

Abstract
While only one Atg4 is present in yeast, there are four Atg4 homologues in human and in mouse with different substrate specificities and catalytic efficiencies. The molecule Atg4 is a type of cysteine protease, and is known for its crucial roles in cleavage of the Atg8 family proteins before they can be conjugated to phospholipids, and also in cleavage of the conjugated Atg8 molecules from the membrane, a process known as deconjugation. Both processes are required for the maximal efficiency in autophagosome biogenesis. Atg4 could thus be a target for intervention of the autophagy process. It is thus important to measure Atg4 activity to determine and to modulate the autophagy function. Here we review the catalytic functions and regulatory mechanisms of human Atg4 proteases, and discuss the methodology for analyzing Atg4 activity in details.
Keywords: Atg4, Atg8, LC3, GATE-16, GABARAP, FRET, enzymatic kinetics, high throughput screening
1. Introduction
Autophagy is an evolutionarily conserved, multi-step process, whereby cellular components and damaged organelles are sequestered within autophagosomes for lysosomal degradation. Among them, autophagosome biogenesis requires two ubiquitin-like conjugation systems: the Atg12-Atg5 and the Atg8-phosphatidylethanolamine (PE) systems. Atg4 is a cysteine protease of the C54 family and plays an important role in the Atg8/LC3 lipid conjugation system (Marino et al., 2003).
Atg4 was first found to physically interact with Atg8 in the yeast Saccharomyces cerevisiae in 1998 (Lang et al., 1998). There is only one single member of Atg4 proteins in yeast, and deletion of Atg4 impairs the autophagy process (Kirisako et al., 2000). In mammals, there are four Atg4 homologues, Atg4A, Atg4B, Atg4C, and Atg4D (Marino et al., 2003). There are eight human Atg8 homologues belonging to two subfamilies: the LC3 (microtubule-associated protein 1 light chain 3) subfamily, which is comprised of LC3A (isoform a and b), LC3B, and LC3C; and the GABARAP(GABAA receptor-associated protein) subfamily, including GABARAP, GABARAPL1/Atg8L/GEC1, GABARAPL2/GATE-16/GEF2 and GABARAPL3 (Le Grand et al., 2011). All substrates have a conserved cleavage site for Atg4 (Fig. 1). The substrate specificity of different Atg4 homologues is not identical. Studies have shown Atg4B is able to cleave most of the human Atg8 homologues tested so far (Hemelaar, Lelyveld, Kessler, & Ploegh, 2003; Kabeya et al., 2004; Li et al., 2011; Tanida et al., 2004). Atg4A is a potent protease for the GABARAP subfamily, but not the LC3 subfamily, whereas Atg4C and Atg4D seem to possess marginal activities until they are activated by the cleavage of their N-termini via a caspase (Li et al., 2011; R. Scherz-Shouval, Sagiv, Shorer, & Elazar, 2003).
Figure 1. The cleavage site of Atg4 substrates.

The sequences around the Atg4 cleavage sites are listed for the LC3B subfamily substrates (LC3Aa, LC3Ab, LC3B, LC3C) and the GABARAP subfamily substrates (GABARAP, GABARAPL1/Atg8L/GEC1, GABARAPL2/GATE-16/GEF2 and GABARAPL3). The sequences are aligned at the cleavage site next to the glycine residue (indicated by the arrow) with the sequence location indicated.
Atg4 cleaves Atg8 at the peptide bond on the glycine residue at the C-terminus, thus allowing the conjugation of Atg8 to phosphatidylethanolamine (PE) with the participation of other autophagy molecules. Atg4 can also serve as a deconjugating enzyme, which cleaves the amide bond of the conjugated Atg8 and releases it from the autophagosomal membrane. The latter step is important for reusing Atg8 upon autophagy completion. Moreover, this deconjugation process may also occur on isolation membrane, which would positively or negatively affect membrane formation (Nair et al., 2012; R. Scherz-Shouval, Shvets, E., Fass, E., Shorer, H., Gil, L. and Elazar, Z., 2007; Yu et al., 2012). Therefore, Atg4-mediated deconjugation of Atg8 from the membrane may regulate the autophagy process.
Autophagic activity is diminished in both Atg4-deficient yeast and Atg4B-deficient mice (Marino et al., 2010; Nakatogawa, Ishii, Asai, & Ohsumi, 2012; Yu et al., 2012), while deletion of Atg4C in mice displayed minimal impact on autophagy (Marino et al., 2007), indicating that Atg4B is likely the principal mammal Atg4 homolog. Inhibition of Atg4A and Atg4B activity by reactive oxygen species (ROS) has been shown in one study to block starvation-induced autophagy (R. Scherz-Shouval et al., 2007), suggesting that the cysteine protease nature of Atg4 can be regulated by redox events. A more recent study found that the yeast Atg4 could be regulated by thioredoxin on the reduction of the disulfide bond formed by Cys338 and Cys394 (Perez-Perez, Zaffagnini, Marchand, Crespo, & Lemaire, 2014). Mutation of the two cysteines resulted in an increased recruitment of Atg8 to the phagophore assembly site. In addition, Akin et al. have shown that Atg4B knockdown in osteosarcoma cell line Saos-2 and breast cancer cell line MDA-MB468 reduced starvation-induced autophagy and that Saos-2 cells lacking Atg4B failed to survive in amino acid-starvation conditions and failed to grow as xenografted tumors in mice (Akin et al., 2014). Atg4 proteins may thus have a role in cancer development. Finally, some studies have suggested a potential link between Atg4D and apoptosis, indicating a putative role for Atg4D at the interface between autophagy and apoptosis (Kaminskyy & Zhivotovsky, 2014; Norman, Cohen, & Bampton, 2010).
It is therefore important to measure Atg4 activity in order to study autophagy function and to modulate the activity of autophagy.
2. The structure and regulatory machinery of Atg4
Atg4 proteases range from 393 to 474 amino acids in size and possess several structural features of cysteine proteases. The crystal structure of Atg4A (PDB ID: 2P82) has been resolved (Fig. 2A). Atg4A shares a similar catalytic triad (Cys77/Asp279/His281) with Atg4B (Marino et al., 2003). Structure studies have shown that all the residues of Atg4B that interact with LC3B are conserved in Atg4A except Leu232. Atg4A possesses Ile233 instead of Leu232 at the corresponding position. When Ile233 is changed to Leu, the mutated Atg4A acquires a notable ability to cleave LC3B (Satoo et al., 2009).
Figure 2. Structures of human Atg4A and Atg4B.
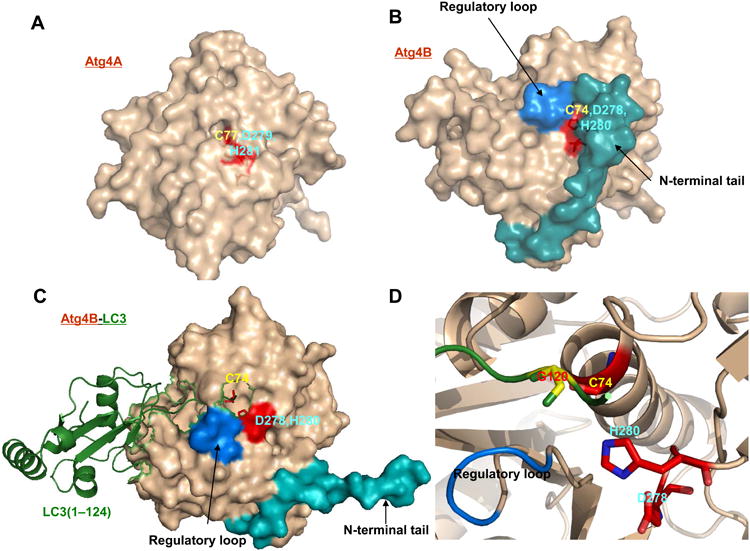
A. Structure of human Atg4A protein with labeled amino acids in the catalytic pocket. B. Structure of human Atg4B protein with labeled amino acids in the catalytic pocket. C. Structure of human Atg4B binding to LC3B. D. Enlarged catalytic pocket of Atg4B interacting with LC3B. The crucial sites, regulatory loop, free N-terminus of Atg4B are highlighted. All structure models were generated using PyMOL.
The crystal structure of human Atg4B (PDB ID: 2CY7) has also been resolved (Fig. 2B). The structure of Atg4B contains a papain-like fold and a small α/β-fold domain, which is thought to be the binding sites for Atg8 homologues. The active site of Atg4B is composed of Cys74, Asp278 and His280. Mutation of these sites is associated with the complete loss of its catalytic activity (Sugawara et al., 2005). The active site of free Atg4B is masked by a regulatory loop (residues 259-262) (Satoo et al., 2009). A large conformational change of Atg4B is induced in the regulatory loop and the N-terminal tail (residues 1-24) when Atg4B interacts with LC3 (Fig. 2C-D). In this process, the regulatory loop masking the entrance of the active site of Atg4B is lifted by LC3 Phe119, resulting in the formation of a groove, into which the LC3 tail can enter, gaining the access to the active site. Besides, the N-terminal tail is originally positioned at the back of the active sites and undergoes a large conformational change as well upon interaction with LC3, which could affect the exit of the cleaved substrates. Thus, deletion of this N-terminal region has been found to increase Atg4B activity (Satoo et al., 2009).
Although the crystal structures of Atg4C and Atg4D are not available, their three dimensional structures can be acquired by homology modeling, using the structure of Atg4B as a template (Zhang, Li, Ouyang, Liu, & Cheng, 2016). In that model, the catalytic triad is conserved (Cys110/Asp345/His347 for Atg4C and Cys134/Asp356/His358 for Atg4D). It is of note that Atg4C and Atg4D have longer sequences than Atg4A and Atg4B, and the catalytic triad may be spatially positioned differently, rendering access to the substrate difficult. Interestingly, deletion of the N-terminal 63 amino acids of Atg4D stimulated its catalytic activity toward Atg8L (Betin & Lane, 2009), indicating that structural changes of Atg4D could improve substrate access to the active sites.
3. Overview of the methods to detect the Atg4 activity in vitro and ex vivo
The classical method to measure Atg4 activity is based on electrophoretic separation of cleaved Atg8 substrates with a C-terminal tag after the cleavage site (Kabeya et al., 2000; Kirisako et al., 2000; Sugawara et al., 2005). This method is described in details below. By measuring the amount of cleaved band of Atg8 migrating at the expected position on SDS-PAGE, one can determine the activityof Atg4. Compared with the SDS-PAGE-based assay, adoption of specific fluorogenic substrates can make the process more automated and convenient. One example is the use of a site-speficic fluorogenic tetrapeptide (Shu et al., 2010), such as acetyl-Gly-Thr-Phe-Gly-AFC (Ac-GTFG-AFC), which is close to the cleavage site (Fig. 1). Release of the fluorogenic AFC following Atg4 cleavage can be detected with a fluorescence spectrometer. A concern on the use of the short 4-amino acid peptide is that the cleavage efficiency by Atg4 is quite low (Shu et al., 2010; Vezenkov et al., 2015) (Li and Yin, unpublished observations). Although different peptide substrates have been optimized with self-immolative linker, which might be convenient for single point read assays, the KM of such peptides are still ten-fold higher than that measured with full-length LC3 (Vezenkov et al., 2015).
Another in vitro system uses the enzyme phospholipase A2, which is inactivated as the result of fusion to the C-terminus of LC3B adjacent to the cleavage site (Ketteler & Seed, 2008; Shu et al., 2010). When the fusion protein is cleaved by Atg4B, PLA2 is released to regain its activity, which can be determined using a separate substrate for PLA2. Although this method is sensitive, quantitative, and suitable for high throughput screening, the Atg4 activity is indirectly measured based on PLA2 activty and thus could be affected by any agents that interfere with the PLA2 reaction.
Methods to measure ex vivo Atg4 activity have been developed. One early study fused G. princeps luciferase (GLUC) and β-actin to the N- and C-terminus of LC3, respectively (Tannous, Kim, Fernandez, Weissleder, & Breakefield, 2005). When expressed in cells, the fusion protein is anchored to the cytoskeleton via β-actin and the soluble cytosol contains few GLUC activity. The increased GLUC activity in the cytosol represents that of Atg4B, which cleaves the fusion protein, releasing it from the cytoskeleton. This method does not interrupt normal metabolism in cells with the overexpression of the substrate. As this is an indirect method, any factors that affect β-actin interaction with the substrates or affect GLUC activity can lead to assay instability, and require multiple parallel controls.
Simple one-reaction designs for ex vivo measurement have been reported, which considerably reduce the complexity and interferences. In one assay (Choi et al., 2011), FITC-labeled Atg4-substrate peptides are conjugated to polymeric nano-particles, which are highly permeable to cells. Once inside the cells, the substrates are cleaved by Atg4, releasing and dequenching the labeled fluorescent dye. The fluorescence is measured and the intensity reflects the Atg4 activity. In another assay, two cell penetrating peptides are used (Ni et al., 2015). One peptide named as AU4S contains the Atg4-recognizable motif ‘GTFG’ and the fluorophore FITC. The other peptide named as AU4R is used as a control, and is not recognized and cleaved by Atg4. The fluorescence ratio defined as ‘F-D value’ between AU4S and AU4R changes as the Atg4 activity. The concern on the ex vivo use of the short 4-amino acid peptide is the same as in the in vitro case, that is, the cleavage efficiency of Atg4 on peptides is quite low (Shu et al., 2010; Vezenkov et al., 2015)(Li and Yin, unpublished observations). Thus the use of full length substrate would be preferred to better reflect Atg4 activity. Toward that end a fluorescence resonance energy transfer (FRET)-based ex vivo assay has been developed (Li, Chen, Ye, Vogt, & Yin, 2012), which is detailed in the following section.
4. Expression and purification of Atg4 and Atg8 proteins for in vitro assays
In vitro analysis using purified Atg4 and/or Atg8 substrates is necessary for the study of enzymatic mechanisms and kinetics, and for determining structure and activity relationship. The assay can also be adapted into the high throughput version for rapid screening of potential chemical modulators that inhibit or enhance the activity of Atg4. In this and subsequent sections, the focus is the methodology and only limited data are shown for illustrative purposes. The readers are referred to the original publications for detailed experimental findings.
4.1. Construct design for recombinant Atg4s and Atg8s
Rationale
Recombinant human Atg8 homologues are prepared for in vitro enzymatic cleavage assays with either purified recombinant Atg4 proteins or cell lysates that contain Atg4 activity. The methods are adapted from Ref (Li et al., 2011).
Methods
The open reading frames of the human Atg4 homologues (Atg4s) and Atg8 homologues (Atg8s) are amplified using RNA extracted from HEK-293A cells. Atg4A (NP_443168), Atg4B (NP_037457), and Atg4C (NP_116241) are cloned into pET-28a (+) in fusion with the N-terminal 6×His tag. Atg4D (NP_116274) is cloned into pGEX-6P-1 in fusion with the N-terminal GST tag for a better expression. For some reasons, Atg4D in fusion with the 6×His tag did not express well in our hands.
Representatives of the human Atg8 homologues, LC3B (NP_073729), GATE16 (NP_009216), GABARAP (NP_009209), and Atg8L (NP_113600) belonging to the two subfamilies are cloned into pET-28a (+) in fusion with the 6×His tag at the N terminus. The DNA fragment encoding GST is then amplified from the pGEX-6P-1 plasmid and inserted in frame at the C termini of the Atg8s in the above constructs. The catalytically-defective mutant Atg4BC74S is constructed using the ExSite in vitro site-directed mutagenesis system. Atg4B with the deletion of the N-terminal first 24 amino acids (Atg4BΔ1–24) is constructed by PCR-based amplification of the corresponding Atg4B sequence. All DNA constructs are confirmed by sequencing.
4.2. Protein expression and purification
Methods
The plasmids are transformed into the BL21 (DE3) strain of E. coli. Protein expression is induced by 0.2-0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 16°C for 16 hours. Atg4D is purified by affinity chromatography using glutathione-Sepharose, whereas all other Atg4s and Atg8s are purified by affinity chromatography using Ni2+-NTA resin and eluted with a 20 to 250 mM imidazole gradient buffer containing150 mM NaCl, 10 mM β-mercaptoethanol, and 20 mM Tris (pH 8.0). All preparations are desalted with a PD-10 desalting column. The purity and the apparent molecular weight of the purified proteins are verified by SDS-PAGE followed by Coomassie Brilliant Blue (CBB) staining (Fig. 3), which also allows for quantification by densitometry.
Figure 3. Expression of Atg4 homologues and Atg8 homologues.
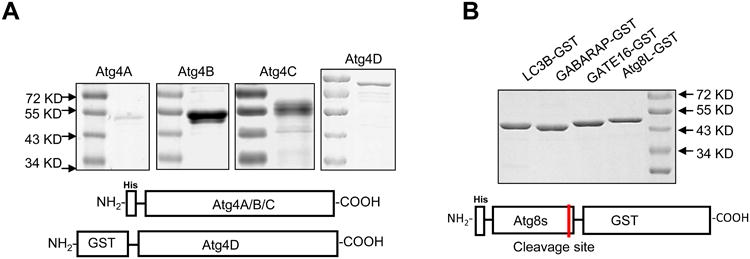
A. His-tagged human Atg4A, Atg4B and Atg4C, and GST-tagged Atg4D were expressed in E. Coli, and purified using Ni2+-NTA resins, or glutathione sepharose, respectively. B. Four human Atg8 homologues were fused with the His tag at the N-terminus and GST at the C-terminus, expressed in E. Coli and purified using Ni2+-NTA resins. Recombinant proteins were separated by SDS-PAGE and stained with CBB for quantification by densitometry.
4.3. Design, expression and purification of FRET substrates
Rationale
Atg8 substrates flanked with CFP and YFP are used in FRET-based assay for a better sensitivity and the ability to be modified for automatic analysis. The methods are adapted from Ref (Li et al., 2012). The illustrated examples are LC3B and GATE-16, representing the LC3 and GABARAP subfamilies of the Atg8-like proteins, respectively.
Methods
full-length LC3B or GATE-16 is first cloned into pET-28a (+) between the BamHI and HindIII restriction sites. The DNA fragments of CFP and YFP are amplified from the commercial vectors pECFP-C1 and pEYFP-C1. CFP is inserted at the N-terminal side of LC3B or GATE-16 at the NdeI and BamHI sites, whereas YFP is inserted at the C-terminal side at the HindIII and XhoI sites.
The expression and purification of the FRET substrates are essentially the same as the non-FRET substrates as described above, except that the E coli strain used is BL21 (DE3)-Codon Plus followed by 0.5 mM IPTG induction at 16°C for 16h.
5. Measurement of Atg4 activity in vitro and ex vivo
5.1. Gel-based assay under in vitro conditions
Purified Atg4s (0.25 μg for Atg4A and Atg4B, 0.5 μg for Atg4C and Atg4D) and Atg8s-GST (5 μg) are admixed with Buffer A (150 mM NaCl, 1mM EDTA, 1 mM DTT, and 50 mM Tris-HCl, pH 7.2) in a total volume of 20 μl. Atg4C and Atg4D are weaker in catalytic efficiency than Atg4A and Atg4B, and more are used in the assay. The mixture is incubated at 37°C for 0-60 minutes. Reactions are stopped by adding the 5× Laemmli sample buffer (5 μl). Samples are boiled at 95°C for 5 minutes and resolved by SDS-PAGE. Recombinant GST alone will be run as a quantification control.
The amounts of substrates (Atg8s-GST) and the cleaved products (Atg8s and GST) (Fig. 4A) were quantified by densitometry based on the standard curve of GST proteins. Higher Atg4 activities will result in a fewer amount of intact Atg8s-GST but a higher amount of cleavage products: GST and Atg8s. The values of the densitometry measurement can be compared to determine the relative activity of Atg4s, and can be used in kinetics analysis (Fig. 4B), which is discussed below in Section 6.
Figure 4. Determination of the kinetics of Atg4A towards GATE-16 using the gel-based assay.
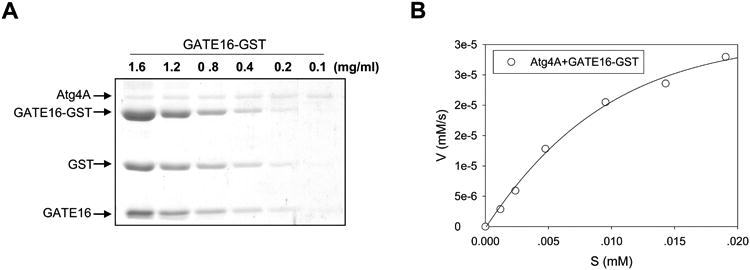
A. Different concentrations (mg/ml) of GATE-16-GST were incubated with Atg4A (0.25 μg) in a 20-μl volume for 1 min. The reaction was then stopped and the proteins were separated by SDS-PAGE, followed by staining with CBB for quantification by standard densitometry. GATE16-GST was quickly cleaved by Atg4A into two fragments representing GST and GATE16. B. The initial velocity (V, y-axis) is defined as the change of the concentration of GST/s ([mM]/s), which is then plotted against the concentration of the substrate (S [mm), x-axis). The curves are fitted using the non-linear regression method.
5.2. FRET-based assay under in vitro conditions
Rationale
Although gel-based method is intuitive and accurate, the sensitivity is limited and the procedure can be labor-intensitve. A larger amount of substrates are needed for the assay, and it is not feasible for high-throughput screening. A number of fluorescence or luminescence-based assays have been developed as discussed above. Provided here in detail is the FRET-based assay that we have developed (Li et al., 2012).
The structures of LC3B and GATE-16 suggest that the N terminus and the C terminus of these proteins are spatially close to each other (Kouno et al., 2005; Paz, Elazar, & Fass, 2000). When LC3B and GATE-16 are fused with CFP and YFP at the N- and C-terminus respectively, the fusion structure would allow FRET to occur, corresponding to a higher fluorescence at the emission wavelength of 527nm (YFP signals). When cleavage occurs, the YFP moiety is separated from the CFP-Atg8 part and FRET signal is lost, leading to a higher fluorescence at the emission wavelength of 477nm (CFP signals). The fluoresence signal ratio of 527nm/477nm is high before the substrates are cleaved, and is low after cleavage.
The FRET-based assay needs a fewer amount of substrate as it is more sensitive than the gel-based assay. The assay is not prone to interferences as the signals are directly coupled with the Atg4 activity without the involvement of a secondary reaction. Small molecules with autofluoresencence may still interfere with the FRET signal in a screening assay, but these can be filtered out easily. The FRET-based assay delivers time- and dose-dependent quantitative responses and has a broad dynamic range sufficient for quantifying and discriminating Atg4 activities under different conditions. The use of a full length molecule, instead of a peptide, as the substrate can ensure maximal specificity and hydrolysis efficiency.
Methods
Purified FRET-LC3B, FRET-LC3BG120A and FRET-GATE-16 (5 μg) are mixed with 0.5 μg of purified Atg4A, Atg4B or Atg4BC74S in Buffer B (150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, pH 7.5). The assay can be laid out on a 96-well plate in a total volume of 100 μl. The plate is placed on a multi-wavelength fluorescence spectrometer (such as TECAN infinite M200pro) at 37°C and assessed at the excitation wavelength of 434 nm and emission wavelength of 477 nm and 527 nm. The fluorescence ratio of 527 nm/477 nm reflects the relative strength of Atg4, with a lower ratio correlating with a higher Atg4 activity (Fig. 5A).
Figure 5. Cleavage of FRET-LC3B by Atg4B using FRET-based assay.
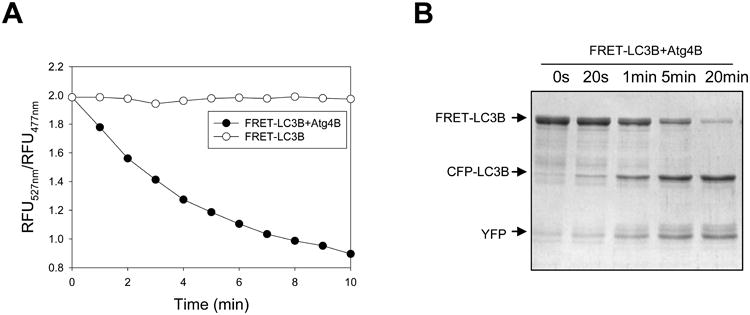
A. FRET-LC3B (5 μg) was incubated with or without Atg4B (0.5 μg) in a volume of 100 μl. The ratio of relative fluorescence unit (RFU) at 477 nm and 527 nm with excitation at 434 nm was recorded at different time. B. FRET-LC3B (5 μg) was incubated with or without Atg4B (0.25 μg) in a 20-μl volume. The reaction was stopped at different times using the SDS-PAGE sample buffer, and separated by SDS-PAGE. Proteins were visualized with CBB staining. Two bands, CFP-LC3B (donor) and YFP (acceptor), were produced after cleavage.
The cleavage can be verified independently by SDS-PAGE. The full-length protein (CFP-LC3B-YFP or CFP-GATE-16-YFP) would be cleaved into CFP-LC3B or CFP-GATE-16, plus YFP by the Atg4A or Atg4B, respectively (Fig. 5B).
5.3. FRET-based high throughput screening assay
High throughput screening (HTS) assays for Atg4B inhibitors have been conducted (Li et al., 2012; Shu CW, 2012; Xu et al., 2016). The method described by Shu et al is based on the Atg4B cleavage of LC3-PLA2 followed by the analysis of PLA2 activity (see Section 3 above). This HTS assay has a Z′ factor of >0.7 and has been validated in pilot screening with the 1280-compound Lopac™ library and 2000-compound Spectrum™ library, with a hit rate of 0.23% and 0.70%, respectively. The HTS developed by Xu et al (Xu et al., 2016) is based on a time-resolved fluorescence resonance energy-transfer (TR-FRET) assay for Atg4B. This HTS assay has an average Z′ factor of approximately 0.9. They validated the assay with a 57,000-compound Roche focus library, which yielded a hit rate of 0.49%.
We have developed the FRET-based HTS for Atg4A and Atg4B with excellent Z′ factors of 0.78 and 0.81, respectively (Li et al., 2012). Pilot screening using FRET-GATE-16 and FRET-LC3B on a 10,000-compound ChemDiv library yielded a hit rate of 0.67%, and 0.7% for Atg4A, and Atg4B, respetively (Li and Yin, unpublished results). The compounds are found to be able to modulate Atg4A and/or Atg4B activity. The basic setup of the HTS assay can be summarized as follows. An amount of 0.5 μg of Atg4A or 0.1 μg of Atg4B are first incubated with different chemicals (final concentration is 10 μM) for 30 min at 37°C in 384-well plates. The stock of compounds are prepared in DMSO at 10 mM. The final concentration of DMSO at the screening is thus 0.1%, which has no effects on Atg4 activity. More Atg4A proteins are used in the assay than Atg4B because Atg4A is weaker in catalytic efficiency. FRET-GATE-16 or FRET-LC3B (3 μg) is then added to a total volume of 50 μl. After 30 min, the fluorescence intensity is recorded. The inhibition effect by a given chemical is calculated as: percentage of inhibition (%) = (RFUX-RFUmin)/(RFUmax– RFUmin)*100%. RFUX is the RFU ratio of 527 nm/477 nm in the presence of a given chemical, RFUmin is the ratio in the absence of the chemical, and RFUmax is the ratio in the absence of Atg4 proteins.
5.4. Detection of Atg4 activity in ex vivo condition
Rationale
Endogenous Atg4 activity in cells can be analyzed by incubating cell lysates with the recombinant Atg8 substrates. To study the regulatory mechanism of Atg4 it is also possible to express a relevant Atg4 homologous in cells and prepare the lysates (or the immunoprecipitates of the specific Atg4 molecules) after proper treatment, which can then be incubated with the substrates to assess any changes in the Atg4 activity. Below is a published example using the gel-based assay (Li et al., 2012). However, by using FRET substrate, the assay can be run with the FRET detection method.
Methods
HEK-293A cells are transfected with Atg4s tagged with FLAG and lysed 24 h later in Buffer C (150 mM NaCl, 1 mM EDTA, 0.1% Triton-100, 50 mM Tris-HCl, pH 8.0) with freshly prepared protease inhibitors. About 20 to 30 μg of the supernatants with overexpressed Atg4s are incubated with 5 μg of Atg8s-GST. The reactions are stopped by SDS-PAGE sample loading buffer for 3 min. The samples are boiled and resolved by SDS-PAGE.
The cleavage assay is characterized for the generation of the cleaved products, GST (Li et al., 2012). Cell lysates with transfected Atg4B can result in increased activities against all four substrates, whereas transfection of Atg4A can result in an increased activity mainly against GATE-16. Transfection of Atg4C, Atg4D, and Atg4BC74S can lead to no more activities than the endogenous background level.
6. Kinetics analysis of Atg4 enzymes
Rationale
One of the most important goals of in vitro assay is to determine the enzymatic kinetics in order to understand the catalytic mechanism and catalytic efficiency. This knowledge will become quite useful in comparing the different Atg4 homologues for the substrate specificity and catalytic efficiency, and in analyzing how chemical modulators work in inhibiting or enhancing the Atg4 enzymes, thus facilitating the better design of such molecules. There are a number of different ways to measure kinetic parameters, depending largely on how enzyme activity is measured (Eleanore Seibert, 2014). Below are the methods we have used when Atg4 enzyme activity is measured by the SDS-PAGE method (Li et al., 2011) or by the FRET method (Li et al., 2012).
Based on these assays, Atg4B is the most potent enzyme among the four Atg4 homologues with a catalytic efficiency ranging from 78,900 to 107, 000 (mol-1Ls-1) for LC3B-GST, GATE-16-GST, GABARAP-GST and Atg8L-GST in the gel-based assay. The results from the gel-based assay and from the FRET-based assay are quite comparable with the same approach for calculation. A different calculation method had been used, which resulted in somewhat different numbers (Shu et al., 2010). However, those data still lead to the same conclusions that GATE-16-GST is the best substrate for both Atg4B and Atg4A and that Atg4B has a broad substrate specificity than Atg4A. In fact, a more detailed analysis indicate that Atg4A has a low activity toward the substrates other than GATE-16 (Li et al., 2011). In addition, Atg4C and Atg4D possess the lowest activity toward the four Atg8 homologues. Furthermore, the catalytic triad of Atg4s has an influence on the catalytic efficiency. The catalytic mutant (C74S) of Atg4B had dramatically reduced the ability of Atg4B to cleave all four substrates. Finally, the deletion of 24 amino acids at N-terminus of Atg4B cam increase the catalytic efficiency (Li et al., 2011).
6.1. Kinetics measurement with the gel-based assay
Purified Atg4s (0.25 μg for Atg4A and Atg4B, 0.5 μg for Atg4C and Atg4D) and Atg8s-GST (5 μg) are admixed together in Buffer A (150 mM NaCl, 1mM EDTA, 1 mM DTT, and 50 mM Tris-HCl, pH 7.2) in a volume of 20 μl at 37°C for 0 to 60 minutes. Reactions are stopped by adding the Laemmli sample buffer and resolved by SDS-PAGE. An example is illustrated in Fig. 4A.
The amounts of uncleaved substrates (Atg8s-GST) and the cleaved products (Atg8s and GST) are quantified by densitometry (OD) based on the standard curve of GST proteins.
The percentage of non-cleaved substrates is equal to ODAtg8s-GST/(ODAtg8s-GST+ODGST+ODAtg8s)*100%. This value is then plotted against the reaction time. The optimal time is determined, at which the reaction is linear in rate under the given experiment conditions.
Atg8s-GST at different amounts (0.1-32 μg) are incubated with a particular type of Atg4 enzyme (0.25-0.5 μg) for an optimal period of time as determined in step 3 above.
The initial velocity is defined as the change in the concentration ([S] in the unit of mM) of GST, one of the cleavage products of GST-Atg8s, per second ([S]/s).
The velocity (mM/s) is plotted against the concentration of the substrate ([S] in the unit of mM) (Fig. 4B). The curve is then fitted using the non-linear regression method, from which the Vmax (mM/s) and KM (Michaelis constant, mM) for each enzyme-substrate reaction are derived using the Michaelis-Menten formula.
The catalytic constant (kcat) is determined by dividing Vmax by the concentration of the enzyme used in the assay. Catalytic efficiency is defined as kcat/KM (mol-1 L s-1).
6.2. Kinetics measurement with the FRET-based assay
The Atg4 enzyme (0.05 μg for Atg4A, 0.02 μg for Atg4B) is incubated with FRET substrates in different amounts (0.1 to 20 μg) in Buffer B (150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, pH 7.5) in a total volume of 20 μl at 37°C for the designated times.
The initial velocity of the cleavage reaction is defined as V = [S]t/t, in which [S]t is the substrate concentration ([S]) in the unit of mM at a given time point (t) (in the unit of second). [S]t is estimated based on the fluorescence change of the substrates at 527 nm ([S]t-527nm) as well as on the fluorescence change of the substrates at 477 nm ([S] t-477nm), so that [S]t = ([S]t-527nm + [S]t-477nm)/2. [S]t-527nm was calculated as [S]*(RFUmax-527nm – RFUt-527nm)/(RFUmax-527nm – RFUmin-527nm), whereas [S]t-477nm= [S]*(RFUt-477nm– RFUmin-477nm)/(RFUmax-477nm – RFUmin-477nm), in which [S] is the initial substrate concentration. RFUmax and RFUmin are the fluorescence intensity at the maximal and minimal levels across the entire reaction period at the corresponding wavelength, and RFUt is the fluorescence intensity at a given time (t).
The initial velocity (V) is plotted against the concentration of the substrate at the given time points ([S]t) and the curve is fitted using the ligand binding method, from which the Vmax ([S]/s, or mM/s) and KM (Michaelis constant, [S] or mM) for each enzyme-substrate reaction are derived based on the Michaelis-Menten formula. The curve-fitting of FRET-GATE-16 cleaved by Atg4B is illustrated (Fig. 6)
The catalytic constant (kcat, s-1) is determined by dividing Vmax by the concentration of the enzyme. The catalytic efficiency is defined as kcat/KM (mol-1Ls-1).
Figure 6. Determination of the kinetics of Atg4B toward GATE-16 using the FRET-based assay.

A. FRET-GATE-16 at different concentrations as indicated by the legends were incubated with 0.02 μg of Atg4B in a 20-μl volume. The consumed substrates were calculated and plotted against the incubation time. The first four minutes were used for initial velocity calculation, which was defined as the increment of the consumed substrate per second. B. The initial velocity (V, y-axis) was plotted against the concentration of the substrate (S [mM], x-axis). The curve was fitted using the ligand-binding method.
7. Summary
Atg4 proteases play a crucial role in preparing Atg8 for conjugation to lipid membranes and for the deconjugation of Atg8 from the autophagosomes (Till & Subramani, 2010). Of the 4 proteases, Atg4B is 1500-fold more catalytically efficient for LC3B activation than the other Atg4s, whereas Atg4A is most selective toward GATE-16 (Li et al., 2011). Atg4B and LC3B are best studied in autophagy and Atg4B seems to be most potent (Nakatogawa, Ichimura, & Ohsumi, 2007; Tanida et al., 2004). These findings suggest that Atg4B could be a potential target for inhibiting autophagy. High throughput screening assays for Atg4B inhibitors have been conducted (Li et al., 2012; Shu CW, 2012; Xu et al., 2016), but it is still a long journey to the availability of practically useful agents with sufficient potency and specificity. We hope that the methods discussed in this chapter will further stimulate studies of Atg4 enzymes and the development of Atg4 modulators.
Acknowledgments
The authors and their relevant works were in part supported by National Institute of Health (R03MH083154, R01CA111456, R01CA83817, R01AA021751) (to X.-M. Y), the Natural Science Foundation of Guangdong Province, China (S2013010015876) (to M. L), and the Specialized Research Fund for the Doctoral Program of Higher Education (20130171120049) (to M.L).
Footnotes
The Conflict-of-Interest and Financial Disclosure Statements: There were no potential conflicts of interest and no significant financial support for this work to disclose
References
- Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, Li M, et al. Dunn WA., Jr A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021–2035. doi: 10.4161/auto.32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betin VMS, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. Journal of Cell Science. 2009;122:2554–2566. doi: 10.1242/jcs.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KM, Nam HY, Na JH, Kim SW, Kim SY, Kim K, et al. Ahn HJ. A monitoring method for Atg4 activation in living cells using peptide-conjugated polymeric nanoparticles. Autophagy. 2011;7:1052–1062. doi: 10.4161/auto.7.9.16451. [DOI] [PubMed] [Google Scholar]
- Eleanore Seibert TST. Fundamentals of Enzyme Kinetics. Methods Mol Biol. 2014;1113:23–35. doi: 10.1007/978-1-62703-758-7_2. [DOI] [PubMed] [Google Scholar]
- Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. Journal of Biological Chemistry. 2003;278:51841–51850. doi: 10.1074/jbc.M308762200. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Uero T, Yamamoto A, Kirisako T, Noda T, et al. Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo Journal. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Kaminskyy VO, Zhivotovsky B. Free Radicals in Cross Talk Between Autophagy and Apoptosis. Antioxidants & Redox Signaling. 2014;21:86–102. doi: 10.1089/ars.2013.5746. [DOI] [PubMed] [Google Scholar]
- Ketteler R, Seed B. Quantitation of autophagy by luciferase release assay. Autophagy. 2008;4:801–806. doi: 10.4161/auto.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. Ohsumi Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–276. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouno T, Mizuguchi M, Tanida I, Ueno T, Kanematsu T, Mori Y, et al. Kawano K. Solution structure of microtubule-associated protein light chain 3 and identification of its functional subdomains. Journal of Biological Chemistry. 2005;280:24610–24617. doi: 10.1074/jbc.M413565200. [DOI] [PubMed] [Google Scholar]
- Lang T, Schaeffeler E, Bernreuther D, Bredschneider M, Wolf DH, Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998;17:3597–3607. doi: 10.1093/emboj/17.13.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand JN, Chakrama FZ, Seguin-Py S, Fraichard A, Delage-Mourroux R, Jouvenot M, Boyer-Guittaut M. GABARAPL1 (GEC1): original or copycat? Autophagy. 2011;7:1098–1107. doi: 10.4161/auto.7.10.15904. [DOI] [PubMed] [Google Scholar]
- Li M, Chen X, Ye QZ, Vogt A, Yin XM. A high-throughput FRET-based assay for determination of Atg4 activity. Autophagy. 2012;8:401–412. doi: 10.4161/auto.18777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hou Y, Wang J, Chen X, Shao ZM, Yin XM. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286:7327–7338. doi: 10.1074/jbc.M110.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Fernandez AF, Cabrera S, Lundberg YW, Cabanillas R, Rodriguez F, et al. Lopez-Otin C. Autophagy is essential for mouse sense of balance. Journal of Clinical Investigation. 2010;120:2331–2344. doi: 10.1172/JCI42601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, Lopez-Otin C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278:3671–3678. doi: 10.1074/jbc.M208247200. [DOI] [PubMed] [Google Scholar]
- Nair U, Yen WL, Mari M, Cao Y, Xie Z, Baba M, et al. Klionsky DJ. A role for Atg8-PE deconjugation in autophagosome biogenesis. Autophagy. 2012;8:780–793. doi: 10.4161/auto.19385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ishii J, Asai E, Ohsumi Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy. 2012;8:177–186. doi: 10.4161/auto.8.2.18373. [DOI] [PubMed] [Google Scholar]
- Ni ZH, Gong Y, Dai XF, Ding W, Wang B, Gong HY, et al. He FT. AU4S: A novel synthetic peptide to measure the activity of ATG4 in living cells. Autophagy. 2015;11:403–415. doi: 10.1080/15548627.2015.1009773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman JM, Cohen GM, Bampton ETW. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042–1056. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- Paz Y, Elazar Z, Fass D. Structure of GATE-16, membrane transport modulator and mammalian ortholog of autophagocytosis factor Aut7p. Journal of Biological Chemistry. 2000;275:25445–25450. doi: 10.1074/jbc.C000307200. [DOI] [PubMed] [Google Scholar]
- Perez-Perez ME, Zaffagnini M, Marchand CH, Crespo JL, Lemaire SD. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy. 2014;10:1953–1964. doi: 10.4161/auto.34396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009;28:1341–1350. doi: 10.1038/emboj.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Sagiv Y, Shorer H, Elazar Z. The COOH terminus of GATE-16, an intra-Golgi transport modulator, is cleaved by the human cysteine protease HsApg4A. Journal of Biological Chemistry. 2003;278:14053–14058. doi: 10.1074/jbc.M212108200. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo Journal. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifi cally regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu CW, L P, Huang CM. High throughput screening for drug discovery of autophagy modulators. Comb Chem High Throughput Screen. 2012;15:721–729. doi: 10.2174/138620712803519734. [DOI] [PubMed] [Google Scholar]
- Shu CW, Drag M, Bekes M, Zhai D, Salvesen GS, Reed JC. Synthetic substrates for measuring activity of autophagy proteases: autophagins (Atg4) Autophagy. 2010;6:936–947. doi: 10.4161/auto.6.7.13075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara K, Suzuki NN, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. Journal of Biological Chemistry. 2005;280:40058–40065. doi: 10.1074/jbc.M509158200. [DOI] [PubMed] [Google Scholar]
- Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem. 2004;279:36268–36276. doi: 10.1074/jbc.M401461200. [DOI] [PubMed] [Google Scholar]
- Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11:435–443. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Till A, Subramani S. A balancing act for autophagin. Journal of Clinical Investigation. 2010;120:2273–2276. doi: 10.1172/JCI43238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezenkov L, Honson NS, Kumar NS, Bosc D, Kovacic S, Nguyen TG, et al. Young RN. Development of fluorescent peptide substrates and assays for the key autophagy-initiating cysteine protease enzyme, ATG4B. Bioorg Med Chem. 2015;23:3237–3247. doi: 10.1016/j.bmc.2015.04.064. [DOI] [PubMed] [Google Scholar]
- Xu D, Xu Z, Han L, Liu C, Zhou Z, Qiu Z, et al. Ding H. Identification of New ATG4B Inhibitors Based on a Novel High-Throughput Screening Platform. J Biomol Screen. 2016 doi: 10.1177/1087057116639202. [DOI] [PubMed] [Google Scholar]
- Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, et al. Xie Z. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012;8:883–892. doi: 10.4161/auto.19652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li J, Ouyang L, Liu B, Cheng Y. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016;373:19–26. doi: 10.1016/j.canlet.2016.01.022. [DOI] [PubMed] [Google Scholar]