

Abstract
The protein kinase Cα (PKCα) enzyme is a member of a broad family of serine/threonine kinases, which are involved in varied cellular signaling pathways. The initial step of PKCα activation involves the C2 subunit docking with the cell membrane, which is followed by interactions of the C1 domains with diacylglycerol (DAG) in the membrane. Notably, the molecular mechanisms of these interactions remain poorly understood, especially what effects, if any, DAG may have on the initial C2 docking. To further understand this process, we have performed a series of conventional molecular dynamics simulations to systematically investigate the interaction between PKCα-C2 domains and lipid bilayers with different compositions to examine the effects of POPS, PIP2, and 1-palmitoyl-2-oleoyl-sn-glycerol (POG) on domain docking. Our results show that the PKCα-C2 domain does not interact with the bilayer surface in the absence of POPS and PIP2. In contrast, the inclusion of POPS and PIP2 to the bilayer resulted in strong domain docking in both perpendicular and parallel orientations, whereas the further inclusion of POG resulted in only parallel domain docking. In addition, lysine residues in the C2 domain formed hydrogen bonds with PIP2 molecule bilayers containing POG. These effects were further explored with umbrella sampling calculations to estimate the free energy of domain docking to the lipid bilayer in the presence of one or two PIP2 molecules. The results show that the binding of one or two PIP2 molecules is thermodynamically favorable, although stronger in bilayers lacking POG. However, in POG-containing bilayers, the binding mode of the C2 domain appears to be more flexible, which may have implications for activation of full-length PKCα. Together, our results shed new insights into the process of C2 bilayer binding and suggest new mechanisms for the roles of different phospholipids in the activation process of PKCα.
Graphical Abstract
INTRODUCTION
Protein kinase Cα (PKCα) is part of a large family of serine/threonine kinases and is involved in regulating a wide array of cellular functions such as proliferation, differentiation, and motility.1–11 As a member of the classical protein kinase C family, it binds to the inner leaflet of the cell and is activated by a combination of calcium, diacylglycerol (DAG), and phosphatidylserine (PS).12–17 Structurally, PKCα is composed of two C1 domains (C1A and C1B), a membrane-targeting C2 domain, and a kinase domain. In full-length PKCα, the kinase domain is inhibited in solution by the C1 domains and it becomes active only through a multistep process of the C2 and C1 domains recognizing and binding to signaling lipids in the membrane.5,17–24
As the initial membrane-targeting region of PKCα, the structure and activation mechanisms of the C2 motif have been the subject of extensive studies.20,25–36 This domain consists of eight β-strands that are organized in an antiparallel β-sandwich (Figure 1).8,19,22,25,37 Of particular note are the three calcium-binding loops (CBLs), which bind three Ca2+ ions, and the lysine-rich cluster that consists of K197, K199, K209, and K211.28,38 Biochemical studies have shown that K209 and K211 are crucial to PIP2 binding, as mutations of either or both of these residues to alanine almost completely abrogate binding of the C2 domain to PIP2.39,40 Furthermore, experiments and simulations have shown that calcium binding to the CBLs alters the electrostatic potential of the C2 domain, which causes it to nonspecifically bind to PS-containing membranes.22,27,41,42 This interaction is reinforced by the formation of hydrogen bonds between residues in the lysine cluster and either one or two PIP2 or PIP3 molecules in the bilayer.32–35,39,43–46 During this docking process, the CBLs insert into the headgroup region and the domain tilts relative to the bilayer. Docking of the C2 module to the bilayer serves to localize the C1 domains to the inner leaflet, where they sequentially insert into the bilayer and are activated by interactions with DAG.16,21,47–50 In particular, 1-palmitoyl-2-oleoyl-sn-glycerol (POG) is highly effective at activating the C1 domains, even in PKCα mutants that are not activated by other DAGs such as DPG.26
Figure 1.

The PKCα-C2 domain structure. The labels highlight the three CBLs (CBL1, CBL2, and CBL3), three Ca2+, and C2 domain four main residues (N189, N206, T214, and R249).
Given that DAG is required for activating C1 and that the C1 and C2 domains are in close proximity to one another, we hypothesized that DAG may also affect the binding structure and dynamics of C2 to the bilayer. To test this, we have performed a series of conventional and free-energy molecular dynamics (MD) simulations of the C2 domain interacting with three model bilayers: pure POPC, POPC/POPS/PIP2, and POPC/POPS/PIP2/POG. Overall, the results show that POPS and PIP2 are critical for C2 domain docking, which occurs in two distinct orientations: perpendicular and parallel. Although direct lysine/PIP2 interactions were observed in only conventional MD (cMD) simulations with POG, umbrella sampling calculations demonstrate that binding of PIP2 molecules is thermodynamically favorable in both bilayers, albeit stronger in bilayers lacking POG. However, the binding mode of the C2 domain appears to be more flexible in POG-containing bilayers, which may have implications for C1/C2 interdomain interactions. Taken together, our results shed new insights into the process of C2 bilayer binding and suggest new mechanisms for the roles of different phospholipids in the activation process of PKCα.
METHODS
System Construction
Three lipid bilayers with different compositions were prepared: one containing pure POPC; one with a mixture of POPC, POPS, and PIP2; and one containing POPC, POPS, PIP2, and POG. When present, the molar concentrations of POPS, PIP2, and POG were fixed at 20, 5, and 10%, respectively (Table 1). The choices of these values were based on experimentally measured concentrations. In mammalian cells, the POPS concentration in the inner leaflet of the plasma membrane is approximately 20%.51,52 PIP2 exists in smaller amounts (about 1 mol %), although local concentrations may be higher.43,53,54 Therefore, we set the PIP2 concentration to 5% to increase the probability of interaction between the C2 domain and PIP2. In addition, Torrecillas et al. observed that PKCα has a maximum activity at 10 mol % of POG.26 Each lipid bilayer system contained a total of 200 lipids and 7866 water molecules and was neutralized with 150 mM NaCl. The POG structure was modified from POPC by replacing the phosphocholine headgroup with a hydroxyl group, and the partial charges were calculated using the force field toolkit in VMD55 and Gaussian.56 The PIP2 structure and force field were obtained from Lupyan et al.57 All lipid bilayer systems were minimized, equilibrated, and simulated for 200 ns at 310 K.
Table 1.
Lipid Concentrations and Leaflet Net Charge of Each Systema
| systems | POPC% | POPS% | PIP2% | POG% | leaflet net charge |
|---|---|---|---|---|---|
| PKCα-C2/POPC | 100 | 0 | 0 | 0 | 0 |
| PKCα-C2/POPC/POPS/PIP2 | 75 | 20 | 5 | 0 | −45 |
| PKCα-C2/POPC/POPS/PIP2/POG | 65 | 20 | 5 | 10 | −45 |
Three systems with different concentrations of POPC, POPS, PIP2, and POG. The net charge is calculated per leaflet, which is the same for the upper and lower leaflets.
The structure of the PKCα-C2 domain was based on the crystal structure from Guerrero-Valero and colleagues (PDB ID 3GPE).37 Figure 1 shows the structure of PKCα-C2 domain and highlights the three main loops. The C2 domain was placed on the surface of the bilayer with tilt angles of 0, 30, and 60° with respect to the bilayer normal, as defined by Landgraf et al.34 (Figure 2). Each system was solvated with TIP3P water molecules,58 and counterions were added to neutralize the systems and simulate an ~150 mM NaCl environment. The CHARMM36 force field was used for all standard protein and lipids parameters.59,60
Figure 2.

Initial orientations of (a) 0°, (b) 30°, and (c) 60°for the the PKCα-C2 domain in the PKCα-C2/POPC bilayer. The PKCα-C2 domain is shown in red, Ca2+ ions in yellow, POPC in silver, and phosphorus and nitrogen atoms are in gold and blue, respectively. The arrow in black from Ca2+ (501) to the Cα atom in the N206 residue represents the long axis of the core β-sandwich. Solvent molecules were removed for clarity.
MD Simulations
All simulations followed a multistage minimization and equilibration protocol. Initially, restraints of 5 kcal/(mol Å2) were applied to the lipid phosphorus and protein Cα atoms, as well as the Ca2+ ions. Systems were energy-minimized in the presence and then the absence of these restraints for 10 000 steps for each minimization process. Next, two stages of NVT simulation were performed in the presence of these restraints to heat the systems to 310 K over 60 ps, and the solvent was then equilibrated for an additional 50 ps. Three 300 ps NPT simulations were then performed to (1) reduce the restraints’ strength from 5 to 1 kcal/(mol Å2), (2) remove the lipid restraints, and (3) remove the protein restraints. The Ca2+ ion restraints were then removed and production simulations of 50 ns for POPC systems and 200 ns for the other systems were performed using the NPT ensemble.
Each protein/lipid system was initiated in three distinct states on the basis of the initial orientation of the C2 domain to the bilayer (Figure 2). To further improve sampling, each configuration was simulated three times with different random seeds, for a total of nine production simulations per system. In each case, the system temperature and pressure were coupled at 310 K and 1 bar, respectively, using Langevin dynamics and Nosé–Hoover Langevin piston pressure control.61,62 Long-range electrostatic interactions were handled with the particle mesh Ewald method,63 using a maximum grid spacing of 1.0 Å and a cubic interpolation. The cutoff distances for Lennard-Jones interactions and electrostatic interactions were set to 1.0 nm. System minimization, equilibration, and simulation were performed using the NAMD 2.9 package.64
Free-Energy Calculations
Umbrella sampling calculations were performed using two reaction coordinates, D1 and D2, that were defined on the basis of the distance between lysine residues K209 and K211 and the phosphorus atom (P5) in PIP2 molecules. Snapshots from the POPC/POPS/PIP2/POG simulations were used as initial window configurations. One of the snapshots was used to create the POPC/POPS/PIP2 system by modifying the POG headgroups to phosphocholine groups. In total, 100 windows per system were used to span the range of 3–9.5 Å along each reaction coordinate. Between 3 and 5 Å, windows were spaced every 0.5 Å and had a harmonic force constant of 40 kcal/(mol Å2), whereas between 5.5 and 9.5 Å, windows were spaced every 1.0 Å and had a harmonic force constant of 5 kcal/(mol Å2). Each window was run for 20 ns, and the last 10 ns were analyzed using the weighted histogram analysis method to calculate the free-energy profile.65 A Jacobian correction of 2kT ln(r) was applied along each coordinate in the resulting potential of mean force (PMF).66 The colvar module was used for all umbrella sampling simulations.67
Simulation Analysis
The PKCα-C2 tilt angle was calculated as the angle between the simulation box normal and a vector defined from Ca2+ ion 501 to the α-carbon in the N206 residue.34 We also defined a vertical distance (Zx) between x and the center of mass of the phosphorus atoms in the lipid headgroups in the upper leaflet of the bilayer. For example, ZCBL3 represents the height of the CBL3 loop above the headgroup region and was calculated as the vertical distance between loop 3 (CBL3) in the PKCα-C2 domain and the center of mass of the phosphorus atoms in the headgroups of the bilayer upper leaflet. To examine the docking effect on the lipid bilayer, we calculated the density of the phosphorus atoms in the lipid headgroups in each leaflet. To remove the effects of protein rotation and translation, the bilayers were centered on the CBL3 loop center of mass and rotated around by an angle defined by a reference vector between residues N189 and R250 and the +x axis. The number of PIP2 (NPIP2) was defined as the number of PIP2 molecules within 5 Å of residues K209 or K211.
RESULTS AND DISCUSSION
Docking of PKCα-C2 Domain
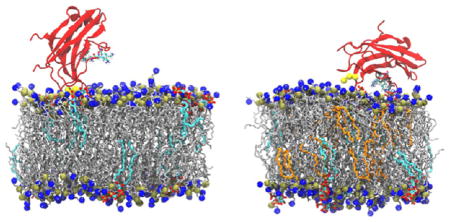
For each bilayer composition, we performed a total of nine, 200 ns MD simulations that were divided among three difierent initial tilt angles (see Method). During simulations, the tilt angles readily converted between difierent initial values; thus, the initial conformations of each individual simulation did not have a significant impact on the final results. In each simulation containing pure POPC bilayers, the PKCα-C2 domain did not dock to the lipid bilayer but instead drifted into solution (Figure S1a–c), adopting final configurations similar to that shown in Figure 3a. In contrast, electrostatic interactions between the negatively charged POPS and PIP2 headgroups and PKCα-C2 resulted in the protein quickly and stably docking to the POPC/POPS/PIP2 bilayer surface (Figures 3b–d and S1d–i). In this bilayer system, PKCα-C2 adopted two distinct conformations. In the parallel orientation (Figure 3b), loop 3 (CBL3) was inserted into the headgroup region and the β-strands were orientated parallel to the membrane surface. In the perpendicular orientation (Figure 3c), the docking surface was localized by insertion of loops 1 and 3 (CBL1 and 3) into the headgroup region and the β-strands were orientated perpendicular to the membrane surface. Systems with POG exhibited two significant difierences from those without. First, only the parallel docking orientation was observed (Figure 3d). Second, lysine residues K209 and K211 in the β4-strand “latched” on the PIP2 molecules by forming hydrogen bonds with their headgroups, an interaction that was not observed in systems lacking POG. In all simulations, the lipid bilayers displayed areas per lipid and bilayer thicknesses that were in accord with previous experiments and simulations and that were largely unaffected by PKCα-C2 binding (see the Supporting Information).
Figure 3.

Snapshots from cMD simulations of the complex composed of a C2 domain and (a) a pure POPC bilayer; (b, c) a POPC/POPS/PIP2 bilayer; and (d) a POPC/POPS/PIP2/POG bilayer. The PKCα-C2 domain is shown in red, Ca2+ ions in yellow, POPC and POPS in silver, PIP2 in cyan, POG in orange, and phosphorus and nitrogen atoms in POPC (and POPS) are shown in gold and blue, respectively. Solvent molecules were removed for clarity.
To quantify the docking and latching characteristics of these systems, configurations from the final 150 ns of each simulation were divided on the basis of three metrics: the domain tilt angle, the heights of loop CBL3 above the headgroups region, and the distance of lysine residues to PIP2 headgroups (Table 2). A domain was considered docked if the CBL3 height (ZCBL3) was negative and latched if a PIP2 molecule was within 5 Å of K209 or K211. Perpendicular orientations were observed to have tilt angles below 45°, whereas parallel orientations had tilt angles above 45°.
Table 2.
PKCα-C2 Bilayer Docking Propertiesa
| systems | dock | latch | tilt angle (degree) | NPIP2 | fraction of simulation time (%) | ZCBL1 (Å) | ZCBL2 (Å) | ZCBL3 (Å) | ZCa2+ (Å) |
|---|---|---|---|---|---|---|---|---|---|
| without POG | perpendicular | no | 38.8 ± 0.6 | 0.0 ± 0.0 | 5.2 | −3.6 ± 0.2 | 3.6 ± 0.2 | −2.0 ± 0.1 | 5.5 ± 0.2 |
| parallel | no | 75.2 ± 1.5 | 0.0 ± 0.0 | 31.9 | 2.3 ± 0.3 | 6.1 ± 0.2 | −2.4 ± 0.1 | 6.3 ± 0.2 | |
| no | no | 58.7 ± 1.8 | 0.0 ± 0.0 | 62.9 | 4.6 ± 0.3 | 5.0 ± 0.2 | 4.5 ± 0.1 | 13.1 ± 0.3 | |
| with POG | parallel | yes | 85.6 ± 2.0 | 1.5 ± 0.0 | 8.3 | 5.9 ± 0.4 | 3.0 ± 0.2 | −2.0 ± 0.1 | 8.6 ± 0.3 |
| parallel | no | 78.4 ± 1.4 | 0.0 ± 0.0 | 15.0 | 5.3 ± 0.3 | 1.9 ± 0.3 | −2.2 ± 0.1 | 9.6 ± 0.2 | |
| no | no | 68.1 ± 2.4 | 0.3 ± 0.0 | 76.7 | 7.8 ± 0.3 | 7.6 ± 0.3 | 6.9 ± 0.3 | 14.1 ± 0.2 |
PKCα-C2 average tilt angle, average number of PIP2’s (NPIP2), fraction of simulation time, and height above the headgroup region for CBL1, CBL2, CBL3, and Ca2+. Error bars represent the standard error of the mean. Negative distances indicate insertion into the bilayer. All values were extracted and averaged over the last 150 ns of the simulation.
In the absence of POG, three states were observed: perpendicular, parallel, and no docking. In the perpendicular orientation, the domain was tilted on average ~39° and both CBL1 and CBL3 were inserted into the bilayer, whereas the CBL2 loop was located outside the headgroup region. The parallel state was observed approximately six times as often as the perpendicular state (32–5% of the simulation time) and was defined by the domain tilting closer to the bilayer surface with an average tilt angle of ~75° and inserting only CBL3 into the headgroup region. When not docked, the domain remained close to the bilayer surface without inserting any of the domain loops into the headgroup region.
In simulations with POG, the docked domain adopted only the parallel orientation. The domain had a conformation similar to that observed without POG, with an average tilt angle of ~78° and the CBL3 loop inserted into the headgroup region. Latching of the lysine residues to the PIP2 molecules increased the domain tilt angle to ~86° and relocated the CBL1 and CBL2 loops to positions slightly further from the bilayer, whereas the Ca2+ ions were located closer to the bilayer surface. When the domain loops were not inserted into the headgroup region, the domain primarily laid on the bilayer surface and rarely latched to PIP2 molecules. Docking and latching were observed in 8.3% of the simulations, whereas the domain docked parallel to the bilayer surface without latching to PIP2 molecules approximately twice as often (15% of the simulation time). Overall, the docking of the domain to the bilayer surface was more likely to occur in the absence of POG, but the PIP2 molecules were more likely to latch to the domain in the presence of POG.
PKCα-C2 Domain Position with Respect to the Bilayer Surface
The average height above the lipid headgroups was calculated for each protein state for a series of residues located throughout the domain that have previously been studied with electron-paramagnetic-resonance (EPR) measurements (Tables 3, S2, and S3).29,34 In each state, residues N206 and T214 were located significantly above the bilayer (>8.5 Å), in agreement with EPR experiments that showed that they are located outside the bilayer.34 Furthermore, these experiments indicated that the side chain of R249 is located within the headgroup regions upon docking, which is also in agreement with our calculations that show that R249 is located within 1–2 Å of the bilayer normal in all docked states. In our simulations, N189 was located inside the headgroup region by ~2 Å when the domain portrayed a perpendicular docking; however, it relocated outside the headgroup region when the domain was in the parallel docked state.
Table 3.
Height above the Headgroup Regiona
| systems | dock | latch | ZN189 (Å) | ZN206 (Å) | ZT214 (Å) | ZR249 (Å) |
|---|---|---|---|---|---|---|
| without POG | perpendicular | no | −2.3 ± 0.2 | 22.4 ± 0.3 | 12.1 ± 0.2 | 2.5 ± 0.2 |
| parallel | no | 5.7 ± 0.4 | 11.2 ± 0.5 | 11.4 ± 0.2 | 3.4 ± 0.2 | |
| no | no | 7.9 ± 0.3 | 24.5 ± 0.6 | 12.2 ± 0.2 | 7.0 ± 0.2 | |
| with POG | parallel | yes | 7.5 ± 0.3 | 9.9 ± 0.6 | 8.7 ± 0.1 | 1.4 ± 0.2 |
| parallel | no | 7.1 ± 0.3 | 14.6 ± 0.4 | 8.5 ± 0.2 | 0.8 ± 0.2 | |
| no | no | 11.5 ± 0.4 | 22.1 ± 0.6 | 15.5 ± 0.4 | 10.6 ± 0.5 |
Height above the headgroup region for residues N189, N206, T214, and R249 in each state. Residues N189, L219, and R249 represent the three main loops (CBL1, CBL2, and CBL3), and residue N206 represents the upper side of the C2 domain long axis. Error bars represent the standard error of the mean. All values were extracted and averaged over the last 150 ns of the simulation.
EPR experiments29,34 in the literature were performed on membranes that lack POG; however, our results suggest that POG may influence the position of C2 domain residues. For example, the addition of POG to the bilayer decreased the height of R249 when the domain docks in the parallel orientation, with heights of 1.4 and 0.8 Å for states with and without PIP2 latching, respectively. In addition, lysine residues K209 and K211, which are in the β4-strand, were located relatively close to the headgroup region when PKCα-C2 docked to POG-containing bilayers, whereas K197 and K199, which are in the β3-strand, were located further above the bilayer. This allowed K209 and K211 to latch to PIP2 molecules, whereas K197 and K199 were located too far from the bilayer to form hydrogen bonds with any bilayer constituent. This is consistent with EPR results, which showed that K197 and K199 are located far from the bilayer surface in the presence of PIP2.34
Headgroup Spacing
Density maps of the lipid phosphorus atoms demonstrate that PKCα-C2 docking significantly disturbs the headgroup region (Figures 4 and 5). In the absence of POG, there was a large disturbance when the domain docked perpendicular to the bilayer surface, which is primarily due to the insertion of CBL1 and CBL3 into the bilayer (Figure 4a). In the parallel docking orientation, there was still a disturbance due to CBL3 insertion, although it was significantly reduced relative to that in the perpendicular orientation as a result of CBL1 not inserting into the bilayer (Figure 4a). In the presence of POG, a large headgroup disturbance was observed when the domain docked parallel to the bilayer surface without PIP2 latching (Figure 5a). Upon latching (Figure 5b), the shape and size of this disturbance changed slightly due to the repositioning of the three domain loops above the location of the loops in the docking case. Whether POG was present or not, bilayer disturbances were nearly eliminated when there was no docking because both loops were located outside of the headgroups (Figures 4c and 5c).
Figure 4.

Density maps of lipid phosphorus atoms in the PKCα-C2/POPC/POPS/PIP2 system for different docking states: (a) perpendicular, (b) parallel, and (c) no docking. The maps represent the phosphorus atoms position density, in POPC, POPS, and PIP2, with respect to the center of mass of CBL3.
Figure 5.

Lipid phosphorus atom density maps with respect to the PKCα-C2/POPC/POPS/PIP2/POG system for different docking cases: (a) docking, (b) docking and latching, and (c) no docking. The maps represent the phosphorus atoms position density, in POPC, POPS, and PIP2, with respect to the center of mass of CBL3.
Similar calculations were performed for the lower leaflets in both systems (Figures S2 and S3). In general, lipids had nearly uniform densities when POG was not present. However, when POG was present and the domain docked to the upper leaflet, there was decreased uniformity between lipids in the lower leaflet, suggesting that disturbances in the upper layer may propagate through the bilayer in the presence of POG.
Umbrella Sampling
In simulations with POG, we observed that lysine residues in the PKCα-C2 domain interacted with either one or two PIP2 molecules, in agreement with previous experiments.23,32,35 Specifically, this occurred through hydrogen bonds between K209 and K211 with the phosphate groups (P5 and P4) in the PIP2 molecules, suggesting that these residues are more critical for specific PIP2 binding than K197 and K199 residues, in agreement with previous experiments.32,34 However, despite extensive sampling, these conventional simulations provided only a qualitative description of PKCα-C2/bilayer interactions and may be subject to significant noise due to their stochastic nature.
To more rigorously examine the free energy of latching to one or two PIP2 molecules, two-dimensional umbrella sampling calculations were performed. Two reaction coordinates, D1 and D2, were defined on the basis of the distance between the nitrogen atoms in lysine residue K211 and K209 side chains and the phosphorus atom (P5) of PIP2 molecules (Figure 6). Potentials of mean force (PMFs) were calculated in both the absence and the presence of POG (Figure 7). PMF convergence was tested using one-dimensional projection of the PMF on each reaction coordinate (Figures S4 and S5), which showed good convergence following 10 ns of equilibration. In the absence of POG, the latching of both PIP2’s to K209 and K211 residues is favorable by ~11 kcal/mol. The PMF displays an asymmetry, as binding to only K211 is favorable by ~7 kcal/mol, whereas binding to only K209 is favorable by ~5 kcal/mol. In addition, there is a slight negative cooperativity, as after K211/PIP2 hydrogen bonds have formed, the free energy of K209 latching is reduced by 1 kcal/mol. In the presence of POG, the overall shape of the PMF is maintained; however, the latching free energy is significantly reduced (Figure 7b). The free-energy gain from latching both PIP2 molecules is ~6 kcal/mol, with a gain of ~5 kcal/mol resulting from latching only K211. Overall, these calculations suggest a mechanism whereby K211 initially forms hydrogen bonds to PIP2, which is followed by K209 reinforcing the PKCα-C2/bilayer interactions with additional hydrogen bonds to a second PIP2 molecule. The latching mechanism appears to be largely unaffected by POG; however, the free energy gained through this process is reduced from nanomolar to micromolar affinity by the addition of POG to the bilayer.
Figure 6.

Snapshots from umbrella sampling simulations. The labels show the reaction coordinates (D1 and D2) in the POG-containing system. The PKCα-C2 domain is shown in red, Ca2+ ions in yellow, POPC and POPS in silver, PIP2 in cyan, POG in orange, and phosphorus and nitrogen atoms are shown in gold and blue, respectively. Solvent molecules were removed for clarity.
Figure 7.

Free-energy profile as a function of the lysine–PIP2 interaction distances D1 and D2 in the absence (a) and presence (b) of POG.
To determine the effects of K209 and K211 latching on the PKCα-C2 domain conformation, the PMFs in Figure 6 were divided into four distinct regions and the last 10 ns of the corresponding trajectories were analyzed to determine the domain tilt angles and residue heights in these states. These regions were defined as follows: (1) K211-only binding (D1 < 4.5 Å and D2 > 4.5 Å), (2) K209-only binding (D1 > 4.5 Å and D2 < 4.5 Å), (3) K211 and K209 binding (D1 < 4.5 Å and D2 < 4.5 Å), and (4) no binding (D1 > 4.5 Å and D2 > 4.5 Å). Measurements were weighted by the Boltzmann factor of the free energies computed in the PMFs (e−G/kT) to determine the values that would be expected from infinitely long simulations of these states. In each latching state, the addition of POG increases the tilt angle distribution, which corresponds to a more flexible docked state (Figure 8). In addition, in both systems, K209 binding increases the tilt angle by 5–6°, regardless of whether or not K211 is bound. This is due to K209’s location being further away from the domain loops than that of K211; therefore, K209 latching pulls the domain closer to the bilayer and increases the tilt angle.
Figure 8.

PKCα-C2 tilt angle in the absence (a) and presence (b) of POG. The PKCα-C2 tilt angle was calculated from the PMF trajectories for the following four regions: K211-only binding (black), K209-only binding (red), K211 and K209 binding (blue), and no binding (green).
We also calculated the free energy weighted height with respect to the headgroup region for selected residues in the PKCα-C2 domain in the absence and presence of POG (Figures S6–S9). In all binding states, residues N189, N206, and T214 are consistently located outside the headgroups, whereas R249 is located partially within the headgroups. This is in agreement with both our cMD results (Table 3) and EPR experiments for systems containing POPS and PIP2.34
To determine the correlation between the domain tilt angle and residue heights above the headgroups, two-dimensional free-energy profiles for residues N189, N206, and R249 were computed on the basis of their joint probability distributions for each binding state as defined above. In all binding cases, N189 and N206, respectively, show positive and negative correlations between the domain tilt angle and their heights above the headgroups: as the domain tilts closer to the bilayer surface, N189 moves further from the headgroup region (Figure S10), whereas N206, which is located on the upper side of the domain axis, tends to reposition closer to the headgroup region (Figure S11). These observations are consistent for systems that contain and lack POG.
In contrast, POG has a larger effect on residue R249 (Figure 9). In the absence of POG, R249 is located outside the headgroups in the K211-only binding case and the domain has a tilt angle of ~100°. When only K209 is bound, R249 is located at the bilayer surface and the domain tilt angle is ~95°. In the presence of POG, R249 is located at nearly the same position when only K209 is bound; however, it is located at the surface of the bilayer when only K211 is bound, with a domain tilt angle of only ~80°. In each binding state, inclusion of POG is synonymous with a significant increase in the accessible phase space, further highlighting the increased flexibility of the PKCα-C2 domains when docked to POG-containing bilayers.
Figure 9.

Domain tilt angle vs height above headgroups for residue R249 in the absence (a) and presence (b) of POG. The two-dimensional free-energy profiles for residues N189, N206, and R249 were computed on the basis of their joint probability distributions for the following binding state: K211-only binding, K209-only binding, K211 and K209 binding, and no binding.
In each set of domain tilt angle/residue height free-energy plots, the “no-binding” case appears to sample more phase space than either of the binding states. To quantify the degree to which K211 or K209 binding alters the motions of PKCα-C2, the Kullback–Leibler divergences (KLDs) of the tilt angle/residue height free-energy profiles of the bound states were calculated relative to those of the unbound state using eq 1 (see Table 4).68
Table 4.
KLD for Each Binding Case with Respect to No Binding for N189, N206, and R249 Residues
| systems | binding | KLDN189 | KLDN206 | KLDR249 |
|---|---|---|---|---|
| without POG | K211 | 2.29 | 2.78 | 2.79 |
| K209 | 1.91 | 2.53 | 2.51 | |
| K211 and K209 | 2.42 | 3.37 | 3.34 | |
| with POG | K211 | 2.11 | 2.34 | 2.21 |
| K209 | 2.79 | 3.34 | 2.84 | |
| K211 and K209 | 3.78 | 4.61 | 4.12 |
| (1) |
The sum was performed over all values where the no-binding case was chosen as the reference (Q(i)). Note that P(i) and Q(i) represent the sampling probabilities of the perturbed and original states and were thus calculated from a Boltzmann weighting of the computed free energies. Higher KLD values represent larger divergence from the reference case (here, the “no-binding state”). In systems without POG, binding to only K211 has a larger impact on the accessible conformational space relative to that in the no-binding state than does K209-only binding. However, the KLD values of K211-only binding are significantly lower than those of K209-only binding in the presence of POG. This difference may partially contribute to the larger asymmetry in free energies between K211 and K209 binding observed in the system with POG than in those without, as in each system, a different entropic difference opposes domain binding. Finally, in both systems, the KLD values are significantly higher for states in which both K211 and K209 are bound than when only one is bound. This increased restriction of conformational space upon binding of K209 and K211 may contribute to the slight negative binding cooperativity observed in the PMFs, as significantly restricted motions relative to the no-binding state would incur an entropic penalty to binding.
CONCLUSIONS
We have performed a series of conventional and free-energy MD simulations to elucidate the mechanism of PKCα-C2 binding and how it is affected by the composition of lipid bilayers. In general, results indicate that the domain does not interact with pure POPC bilayer surfaces. In contrast, the domain strongly docks to lipid bilayers in both perpendicular and parallel orientations in the presence of POPS and PIP2. The parallel orientation was observed more frequently, which is likely due in part to the perpendicular orientation requiring a significant rearrangement of the headgroup region. In addition, hydrogen bonds were formed between K209 and K211 and the phosphate groups in PIP2 molecules within the bilayer. The effects of these interactions were explored with umbrella sampling calculations, and it was found that K211 latching is stronger than that of K209. Furthermore, the addition of POG to bilayers weakens the overall free energy of these hydrogen bonds but allows for more flexibility of the domain relative to that of the bilayer.
The C2 domain studied here is only one portion of the much larger PKCα enzyme, which is regulated not only by the C2 domain but also by protein/lipid interactions in the C1 domains.1,5,6,8,14,17 In particular, it has been shown that diaclyglycerol-containing lipids are required for activating the C1 domains, and it has been suggested that the regulation of the C1 and C2 domains may be coupled due to bridging interactions between these regions.36,69–71 Given the effects of POG on PKCα-C2/PIP2 interactions observed here, it is tempting to speculate that the increased flexibility of the C2 domain imparted by POG may alter the conformational space accessible to the C1 domains in full-length PKCα and thus may have an additional indirect influence on the activity of the kinase. If this is the case, the increased binding affinity for PIP2 computed in our umbrella sampling simulations for bilayers lacking POG may not manifest itself in the activation process of full-length PKCα, and in fact, an opposite effect could be observed as the increase in rigidity of the C2 domain may counteract the native affinity of the C1 domains for diaclyglycerol.
PKCα is one of a large class of protein modules that are utilized by various proteins to bind to diverse membranes.2,3,7,17,72 Therefore, the binding mechanisms observed in our simulations are likely to have implications for similar domains in proteins involved in processes such as vesicular transport, lipid modification, and GTPase regulation. For example, it is likely that many C2 domains possess the capability of interacting with bilayers in both parallel and perpendicular orientations, although differences in the CBLs may alter the propensity for these states. Additionally, the observation that K209 is highly conserved among C2 domains whereas K211 is not suggests that some properties of PIP2 binding may be consistent among C2 domains, whereas others, such as the ability to bind two PIP2’s, may potentially be a unique feature of PKCα-C2.72 To determine which of these mechanistic details are shared among C2 domains and which uniquely evolved in PKCα-C2 will require further computational and experimental studies.
Supplementary Material
Acknowledgments
The authors wish to thank Samuel Bowerman and Amy Rice for critical comments concerning this manuscript. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. Research reported in this publication was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119647. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.6b10188.
Description of lipid binding characteristics, details on the height of the domain and specific residues above the headgroup region versus time, lipid phosphorous density maps for the lower leaflets, PMF convergence plots, and domain tilt angle versus height above headgroups for N189 and N206 (PDF)
References
- 1.Nishizuka Y. The Molecular Heterogeneity of Protein Kinase C and Its Implications for Cellular Regulation. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 2.Bell RM, Burns DJ. Lipid Activation of Protein Kinase C. J Biol Chem. 1991;266:4661–4664. [PubMed] [Google Scholar]
- 3.Newton AC. Protein Kinase C: Seeing Two Domains. Curr Biol. 1995;5:973–976. doi: 10.1016/s0960-9822(95)00191-6. [DOI] [PubMed] [Google Scholar]
- 4.Nishizuka Y. Protein Kinase C and Lipid Signaling for Sustained Cellular Responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 5.Mellor H, Parker PJ. The Extended Protein Kinase C Superfamily. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein Kinase C Isozymes and the Regulation of Diverse Cell Responses. Am J Physiol: Lung Cell Mol Physiol. 2000;279:L429–L438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 7.Nakashima S. Protein Kinase C Alpha (PKC Alpha): Regulation and Biological Function. J Biochem. 2002;132:669–675. doi: 10.1093/oxfordjournals.jbchem.a003272. [DOI] [PubMed] [Google Scholar]
- 8.Corbalán-García S, Gómez-Fernández JC. Protein Kinase C Regulatory Domains: The Art of Decoding Many Different Signals in Membranes. Biochim Biophys Acta, Mol Cell Biol Lipids. 2006;1761:633–654. doi: 10.1016/j.bbalip.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 9.Griner EM, Kazanietz MG. Protein Kinase C and Other Diacylglycerol Effectors in Cancer. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 10.Evans JH, Falke JJ. Ca2+ Influx Is an Essential Component of the Positive-Feedback Loop That Maintains Leading-Edge Structure and Activity in Macrophages. Proc Natl Acad Sci US A. 2007;104:16176–16181. doi: 10.1073/pnas.0707719104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallegos LL, Newton AC. Spatiotemporal Dynamics of Lipid Signaling: Protein Kinase C as a Paradigm. IUBMB Life. 2008;60:782–789. doi: 10.1002/iub.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kishimoto A, Takai Y, Mori T, Kikkawa U, Nishizuka Y. Activation of Calcium and Phospholipid-Dependent Protein Kinase by Diacylglycerol, Its Possible Relation to Phosphatidylinositol Turnover. J Biol Chem. 1980;255:2273–2276. [PubMed] [Google Scholar]
- 13.Ono Y, Fujii T, Igarashi K, Kuno T, Tanaka C, Kikkawa U, Nishizuka Y. Phorbol Ester Binding to Protein Kinase C Requires a Cysteine-Rich Zinc-Finger-like Sequence. Proc Natl Acad Sci US A. 1989;86:4868–4871. doi: 10.1073/pnas.86.13.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MH, Bell RM. Mechanism of Protein Kinase C Activation by Phosphatidylinositol 4,5-Bisphosphate. Biochemistry. 1991;30:1041–1049. doi: 10.1021/bi00218a023. [DOI] [PubMed] [Google Scholar]
- 15.Giorgione J, Hysell M, Harvey DF, Newton AC. Contribution of the C1A and C1B Domains to the Membrane Interaction of Protein Kinase C. Biochemistry. 2003;42:11194–11202. doi: 10.1021/bi0350046. [DOI] [PubMed] [Google Scholar]
- 16.Gómez-Fernández JC, Corbalán-García S. Diacylglycerols, Multivalent Membrane Modulators. Chem Phys Lipids. 2007;148:1–25. doi: 10.1016/j.chemphyslip.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Newton AC. Lipid Activation of Protein Kinases. J Lipid Res. 2009;50:S266–S271. doi: 10.1194/jlr.R800064-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newton AC. Regulation of Protein Kinase C. Curr Opin Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 19.Sutton RB, Sprang SR. Structure of the Protein Kinase Cbeta Phospholipid-Binding C2 Domain Complexed with Ca2+ Structure. 1998;6:1395–1405. doi: 10.1016/s0969-2126(98)00139-7. [DOI] [PubMed] [Google Scholar]
- 20.Oancea E, Meyer T. Protein Kinase C as a Molecular Machine for Decoding Calcium and Diacylglycerol Signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- 21.Slater SJ, Milano SK, Stagliano BA, Gergich KJ, Ho CJ, Mazurek A, Taddeo FJ, Kelly MB, Yeager MD, Stubbs CD. Synergistic Activation of Protein Kinase C Alpha, -Beta I, and -Gamma Isoforms Induced by Diacylglycerol and Phorbol Ester: Roles of Membrane Association and Activating Conformational Changes. Biochemistry. 1999;38:3804–3815. doi: 10.1021/bi982778r. [DOI] [PubMed] [Google Scholar]
- 22.Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gómez-Fernández JC. Ca(2+) Bridges the C2 Membrane-Binding Domain of Protein Kinase Calpha Directly to Phosphatidylserine. EMBO J. 1999;18:6329–6338. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corbin JA, Evans JH, Landgraf KE, Falke JJ. Mechanism of Specific Membrane Targeting by C2 Domains: Localized Pools of Target Lipids Enhance Ca2+ Affinity. Biochemistry. 2007;46:4322–4336. doi: 10.1021/bi062140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corbalán-García S, Gómez-Fernández JC. The C2 Domains of Classical and Novel PKCs as Versatile Decoders of Membrane Signals. BioFactors. 2010;36:1–7. doi: 10.1002/biof.68. [DOI] [PubMed] [Google Scholar]
- 25.Shao X, Davletov BA, Sutton RB, Südhof TC, Rizo J. Bipartite Ca2+-Binding Motif in C2 Domains of Synaptotagmin and Protein Kinase C. Science. 1996;273:248–251. doi: 10.1126/science.273.5272.248. [DOI] [PubMed] [Google Scholar]
- 26.Torrecillas A, Corbalán-García S, de Godos A, Gómez-Fernández JC. Activation of Protein Kinase C Alpha by Lipid Mixtures Containing Different Proportions of Diacylglycerols. Biochemistry. 2001;40:15038–15046. doi: 10.1021/bi015599+. [DOI] [PubMed] [Google Scholar]
- 27.Murray D, Honig B. Electrostatic Control of the Membrane Targeting of C2 Domains. Mol Cell. 2002;9:145–154. doi: 10.1016/s1097-2765(01)00426-9. [DOI] [PubMed] [Google Scholar]
- 28.Kohout SC, Corbalán-Garcáa S, Torrecillas A, Goméz-Fernandéz JC, Falke JJ. C2 Domains of Protein Kinase C Isoforms A, B, and γ: Activation Parameters and Calcium Stoichiometries of the Membrane-Bound State. Biochemistry. 2002;41:11411–11424. doi: 10.1021/bi026041k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohout SC, Corbalán-García S, Gómez-Fernández JC, Falke JJ. C2 Domain of Protein Kinase Cα: Elucidation of the Membrane Docking Surface by Site-Directed Fluorescence and Spin Labeling. Biochemistry. 2003;42:1254–1265. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolsover SR, Gomez-Fernandez JC, Corbalan-Garcia S. Role of the Ca2+/phosphatidylserine Binding Region of the C2 Domain in the Translocation of Protein Kinase Calpha to the Plasma Membrane. J Biol Chem. 2003;278:10282–10290. doi: 10.1074/jbc.M212145200. [DOI] [PubMed] [Google Scholar]
- 31.Torrecillas A, Corbalan-Garcia S, Gomez-Fernandez JC. An Infrared Spectroscopic Study of the Secondary Structure of Protein Kinase C Alpha and Its Thermal Denaturation. Biochemistry. 2004;43:2332–2344. doi: 10.1021/bi035128i. [DOI] [PubMed] [Google Scholar]
- 32.Evans JH, Murray D, Leslie CC, Falke JJ. Specific Translocation of Protein Kinase Calpha to the Plasma Membrane Requires Both Ca2+ and PIP2 Recognition by Its C2 Domain. Mol Biol Cell. 2006;17:56–66. doi: 10.1091/mbc.E05-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guerrero-Valero M, Marín-Vicente C, Gómez-Fern ández JC, Corbalán-García S. The C2 Domains of Classical PKCs Are Specific PtdIns(4,5)P2-Sensing Domains with Different Affinities for Membrane Binding. J Mol Biol. 2007;371:608–621. doi: 10.1016/j.jmb.2007.05.086. [DOI] [PubMed] [Google Scholar]
- 34.Landgraf KE, Malmberg NJ, Falke JJ. Effect of PIP2 Binding on the Membrane Docking Geometry of PKCα C2 Domain: An EPR Site-Directed Spin-Labeling and Relaxation Study. Biochemistry. 2008;47:8301–8316. doi: 10.1021/bi800711t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai CL, Landgraf KE, Voth GA, Falke JJ. Membrane Docking Geometry and Target Lipid Stoichiometry of Membrane-Bound PKCα C2 Domain: A Combined Molecular Dynamics and Experimental Study. J Mol Biol. 2010;402:301–310. doi: 10.1016/j.jmb.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziemba BP, Li J, Landgraf KE, Knight JD, Voth GA, Falke JJ. Single-Molecule Studies Reveal a Hidden Key Step in the Activation Mechanism of Membrane-Bound Protein Kinase C-α. Biochemistry. 2014;53:1697–1713. doi: 10.1021/bi4016082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerrero-Valero M, Ferrer-Orta C, Querol-Audí J, Marin-Vicente C, Fita I, Gómez-Fernández JC, Verdaguer N, Corbalán-García S. Structural and Mechanistic Insights into the Association of PKCalpha-C2 Domain to PtdIns(4,5)P2. Proc Natl Acad Sci US A. 2009;106:6603–6607. doi: 10.1073/pnas.0813099106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torrecillas A, Laynez J, Menéndez M, Corbalán-García S, Gómez-Fernández JC. Calorimetric Study of the Interaction of the C2 Domains of Classical Protein Kinase C Isoenzymes with Ca2+ and Phospholipids. Biochemistry. 2004;43:11727–11739. doi: 10.1021/bi0489659. [DOI] [PubMed] [Google Scholar]
- 39.Sánchez-Bautista S, Marín-Vicente C, Gómez-Fernández JC, Corbalán-García S. The C2 Domain of PKCalpha Is a Ca2+-Dependent PtdIns(4,5)P2 Sensing Domain: A New Insight into an Old Pathway. J Mol Biol. 2006;362:901–914. doi: 10.1016/j.jmb.2006.07.093. [DOI] [PubMed] [Google Scholar]
- 40.Corbalán-García S, García-García J, Rodríguez-Alfaro JA, Gómez-Fernández JC. A New Phosphatidylinositol 4,5-Bisphosphate-Binding Site Located in the C2 Domain of Protein Kinase Cα. J Biol Chem. 2003;278:4972–4980. doi: 10.1074/jbc.M209385200. [DOI] [PubMed] [Google Scholar]
- 41.Bolsover SR, Gómez-Fernández JC, Corbalan-Garcia S. Role of the Ca2+/phosphatidylserine Binding Region of the C2 Domain in the Translocation of Protein Kinase Calpha to the Plasma Membrane. J Biol Chem. 2003;278:10282–10290. doi: 10.1074/jbc.M212145200. [DOI] [PubMed] [Google Scholar]
- 42.Stahelin RV, Rafter JD, Das S, Cho W. The Molecular Basis of Differential Subcellular Localization of C2 Domains of Protein Kinase C-Alpha and Group IVa Cytosolic Phospholipase A2. J Biol Chem. 2003;278:12452–12460. doi: 10.1074/jbc.M212864200. [DOI] [PubMed] [Google Scholar]
- 43.McLaughlin S, Murray D. Plasma Membrane Phosphoinositide Organization by Protein Electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 44.Marín-Vicente C, Gómez-Fernández JC, Corbalán-García S. The ATP-Dependent Membrane Localization of Protein Kinase Calpha Is Regulated by Ca2+ Influx and Phosphatidylinositol 4,5-Bisphosphate in Differentiated PC12 Cells. Mol Biol Cell. 2005;16:2848–2861. doi: 10.1091/mbc.E05-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marín-Vicente C, Nicolás FE, Gómez-Fernández JC, Corbalán-García S. The PtdIns(4,5)P2 Ligand Itself Influences the Localization of PKCα in the Plasma Membrane of Intact Living Cells. J Mol Biol. 2008;377:1038–1052. doi: 10.1016/j.jmb.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 46.Montaville P, Coudevylle N, Radhakrishnan A, Leonov A, Zweckstetter M, Becker S. The PIP2 Binding Mode of the C2 Domains of Rabphilin-3A. Protein Sci. 2008;17:1025–1034. doi: 10.1110/ps.073326608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slater SJ, Kelly MB, Taddeo FJ, Rubin E, Stubbs CD. Evidence for Discrete Diacylglycerol and Phorbol Ester Activator Sites on Protein Kinase C. Differences in Effects of 1-Alkanol Inhibition, Activation by Phosphatidylethanolamine and Calcium Chelation. J Biol Chem. 1994;269:17160–17165. [PubMed] [Google Scholar]
- 48.Slater SJ, Ho C, Kelly MB, Larkin JD, Taddeo FJ, Yeager MD, Stubbs CD. Protein Kinase Cα Contains Two Activator Binding Sites That Bind Phorbol Esters and Diacylglycerols with Opposite Affinities. J Biol Chem. 1996;271:4627–4631. doi: 10.1074/jbc.271.9.4627. [DOI] [PubMed] [Google Scholar]
- 49.Medkova M, Cho W. Interplay of C1 and C2 Domains of Protein Kinase C-Alpha in Its Membrane Binding and Activation. J Biol Chem. 1999;274:19852–19861. doi: 10.1074/jbc.274.28.19852. [DOI] [PubMed] [Google Scholar]
- 50.Slater SJ, Seiz JL, Cook AC, Buzas CJ, Malinowski SA, Kershner JL, Stagliano BA, Stubbs CD. Regulation of PKCα Activity by C1–C2 Domain Interactions. J Biol Chem. 2002;277:15277–15285. doi: 10.1074/jbc.M112207200. [DOI] [PubMed] [Google Scholar]
- 51.Vance JE, Steenbergen R. Metabolism and Functions of Phosphatidylserine. Prog Lipid Res. 2005;44:207–234. doi: 10.1016/j.plipres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 52.Manna D, Bhardwaj N, Vora MS, Stahelin RV, Lu H, Cho W. Differential Roles of Phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in Plasma Membrane Targeting of C2 Domains: Molecular Dynamics Simulation, Membrane Binding, and Cell Translocation Studies of the PKCalpha C2 Domain. J Biol Chem. 2008;283:26047–26058. doi: 10.1074/jbc.M802617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. Recruitment and Regulation of Phosphatidylinositol Phosphate Kinase Type 1γ by the FERM Domain of Talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- 54.McLaughlin S, Wang J, Gambhir A, Murray D. PIP2 and Proteins: Interactions, Organization, and Information Flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 55.Mayne CG, Saam J, Schulten K, Tajkhorshid E, Gumbart JC. Rapid Parameterization of Small Molecules Using the Force Field Toolkit. J Comput Chem. 2013;34:2757–2770. doi: 10.1002/jcc.23422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 57.Lupyan D, Mezei M, Logothetis DE, Osman R. A Molecular Dynamics Investigation of Lipid Bilayer Perturbation by PIP2. Biophys J. 2010;98:240–247. doi: 10.1016/j.bpj.2009.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926. [Google Scholar]
- 59.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone Φ, ψ and Side-Chain χ1 and χ 2 Dihedral Angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martyna GJ, Tobias DJ, Klein ML. Constant Pressure Molecular Dynamics Algorithms. J Chem Phys. 1994;101:4177. [Google Scholar]
- 62.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J Chem Phys. 1995;103:4613. [Google Scholar]
- 63.Darden T, York D, Pedersen L. Particle Mesh Ewald: An N.log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089. [Google Scholar]
- 64.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grossfield A. WHAM: The Weighted Histogram Analysis Method. University of Rochester; Rochester: 2013. [Google Scholar]
- 66.Trzesniak D, Kunz APE, van Gunsteren WF. A Comparison of Methods to Compute the Potential of Mean Force. Chemphyschem. 2007;8:162–169. doi: 10.1002/cphc.200600527. [DOI] [PubMed] [Google Scholar]
- 67.Fiorin G, Klein ML, Hénin J. Using Collective Variables to Drive Molecular Dynamics Simulations. Mol Phys. 2013;111:3345–3362. [Google Scholar]
- 68.Kullback S, Leibler RA. On information and sufficiency. Ann Math Stat. 1951;22:79–86. [Google Scholar]
- 69.Xu RX, Pawelczyk T, Xia TH, Brown SC. NMR Structure of a Protein Kinase C-γ Phorbol-Binding Domain and Study of Protein-Lipid Micelle Interactions. Biochemistry. 1997;36:10709–10717. doi: 10.1021/bi970833a. [DOI] [PubMed] [Google Scholar]
- 70.Conesa-Zamora P, Gómez-Fernández JC, Corbalán-García S. The C2 Domain of Protein Kinase Cα Is Directly Involved in the Diacylglycerol-Dependent Binding of the C1 Domain to the Membrane. Biochim Biophys Acta, Mol Cell Biol Lipids. 2000;1487:246–254. doi: 10.1016/s1388-1981(00)00099-8. [DOI] [PubMed] [Google Scholar]
- 71.Leonard TA, Różycki B, Saidi LF, Hummer G, Hurley JH. Crystal Structure and Allosteric Activation of Protein Kinase C Beta Ll. Cell. 2011;144:55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corbalan-Garcia S, Gómez-Fernández JC. Signaling through C2 Domains: More than One Lipid Target. Biochim Biophys Acta, Biomembr. 2014;1838:1536–1547. doi: 10.1016/j.bbamem.2014.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.