

Abstract
Triple-negative breast cancer (TNBC) remains an aggressive disease without effective targeted therapies. In this study, we addressed this challenge by testing 128 FDA-approved or investigational drugs as either single agents or in 768 pairwise drug combinations in TNBC cell lines to identify synergistic combinations tractable to clinical translation. Medium-throughput results were scrutinized and extensively analyzed for sensitivity patterns, synergy, anticancer activity and validation in low-throughput experiments. Principal component analysis revealed that a fraction of all upregulated or downregulated genes of a particular targeted pathway could partly explain cell sensitivity towards agents targeting that pathway. Combination therapies deemed immediately tractable to translation included ABT-263/crizotinib, ABT-263/paclitaxel, paclitaxel/JQ1, ABT-263/XL184 and paclitaxel/nutlin-3, all of which exhibited synergistic antiproliferative and apoptotic activity in multiple TNBC backgrounds. Mechanistic investigations of the ABT-263/crizotinib combination offering a potentially rapid path to clinic demonstrated RTK blockade, inhibition of mitogenic signaling and pro-apoptotic signal induction in basal and mesenchymal stem-like TNBC. Our findings provide preclinical proof of concept for several combination treatments of TNBC which offer near-term prospects for clinical translation.
INTRODUCTION
The prevailing standard treatment for triple negative breast cancer (TNBC) is cytotoxic chemotherapy, but more than 70% of the patients have a partial response only and have significantly higher relapse and mortality rates compared to non-TNBC patients (1). Non-TNBC patients greatly benefit from targeted therapies, such as trastuzumab and tyrosine kinase inhibitors, but effective targeted therapies are not available for TNBC. TNBCs are transcriptionally distinct compared to other breast cancer subtypes, and often have active EGFR, high activity of PI3K pathway, frequent functional loss of RB pathway activity and a very high rate of TP53 mutations (2). Yet, EGFR inhibitors have not proven effective in clinical trials and synthetic lethality strategies targeting DNA repair pathway deficiency have failed to improve cure rates in TNBCs (3,4).
To help generate genetic predictors of drug response, the Cancer Cell Line Encyclopedia (CCLE) study compared pharmacological profiles of 24 anticancer drugs across 479 cancer cell lines, including 56 breast cell lines (5), but only few predictors emerged, including SLFN11 expression that predicted sensitivity to irinotecan and topotecan. Genome-wide shRNA screen in 77 breast cancer cell lines coupled with genomic and proteomic data revealed that basal A cells are preferentially sensitive to CAND1-NEDD8 depletion and TNBC patients may benefit from NEDD8 inhibitors (6). A screen testing 130 drugs in 639 cell lines from different cancers reported that single gene-drug associations rarely explain drug sensitivities (7). Drug screen in lymphoma and melanoma have discovered multiple combinations for lymphoma and mutant BRAF melanomas that could be selected for further clinical evaluation (8–10). Although broad combinatorial screens are crucial to identify and prioritize effective drug combinations for a thorough investigation prior to clinical evaluation, only few such screens have been reported for cancers.
To address these limitations, we undertook a comprehensive medium-throughput drug combination screen in TNBC cell lines with different and overlapping genetic backgrounds to identify novel effective combination therapies for TNBC. In order to fast-track clinical testing of potentially promising combinations, we systematically assessed the impact of 128 single agents in combinations with each of six drugs already approved by the US-FDA, resulting in 768 pairwise drug combinations covering a wide range of targets and processes implicated in cancer biology. We report anticancer activity of drug combinations based on overall growth inhibition and synergistic drug effects in six TNBC cell lines. Finally, we used transcriptional profiles, protein expression and activity and mutational data to characterize the drug sensitivity patterns and mechanisms of action of the most promising drug combinations.
METHODS
Cell lines and selected drug panels
TNBC cell lines (BT-20, MDA-MB-231, MDA-MB-468, BT-549, MDA-MB-436, HCC-38, HCC-70, HCC1500, MDA-MB-157) harboring genetic abnormalities commonly observed in TNBC patients [15, 16] were purchased from the American Type Culture Collection (Manassas, VA) where all cell lines were authenticated by short tandem repeat profiling, karyotyping, morphology and cytochrome C oxidase I testing. Cell lines were obtained between 2013 and 2015, and used at passages 3–9, and cultured less than 3 months after resuscitation. For details, see Supplementary Data.
Medium throughput screen protocol (MTS)
On the day of the experiment, cells were trypsinized, harvested, counted using Countess (Invitrogen) and deposited into 384-well ViewPlate-384F microtiter plates (Perkin Elmer) at 750 cells/well in 16 μL respective medium using a multidrop dispenser (Thermo) and allowed to attach overnight (Figure 1). The following day, single-dose 5× stocks of the six Panel A drugs were prepared in medium, and 4 μL of respective drug-containing medium was added to each well (4 plates/drug) using the multidrop dispenser. For Panel B drug addition, a PlateMate Plus automated instrument (MatrixTechCorp) was used for pin transfer of 20 nL drug volume from 1000× drug stock plates into 384-well microtiter cell plates. For details, see Supplementary Data.
Figure 1.
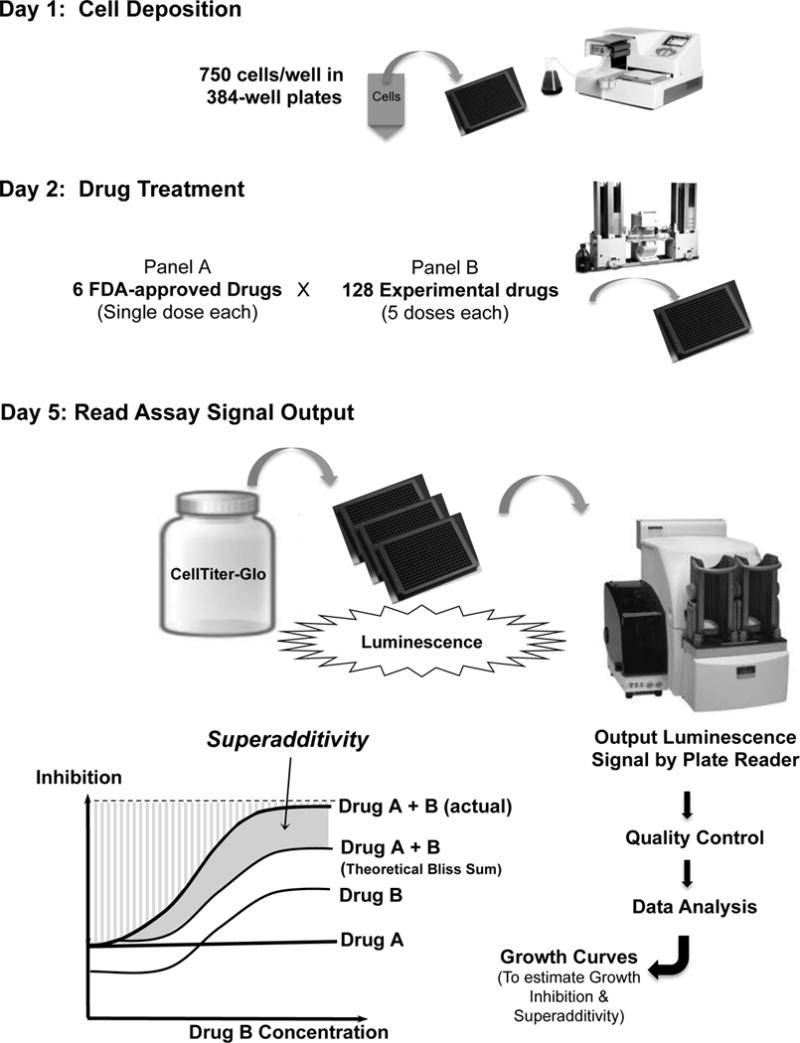
Diagram of medium-throughput drug screen in TNBC.
Synergy assessment
Synergy was assessed as the deviation from the Bliss non-interaction model (11) as well as by Chou and Talalay isobologram analysis (12). Best combinations screened by MTS were validated in low-throughput format. See Supplementary Data for details.
Flow-cytometry
Cells were plated at 1×106 cells/well in 100mm culture plates and allowed to adhere overnight. Cell lines were exposed to respective control or treatment media for 24h. Cells were stained using the BD Pharmingen Apoptosis Detection Kit II according to the manufacturer’s protocol (BD Biosciences, San Jose, CA). See Supplementary Data for details.
Reverse phase protein arrays (RPPA)
Cells were plated in 100 mm culture plates (4 plates/cell line) that were exposed to DMSO control, 1μM crizotinib, 1μM ABT-263 or their combination. The experiment was run in duplicate. After 24h of treatment exposure, cells were collected and washed with PBS and 48 cell pellets corresponding to all samples were shipped to RPPA Core Facility at the MD Anderson Cancer Center (Houston, TX) for analysis.
Western blot and immunoprecipitation (IP)
Whole protein lysates were prepared and protein concentrations determined by the Bradford assay. Phospho-tyrosine immunoprecipitations and standard blotting procedures for gel electrophoresis and immunoblotting using PVDF membranes were performed as described previously (13). See Supplementary Data for details.
RESULTS
Growth inhibitory effect of drugs as single agents
Paclitaxel (microtubule inhibitor), erlotinib (EGFR inhibitor), everolimus (mTOR inhibitor), vismodegib (SMO/Hedgehog pathway inhibitor), XL-184 (VEGFR2/MET inhibitor) and crizotinib (ALK/MET inhibitor) are the six US FDA-approved drugs (Panel A drugs) that we used in the MTS along with 128 Panel B drugs in basal-like (MDA-MB-468, HCC-38, BT-20), mesenchymal stem-like (MDA-MB-231, MDA-MB-436), and mesenchymal (BT-549) TNBC cell lines (14). The dose response of Panel A drugs in cell lines was first characterized in low-throughput screening experiments (Supplementary Figure 1) to select a single dose of each drug. Single dose was selected as a highest concentration that was still ≤ IC50 across all cell lines and was also well tolerated clinically (Supplementary Table 1). This single dose was combined with five doses each of 128 Panel B drugs (Supplementary Table 2) for MTS. Drugs were tested singly or as combination treatments in 384-well plates to assess growth inhibition and superadditivity as described in Methods (Figure 1). Growth inhibition of TNBC cells by a single dose of each of the six Panel A drugs in the MTS format was moderate (Figure 2A), as expected for these doses (<IC50). The growth inhibitory effect of the 128 secondary drugs (Panel B), estimated by the area under the dose response curve (AUC) for each drug, indicated cell growth suppression by most drugs in all cell lines (Figure 2B). TNBC cells were more sensitive to genotoxins (GTOX) and apoptosis regulators (APOP), as indicated by higher AUC (red color) of most single agents in these drug categories (Figure 3A and Supplementary Figure 2A). Single agents that suppressed growth with AUC > 75% across TNBC cells clearly show enrichment in GTOX and APOP categories (Figure 3B and Supplementary Figure 2B). Basal-like subtype of TNBC reportedly exhibit higher expression of cell cycle and DNA damage response genes and a higher sensitivity to DNA-damaging agents (14), while we have reported resistance of mesenchymal stem-like (MSL) cells to EGFR inhibitors including lapatinib, and BEZ-235, a PI3K-mTOR inhibitor (15). Indeed, basal like-1 HCC-38 and MDA-MB-468 cells were most sensitive while MSL MDA-MB-436 and MDA-MB-231 were least sensitive to these drugs. P53, a prime regulator of apoptosis, is mutated in most TNBCs (16). All TNBC cell lines in our screen carried a TP53 mutation, and interestingly were highly sensitive to APOP drugs (Figure 3B and Supplementary Figure 2B). Specifically, APOP drugs YM155, bortezomib, and carfilzomib displayed most potent anticancer activity as single agents across all TNBC lines, as indicated by higher AUC (red color) in Figure 3A. YM155 is a selective suppressant of survivin, a member of the inhibitor of antiapoptosis (IAP) family that has higher nuclear activity in TNBC compared to other subtypes (17). Interestingly, MDA-MB-231 TNBC cells were more sensitive to YM155 induced growth inhibition than SKBR3 or MCF7 non-TNBC cell lines (18). AKT inhibitor KP372-1, dactinomycin, digoxin and triptolide were a few other highly potent drugs that similarly inhibited growth of all TNBCs.
Figure 2.
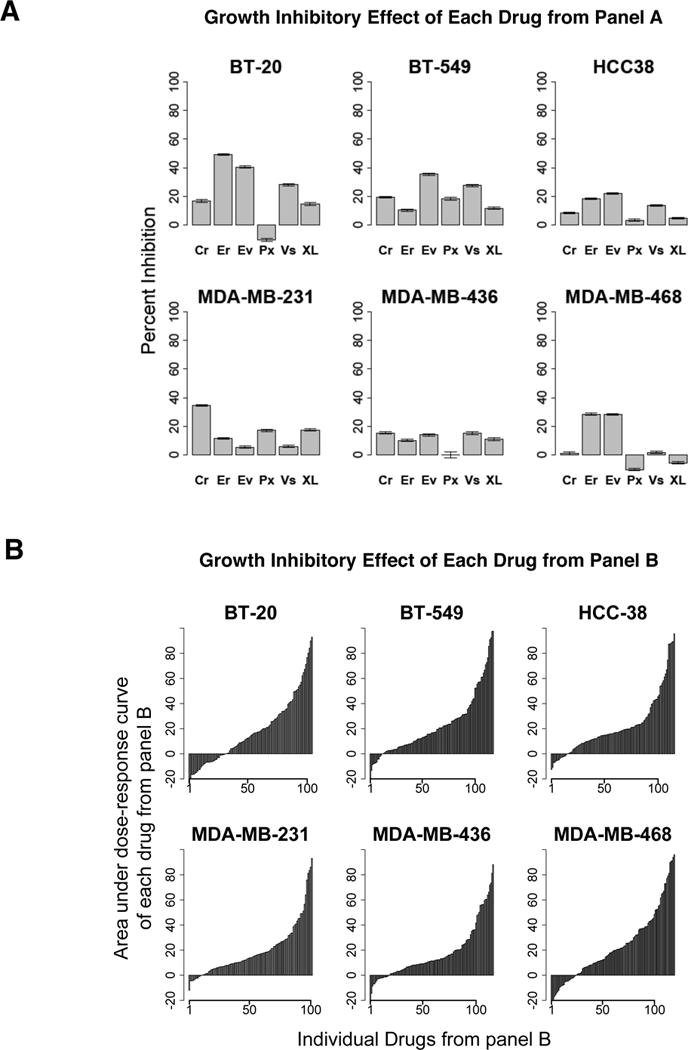
Effect of drugs from panel A and panel B on the growth of TNBC cell lines. A: Percent growth inhibition of TNBC cell lines by single dose of each of the six panel A drugs (Cr=Crizotinib 1μM, Er=Erlotinib 3μM, Ev=Everolimus 30nM, Px=Paclitaxel 3nM, Vs=Vismodegib 10μM, XL=XL-184 3μM) from the medium-throughput drug screen. B: Growth inhibition by 128 panel B drugs as single treatments (2.4nM-10μM) in TNBC cell lines; vertical bars indicate the area under the individual dose response curve (AUC) for each drug.
Figure 3.
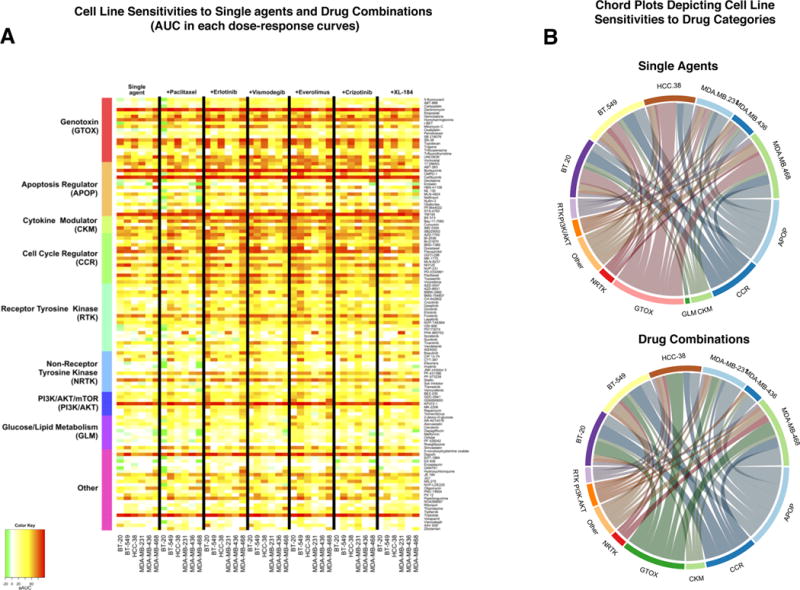
Patterns of activity of drugs grouped in different categories across cell lines. A: Heatmap summarizing the results of MTS by the area under the curve (AUC) for each dose-response curve for single agents or pairwise drug combinations. AUC gives a single estimate of growth inhibition over multiple doses for each curve, with higher red color intensity indicating higher AUC. The first column set indicates response of six cell lines to 128 single agents (panel B) as rows, arranged in categories according to the drug’s target or mechanism of action. The subsequent six column-sets indicate growth inhibitory response when single agents are combined with either paclitaxel, everolimus, vismodegib, everolimus, crizotinib or XL-184 (panel A) respectively in the six TNBC cell lines. B: Chord plots display the drugs in each category that suppressed growth with AUC > 75% as single agents (top) or drug combinations (bottom) for each of the cell lines. In each chord plot, the upper semicircle reflects the relative sensitivities of cell lines toward various drug categories, while the lower semicircle reflects the relative efficacy of drug categories: circumference length indicates the number of drugs achieving AUC > 75% in that category.
Growth inhibitory effect of pairwise drug combinations
The inhibitory effect of 128 drugs individually and in combination with paclitaxel, erlotinib, vismodegib, everolimus, crizotinib, or XL-184 was assessed by the AUC for each dose response curve and is shown across cell lines in the heatmaps in Figure 3A and Supplementary Figure 2A. More than 90% of the combinations tested exert antiproliferative activity while the remaining have either no effect on cell growth or a slight pro-proliferative effect (Supplementary Figure 3). Global comparison between cell line sensitivities in this screen reveal overall lower sensitivity of MSL MDA-MB-436 and MDA-MB-231 versus basal-like TNBC cell lines. (Supplementary Figure 2A, B). Since mesenchymal and MSL TNBC cells often have downregulated RTK expression, we examined the expression of RTKs in the CCLE portal (http://www.broadinstitute.org/ccle) and observed that TNBC cell lines express very low levels of many RTK genes, RET, TEK, PDGFRA, PDGFRB, FLT1, FLT3, FLT4, and ALK, but express MET, KIT, EGFR, ERBB3, MST1R, AXL. AXL expression is variable in each TNBC category while KIT, EGFR, ERBB3, and MST1R are downregulated in MSL and mesenchymal cell lines, which may contribute to their lower sensitivity to drugs. Interestingly, MET is expressed by all TNBC categories.
Combinations with erlotinib were particularly effective in BT-20 and MDA-MB-468 cell lines, while MSL MDA-MB-231 and MDA-MB-436 cells were the least sensitive (Figures 3A and 4B). We have previously reported that MSL cells downregulate EGFR and become erlotinib resistant (15). MDA-MB-468 cells express the highest levels of EGFR followed by BT-20 (data not shown), which may partly explain their higher sensitivity to drug combinations involving EGFR inhibitor erlotinib. The effects of drug combinations grouped by categories and visualized in the chord plot of Figure 3B also suggest drugs in the GTOX and APOP categories as most potent suppressors of TNBC cell growth, while drugs in the glucose/lipid metabolism (GLM) category have the lowest potency overall (Figure 3B and Supplementary Figure 2B). Cell lines displayed differential sensitivity towards GTOX drugs with basal-like HCC-38 and MDA-MB-468 being most sensitive and MSL MDA-MB-436 cells least sensitive, but displayed similar sensitivity to APOP drug combinations. However, combinations with GLM were least effective and therefore absent from the chord plot for drug combinations with AUC >75% (Figure 3B, Supplementary Figure 2B).
Figure 4.
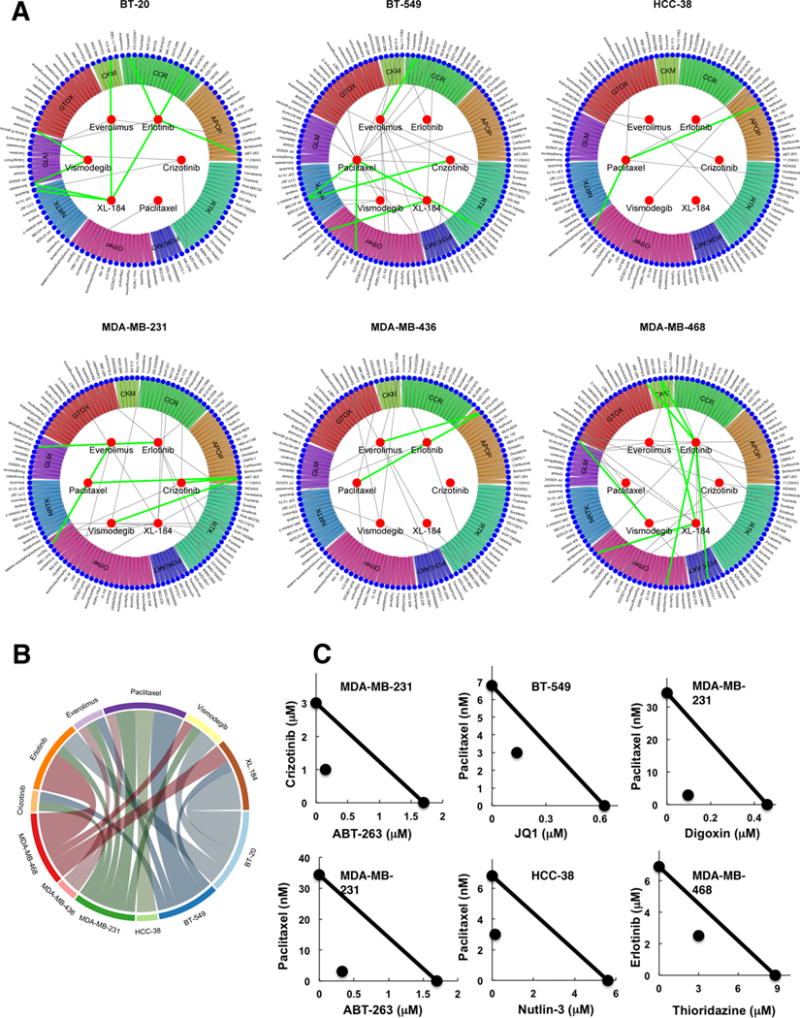
Synergistic drug combinations against TNBC cells. A: Circular plots show the six drugs inside the inner circle (panel A) that were combined with 128 drugs (panel B) shown in the periphery grouped in drug categories. Green lines link the best while grey lines link the second best synergistic combinations, as described in the results. B: Top synergistic combinations are summarized in a chord plot by cell line and panel A drug. C: Isobologram analysis depicting pharmacological interaction between two drugs at the IC50 (except Nutlin-3 where IC10 was used) level of individual drugs and their combination. The straight line in each isobologram joins the individual IC50 doses of two drugs on the x- and y-axes. The data point in each isobologram, corresponding to IC50 dose of drugs given in combination, lies below the line indicating synergism.
Association between drug-sensitivities of cell lines and expression of drug targets
To explore potential links between the drug sensitivities of cell lines and their genetic background, we compared the overall response of cell lines to drugs with expression levels obtained from CCLE portal (5) of the 170 genes that are direct or indirect targets of the 128 drugs included in the MTS (Supplementary Figure 4). Although sensitivities differed between cell lines, the expression patterns of drug targets appeared broadly similar across cell lines (Supplementary Figure 4). We evaluated these effects more systematically at the cellular pathway level by grouping the proteins targeted by drugs in each of the 9 categories into “targeted pathways” and assessing the expression of genes in each targeted pathway across cell lines (Supplementary Table 3). Specifically, the degree of upregulation or downregulation of a targeted pathway was determined for each cell line as the percentage of the pathway genes found in the upper or lower tertiles of expression relative to all CCLE breast cancer cell lines. We then performed unsupervised principal component analysis (PCA) to segregate the cell lines according to the extent of upregulation or downregulation of all targeted pathways (Supplementary Figure 5A). Interestingly, pathways targeted by drugs in the RTK and NRTK categories appear to be highly upregulated in HCC-38 cells (Supplementary Figure 5A), and these drugs appear to be largely ineffective against these cells, either when used alone or in combination with other drugs (Supplementary Figures 5B). Lack of upregulation of the same pathways in BT-549 cells appears to be associated with sensitivity to these agents. Upregulation of GLM targets is associated with greater sensitivity to GLM drugs in BT-20 while and downregulation of RTK genes was associated with reduced sensitivity to RTK, NRTK and cytokine modulator (CKM) drugs in MDA-MB-436 cells (Supplementary Figure 5A, B). Accounting for drug-to-drug variability over drugs within the same category, the observed patterns suggest that upregulation of the targeted pathway as determined by a high proportion of highly expressed targeted genes, is generally but not always associated with sensitivity to the targeted treatment in these TNBC cell lines. Collectively, these results suggest that the fraction of all upregulated or downregulated genes of a particular targeted pathway as a surrogate index of overall pathway activity may partly explain cell sensitivity towards agents targeting that pathway.
Top Synergistic Combinations
Drug combinations where two agents at lower doses act synergistically are of particular interest owing to their potential to improve efficacy and moderate the toxicity associated with high doses of single agents in the clinic. We used the Bliss model of synergy (19,20) to assess superadditivity. Sham combinations in which the same drug was present in both drug panels were evaluated in the same MTS protocol as controls and yielded almost zero superadditivity, as expected (Supplementary Figure 6A). Most drug combinations tested were antiproliferative, but only a minority displayed superadditive effects (Supplementary Figure 6B). We selected top synergistic combinations as those having superadditivity (area between combination and Bliss dose-response curves) ≥ 10%, inhibitory potential (area above the curve) ≤ 75%, estimable EC10 concentrations for the combination and theoretical Bliss curves, maximum growth inhibition by drug combination ≥ 80%, and superadditivity observed for at least 3 consecutive doses of the panel B drug used in the combination. These top combinations are indicated by green lines in the circular synergy plots in Figure 4A (listed in Supplementary Table 4), while grey lines indicate second best combinations where superadditivity was observed for at least 2 consecutive panel B drug concentrations and had maximum growth inhibition ≥ 50%. These represent novel synergistic drug combinations that can be potentially effective in TNBC and therefore merit further investigation. Summary of the top combinations (green lines) by Panel A drugs and cell lines indicate higher number of synergistic combinations in MDA-MB-468 and BT-20, frequently containing erlotinib or XL-184 panel A drugs (Figure 4B). Isobologram-based synergy analyses (12,21) of representative top combinations also demonstrated synergistic growth inhibitory effects of combinations (Figure 4C). Combination index (CI) values were less than 1, indicating a high level of synergism that resulted from combined treatment (Supplementary Table 5). The dose reduction index (DRI) confirmed a high level of synergism that resulted from combined treatment, e.g., the dose of nutlin-3 or paclitaxel could be respectively reduced 34- and 2-fold and still produce the same antiproliferative effect in HCC-38 cells if combined together (Supplementary Table 5). As the cell lines used represent distinct mutational and expression backgrounds, it is not surprising that none of the drug combinations was highly synergistic across all cell lines. However, it is worth noting that top combinations indicated in the circular plots may also be synergistic in multiple cell lines albeit to a lesser degree but falling below the threshold for top hits in circular plots. Combinations of NFkB inhibitor Bay-11-7082 and erlotinib, and WEE1 kinase inhibitor MK-1775 and everolimus had mild synergistic activity in all TNBC lines. Superadditivity between Bay-11-7082 and erlotinib was high enough to be indicated by grey lines in BT-549, MDA-MB-436 and MDA-MB-468 circular plots (Figure 4).
A striking observation was that most synergistic combinations contained drugs inducing apoptosis (APOP) or receptor tyrosine kinases (RTKs) or both, suggesting apoptotic and RTK signaling as two major cellular signaling epicenters critical to survival and proliferation of TNBC cells. Alternatively, RTKs could also be activated as part of resistance mechanisms in TNBCs. ABT-263, a BCL2 inhibitor (apoptosis inducer) synergized with RTKs erlotinib and crizotinib. Erlotinib also synergized with 5-FU, an antimetabolite commonly administered systemically in many cancers. Apoptosis inducers, ABT-263, Nutlin-3 (MDM2 inhibitor), STA-4783 (oxidative stress inducer) and JQ1 (bromodomain inhibitor) also synergized with paclitaxel, which is a part of combined chemotherapy regimen with cyclophosphamide and adriamycin in breast cancer. Nutlin-3 inhibits the interaction between MDM2 and P53, stabilizing P53 and inducing apoptosis, and has shown potent preclinical activity in several cancers (22). STA-4783 or elesclomol triggers apoptosis in cancer cells by inducing oxidative stress, and is being tested in trials for various cancers (23,24). JQ1 is a bromodomain (BRD) inhibitor. BRD induced oncogenic signaling appears to be pervasive in all breast cancer subtypes (25); disrupting BRD function in basal-like breast cancer suppressed tumorigenesis (26), and TNBCs appear to be preferentially sensitive to BRD inhibition (27). We have recently reported anticancer activity of JQ1 in drug combinations in vitro and in vivo (28). Interestingly, JQ1 was also found to synergize with RTK inhibitors, erlotinib, XL-184, and crizotinib.
YM155, bortezomib, carfilzomib (APOP), KP372-1, and triptolide resulted in complete growth inhibition of all TNBC cell lines at low nanomolar concentrations as single agents, and also in combinations. The lowest two or three doses of these drugs already achieved 100% growth inhibition but the data points were inadequate to reliably assess synergy. Nevertheless, our observations recognize and emphasize the potential of these drugs to benefit TNBC patients when given alone or in combination with other agents.
Validation of MTS results in low-throughput experiments
A large number of variables can affect high/medium-throughput assays, and lack of standardized protocols adds to the likelihood of discordant and misleading results (29). Our MTS was conducted in duplicate plates following rigorous quality control and data filtering steps as described in Methods. Yet, to assess the reproducibility of MTS results, we re-tested two of the top synergistic combinations, ABT-263 with crizotinib or paclitaxel, in 96-well format across all cell lines and compared to the medium-throughput results (Figure 5A). 3nM paclitaxel or 1μM crizotinib inhibited <50% of cell growth, and 2.4nM-10μM ABT-263 when given alone generated a sigmoidal dose-response curve in sensitive cell lines or a flatter curve in resistant cell lines similarly in both formats. Also, superadditive inhibitory effects of combinations were observed, as indicated by the dose-response curve for the combination (black line) being above the theoretical Bliss sum (green line) in same cell lines in MTS as well as low-throughput experiments. Broadly, growth inhibition and synergy patterns observed with two combinations across cell lines in 96-well plate low-throughput experiments with 4 replicates per group were similar to MTS results with 2 replicates per group, validating medium-throughput findings. The entire MTS dataset used for analysis is provided as Supplementary Table 6, while the low-throughput validation dataset is provided as Supplementary Table 7.
Figure 5.
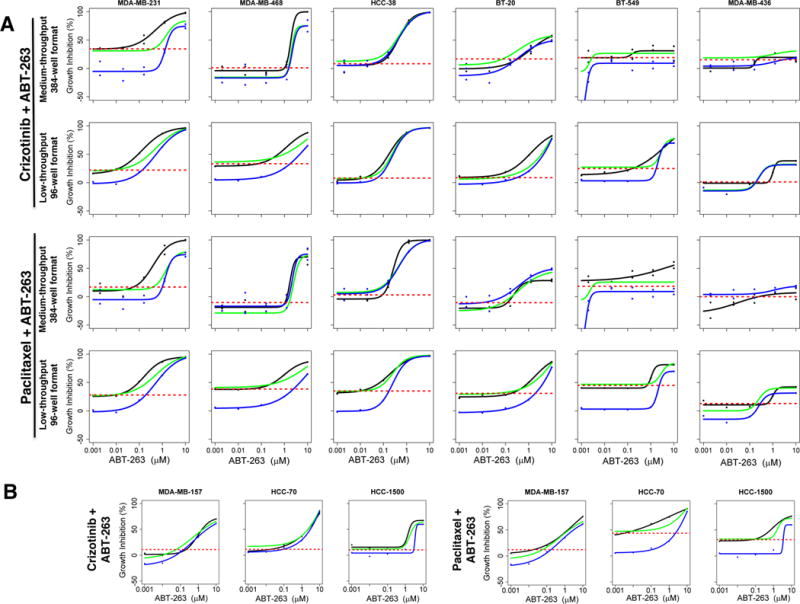
Validation of MTS results in low-throughput format. A: Experiments with Crizotinib-ABT-263 and paclitaxel-ABT-263 were repeated across cell lines in low-throughput 96-well format (4 replicates/group) as opposed to 384-well medium-throughput format (2 replicates/group). Each graph shows growth response curves for individual drugs and their combination. Growth inhibition by a single dose of panel A drugs (1μM crizotinib or 3nM paclitaxel) is indicated by broken red line, while the dose response curves for ABT-263 (panel B drug) alone, theoretical bliss (additive effect), and drug combinations are indicated by blue, green and black lines respectively. B: The two drug combinations were tested in additional basal (HCC-70, HCC-1500) and mesenchymal stem-like (MDA-MB-157) cell lines in 96-well plate format.
These drug combinations were also tested in additional mesenchymal and stem-like (MDA-MB-157) and basal (HCC-1500, HCC-70) cell lines. ABT-263 elicited dose-dependent growth inhibition alone and in combination with paclitaxel or crizotinib in all these cell lines. Synergy was observed with paclitaxel-ABT-263 in all these cell lines; and with crizotinib-ABT-263 in HCC-1500 cells (Figure 5B). Intriguingly, XL-184, another MET inhibitor, exhibited very similar response to crizotinib when combined with ABT-263 across all cell lines. (Supplementary Figure 7A).
Mechanistic effects of ABT-263 and crizotinib
To assess the biological mechanism of crizotinib/ABT-263, we looked at the mRNA expression of their targets in CCLE. Targets of ABT-263 are BCL-xL and BCL2, while crizotinib inhibits activity of anaplastic lymphoma kinase (ALK), MET and AXL, and is approved by the US-FDA for treatment of ALK positive non-small cell lung cancer. However, ALK expression is generally low and ALK rearrangement or mutations are very rare in breast cancer, and none of the cell lines expressed ALK or had any ALK gene aberration. Interestingly, MDA-MB-231 cells express higher levels of BCL-xL, MET and AXL mRNA, and are most sensitive, while MDA-MB-436 cells with the lowest expression of BCL-xL are least senstitive to the drug combination. However, expression of BCL-xL, AXL and of about 26 direct and indirect targets of these two drugs does not explain sensitivities of all cell lines (Supplementary Figure 7B).
To directly assess the mechanistic effects, we measured the basal and activated levels of 291 signaling proteins, including the direct drug targets, before and after crizotinib and ABT-263 treatment by reverse phase protein array (RPPA) analysis. Table 1 describes the RPPA results as fold-changes of the most affected proteins in each cell line, as compared to respective vehicle treated control cells. Proteins with fold changes differing in multiple cell lines but with same directionality are also listed in this table. ABT-263 treatment increased the expression of cleaved-caspase-7, cleaved caspase-3, Poly(ADP-ribose) polymer (PAR), and slightly decreased BCL-xL, while crizotinib decreased activated levels of phosphorylated EGFR or MAP kinases in different cell lines, and their combination produced enhanced alterations in these proteins. Intriguingly however, combined treatment with ABT-263 and crizotinib predominantly altered the levels of histone H3, dimethyl histone H3, plasminogen activator inhibitor 1 (PAI1), Collagen IV, and lactate dehydrogenase A (LDHA) across most TNBC cell lines in addition to alterations in phosphorylated kinases in MAPK pathway (p44/42, pT202/204, p180/182 p38, p235/236 and p240/244 S6), BCL-xL, PAR, and cleaved caspases. Whether these effects on H3, PAI1, collagen IV and LDHA are bystander changes caused by ABT-263-crizotinib, or if they play a critical role in TNBC growth needs further investigation. Highest fold changes in proteins were observed in HCC-38 cells that were most sensitive to this treatment (Table 1). Overall, crizotinib alone modestly altered protein expression, ABT-263 alone produced relatively greater fold changes, while the combination produced the highest fold changes in the above proteins. In contrast to more sensitive HCC38, MDA-MB-468, MDA-MB-231 and BT-20 cell lines in which MAPK pathway was suppressed, no decrease in pathway activity was seen in BT-549 or MDA-MB-436 cells that were least sensitive. These results suggest a pro-apoptotic program turned on by ABT-263 and inhibition of mitogenic signaling by crizotinib blockade of RTKs resulting in growth inhibition and apoptosis of cells.
Table 1.
Proteins altered by ABT-263-crizotinib as assessed by RPPA. TNBC cell lines were exposed to 1μM crizotinib or 1μM ABT-263 alone or their combination in duplicate for 24h, and the protein extracts were assayed for the expression of basal and activated levels of 291 proteins at the RPPA Core Facility at the MD Anderson Cancer Center (Houston, TX). Top altered proteins are listed and their fold change compared with respective vehicle treated controls is shown in brackets. Blue indicates proteins upregulated while red indicates proteins downregulated by the drug combination. Interesting candidate proteins are represented in bold.
| Crizotinib (Protein FC relative to control) |
ABT-263 (Protein FC relative to control) |
Combination (Protein FC relative to control) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HCC38 | ||||||||||||||
| DM Histone H3 (1.11) Histone H3 (1.18) PAI1 (1.19) PAR (1.01) Collagen IV (1.3) Cleaved Caspase-7 (1.03) Cleaved Caspase-3 (1.04) p27 Kip1 (1.03) Bcl-xL (0.99) p44/42 MAPK (0.99) p38 p180_pY182 (0.95) LDHA (1.04) |
DM Histone H3 (2.09) Histone H3 (2.26) PAI1 (1.45) PAR (4.35) Collagen IV (1.86) Cleaved Caspase-7 (1.58) Cleaved Caspase-3 (1.23) p27 Kip1 (1.03) Bcl-xL (0.91) p44/42 MAPK (0.85) p38 p180_pY182 (0.65) LDHA (0.47) |
|
||||||||||||
| MDA-MB-468 | ||||||||||||||
| PAI1 (1.41) PAR (1.29) Collagen IV (1.05) NDRG1_pT346 (1.55) Bcl-xL (1.06) MAPK_pT202_Y204 (0.91) EGFR_pY1068 (0.84) LDHA (1.05) B7H4 (0.83) X14.3.3.zeta (0.77) |
PAI1 (1.45) PAR (1.33) Collagen IV (1.08) NDRG1_pT346 (1.54) Bcl-xL (0.91) MAPK_pT202_Y204 (0.82) EGFR_pY1068 (0.92) LDHA (0.79) B7H4 (0.92) X14.3.3.zeta (0.98) |
|
||||||||||||
| MDA-MB-231 | ||||||||||||||
| Collagen IV (1.16) PAR (1.05) S6_pS235_S236 (0.95) S6_pS240_S244 (0.87) Bcl-xL (0.95) LDHA (0.95) B7H4 (0.94) Bad_pS112 (0.97) |
Collagen IV (1.11) PAR (1.21) S6_pS235_S236 (0.75) S6_pS240_S244 (0.73) Bcl-xL (0.82) LDHA (0.73) B7H4 (0.77) Bad_pS112 (0.88) |
|
||||||||||||
| BT-20 | ||||||||||||||
| DM Histone H3 (1.04) Histone H3 (0.98) Collagen IV (1.07) PAR (1.08) Mcl1 (1.00) PDL1 (1.02) p21 (0.95) Bcl-xL (1.00) LDHA (1.10) SDHA (0.99) NDRG1_pT346 (0.98) MAPK_pT202_Y204 (1.18) |
DM Histone H3 (1.63) Histone H3 (1.67) Collagen IV (1.07) PAR (3.34) Mcl1 (1.74) PDL1 (1.70) p21 (1.28) Bcl-xL (0.78) LDHA (0.64) SDHA (0.54) NDRG1_pT346 (0.56) MAPK_pT202_Y204 (0.67) |
|
||||||||||||
| BT-549 | ||||||||||||||
| DM Histone H3 (1.23) Histone H3 (1.28) Mcl-1 (1.31) PAR (1.39) S6_pS235_S236 (0.95) S6_pS240_S244 (0.92) Bcl-xL (1.00) LDHA (0.688) |
DM Histone H3 (1.05) Histone H3 (1.28) Mcl-1 (1.31) PAR (1.39) S6_pS235_S236 (1.23) S6_pS240_S244 (1.08) Bcl-xL (0.92) LDHA (0.688) |
|
||||||||||||
| MDA-MB-436 | ||||||||||||||
| Mcl1 (1.10) DUSP4 (0.99) Histone H3 (1.01) DM Histone H3 (1.04) PAI1 (1.09) PAR (0.91) Collagen IV (0.96) Bcl-xL (0.97) |
Mcl1 (1.46) DUSP4 (1.45) Histone H3 (1.36) DM Histone H3 (1.33) PAI1 (1.20) PAR (1.34) Collagen IV (1.07) Bcl-xL (1.04) |
|
The effect of ABT-263/crizotinib on programmed cell death was further corroborated by Annexin V labeling. Exposure to crizotinib across cell lines resulted in maximum apoptotic/dead cells in BT-20 (21%) while ABT-263 alone resulted in maximum cell death in HCC-38 (73%) as indicated by the sum of two upper quadrants in each dot plot in Supplementary Figure 8. However, combined treatment with ABT-263 and crizotinib resulted in maximum cell death in HCC-38 cells (82%). Apoptosis was greater in HCC-38, MDA-MB-468 and MDA-MB-231 cell lines, consistent with their higher sensitivity in growth assays, while BT-549 and MDA-MB-436 were least sensitive to drug induced apoptosis (Supplementary Figure 8).
To confirm RPPA and flow-cytometry results, we performed immunoblotting using sensitive MDA-MB-231 and relatively resistant MDA-MB-436 cell lines. We first looked at the effect of crizotinib on the total and activated levels of its targets, MET and AXL, when given alone or in combination with ABT-263. Crizotinib blocked MET phosphorylation in both cell lines, when given alone or in combination, but had no effect on the phosphorylated AXL levels after 1h treatment exposure (Figure 6A). Basal MET and AXL levels were both higher in MDA-MB-231 cells than MDA-MB-436. Total MET levels remained unaltered, and total AXL was slightly increased by crizotinib, while ABT-263 had no effect on the total or active levels of AXL or MET. Next, we determined the effect of this combination on downstream AKT and MAPK activity. Consistent with the RPPA results, there was little or no decrease in the phosphorylated levels of AKT and ERK1/2 upon drug combination treatment in these cell lines (Figure 6B). It is noteworthy that although MDA-MB-436 cells express lower levels of AKT than MDA-MB-231, they express conspicuously higher levels of phosphorylated (active) AKT, indicating a hyperactive AKT signaling in this cell line which may contribute to its lower sensitivity to the drug combination. Indeed, MDA-MB-436 cells were more sensitive than MDA-MB-231 to AKT inhibitor KP372-1 that resulted in 100% growth inhibition of MDA-MB-436 cells in nanomolar doses when given alone or in combination in our screen. On the other hand, we looked at the effect of ABT-263 on caspase-3 and PARP cleavage. As shown in Figure 6C, ABT-263 induces cleavage of caspase-3 and PARP which is further enhanced by combination treatment with crizotinib, confirming apoptosis. BCL-xL protein expression was higher in MDA-MB-231 than in MDA-MB-436 cells, but neither the levels of BCL-xL nor its pro-apoptotic counterpart BAD changed upon drug treatment in 24h. Interestingly though, we observed a slight decline in BCL-xL levels after 72h of ABT-263 treatment in additional experiments (data not shown). Among the two MSL cell lines, MDA-MB-436 cells express very low levels of basal and active levels of EGFR. Although drug treatment alone had no effect on EGFR, combined treatment slightly decreased phosphorylated EGFR levels in MDA-MB-231 cells. Taken together, ABT-263 induces apoptosis and exerts synergistic anticancer activity in combination with MET inhibitor crizotinib that suppresses MET kinase activity in TNBCs, as summarized in Figure 6D.
Figure 6.
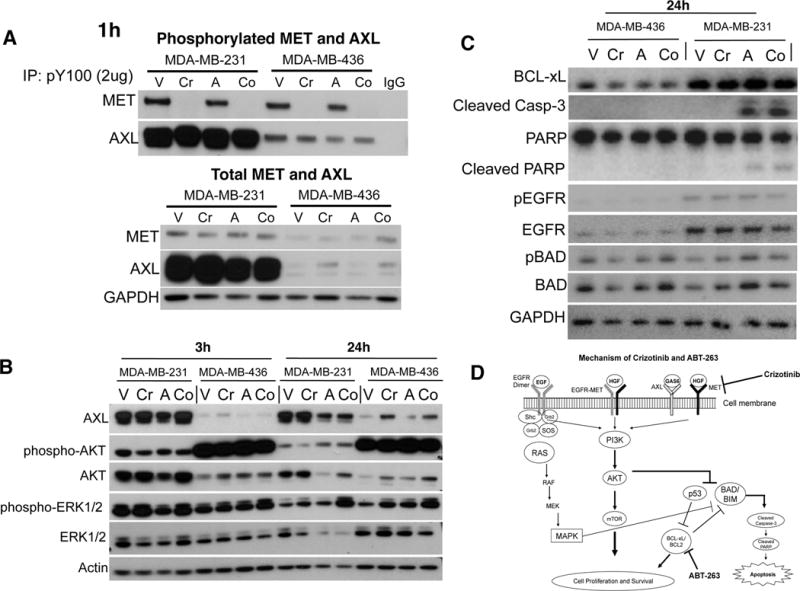
Mechanism of action of ABT-263 and crizotinib. MDA-MB-231 cells in which crizotinib-ABT-263 elicited highest synergistic response and MDA-MB-436 cells that were least sensitive were treated with either vehicle (V), 1μM crizotinib (Cr), 1μM ABT-263 (A), or their combination (Co) for 1, 3 or 24h. A: To determine phosphorylated MET and AXL levels, phosphorylated proteins were immunoprecipitated with anti-phospho-tyrosine (P-Tyr-100), using IgG as control, and then immunoblotted with anti-MET and anti-AXL antibodies. Total MET and AXL were directly immunoblotted using respective antibodies using GAPDH as internal control. B: Effect of treatments on the downstream activated (phosphorylated) levels of AKT and ERK1/2 were determined at 3h and 24h, using actin as internal control. C: To determine the effects of drug treatments on apoptotic markers and EGFR activity, relative levels of BCL-xL, cleaved caspase-3 and PARP, total and phosphorylated BAD (pBAD) and EGFR were determined by immunoblotting after 24h treatment exposure. D: Proposed mechanism of action of the crizotinib-ABT-263 combination. Crizotinib inhibits MET activity and MET dependent RTK activity and downstream signaling. In contrast, ABT-263 inhibits BCL-xL and BCL2, thereby enabling proapoptotic proteins to trigger apoptosis indicated by cleavage of caspase-3 and PARP. When combined, crizotinib and ABT-263 act synergistically resulting in enhanced inhibition of TNBC cell growth and apoptosis.
DISCUSSION
We chose six US FDA-approved drugs and combined them with 128 agents to identify highly synergistic and effective combination treatments in TNBC. This is a novel guided high-throughput drug screening design that allows to identify effective combinations that can be translated quickly to the clinic. The broadly selected drug panel allowed us to systematically evaluate associations of cell line sensitivities with drug categories, and integrating genomic, transcriptomic, and proteomic data to provide additional biological insights for the effective combinations. This could enable making informed predictions as to which drug combinations are likely to have the highest impact in clinical trials. We discovered specific promising drug combinations that could be tested in clinical trials owing to their potent synergistic anticancer activity in culture.
The cost and time needed to develop a new chemical entity is often prohibitive (30). Therefore, repurposing approved and partially developed drugs for cancer creates an opportunity to rapidly advance to patients better drug therapies by capitalizing on existing data and experience (31,32). Furthermore, rationally selecting and testing such drugs in large combination screens is critical to find novel effective therapies, but such screens have been rather limited (8). We present the first guided medium-throughput combination screen for TNBC to test efficacy and synergy of 768 unique drug combinations.
One of the effective synergistic drug combinations identified in this screen was ABT-263 (BCL-xL/BCL2 inhibitor) and crizotinib. Targeting the BCL2 apoptotic pathway has been proposed as a therapeutic strategy in TNBC (16,33). ABT-263 or navitoclax was well tolerated and showed efficacy in chronic lymphocytic leukaemia (CLL) in Phase I and Phase II trials (34,35) but thrombocytopenia was the dose limiting toxicity. Recently, ABT-199, a more specific BCL2 inhibitor, was granted priority review by US-FDA for CLL. However, we found ABT-263 to be considerably more potent than ABT-199 in inhibiting TNBC cell growth. If combined with standard cytotoxics or targeted therapy at lower doses, ABT-263 may enhance sensitivity of solid tumors and prevent high-dose toxicity.
ABT-263 synergized with crizotinib (MET/AXL inhibitor). We identified MET as one of the top 681 overexpressed genes in TNBCs versus non-TNBCs in three independent breast cancer clinical datasets. RTKs including MET and EGFR have been suggested as potential targets in triple-negative breast and in other cancers (36–38). However, targeting RTKs with EGFR inhibitors in TNBC has shown limited clinical efficacy. MET interaction with AXL and EGFR family has been suggested as a possible resistance mechanism (39). Although AXL was not overexpressed, MET expression was clearly high in TNBC subtype in TCGA samples (Supplementary Figure 9). Similarly, most TNBC cell lines from CCLE express MET but only a minority express AXL. Crizotinib and ABT-263 displayed synergistic and potent antiproliferative and apoptotic activity in both MSL and basal TNBCs. The same combination also demonstrated anticancer activity in additional TNBC cell lines from each category. We evaluated the results of another MET inhibitor XL-184 in combination with ABT-263 from our screen, and found strikingly similar response with crizotinib/ABT-263 in all cell lines (Supplementary Figure 7A). ABT-263 has also been shown to synergize with other RTK inhibitors (40). ABT-263 with the EGFR inhibitor AZD9291 will be tested in clinical trial (ClinicalTrials.gov study NCT02520778) in EGFR inhibitor-resistant lung cancer. Since TNBCs do not respond well to EGFR inhibitors in clinic, some additional interesting combinations that emerge from this study are erlotinib with 5-fluorouracil or ABT-263 that could be tested.
YM155, Bortezomib, and carfilzomib showed remarkable anticancer activity alone and also, in combinations across all cell lines tested. A recently concluded Phase II randomized study of YM155 and docetaxel advocates rational selection of breast cancer subpopulation with higher survivin activity (41), while LCL161, another inhibitor of IAP, has shown promising efficacy with paclitaxel in a phase II, neoadjuvant, randomized study in TNBC patients (ClinicalTrials.gov study NCT01617668). Bortezomib and carfilzomib are exciting protease inhibitors recently approved by US-FDA for multiple myeloma and are in clinical trials for various cancers including breast (42,43).
ABT-263, nutlin-3, and JQ1 combined synergistically with paclitaxel in our screen. ABT-263-paclitaxel combination has demonstrated preclinical efficacy in NSCLC, ovarian and prostate cancer (44–46). Phase I study of ABT-263 in combination with carboplatin/paclitaxel revealed modest anti-tumor activity in solid tumors, however, only one breast cancer patient participated in this study (47); hence, ABT-263-paclitaxel combination may be worth exploring in TNBC patients. Nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in NSCLC (48). However, nutlins alone at higher doses have off-target effects, and therefore are being studied in combination with other drugs (48). Thus, ABT-463, nutlin-3 or its derivatives, and JQ1 emerge as obvious candidates to test in combination with paclitaxel in clinic. Since paclitaxel is an already integral component of standard chemotherapy in TNBC, such trials can be designed and conducted faster and if successful, these new drugs can be directly introduced to the existing therapeutic regimen.
Our study design tested FDA approved drugs with selected anticancer agents in a medium-throughput screen whose output raw signal was scrutinized for systematic errors, filtered, and extensively analyzed. Overall, MSL cells were relatively less sensitive, while genotoxins, apoptosis and cell cycle regulators were more effective drug classes in TNBC. Drug sensitivities could be explained better by the proportion of up- and down-regulated genes rather than the expression of all or specific genes of a targeted pathway. Results were validated systematically in addition to initial QC steps and internal controls to assess synergy, critical to reliably interpret MTS results. The dataset generated in this study is made publicly available to help translational researchers by providing a roadmap of effective or sub-effective, synergistic or sub-synergistic drug combinations in TNBC, and to possibly help train predictors of effective drug combinations from single agent data. Our results revealed distinct patterns between TNBC backgrounds and cell line sensitivities, and proposed several promising novel drug combinations including ABT-263-crizotinib that could be tested in the clinic. All TNBC types express MET unlike most other RTKs, and therefore, targeting MET with crizotinib may achieve broader activity across TNBCs. Combining crizotinib with ABT-263 at lower doses may provide therapeutic benefits in TNBC patients while avoiding high-dose toxicity associated with ABT-263 monotherapy.
Supplementary Material
Acknowledgments
This work was supported by an award from the Breast Cancer Research Foundation to L.P.
Footnotes
CONFLICTS OF INTEREST
Authors have no conflict of interest to disclose.
References
- 1.Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275–81. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 2.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, et al. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin Cancer Res. 2010;16:4702–10. doi: 10.1158/1078-0432.CCR-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30:2615–23. doi: 10.1200/JCO.2010.34.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell. 2016;164:293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A. 2014;111:2349–54. doi: 10.1073/pnas.1311846111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Errico A. Screening: high-throughput screen identifies a roadmap for combination drug trials. Nat Rev Clin Oncol. 2014;11:124. doi: 10.1038/nrclinonc.2014.21. [DOI] [PubMed] [Google Scholar]
- 10.Held MA, Langdon CG, Platt JT, Graham-Steed T, Liu Z, Chakraborty A, et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov. 2013;3:52–67. doi: 10.1158/2159-8290.CD-12-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47:331–85. [PubMed] [Google Scholar]
- 12.Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 13.Langdon CG, Held MA, Platt JT, Meeth K, Iyidogan P, Mamillapalli R, et al. The broad-spectrum receptor tyrosine kinase inhibitor dovitinib suppresses growth of BRAF-mutant melanoma cells in combination with other signaling pathway inhibitors. Pigment Cell Melanoma Res. 2015;28:417–30. doi: 10.1111/pcmr.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colavito SA, Zou MR, Qin Y, Nguyen DX, Stern DF. Significance of glioma-associated oncogene homolog 1 (GLI1)expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res [Internet] 2014:16. doi: 10.1186/s13058-014-0444-4. Available from: http://dx.doi.org/10.1186/s13058-014-0444-4. [DOI] [PMC free article] [PubMed]
- 16.O’Toole SA, Beith JM, Millar EKA, West R, McLean A, Cazet A, et al. Therapeutic targets in triple negative breast cancer. J Clin Pathol. 2013;66:530–42. doi: 10.1136/jclinpath-2012-201361. [DOI] [PubMed] [Google Scholar]
- 17.Sarti M, Pinton S, Limoni C, Carbone GM, Pagani O, Cavalli F, et al. Differential expression of testin and survivin in breast cancer subtypes. Oncol Rep. 2013;30:824–32. doi: 10.3892/or.2013.2502. [DOI] [PubMed] [Google Scholar]
- 18.Cheng SM, Chang YC, Liu CY, Lee JYC, Chan HH, Kuo CW, et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br J Pharmacol. 2015;172:214–34. doi: 10.1111/bph.12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan X, Hu L, Luquette LJ, 3rd, Gao G, Liu Y, Qu H, et al. Systematic identification of synergistic drug pairs targeting HIV. Nat Biotechnol. 2012;30:1125–30. doi: 10.1038/nbt.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–66. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- 21.Wali VB, Sylvester PW. Synergistic antiproliferative effects of gamma-tocotrienol and statin treatment on mammary tumor cells. Lipids. 2007;42:1113–23. doi: 10.1007/s11745-007-3102-0. [DOI] [PubMed] [Google Scholar]
- 22.Khoo KH, Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–36. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 23.Hedley D, Shamas-Din A, Chow S, Sanfelice D, Schuh AC, Brandwein JM, et al. A phase I study of elesclomol sodium in patients with acute myeloid leukemia. Leuk Lymphoma. 2016:1–4. doi: 10.3109/10428194.2016.1138293. [DOI] [PubMed] [Google Scholar]
- 24.O’Day SJ, Eggermont AMM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol. 2013;31:1211–8. doi: 10.1200/JCO.2012.44.5585. [DOI] [PubMed] [Google Scholar]
- 25.Fu L-L, Tian M, Li X, Li J-J, Huang J, Ouyang L, et al. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget. 2015;6:5501–16. doi: 10.18632/oncotarget.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210–25. doi: 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–7. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langdon CG, Wiedemann N, Held MA, Mamillapalli R, Iyidogan P, Theodosakis N, et al. SMAC mimetic Debio 1143 synergizes with taxanes, topoisomerase inhibitors and bromodomain inhibitors to impede growth of lung adenocarcinoma cells. Oncotarget. 2015;6:37410–25. doi: 10.18632/oncotarget.6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatzis C, Bedard PL, Birkbak NJ, Beck AH, Aerts HJWL, Stem DF, et al. Enhancing reproducibility in cancer drug screening: how do we move forward? Cancer Res. 2014;74:4016–23. doi: 10.1158/0008-5472.CAN-14-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 31.Weir SJ, DeGennaro LJ, Austin CP. Repurposing approved and abandoned drugs for the treatment and prevention of cancer through public-private partnership. Cancer Res. 2012;72:1055–8. doi: 10.1158/0008-5472.CAN-11-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A, et al. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3:80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodwin CM, Rossanese OW, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015;22:2098–106. doi: 10.1038/cdd.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–9. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim YJ, Choi J-S, Seo J, Song J-Y, Lee SE, Kwon MJ, et al. MET is a potential target for use in combination therapy with EGFR inhibition in triple-negative/basal-like breast cancer. Int J Cancer. 2014;134:2424–36. doi: 10.1002/ijc.28566. [DOI] [PubMed] [Google Scholar]
- 37.Dietrich MF, Yan SX, Schiller JH. Response to Crizotinib/Erlotinib Combination in a Patient with a Primary EGFR-Mutant Adenocarcinoma and a Primary c-met-Amplified Adenocarcinoma of the Lung. J Thorac Oncol. 2015;10:e23–5. doi: 10.1097/JTO.0000000000000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaule PB, Crown J, O’Donovan N, Duffy MJ. cMET in triple-negative breast cancer: is it a therapeutic target for this subset of breast cancer patients? Expert Opin Ther Targets. 2014;18:999–1009. doi: 10.1517/14728222.2014.938050. [DOI] [PubMed] [Google Scholar]
- 39.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:ra66. doi: 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med [Internet] 2016 doi: 10.1038/nm.4040. Available from: http://dx.doi.org/10.1038/nm.4040. [DOI] [PMC free article] [PubMed]
- 41.Clemens MR, Gladkov OA, Gartner E, Vladimirov V, Crown J, Steinberg J, et al. Phase II, multicenter, open-label, randomized study of YM155 plus docetaxel as first-line treatment in patients with HER2-negative metastatic breast cancer. Breast Cancer Res Treat. 2015;149:171–9. doi: 10.1007/s10549-014-3238-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falchook GS, Wheler JJ, Naing A, Jackson EF, Janku F, Hong D, et al. Targeting hypoxia-inducible factor-1α (HIF-1α) in combination with antiangiogenic therapy: a phase I trial of bortezomib plus bevacizumab. Oncotarget. 2014;5:10280–92. doi: 10.18632/oncotarget.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papadopoulos KP, Burris HA, 3rd, Gordon M, Lee P, Sausville EA, Rosen PJ, et al. A phase I/II study of carfilzomib 2–10-min infusion in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;72:861–8. doi: 10.1007/s00280-013-2267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamelos VA, Robinson E, Redman CW, Richardson A. Navitoclax augments the activity of carboplatin and paclitaxel combinations in ovarian cancer cells. Gynecol Oncol. 2013;128:377–82. doi: 10.1016/j.ygyno.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 45.Wang C, Huang S-B, Yang M-C, Lin Y-T, Chu I-H, Shen Y-N, et al. Combining paclitaxel with ABT-263 has a synergistic effect on paclitaxel resistant prostate cancer cells. PLoS One. 2015;10:e0120913. doi: 10.1371/journal.pone.0120913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan N, Malek M, Zha J, Yue P, Kassees R, Berry L, et al. Navitoclax enhances the efficacy of taxanes in non-small cell lung cancer models. Clin Cancer Res. 2011;17:1394–404. doi: 10.1158/1078-0432.CCR-10-2353. [DOI] [PubMed] [Google Scholar]
- 47.Vlahovic G, Karantza V, Wang D, Cosgrove D, Rudersdorf N, Yang J, et al. A phase I safety and pharmacokinetic study of ABT-263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors. Invest New Drugs. 2014;32:976–84. doi: 10.1007/s10637-014-0116-3. [DOI] [PubMed] [Google Scholar]
- 48.Deben C, Wouters A, Op de Beeck K, van Den Bossche J, Jacobs J, Zwaenepoel K, et al. The MDM2-inhibitor Nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget. 2015;6:22666–79. doi: 10.18632/oncotarget.4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.