubiquitination remains largely unclear, several key proteins have been shown in recent years to be involved in the likely mechanisms of ubiquitin-dependent autophagic clearance. First, the autophagy adaptors, such as p62, NDP52, NBR1, and optineurin (OPTN), are recruited to the ubiquitin-associated bacteria and then are attached to LC3 to target the intracellular bacteria to autophagosomes for degradation (34). Parkin, an E3 ubiquitin ligase, targets Mtb and Mtb-associated structures to autophagosomes and promotes autophagy-mediated host resistance to TB (82). Ubiquilin-1 (UBQLN1), a member of a protein family that contains an ubiquitin-like domain, an ubiquitin-associated domain, and stress-inducible protein-1 (STI1) motifs, recruits ubiquitin, p62, and LC3 to Mtb-containing vacuoles and hampers bacterial growth and replication (112). Another such protein, Smurf1, which is an E3 ubiquitin ligase, catalyzes the ubiquitination of substrates for subsequent proteasomal degradation. Recently, Franco et al. demonstrated that macrophages lacking Smurf1 are unable to recruit polyubiquitin, the proteasome, ubiquitin-binding autophagy adaptor NBR1, autophagy protein LC3, and lysosomal marker lysosomal associated membrane protein-1 (LAMP1) to Mtb-associated structures (30). They further found that the mice lacking the Smurf1 gene had higher loads of Mtb, exaggerated lung inflammation, and poorer survival than the control mice (30).
Nevertheless, mycobacteria have evolved several mechanisms to inhibit, modulate, or exploit the autophagy response of the host. Certain mycobacteria, such as M. avium complex (MAC), have evolved mechanisms to evade both apoptosis and autophagy, leaving the bacteria free to infect nearby macrophages in the process of spreading (26). Furthermore, the virulent Mtb H37Rv has been known to strategically upregulate IL-6 production to combat innate immunity (25). The production of IL-6 by the Mtb-infected macrophages selectively inhibits IFN-γ-induced autophagosome biogenesis by attenuating the ATG12-ATG5 complex (91). In fact, infection with Mtb time-dependently increases IL-6 production, and the neutralization of IL-6 by an anti-IL-6 antibody significantly enhances the IFN-γ-mediated killing of the intracellular bacteria (25).
In addition, the virulent Mtb H37Rv possesses a unique gene called the “enhanced intracellular survival” (Eis) gene. The protein product of Eis is a unique 42-kDa protein that has been shown to enhance the survival of Mtb during repeated passages through human macrophages (137). Shin et al. demonstrated that the Mtb EIS modulates macrophage autophagy, inflammation, and cell death (122). They found that the macrophages infected with an Mtb Eis deletion-mutant H37Rv (Mtb-Δeis) had markedly increased autophagic vacuoles and autophagosome formation (122). Duan et al. showed that EIS inhibits autophagy in macrophages by upregulating IL-10 gene expression and increasing Akt/mTOR/p70S6K pathway activity (24). They further found that EIS increased the acetylation of H3 histones in the IL-10 gene promoter sequence, thereby increasing IL-10 expression (24). Furthermore, Mtb also inhibits Ras-related protein Rab-7a, an endosomal marker that is also involved in the maturation of Mtb-containing autophagosomes into autolysosomes; thus Mtb selectively modulates autophagy flux in macrophages (12).
Mtb can live in macrophages and evade immune attack by also regulating microRNAs (miRNAs). miRNAs are a class of noncoding small single-strand RNA molecules (~22 nucleotides in length) that play a critical role in macrophage function. miRNAs bind to the 3′ untranslated region of targeted mRNAs and regulate gene expression posttranscriptionally (127). It is well known that miRNAs regulate immune responses through TLR signaling (97), play a critical role in autophagy (147), and regulate host immunity during Mtb infection (84). In a recent publication, Gu et al. demonstrated that the infection of macrophages by Mtb upregulated the expression of miR-23a-5p in a time- and dose-dependent manner (36). Furthermore, they found that the increased expression of miR-23a-5p increased Mtb survival, whereas the inhibition of miR-23a-5p attenuated Mtb survival in the cells (36). Interestingly, miR-23a-5p overexpression prevented the activation of autophagy by the Mtb through TLR2-mediated signaling, suggesting an important role of miR-23a-5p in influencing the innate immune response by inhibiting autophagy and enhancing Mtb survival (36). In another study, Guo et al. showed that BCG infection of macrophages increased the expression of miR-20a, which inhibits autophagic processes by targeting ATG7 and ATG16L1 mRNAs (37). miR-20a also decreased the levels of LC3-II in macrophages and promoted BCG survival (37). In addition, it was found that by inducing miR-33, Mtb inhibited integrated pathways involving autophagy, lysosomal function, and fatty acid oxidation to support bacterial survival and replication (99).
The clinical relevance of autophagy in Mtb infection has been investigated by Li et al. in a recent publication. The authors examined the relationship of autophagy induction by clinically isolated Mtb strains with the disease outcomes of patients with TB (69). They collected sputum, urine, or cerebrospinal fluid samples from 185 patients and found that most of the clinical isolates of Mtb were able to induce autophagosome formation in macrophages; however, the autophagy-inducing ability varied significantly among different isolates (69). They further deduced that the patients infected by Mtb with poor autophagy-inducing ability had more severe disease and worse outcomes (69). The study also suggests that through intimate and persistent interactions with its human host, Mtb has evolved counterstrategies to negate the host antibacterial effects of autophagy. Collectively, it is evident that autophagy represents an important immunologically regulated process in Mtb infection (Fig. 2), and a better understanding of the process will have greater implications in the management of TB infection.
Fig. 2.
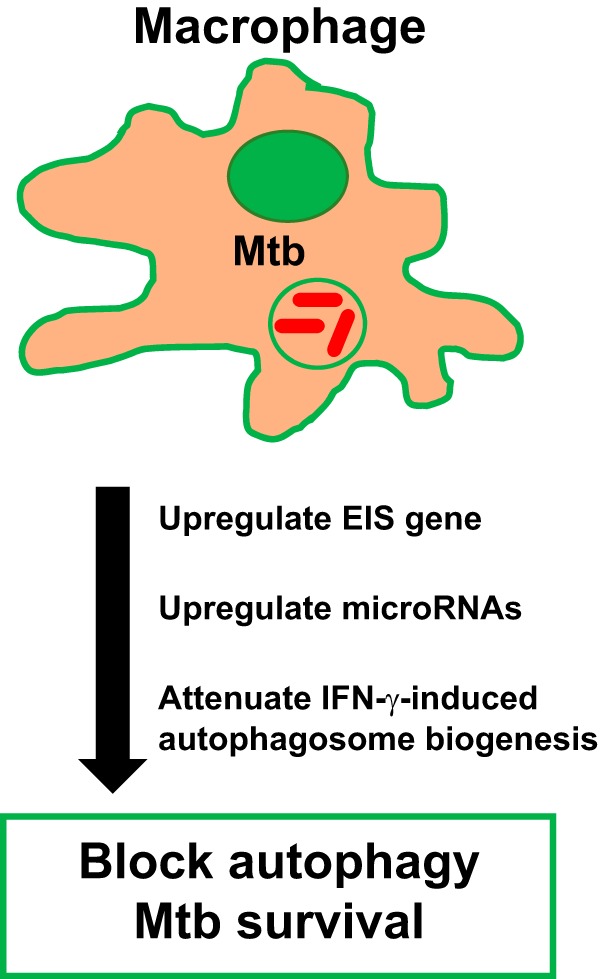
Strategies to inhibit autophagy by mycobacterial tuberculosis (Mtb). The major strategies adopted by Mtb to combat bacterial death by autophagy are summarized. Mtb has evolved techniques to evade killing by autophagy, allowing unchecked bacterial growth. Macrophages infected with Mtb have upregulated IL-6 production, which selectively inhibits IFN-γ-induced autophagosome biogenesis. In addition, Mtb possess the “enhanced intracellular survival” (Eis) gene, which attenuates autophagy and improves Mtb survival. Finally, Mtb upregulates the microRNAs miR-23a-5p, miR-20a, and miR-33, which prevent the activation of autophagy by Mtb. Currently, there is not enough data in the literature to compare the role of autophagy in avirulent and virulent Mtb strains, as mentioned in Fig. 1 for apoptosis.
Summary of autophagy in tuberculosis.
First, autophagy is a homeostatic lysosomal process that involves the degradation of cellular components to their basic elements. Second, stimulation of autophagy increases Mtb killing, whereas suppression of autophagy increases bacterial survival in macrophages. Third, impaired autophagy is associated with severe disease and poor outcomes in humans infected with Mtb. Fourth, the upregulation of Lamp2, Rab7, and Tfeb genes by PPARα plays a significant role in autophagy and lysosomal biogenesis and host antimycobacterial responses. Fifth, increased production of IFN-γ by Mtb activates macrophages to induce autophagy and traffic Mtb to lysosomes for degradation. This process requires a family of proteins including LC3, LAMP1, and Smurf1. Finally, the strategies incorporated by Mtb to evade autophagy include 1) upregulation of the Eis gene, 2) inhibition of IFN-γ by IL-6 produced by Mtb, 3) inhibition of Rab-7a-dependent maturation of Mtb-containing autophagosomes into autolysosomes, and 4) upregulation of specific microRNAs that attenuate TLR signaling and autophagy.
Potential Therapeutics and Concluding Remarks
A nonnaturally occurring type I IFN, called IFN alfacon-1 or Infergen (IFG), has been recently approved against the hepatitis C virus (18, 130). Importantly, IFG has been recently shown to enhance the maturation and activation of macrophages (58). IFG improved bactericidal activity through autophagy and attenuated the survival of Mtb in human macrophages (58). Therefore, immunotherapy with IFG may provide a novel adjuvant treatment strategy in targeting autophagy in Mtb infection (58). Similarly, PPARα agonists, which regulate energy homeostasis and inflammation, are currently approved to be used therapeutically for cholesterol and triglyceride disorders. As mentioned in the review above, these PPARα agonists promote autophagy, lysosomal biogenesis, phagosomal maturation, and antimicrobial defense against Mtb and BCG (59) and therefore can be used in the future to manage TB. Furthermore, loperamide, an antidiarrheal drug, which increases the degradation of long-lived cellular proteins (76), has been shown to induce autophagy in macrophages and reduce Mtb growth and burden (49). Additionally, it was shown that vitamin D supplementation in patients with pulmonary TB without cavitation could enhance autophagy in macrophages and improve innate immune function and therefore may help to control the intracellular growth of mycobacteria (1). Interestingly, it was also noticed that adjunct therapy employing CD40 and TLR4 agonists (C40.T4) significantly enhanced the bactericidal potency of anti-TB drugs by increasing autophagy (58), thus providing a novel therapeutic avenue to target drug resistance in anti-TB therapy.
In conclusion, despite advances in understanding the disease, TB remains a global health challenge and a major cause of morbidity and mortality throughout the world. Rising rates of drug resistance coupled with frequently inappropriate treatment regimens have impeded efforts to control TB worldwide. Therefore, a better understanding of Mtb pathology and the development of novel therapies based on that knowledge are urgently needed. In this regard, targeting molecular pathways controlling apoptosis and autophagy (93, 96) may play a significant role in combating TB.
GRANTS
This work was supported by the Countermeasures Against Chemical Threats (CounterACT) program, National Institutes of Health Office of the Director, National Institute of Neurological Disorders and Stroke, and National Institute of Environmental Health Sciences, Grants 5U01-ES-026458-02 and 1U01-ES-027697-01.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.A. prepared figures; A.L., R.P., C.M.G., V.S., and S.A. drafted manuscript; A.L., L.A.R., V.S., and S.A. edited and revised manuscript; V.S. and S.A. approved final version of manuscript; S.A. conceived and designed research.
ACKNOWLEDGMENTS
We thank Dr. Sadis Matalon, Department of Anesthesiology and Perioperative Medicine, University of Alabama at Birmingham, for support in the generation of the manuscript.
REFERENCES
- 1.Afsal K, Selvaraj P. Effect of 1,25-dihydroxyvitamin D3 on the expression of mannose receptor, DC-SIGN and autophagy genes in pulmonary tuberculosis. Tuberculosis (Edinb) 99: 1–10, 2016. doi: 10.1016/j.tube.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol 311: L433–L452, 2016. doi: 10.1152/ajplung.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Orainey IO. Diagnosis of latent tuberculosis: can we do better? Ann Thorac Med 4: 5–9, 2009. doi: 10.4103/1817-1737.44778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol 161: 2636–2641, 1998. [PubMed] [Google Scholar]
- 5.Barnes PF, Lu S, Abrams JS, Wang E, Yamamura M, Modlin RL. Cytokine production at the site of disease in human tuberculosis. Infect Immun 61: 3482–3489, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu S, Pathak SK, Banerjee A, Pathak S, Bhattacharyya A, Yang Z, Talarico S, Kundu M, Basu J. Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J Biol Chem 282: 1039–1050, 2007. doi: 10.1074/jbc.M604379200. [DOI] [PubMed] [Google Scholar]
- 7.Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, Divangahi M, Remold HG. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol 4: 279–287, 2011. doi: 10.1038/mi.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellamy R, Ruwende C, Corrah T, McAdam KP, Whittle HC, Hill AV. Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N Engl J Med 338: 640–644, 1998. doi: 10.1056/NEJM199803053381002. [DOI] [PubMed] [Google Scholar]
- 9.Blackwell JM, Searle S, Goswami T, Miller EN. Understanding the multiple functions of Nramp1. Microbes Infect 2: 317–321, 2000. doi: 10.1016/S1286-4579(00)00295-1. [DOI] [PubMed] [Google Scholar]
- 10.Brudney K, Dobkin J. Resurgent tuberculosis in New York City. Human immunodeficiency virus, homelessness, and the decline of tuberculosis control programs. Am Rev Respir Dis 144: 745–749, 1991. doi: 10.1164/ajrccm/144.4.745. [DOI] [PubMed] [Google Scholar]
- 11.Cervino AC, Lakiss S, Sow O, Hill AV. Allelic association between the NRAMP1 gene and susceptibility to tuberculosis in Guinea-Conakry. Ann Hum Genet 64: 507–512, 2000. doi: 10.1046/j.1469-1809.2000.6460507.x. [DOI] [PubMed] [Google Scholar]
- 12.Chandra P, Ghanwat S, Matta SK, Yadav SS, Mehta M, Siddiqui Z, Singh A, Kumar D. Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci Rep 5: 16320, 2015. doi: 10.1038/srep16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123, 2009. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi HH, Shin DM, Kang G, Kim KH, Park JB, Hur GM, Lee HM, Lim YJ, Park JK, Jo EK, Song CH. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett 584: 2445–2454, 2010. doi: 10.1016/j.febslet.2010.04.050. [DOI] [PubMed] [Google Scholar]
- 15.Choi JA, Lim YJ, Cho SN, Lee JH, Jeong JA, Kim EJ, Park JB, Kim SH, Park HS, Kim HJ, Song CH. Mycobacterial HBHA induces endoplasmic reticulum stress-mediated apoptosis through the generation of reactive oxygen species and cytosolic Ca2+ in murine macrophage RAW 264.7 cells. Cell Death Dis 4: e957, 2013. doi: 10.1038/cddis.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christofidou-Solomidou M, Pietrofesa RA, Arguiri E, Schweitzer KS, Berdyshev EV, McCarthy M, Corbitt A, Alwood JS, Yu Y, Globus RK, Solomides CC, Ullrich RL, Petrache I. Space radiation-associated lung injury in a murine model. Am J Physiol Lung Cell Mol Physiol 308: L416–L428, 2015. doi: 10.1152/ajplung.00260.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciaramella A, Cavone A, Santucci MB, Garg SK, Sanarico N, Bocchino M, Galati D, Martino A, Auricchio G, D’Orazio M, Stewart GR, Neyrolles O, Young DB, Colizzi V, Fraziano M. Induction of apoptosis and release of interleukin-1 beta by cell wall-associated 19-kDa lipoprotein during the course of mycobacterial infection. J Infect Dis 190: 1167–1176, 2004. doi: 10.1086/423850. [DOI] [PubMed] [Google Scholar]
- 18.Clark V, Nelson DR. Novel interferons for treatment of hepatitis C virus. Clin Liver Dis 13: 351–363, 2009. doi: 10.1016/j.cld.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Cooper AM, Segal BH, Frank AA, Holland SM, Orme IM. Transient loss of resistance to pulmonary tuberculosis in p47(phox−/−) mice. Infect Immun 68: 1231–1234, 2000. doi: 10.1128/IAI.68.3.1231-1234.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daniel TM, Iversen PA. Hippocrates and tuberculosis. Int J Tuberc Lung Dis 19: 373–374, 2015. doi: 10.5588/ijtld.14.0736. [DOI] [PubMed] [Google Scholar]
- 21.Davies PD, Brown RC, Woodhead JS. Serum concentrations of vitamin D metabolites in untreated tuberculosis. Thorax 40: 187–190, 1985. doi: 10.1136/thx.40.3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denis M. Killing of Mycobacterium tuberculosis within human monocytes: activation by cytokines and calcitriol. Clin Exp Immunol 84: 200–206, 1991. doi: 10.1111/j.1365-2249.1991.tb08149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dheda K, Barry CE 3rd, Maartens G. Tuberculosis. Lancet 387: 1211–1226, 2016. doi: 10.1016/S0140-6736(15)00151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duan L, Yi M, Chen J, Li S, Chen W. Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3. Biochem Biophys Res Commun 473: 1229–1234, 2016. doi: 10.1016/j.bbrc.2016.04.045. [DOI] [PubMed] [Google Scholar]
- 25.Dutta RK, Kathania M, Raje M, Majumdar S. IL-6 inhibits IFN-γ induced autophagy in Mycobacterium tuberculosis H37Rv infected macrophages. Int J Biochem Cell Biol 44: 942–954, 2012. doi: 10.1016/j.biocel.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 26.Early J, Fischer K, Bermudez LE. Mycobacterium avium uses apoptotic macrophages as tools for spreading. Microb Pathog 50: 132–139, 2011. doi: 10.1016/j.micpath.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ernst JD. Macrophage receptors for Mycobacterium tuberculosis. Infect Immun 66: 1277–1281, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 24: 24–41, 2014. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox GJ, Mitnick CD, Benedetti A, Chan ED, Becerra M, Chiang CY, Keshavjee S, Koh WJ, Shiraishi Y, Viiklepp P, Yim JJ, Pasvol G, Robert J, Shim TS, Shin SS, Menzies D; Collaborative Group for Meta-Analysis of Individual Patient Data in MDR-TB . Surgery as an adjunctive treatment for multidrug-resistant tuberculosis: an individual patient data metaanalysis. Clin Infect Dis 62: 887–895, 2016. doi: 10.1093/cid/ciw002. [DOI] [PubMed] [Google Scholar]
- 30.Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU, Levine B. The ubiquitin ligase Smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe 21: 59–72, 2017. doi: 10.1016/j.chom.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fratazzi C, Arbeit RD, Carini C, Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Macrophage apoptosis in mycobacterial infections. J Leukoc Biol 66: 763–764, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Gaynor CD, McCormack FX, Voelker DR, McGowan SE, Schlesinger LS. Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol 155: 5343–5351, 1995. [PubMed] [Google Scholar]
- 33.Gercken J, Pryjma J, Ernst M, Flad HD. Defective antigen presentation by Mycobacterium tuberculosis-infected monocytes. Infect Immun 62: 3472–3478, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell 54: 224–233, 2014. doi: 10.1016/j.molcel.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Gordon S. Alternative activation of macrophages. Nat Rev Immunol 3: 23–35, 2003. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 36.Gu X, Gao Y, Mu DG, Fu EQ. MiR-23a-5p modulates mycobacterial survival and autophagy during Mycobacterium tuberculosis infection through TLR2/MyD88/NF-κB pathway by targeting TLR2. Exp Cell Res 354: 71–77, 2017. doi: 10.1016/j.yexcr.2017.03.039. [DOI] [PubMed] [Google Scholar]
- 37.Guo L, Zhao J, Qu Y, Yin R, Gao Q, Ding S, Zhang Y, Wei J, Xu G. MicroRNA-20a inhibits autophagic process by targeting ATG7 and ATG16L1 and favors mycobacterial survival in macrophage cells. Front Cell Infect Microbiol 6: 134, 2016. doi: 10.3389/fcimb.2016.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta I, Guin P. Communicable diseases in the South-East Asia Region of the World Health Organization: towards a more effective response. Bull World Health Organ 88: 199–205, 2010. doi: 10.2471/BLT.09.065540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766, 2004. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 40.He C, Levine B. The beclin 1 interactome. Curr Opin Cell Biol 22: 140–149, 2010. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J Immunol 159: 635–643, 1997. [PubMed] [Google Scholar]
- 42.Herzog H. History of tuberculosis. Respiration 65: 5–15, 1998. [DOI] [PubMed] [Google Scholar]
- 43.Hirsch CS, Toossi Z, Johnson JL, Luzze H, Ntambi L, Peters P, McHugh M, Okwera A, Joloba M, Mugyenyi P, Mugerwa RD, Terebuh P, Ellner JJ. Augmentation of apoptosis and interferon-gamma production at sites of active Mycobacterium tuberculosis infection in human tuberculosis. J Infect Dis 183: 779–788, 2001. doi: 10.1086/318817. [DOI] [PubMed] [Google Scholar]
- 44.Hirsch CS, Toossi Z, Vanham G, Johnson JL, Peters P, Okwera A, Mugerwa R, Mugyenyi P, Ellner JJ. Apoptosis and T cell hyporesponsiveness in pulmonary tuberculosis. J Infect Dis 179: 945–953, 1999. doi: 10.1086/314667. [DOI] [PubMed] [Google Scholar]
- 45.Im J, Hergert P, Nho RS. Reduced FoxO3a expression causes low autophagy in idiopathic pulmonary fibrosis fibroblasts on collagen matrices. Am J Physiol Lung Cell Mol Physiol 309: L552–L561, 2015. doi: 10.1152/ajplung.00079.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Itakura E, Mizushima N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 192: 17–27, 2011. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, Zong WX. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci USA 109: 2003–2008, 2012. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juárez E, Carranza C, Hernández-Sánchez F, León-Contreras JC, Hernández-Pando R, Escobedo D, Torres M, Sada E. NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol 42: 880–889, 2012. doi: 10.1002/eji.201142105. [DOI] [PubMed] [Google Scholar]
- 49.Juárez E, Carranza C, Sánchez G, González M, Chávez J, Sarabia C, Torres M, Sada E. Loperamide restricts intracellular growth of Mycobacterium tuberculosis in lung macrophages. Am J Respir Cell Mol Biol 55: 837–847, 2016. doi: 10.1165/rcmb.2015-0383OC. [DOI] [PubMed] [Google Scholar]
- 50.Jurkuvenaite A, Benavides GA, Komarova S, Doran SF, Johnson M, Aggarwal S, Zhang J, Darley-Usmar VM, Matalon S. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med 85: 83–94, 2015. doi: 10.1016/j.freeradbiomed.2015.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117: 2805–2812, 2004. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 52.Kaneko H, Yamada H, Mizuno S, Udagawa T, Kazumi Y, Sekikawa K, Sugawara I. Role of tumor necrosis factor-alpha in Mycobacterium-induced granuloma formation in tumor necrosis factor-alpha-deficient mice. Lab Invest 79: 379–386, 1999. [PubMed] [Google Scholar]
- 53.Kang R, Zeh HJ, Lotze MT, Tang D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18: 571–580, 2011. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2: e00772, 2013. doi: 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, Kornfeld H. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun 65: 298–304, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol 164: 2016–2020, 2000. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 57.Kersten S. Integrated physiology and systems biology of PPARα. Mol Metab 3: 354–371, 2014. doi: 10.1016/j.molmet.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. Stimulation through CD40 and TLR-4 is an effective host directed therapy against Mycobacterium tuberculosis. Front Immunol 7: 386, 2016. doi: 10.3389/fimmu.2016.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim YS, Lee HM, Kim JK, Yang CS, Kim TS, Jung M, Jin HS, Kim S, Jang J, Oh GT, Kim JM, Jo EK. PPAR-α activation mediates innate host defense through induction of TFEB and lipid catabolism. J Immunol 198: 3283–3295, 2017. doi: 10.4049/jimmunol.1601920. [DOI] [PubMed] [Google Scholar]
- 60.Knapp S, Leemans JC, Florquin S, Branger J, Maris NA, Pater J, van Rooijen N, van der Poll T. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med 167: 171–179, 2003. doi: 10.1164/rccm.200207-698OC. [DOI] [PubMed] [Google Scholar]
- 61.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131: 1149–1163, 2007. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 62.Labonte AC, Tosello-Trampont AC, Hahn YS. The role of macrophage polarization in infectious and inflammatory diseases. Mol Cells 37: 275–285, 2014. doi: 10.14348/molcells.2014.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lai YM, Mohammed KA, Nasreen N, Baumuratov A, Bellew BF, Antony VB. Induction of cell cycle arrest and apoptosis by BCG infection in cultured human bronchial airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 293: L393–L401, 2007. doi: 10.1152/ajplung.00392.2006. [DOI] [PubMed] [Google Scholar]
- 64.Lammas DA, Stober C, Harvey CJ, Kendrick N, Panchalingam S, Kumararatne DS. ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity 7: 433–444, 1997. doi: 10.1016/S1074-7613(00)80364-7. [DOI] [PubMed] [Google Scholar]
- 65.Lawn SD, Zumla AI. Tuberculosis. Lancet 378: 57–72, 2011. doi: 10.1016/S0140-6736(10)62173-3. [DOI] [PubMed] [Google Scholar]
- 66.Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J 50: 1–11, 2009. doi: 10.3349/ymj.2009.50.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee J, Repasy T, Papavinasasundaram K, Sassetti C, Kornfeld H. Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PLoS One 6: e18367, 2011. doi: 10.1371/journal.pone.0018367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, van Deventer SJ, van der Poll T. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J Immunol 166: 4604–4611, 2001. doi: 10.4049/jimmunol.166.7.4604. [DOI] [PubMed] [Google Scholar]
- 69.Li F, Gao B, Xu W, Chen L, Xiong S. The defect in autophagy induction by clinical isolates of Mycobacterium tuberculosis is correlated with poor tuberculosis outcomes. PLoS One 11: e0147810, 2016. doi: 10.1371/journal.pone.0147810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Yu G, Yuan S, Tan C, Xie J, Ding Y, Lian P, Fu L, Hou Q, Xu B, Wang H. 14,15-Epoxyeicosatrienoic acid suppresses cigarette smoke condensate-induced inflammation in lung epithelial cells by inhibiting autophagy. Am J Physiol Lung Cell Mol Physiol 311: L970–L980, 2016. doi: 10.1152/ajplung.00161.2016. [DOI] [PubMed] [Google Scholar]
- 71.Lim YJ, Choi HH, Choi JA, Jeong JA, Cho SN, Lee JH, Park JB, Kim HJ, Song CH. Mycobacterium kansasii-induced death of murine macrophages involves endoplasmic reticulum stress responses mediated by reactive oxygen species generation or calpain activation. Apoptosis 18: 150–159, 2013. doi: 10.1007/s10495-012-0792-4. [DOI] [PubMed] [Google Scholar]
- 72.Lim YJ, Choi JA, Choi HH, Cho SN, Kim HJ, Jo EK, Park JK, Song CH. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS One 6: e28531, 2011. doi: 10.1371/journal.pone.0028531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lim YJ, Choi JA, Lee JH, Choi CH, Kim HJ, Song CH. Mycobacterium tuberculosis 38-kDa antigen induces endoplasmic reticulum stress-mediated apoptosis via toll-like receptor 2/4. Apoptosis 20: 358–370, 2015. doi: 10.1007/s10495-014-1080-2. [DOI] [PubMed] [Google Scholar]
- 74.Lim YJ, Yi MH, Choi JA, Lee J, Han JY, Jo SH, Oh SM, Cho HJ, Kim DW, Kang MW, Song CH. Roles of endoplasmic reticulum stress-mediated apoptosis in M1-polarized macrophages during mycobacterial infections. Sci Rep 6: 37211, 2016. doi: 10.1038/srep37211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci 10: 520–529, 2014. doi: 10.7150/ijbs.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loetchutinat C, Saengkhae C, Marbeuf-Gueye C, Garnier-Suillerot A. New insights into the P-glycoprotein-mediated effluxes of rhodamines. Eur J Biochem 270: 476–485, 2003. doi: 10.1046/j.1432-1033.2003.03403.x. [DOI] [PubMed] [Google Scholar]
- 77.López M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol 170: 2409–2416, 2003. doi: 10.4049/jimmunol.170.5.2409. [DOI] [PubMed] [Google Scholar]
- 78.Luciw PA, Oslund KL, Yang XW, Adamson L, Ravindran R, Canfield DR, Tarara R, Hirst L, Christensen M, Lerche NW, Offenstein H, Lewinsohn D, Ventimiglia F, Brignolo L, Wisner ER, Hyde DM. Stereological analysis of bacterial load and lung lesions in nonhuman primates (rhesus macaques) experimentally infected with Mycobacterium tuberculosis. Am J Physiol Lung Cell Mol Physiol 301: L731–L738, 2011. doi: 10.1152/ajplung.00120.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Malik ZA, Iyer SS, Kusner DJ. Mycobacterium tuberculosis phagosomes exhibit altered calmodulin-dependent signal transduction: contribution to inhibition of phagosome-lysosome fusion and intracellular survival in human macrophages. J Immunol 166: 3392–3401, 2001. doi: 10.4049/jimmunol.166.5.3392. [DOI] [PubMed] [Google Scholar]
- 80.Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci 61: 393–416, 2004. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25: 677–686, 2004. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 82.Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501: 512–516, 2013. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martinson NA, Barnes GL, Moulton LH, Msandiwa R, Hausler H, Ram M, McIntyre JA, Gray GE, Chaisson RE. New regimens to prevent tuberculosis in adults with HIV infection. N Engl J Med 365: 11–20, 2011. doi: 10.1056/NEJMoa1005136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maudet C, Mano M, Eulalio A. MicroRNAs in the interaction between host and bacterial pathogens. FEBS Lett 588: 4140–4147, 2014. doi: 10.1016/j.febslet.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 85.McDonald LC, Archibald LK, Rheanpumikankit S, Tansuphaswadikul S, Eampokalap B, Nwanyanawu O, Kazembe P, Dobbie H, Reller LB, Jarvis WR. Unrecognised Mycobacterium tuberculosis bacteraemia among hospital inpatients in less developed countries. Lancet 354: 1159–1163, 1999. doi: 10.1016/S0140-6736(98)12325-5. [DOI] [PubMed] [Google Scholar]
- 86.McNerney R, Maeurer M, Abubakar I, Marais B, McHugh TD, Ford N, Weyer K, Lawn S, Grobusch MP, Memish Z, Squire SB, Pantaleo G, Chakaya J, Casenghi M, Migliori GB, Mwaba P, Zijenah L, Hoelscher M, Cox H, Swaminathan S, Kim PS, Schito M, Harari A, Bates M, Schwank S, O’Grady J, Pletschette M, Ditui L, Atun R, Zumla A. Tuberculosis diagnostics and biomarkers: needs, challenges, recent advances, and opportunities. J Infect Dis 205, Suppl 2: S147–S158, 2012. doi: 10.1093/infdis/jir860. [DOI] [PubMed] [Google Scholar]
- 87.Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, Tsai MM, Flynn JL, Chan J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun 69: 1847–1855, 2001. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Molloy A, Laochumroonvorapong P, Kaplan G. Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guérin. J Exp Med 180: 1499–1509, 1994. doi: 10.1084/jem.180.4.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mosser DM. The many faces of macrophage activation. J Leukoc Biol 73: 209–212, 2003. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 90.Mulugeta S, Maguire JA, Newitt JL, Russo SJ, Kotorashvili A, Beers MF. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am J Physiol Lung Cell Mol Physiol 293: L720–L729, 2007. doi: 10.1152/ajplung.00025.2007. [DOI] [PubMed] [Google Scholar]
- 91.Nagabhushanam V, Solache A, Ting LM, Escaron CJ, Zhang JY, Ernst JD. Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-gamma. J Immunol 171: 4750–4757, 2003. doi: 10.4049/jimmunol.171.9.4750. [DOI] [PubMed] [Google Scholar]
- 92.Nagata S. Apoptosis by death factor. Cell 88: 355–365, 1997. doi: 10.1016/S0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 93.Nakahira K, Choi AM. Autophagy: a potential therapeutic target in lung diseases. Am J Physiol Lung Cell Mol Physiol 305: L93–L107, 2013. doi: 10.1152/ajplung.00072.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen H, Uhal BD. The unfolded protein response controls ER stress-induced apoptosis of lung epithelial cells through angiotensin generation. Am J Physiol Lung Cell Mol Physiol 311: L846–L854, 2016. doi: 10.1152/ajplung.00449.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol 160: 5448–5454, 1998. [PubMed] [Google Scholar]
- 96.O’Dwyer DN, Ashley SL, Moore BB. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am J Physiol Lung Cell Mol Physiol 311: L590–L601, 2016. doi: 10.1152/ajplung.00221.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol 11: 163–175, 2011. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 98.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13: 715–725, 2000. doi: 10.1016/S1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 99.Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, Karunakaran D, Portal-Celhay C, Sheedy FJ, Ray TD, Cecchini K, Zamore PD, Rayner KJ, Marcel YL, Philips JA, Moore KJ. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol 17: 677–686, 2016. doi: 10.1038/ni.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11: 381–389, 2004. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 101.Pai M, Kalantri S, Aggarwal AN, Menzies D, Blumberg HM. Nosocomial tuberculosis in India. Emerg Infect Dis 12: 1311–1318, 2006. doi: 10.3201/eid1209.051663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, Higgins DE, Daly MJ, Bloom BR, Kramnik I. Ipr1 gene mediates innate immunity to tuberculosis. Nature 434: 767–772, 2005. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pancholi P, Mirza A, Bhardwaj N, Steinman RM. Sequestration from immune CD4+ T cells of mycobacteria growing in human macrophages. Science 260: 984–986, 1993. doi: 10.1126/science.8098550. [DOI] [PubMed] [Google Scholar]
- 104.Pervin N, Akram S, Hudali T, Bhattarai M, Waqar S. Disseminated tuberculosis presenting as Baker’s cyst infection. Case Rep Infect Dis 2017: 6527675, 2017. doi: 10.1155/2017/6527675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Riendeau CJ, Kornfeld H. THP-1 cell apoptosis in response to mycobacterial infection. Infect Immun 71: 254–259, 2003. doi: 10.1128/IAI.71.1.254-259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rivera-Marrero CA, Schuyler W, Roser S, Ritzenthaler JD, Newburn SA, Roman J. M. tuberculosis induction of matrix metalloproteinase-9: the role of mannose and receptor-mediated mechanisms. Am J Physiol Lung Cell Mol Physiol 282: L546–L555, 2002. doi: 10.1152/ajplung.00175.2001. [DOI] [PubMed] [Google Scholar]
- 107.Rockett KA, Brookes R, Udalova I, Vidal V, Hill AV, Kwiatkowski D. 1,25-Dihydroxyvitamin D3 induces nitric oxide synthase and suppresses growth of Mycobacterium tuberculosis in a human macrophage-like cell line. Infect Immun 66: 5314–5321, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rojas M, Barrera LF, García LF. Induction of apoptosis in murine macrophages by Mycobacterium tuberculosis is reactive oxygen intermediates-independent. Biochem Biophys Res Commun 247: 436–442, 1998. doi: 10.1006/bbrc.1998.8802. [DOI] [PubMed] [Google Scholar]
- 109.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res 24: 42–57, 2014. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sagar T, Gupta K, Rani M, Kaur IR. Disseminated tuberculosis in a newborn infant. J Family Med Prim Care 5: 695–697, 2016. doi: 10.4103/2249-4863.197301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saha B, Das G, Vohra H, Ganguly NK, Mishra GC. Macrophage-T cell interaction in experimental mycobacterial infection. Selective regulation of co-stimulatory molecules on Mycobacterium-infected macrophages and its implication in the suppression of cell-mediated immune response. Eur J Immunol 24: 2618–2624, 1994. doi: 10.1002/eji.1830241108. [DOI] [PubMed] [Google Scholar]
- 112.Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, Maurer K, Cadwell K, Philips JA. Ubiquilin 1 promotes IFN-γ-induced xenophagy of Mycobacterium tuberculosis. PLoS Pathog 11: e1005076, 2015. doi: 10.1371/journal.ppat.1005076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sanchez A, Espinosa P, Esparza MA, Colon M, Bernal G, Mancilla R. Mycobacterium tuberculosis 38-kDa lipoprotein is apoptogenic for human monocyte-derived macrophages. Scand J Immunol 69: 20–28, 2009. doi: 10.1111/j.1365-3083.2008.02193.x. [DOI] [PubMed] [Google Scholar]
- 114.Saquib NM, Jamwal S, Midha MK, Verma HN, Manivel V. Quantitative proteomics and lipidomics analysis of endoplasmic reticulum of macrophage infected with Mycobacterium tuberculosis. Int J Proteomics 2015: 270438, 2015. doi: 10.1155/2015/270438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sasindran SJ, Torrelles JB. Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front Microbiol 2: 2, 2011. doi: 10.3389/fmicb.2011.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, Modlin RL, Brinkmann V, Kaufmann SH. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med 9: 1039–1046, 2003. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- 117.Schlesinger LS. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol 150: 2920–2930, 1993. [PubMed] [Google Scholar]
- 118.Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta 1302: 93–109, 1996. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 119.Seimon TA, Kim MJ, Blumenthal A, Koo J, Ehrt S, Wainwright H, Bekker LG, Kaplan G, Nathan C, Tabas I, Russell DG. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS One 5: e12772, 2010. doi: 10.1371/journal.pone.0012772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Seto S, Tsujimura K, Horii T, Koide Y. Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome formation in response to Mycobacterium tuberculosis infection in dendritic cells. PLoS One 8: e86017, 2013. doi: 10.1371/journal.pone.0086017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433, 2011. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, Lee SH, Lee ZW, Cho SN, Kim JM, Friedman RL, Jo EK. Mycobacterium tuberculosis Eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog 6: e1001230, 2010. doi: 10.1371/journal.ppat.1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol 170: 430–437, 2003. doi: 10.4049/jimmunol.170.1.430. [DOI] [PubMed] [Google Scholar]
- 124.Sohn H, Kim JS, Shin SJ, Kim K, Won CJ, Kim WS, Min KN, Choi HG, Lee JC, Park JK, Kim HJ. Targeting of Mycobacterium tuberculosis heparin-binding hemagglutinin to mitochondria in macrophages. PLoS Pathog 7: e1002435, 2011. doi: 10.1371/journal.ppat.1002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Srinivasan L, Ahlbrand S, Briken V. Interaction of Mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb Perspect Med 4: a022459, 2014. doi: 10.1101/cshperspect.a022459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Surolia R, Karki S, Wang Z, Kulkarni T, Li FJ, Vohra S, Batra H, Nick JA, Duncan SR, Thannickal VJ, Steyn AJ, Agarwal A, Antony VB. Attenuated heme oxygenase-1 responses predispose the elderly to pulmonary nontuberculous mycobacterial infections. Am J Physiol Lung Cell Mol Physiol 311: L928–L940, 2016. doi: 10.1152/ajplung.00397.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Taganov KD, Boldin MP, Baltimore D. MicroRNAs and immunity: tiny players in a big field. Immunity 26: 133–137, 2007. doi: 10.1016/j.immuni.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 128.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36: 2503–2518, 2004. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 302: L721–L729, 2012. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thitinan S, McConville JT. Interferon alpha delivery systems for the treatment of hepatitis C. Int J Pharm 369: 121–135, 2009. doi: 10.1016/j.ijpharm.2008.11.027. [DOI] [PubMed] [Google Scholar]
- 131.Thurnher M, Ramoner R, Gastl G, Radmayr C, Böck G, Herold M, Klocker H, Bartsch G. Bacillus Calmette-Guérin mycobacteria stimulate human blood dendritic cells. Int J Cancer 70: 128–134, 1997. doi: 10.1002/(SICI)1097-0215(19970106)70:1<128::AID-IJC19>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 132.van Crevel R, Ottenhoff TH, van der Meer JW. Innate immunity to Mycobacterium tuberculosis. Clin Microbiol Rev 15: 294–309, 2002. doi: 10.1128/CMR.15.2.294-309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de Waal-Malefyt R, Ottenhoff TH. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA 101: 4560–4565, 2004. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang J, Yang K, Zhou L, Wu MH, Wu Y, Zhu M, Lai X, Chen T, Feng L, Li M, Huang C, Zhong Q, Huang X. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog 9: e1003697, 2013. doi: 10.1371/journal.ppat.1003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang JY, Hsueh PR, Wang SK, Jan IS, Lee LN, Liaw YS, Yang PC, Luh KT. Disseminated tuberculosis: a 10-year experience in a medical center. Medicine (Baltimore) 86: 39–46, 2007. doi: 10.1097/MD.0b013e318030b605. [DOI] [PubMed] [Google Scholar]
- 136.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150: 803–815, 2012. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wei J, Dahl JL, Moulder JW, Roberts EA, O’Gaora P, Young DB, Friedman RL. Identification of a Mycobacterium tuberculosis gene that enhances mycobacterial survival in macrophages. J Bacteriol 182: 377–384, 2000. doi: 10.1128/JB.182.2.377-384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730, 2001. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weikert LF, Lopez JP, Abdolrasulnia R, Chroneos ZC, Shepherd VL. Surfactant protein A enhances mycobacterial killing by rat macrophages through a nitric oxide-dependent pathway. Am J Physiol Lung Cell Mol Physiol 279: L216–L223, 2000. [DOI] [PubMed] [Google Scholar]
- 140.Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev 264: 182–203, 2015. doi: 10.1111/imr.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wilkinson RJ, Llewelyn M, Toossi Z, Patel P, Pasvol G, Lalvani A, Wright D, Latif M, Davidson RN. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case-control study. Lancet 355: 618–621, 2000. doi: 10.1016/S0140-6736(99)02301-6. [DOI] [PubMed] [Google Scholar]
- 142.Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, Sandhoff K, Brinkmann V, Kaufmann SH, Schaible UE. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24: 105–117, 2006. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 143.Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79: 155–169, 2000. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 144.World Health Organization Global Tuberculosis Report 2016. Geneva: WHO Global TB Programme, http://www.who.int/tb/publications/global_report/en/ [April 10, 2017]. [Google Scholar]
- 145.Yang J, Xiang F, Cai PC, Lu YZ, Xu XX, Yu F, Li FZ, Greer PA, Shi HZ, Zhou Q, Xin JB, Ye H, Su Y, Ma WL. Activation of calpain by renin-angiotensin system in pleural mesothelial cells mediates tuberculous pleural fibrosis. Am J Physiol Lung Cell Mol Physiol 311: L145–L153, 2016. doi: 10.1152/ajplung.00348.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ye Y, Li X, Wang W, Ouedraogo KC, Li Y, Gan C, Tan S, Zhou X, Wu M. Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am J Physiol Lung Cell Mol Physiol 307: L355–L363, 2014. doi: 10.1152/ajplung.00046.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhai H, Fesler A, Ju J. MicroRNA: a third dimension in autophagy. Cell Cycle 12: 246–250, 2013. doi: 10.4161/cc.23273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 454: 455–462, 2008. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol Lung Cell Mol Physiol 307: L173–L185, 2014. doi: 10.1152/ajplung.00083.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhou JS, Zhao Y, Zhou HB, Wang Y, Wu YF, Li ZY, Xuan NX, Zhang C, Hua W, Ying SM, Li W, Shen HH, Chen ZH. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol Lung Cell Mol Physiol 310: L1042–L1052, 2016. doi: 10.1152/ajplung.00418.2015. [DOI] [PubMed] [Google Scholar]
- 151.Zink AR, Sola C, Reischl U, Grabner W, Rastogi N, Wolf H, Nerlich AG. Characterization of Mycobacterium tuberculosis complex DNAs from Egyptian mummies by spoligotyping. J Clin Microbiol 41: 359–367, 2003. doi: 10.1128/JCM.41.1.359-367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zumla A. Current trends and newer concepts on diagnosis, management and prevention of respiratory tract infections. Curr Opin Pulm Med 19: 189–191, 2013. doi: 10.1097/MCP.0b013e32835f8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zwilling BS, Kuhn DE, Wikoff L, Brown D, Lafuse W. Role of iron in Nramp1-mediated inhibition of mycobacterial growth. Infect Immun 67: 1386–1392, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]