

ABSTRACT
The production of biofuels from lignocellulose yields a substantial lignin by-product stream that currently has few applications. Biological conversion of lignin-derived compounds into chemicals and fuels has the potential to improve the economics of lignocellulose-derived biofuels, but few microbes are able both to catabolize lignin-derived aromatic compounds and to generate valuable products. While Escherichia coli has been engineered to produce a variety of fuels and chemicals, it is incapable of catabolizing most aromatic compounds. Therefore, we engineered E. coli to catabolize protocatechuate, a common intermediate in lignin degradation, as the sole source of carbon and energy via heterologous expression of a nine-gene pathway from Pseudomonas putida KT2440. We next used experimental evolution to select for mutations that increased growth with protocatechuate more than 2-fold. Increasing the strength of a single ribosome binding site in the heterologous pathway was sufficient to recapitulate the increased growth. After optimization of the core pathway, we extended the pathway to enable catabolism of a second model compound, 4-hydroxybenzoate. These engineered strains will be useful platforms to discover, characterize, and optimize pathways for conversions of lignin-derived aromatics.
IMPORTANCE Lignin is a challenging substrate for microbial catabolism due to its polymeric and heterogeneous chemical structure. Therefore, engineering microbes for improved catabolism of lignin-derived aromatic compounds will require the assembly of an entire network of catabolic reactions, including pathways from genetically intractable strains. Constructing defined pathways for aromatic compound degradation in a model host would allow rapid identification, characterization, and optimization of novel pathways. We constructed and optimized one such pathway in E. coli to enable catabolism of a model aromatic compound, protocatechuate, and then extended the pathway to a related compound, 4-hydroxybenzoate. This optimized strain can now be used as the basis for the characterization of novel pathways.
KEYWORDS: lignin, protocatechuic acid, ortho-cleavage pathway, experimental evolution, synthetic biology, ligninolysis, metabolic engineering, ortho-cleavage
INTRODUCTION
Biofuels derived from lignocellulose will likely play an important role in the transition to a sustainable and carbon-neutral economy (1). In a typical biotransformation, carbohydrate-rich cellulose and hemicellulose are extracted and fermented to yield the desired biofuel, such as bioethanol. The lignin, comprising up to 25% of the dry biomass, is generally then burned or diverted to other low-value uses (2). Lignin is a challenging feedstock for chemical or biochemical transformations due to its inherent heterogeneity, which varies widely depending on the biomass source and pretreatment strategy (3–5). In a future biorefinery setting, thermochemical or catalytic approaches will likely be used to depolymerize lignin into low-molecular-weight aromatic compounds suitable for biological upgrading by engineered microbes (6).
Numerous microorganisms have been isolated based on their ability to catabolize aromatic compounds derived from lignin (7). Using specific pathways to funnel the products of lignin depolymerization into a few central metabolic intermediates, microbes are capable of converting complex lignin-rich feedstocks into valuable bioproducts (8, 9). However, lignin deconstruction results in a vast slate of aromatic compounds, and even organisms that grow well on aromatic compounds such as Pseudomonas putida and Rhodococcus opacus are not capable of catabolizing them all (8). Therefore, a platform is needed for the discovery of new enzymatic reactions to funnel these diverse aromatic compounds into central metabolism.
Escherichia coli is a major platform organism for metabolic engineering and synthetic biology due to the availability of extensive genetic, biochemical, and physiological information and the relative ease of genetic modification. E. coli is routinely used to prototype metabolic pathways for the production of fuels and chemicals. However, E. coli is capable of catabolizing only a limited set of aromatic compounds, such as phenylpropanoic acids (10–12), limiting its utility for the bioconversion of lignin-derived aromatics. In order to use E. coli for aromatic pathway discovery or for bioconversion, it must first be engineered to robustly catabolize major metabolic intermediate compounds in the aromatic funneling pathways.
For many organisms that are capable of catabolizing aromatic compounds, protocatechuic acid (PCA) is a key metabolic intermediate in this pathway. Complex lignin-derived compounds are first metabolized into PCA before being assimilated into central metabolism (13). Three pathways for degrading PCA have been characterized, differing in the location of the initial ring-opening oxidation. Representatives from each class of pathway have been expressed from plasmids in E. coli, with various levels of success. The 4,5-cleavage pathway has been best characterized in Sphingobium sp. strain SYK-6 (14), and heterologous expression in E. coli of a pathway variant from Pseudomonas ochraceae NGJ1 allowed colony formation after 2 days (15). The 2,3-cleavage pathway has been studied in Paenibacillus sp. strain JJ-1b, and heterologous expression of this pathway in E. coli enabled colony formation after a 10-day incubation (16). Finally, the most widely distributed pathway begins through 3,4-cleavage, as typified by Pseudomonas putida (13, 17) (Fig. 1A). Expression of the 3,4-cleavage pathway from Acinetobacter calcoaceticus in E. coli quantitatively produced an intermediate, β-ketoadipate, but allowed no growth (18). Since PCA degradation is the core pathway involved in converting many lignin-derived aromatic compounds into central metabolites, any inefficiencies in this pathway will limit all future efforts if they are not identified and ameliorated.
FIG 1.

(A) The 3,4-cleavage pathway of PCA was transferred from P. putida KT2440 to E. coli, allowing growth with PCA as the sole source of carbon and energy. Abbreviations: PcaHG, protocatechuate 3,4-dioxygenase; PcaB, 3-carboxymuconate cycloisomerase; PcaC, 4-carboxymuconolactone decarboxylase; PcaD, β-ketoadipate enol-lactone hydrolase; PcaIJ, β-ketoadipate:succinyl coenzyme A (CoA) transferase; PcaF, β-ketoadipyl-CoA thiolase. (B) Strain AG978 (ΔompT::pcaHGBDC ΔpflB::pcaIJFK) was grown in minimal medium containing PCA as the sole source of carbon and energy. The control strain, AG977 (ΔompT ΔpflB), showed a slow increase in absorbance due to oxidation of the substrate.
When microbes grow poorly on a particular substrate, experimental evolution can be used to optimize inefficient metabolic pathways, whether native (19, 20) or engineered (21–23). Serial propagation under conditions where the relevant pathway is necessary for growth selects for mutants with improved pathway function. Genome resequencing can then be used to identify putative causal mutations. Reconstructing these mutations in the parental strain verifies their effects. By studying the mutations that improve pathway function, we can deduce the factors that were initially limiting pathway effectiveness and the biochemical solutions that overcame these limitations.
In this work, we combined rational and evolutionary approaches to construct and optimize a heterologous pathway for PCA catabolism in E. coli. We first transferred the 3,4-cleavage pathway from P. putida KT2440 into E. coli and demonstrated that the engineered strain can grow with PCA as the sole source of carbon and energy. Next, we used experimental evolution to identify a single mutation that increased the growth rate with PCA while maintaining basal growth rates with glucose. Finally, we showed that the optimized PCA degradation pathway enables further extension of the catabolic network to the related compound 4-hydroxybenzoate (4-HB). This optimized strain will serve as a platform for further reconstruction of catabolic pathways for lignin-derived aromatic compounds.
RESULTS AND DISCUSSION
PCA 3,4-cleavage pathway design and construction.
The eight genes of the PCA 3,4-cleavage pathway from Pseudomonas putida KT2440 (pcaHGBCDIJF) and the PCA transporter gene pcaK were codon optimized for E. coli and combined into two operons, each driven by an inducible T5-lac promoter. These operons were synthesized de novo and inserted into the chromosome of E. coli BW25113 at the ompT and pflB loci, yielding strain AG978 (ΔompT::pcaHGBDC ΔpflB::pcaIJFK).
Expression of the PCA pathway allowed growth with 1 g/liter PCA as the sole source of carbon and energy in M9 minimal medium, with a growth rate of 0.16 h−1, corresponding to a doubling time of 260 min (Fig. 1B). A control strain lacking the pathway genes showed a slow increase in optical density at 600 nm (OD600) due to the appearance of red coloring, likely caused by oxidation of PCA, but no increase in turbidity or cell count was observed.
Evolution improved function of the heterologous PCA pathway.
While strain AG978 grows in the presence of PCA, its growth rate is roughly one-quarter that of P. putida grown under similar conditions (24). To optimize this pathway, we used experimental evolution to select for mutants with increased fitness during growth with PCA. Three replicate cultures of AG978 were grown in minimal medium with 1 g/liter PCA as the sole source of carbon and energy. When each culture reached saturation, it was diluted 128-fold into fresh medium and allowed to regrow. After 500 generations, individual mutants were isolated and characterized. Each of the evolved isolates grew significantly faster than the parent, with improvements of between 2.2-fold and 2.4-fold (Fig. 2). We identified mixed phenotypes in two of the evolved populations. Replicate population A yielded strains JME1 and JME2, and replicate population B yielded strains JME3 and JME4, but all of the isolates from replicate population C showed the same phenotype as JME6. While all of the isolates had similarly improved growth rates, JME1, JME4, and JME6 reached a substantially lower final OD, even compared to the unevolved strain.
FIG 2.
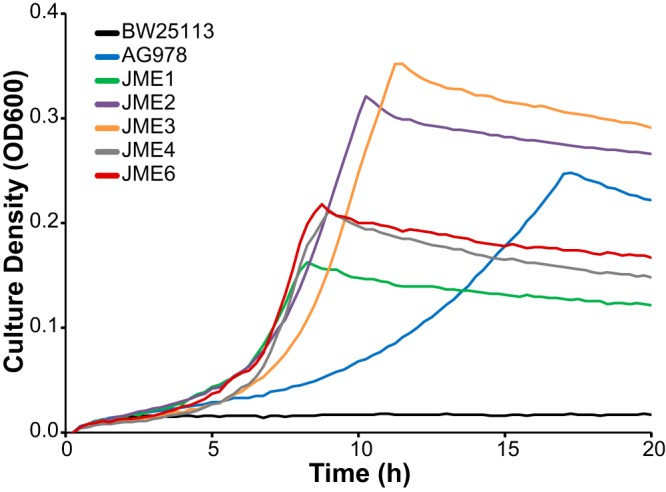
Experimental evolution selects for improved growth with PCA. Five strains, isolated from three replicate evolution experiments, were compared to the wild-type BW25113 strain and the engineered parent AG978 strain during growth in minimal medium containing 1 g/liter PCA.
Genome resequencing and reconstruction identify causal mutations.
We resequenced the genomes of all five isolates to identify mutations relative to the parental strain. The isolates had between five and eight mutations, including several large insertion sequence (IS)-mediated deletions and rearrangements (see Table S4 in the supplemental material). Several strains shared independent mutations in the same genes or genomic regions, even across replicate cultures.
All five strains had mutations predicted to affect the pcaH gene, which catalyzes the first step in PCA oxidation (Fig. 1A; see also Fig. S1 in the supplemental material). Strains JME1, JME2, and JME3 had IS-mediated duplications of a 118-kb region containing the introduced pcaHGBDC operon. Strain JME4 had a single nucleotide substitution in the predicted ribosomal binding site (RBS) for pcaH (Fig. S1 in the supplemental material). Using the RBS Calculator, the RBS mutation in JME4 is predicted to increase the strength of the pcaH RBS by 2.2-fold (25). Strain JME6 contains an IS element inserted between the promoter and RBS of pcaH that is likely to increase expression of the PcaH operon (26). Direct proteomic measurements showed that the abundances of PcaH and PcaG were increased by at least 7-fold in each of JME1, JME4, and JME6, while other heterologous proteins showed variable or decreasing expression (see Fig. 4). Notably, the promoter and RBS mutations increased expression of the genes encoding both PcaH and PcaG but not expression of the genes further downstream that encode PcaBDC. In combination, these results demonstrate that there are multiple routes to increase PcaHG expression, including through changes in copy number, the acquisition of a new promoter, or an RBS mutation. Parallelism in both mutations and expression levels suggests that these changes are highly beneficial during growth with PCA.
FIG 4.

Diverse mutations produce similar changes in protein abundance. In addition to other mutations described in Table S1 in the supplemental material, strain JME1 has a duplication of the pcaHGBDC operon, strain JME4 has a mutation to the pcaH RBS, and strain JME6 has a new promoter inserted upstream of pcaHGBDC. Strain JME17 has only the single RBS mutation also present in JME4. Error bars show the standard deviations for data calculated from three biological replicates.
Reconstructing IS-mediated rearrangements is challenging, so to test this hypothesis we introduced the single nucleotide mutation from JME4 into the parental AG978 strain, yielding strain JME17. This single mutation recapitulated the majority of the evolutionary improvement in growth with PCA, raising the growth rate from 0.16 h−1 to 0.41 h−1 (Fig. 3A), decreasing the doubling time from 260 min to 100 min, and producing a comparable increase in expression of PcaHG (Fig. 4). Comparing JME17 to the evolved isolate JME3, JME3 grew 24% faster with PCA and 20% faster with glucose (Fig. 3B). Since JME3 displayed an improvement in growth similar to that seen with JME17 when grown with either glucose or PCA, the unexamined mutations in JME3 likely reflect general adaptations to growth in minimal medium under these experimental conditions rather than any PCA-specific adaptation. We placed the two operons under the control of IPTG (isopropyl β-d-1-thiogalactopyranoside)-inducible promoters out of concern that pathway expression would place a substantial burden on the host. However, the engineered strains grew as well with glucose as the unmodified parent (Fig. 3B), likely due to the reduced levels of enzymes that result from chromosomal integration compared to expression from a plasmid, indicating that this concern was unfounded. Additionally, our results obtained with PcaH are consistent with previous studies that have shown large changes in growth rates resulting from small changes in gene expression of metabolic enzymes (21, 27).
FIG 3.
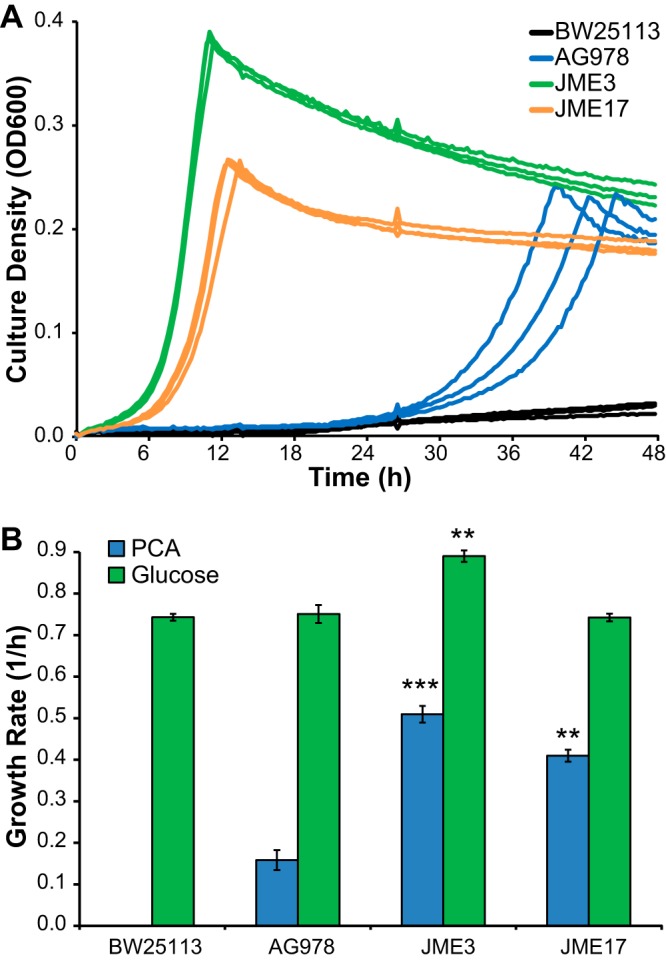
A mutation in the RBS of pcaH is sufficient for reproduction of the increase in the PCA-specific growth rate. (A) The growth of evolved isolate JME3 is compared with that of the reconstructed JME17 strain and the parent AG978 strain during growth with PCA. JME17 differs from AG978 by a single point mutation in the RBS of pcaH. (B) Comparing growth rates in minimal medium containing 1 g/liter PCA or 2 g/liter glucose, JME3 had a moderate growth advantage relative to JME17 under both conditions. Error bars show 1 standard deviation for data calculated from three biological replicates. **, statistically significant relative to AG978, P < 0.001; ***, statistically significant relative to AG978, P < 0.0001; 2-tailed t test.
Three of the strains, JME1, JME4, and JME6, reached saturation at relatively low optical densities (Fig. S2A). The only mutational target common to these three strains, but not to JME2 or JME3, is the Ag43 surface antigen encoded by flu. JME1 contains a nonsynonymous mutation in flu, while JME4 and JME6 have nonoverlapping 156-nucleotide (nt) and 261-nt deletions, respectively. Proteomic measurements show a 60-fold to 190-fold increase in expression of Ag43 in the three evolved strains relative to the AG978 parent (Table S2). We introduced the 261-nt deletion from JME6 into the parental strain and into the strain carrying the pcaH RBS mutation, yielding strains JME15 and JME16. In both cases, the flu deletion reduced the maximum optical density without significantly affecting the growth rate (Fig. S2B). The replicate evolution of flu mutations across all three populations strongly suggests that the mutation was beneficial, and a similar mutation was seen in a previous evolution experiment looking at adaptation to acid stress (28). All of the mutations observed in this experiment increase expression of modified forms of Ag43, rather than abolishing expression or producing frameshifts. Ag43 mediates cell aggregation (29), and we hypothesize that aggregation due to increased Ag43 levels may increase cell survival under acid-stressed conditions such as those represented by minimal medium containing PCA.
Targeted RBS mutation replaced large-scale gene duplication.
In replicate population B, isolates JME3 and JME4 demonstrated two different mechanisms to increase PcaH expression. JME3 contains a 118-kb duplication that spans the entire heterologous gene cluster containing pcaH. In contrast, JME4 has only a single copy of that region and, instead, has a single nucleotide mutation in the RBS of pcaH. The RBS mutation in JME4 and the similar duplication in JME1 have similar effects on PcaHG expression (Fig. 4). Quantitative PCR (qPCR) results showed that the population-averaged copy number of pcaH increased to more than three copies per cell before declining between generations 400 and 500 (Fig. S3A). Conversely, targeted Sanger sequencing showed that the RBS mutation seen in JME4 rose to an observable frequency between generations 400 and 500 (Fig. S3B). In combination, these results suggest that the tandem duplication evolved first and was then in the process of being replaced by the RBS mutation between generations 400 and 500. Due to possible clonal interference, the replacement of the JME3 lineage with the JME4 lineage cannot be attributed solely to the different mechanisms for overexpressing PcaH since the JME4 lineage may have additional beneficial mutations. However, similar evolutionary trajectories of transient duplication and refinement have been observed in evolving populations of E. coli and Saccharomyces cerevisiae (30, 31).
Additional increases in pcaH RBS strength do not improve growth.
To further explore the importance of the pcaH RBS for improved growth, we designed three additional ribosome binding sites with predicted 2-, 4-, and 8-fold increases in RBS strength relative to JME17 (25). Proteomic measurements demonstrate that the increase in PcaHG expression was smaller than predicted, with a maximum increase in PcaH expression of 2-fold relative to JME17 (Fig. S3C). These strains showed growth rates similar to that of the JME17 engineered mutant (Fig. S3C). Since the engineered RBSs did not increase the growth rate relative to JME17, the increase due to the single nucleotide mutation in JME17 was sufficient to relieve any growth restriction due to limited PcaHG expression.
Improved PCA catabolism supports further pathway elaboration.
A single enzyme, 4-hydroxybenzoate 3-monooxygenase, can convert 4-HB into PCA. This enzymatic activity can be provided by PraI from Paenibacillus sp. strain JJ-1B (16). We introduced the corresponding gene into the AG978 parental strain and the JME17 engineered strain, yielding strains JME7 and JME50, respectively (Fig. 5A). Cultured with 4-HB as the sole source of carbon and energy, JME7 grew at a rate of 0.027 ± 0.005 h−1, while JME50 grew at 0.259 ± 0.002 h−1 (Fig. 5B), corresponding to doubling times of 1,540 min and 160 min, respectively. The RBS mutation in pcaH was the only difference between JME7 and JME50; while this mutation increased growth with PCA by roughly 2.5-fold, it increased growth with 4-HB by nearly 10-fold. Further extension of the catabolic network to include new growth substrates will require careful optimization of each new pathway.
FIG 5.
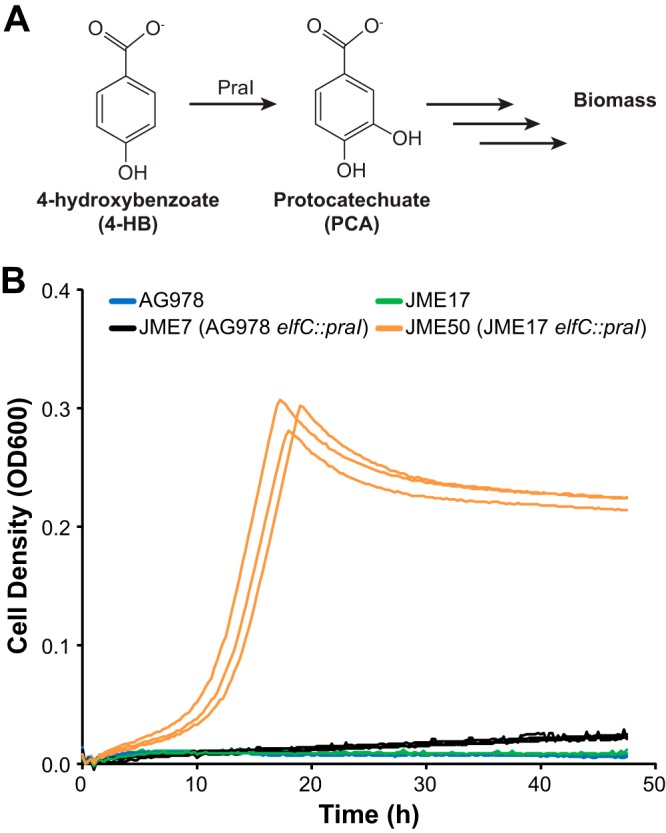
Evolutionary optimization of the core PCA catabolism pathway enables elaboration of the catabolic network. (A) The 4-hydroxybenzoate 3-monooxygenase PraI converts 4-HB into PCA. (B) The monooxygenase gene, praI, is integrated in both JME7 (black) and JME50 (orange). The difference between JME7 and JME50 is a single point mutation in the RBS of pcaH. All strains were grown in minimal medium containing 1 g/liter 4HB as the sole source of carbon and energy.
Conclusion.
Using a combination of engineering and evolution, we have constructed strains of E. coli that are capable of growing with the model aromatic compounds PCA and 4-HB at rates approaching growth with glucose. Initially, expression of the ring-cleaving PcaHG dioxygenase was limiting, and we demonstrated that three different types of mutations produced similar changes in both enzyme expression and growth rates with PCA. Chromosomal integration of the heterologous pathway, while technically more challenging than expression from a plasmid, avoided any measurable burden on the host resulting from introduction of the new pathway. We anticipate that these engineered strains will be useful for the identification, characterization, and optimization of additional pathways for the conversion of lignin-derived aromatic compounds into valuable bioproducts.
MATERIALS AND METHODS
Strains, plasmids, and primers.
The strains, plasmids, and primers used in this work are listed in Table 1, Table 2, and Table 3, respectively.
TABLE 1.
Strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| BW25113 | F− Δ(araD-araB)567 lacZ4787(del)::rrnB-3 λ− rph-1 Δ(rhaD-rhaB)568 hsdR514 | 33 |
| AG977 | BW25113 ΔompT ΔpflB | This study |
| AG978 | BW25113 ompT::pcaHGBDC pflB::pcaIJFK | This study |
| JME1 | Evolved variant of AG978 | This study |
| JME2 | Evolved variant of AG978 | This study |
| JME3 | Evolved variant of AG978 | This study |
| JME4 | Evolved variant of AG978 | This study |
| JME6 | Evolved variant of AG978 | This study |
| JME7 | AG978 elfC::praI | This study |
| JME15 | AG978 flu(Δ6) | This study |
| JME16 | AG978 flu(Δ6) pcaH(R4) | This study |
| JME17 | AG978 pcaH(R4) | This study |
| JME50 | JME17 elfC::praI | This study |
TABLE 2.
Plasmids used in this study
| Plasmid | Descriptiona | Reference or source |
|---|---|---|
| pCas | Expresses Cas9 and λ-RED | 34 |
| pTarget | Expresses sgRNA | 34 |
| pKD46 | PBAD λ-RED, Ampr | 33 |
| pKD46-I-SceI | PBAD λ-RED, I-SceI, Ampr | This study |
| pET30a | LacI+, Kanr | EMD Millipore, Billerica, MA |
| pET30a-dest | MCS, Kanr | This study |
| pET30a-LP-pcaIJFK | pcaIJFK integration, Kanr | This study |
| pET30a-LP-pcaHGBDC | pcaHGBDC integration, Kanr | This study |
| pET30a-LP-control | Gene deletion, Kanr | This study |
| pJM157 | Expresses elfC sgRNA | This study |
| pJM160 | Contains praI expression construct with elfC homology | This study |
| pJM179 | Expresses flu sgRNA | This study |
| pJM180 | Expresses pcaH sgRNA #1 | This study |
| pJM182 | Expresses pcaH sgRNA #2 | This study |
Ampr, ampicillin resistance; MCS, multiple-cloning site; sgRNA, single-guide RNA.
TABLE 3.
Primers used in this study
| Primer | Sequence | Purpose |
|---|---|---|
| dest-FWD | TAGGGATAACAGGGTAATCAGTCGTGTCATCTGATTACCTGGCGGAAATTAAACTAAGAGAGAGCTCTAAATCATGAAAAATTTATTTGCTTTGTG | Construction of pET30a-dest |
| dest-REV | CGAATTAGCCTGCCAGCCCTGTTTTTATTAGTGCATTTTGCGCGAGGTCAAAATAAATTTTTCATGATTT | Construction of pET30a-dest |
| I-SceI-LP-T5-FWD | TCCGATTAGGGATAACAGGGTAATGTGTAGGCTGGAGCTGCTTCTACGGCCCCAAGGTCCAAACGGTGAAAATCATGAAAAATTTATTTGCTTTGTG | Construction of pET30a-LP-pcaIJFK, pET30a-LP-control |
| LP-IJFK-REV | ACAAGGCCATGGCTGATCATATGAATATCCTCCTTAGTTGGCTTCAGGGATGAGGCGCCATCCTGGGCGGTTCTGATAACGA | Construction of pET30a-LP-pcaIJFK |
| LP-HGBDC-REV | ACAAGGCCATGGCTGATCATATGAATATCCTCCTTAGTTGGCTTCAGGGATGAGGCGCCATCTTTGCCTGGCGGCAGTAG | Construction of pET30a-LP-pcaHGBDC |
| LP-control-REV | ACAAGGCCATGGCTGATCATATGAATATCCTCCTTAGTTGGCTTCAGGGATGAGGCGCCATCAAATAAATTTTTCATGATTT | Construction of pET30a-LP-control |
| ompT-LP-up | ATATAAAAAATACATATTCAATCATTAAAACGATTGAATGGAGAACTTTTCATATGAATATCCTCCTTAGTTGGCTTCAG | I-SceI, landing pad and CmR cassette integration at ompT |
| ompT-LP-down | AGTTATTCCCCGGGGCGATTTTCACCTCGGGGAAATTTTAGTTGGCGTTCGTGTAGGCTGGAGCTGCTTC | I-SceI, landing pad and CmR cassette integration at ompT |
| pflB-LP-up | TTTTACTGTACGATTTCAGTCAAATCTAATTACATAGATTGAGTGAAGGTGTGTAGGCTGGAGCTGCTTC | I-SceI, landing pad and CmR cassette integration at pflB |
| pflB-LP-down | CGAAGTACGCAGTAAATAAAAAATCCACTTAAGAAGGTAGGTGTTACATGCATATGAATATCCTCCTTAG | I-SceI, landing pad and CmR cassette integration at pflB |
| ompT-FWD | TAAACAAAATATAAACAGTGGAGC | PCR screen at ompT |
| ompT-REV | CTATAATTGATGGTTTTCTATGTG | PCR screen at ompT |
| pflB-FWD | TTTTACTGTACGATTTCAGTCAAAT | PCR screen at pflB |
| pflB-REV | CGAAGTACGCAGTAAATAAAAAATC | PCR screen at pflB |
| pTarget FWD | GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAG | Inverse PCR of pTarget |
| pTarget REV | ACTAGTATTATACCTAGGACTGAG | Inverse PCR of pTarget |
| elfC N20F | TTCCTTATGCCACTTCTTATGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | Construction of elfC sgRNA |
| elfC N20R | ATAAGAAGTGGCATAAGGAAACTAGTATTATACCTAGGACTGAGCTAGCTGTCAAGGATC | Construction of elfC sgRNA |
| pcaH N20F1 | TTTTACCTCCTCTCTAGAAGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | Construction of pcaH sgRNA |
| pcaH N20R1 | CTTCTAGAGAGGAGGTAAAAACTAGTATTATACCTAGGACTGAGCTAGCTGTCAAGGATC | Construction of pcaH sgRNA |
| pcaH N20F2 | AGAGGAGGTAAAACATATGCGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | Construction of pcaH sgRNA |
| pcaH N20R2 | GCATATGTTTTACCTCCTCTACTAGTATTATACCTAGGACTGAGCTAGCTGTCAAGGATC | Construction of pcaH sgRNA |
| flu N20F | TACGAACTTCCCTGTGCGGGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | Construction of flu sgRNA |
| flu N20R | CCCGCACAGGGAAGTTCGTAACTAGTATTATACCTAGGACTGAGCTAGCTGTCAAGGATC | Construction of flu sgRNA |
Media and chemicals.
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Fairlawn, NJ) and were molecular grade. All oligonucleotides were ordered from IDT (Coralville, IA). E. coli strains were routinely cultivated at 37°C in LB broth (Miller; MilliporeSigma, Billerica, MA) or M9 medium containing the necessary antibiotics (50 mg/liter kanamycin [Kan], 50 mg/liter carbenicillin [Carb], 15 mg/liter chloramphenicol, or 50 mg/liter spectinomycin). Growth assays with PCA and 4-HB were performed in M9 salts medium (Sigma-Aldrich) containing 300 mg/liter thiamine and 1 mM IPTG. PCA and 4-HB were dissolved in water at 5 g/liter, filter sterilized, and added at a final concentration of 1 g/liter. The pH of the substrates was not controlled, as PCA oxidation occurred more rapidly at neutral pH (32).
Plasmid construction.
The eight genes of the PCA 3,4-degradation pathway from Pseudomonas putida KT2440 (pcaHGBCDIJF) and the PCA transporter gene (pcaKA) were designed as four constructs using GeneDesigner software and ordered through DNA2.0 (Menlo Park, CA). The genes were codon optimized for E. coli, placed behind an inducible T5-lac promoter, and flanked by BioBrick restriction enzyme sites for cloning. Constructs included ribosome binding sites and reinitiation spacer sequences between genes.
Plasmids for chromosomal integration of the PCA 3,4-degradation pathway were constructed in pET30a (EMD Millipore, Billerica, MA). Plasmid pET30a-dest was created by blunt-end cloning of the PCR product created with primers dest-FWD/dest-REV into pET30a. Ligation products were transformed into CopyCutter EPI400 E. coli (Epicentre, Madison, WI), and plasmids were verified by PCR with primers T7 promoter/T7 terminator. The final plasmids used for integration were constructed in pET30a-dest using standard methods to insert PCR products generated using I-SceI-LP-T5-FWD and LP-IJFK-REV, LP-HGBDC-REV, or LP-control REV, resulting in pET30a-LP-control, pET30a-LP-pcaIJFK, and pET30a-LP-pcaHGBDC.
A plasmid containing both the λ Red recombinase system and the homing endonuclease I-SceI was used for chromosomal insertion of the PCA 3,4-degradation pathway. The I-SceI gene was codon optimized for E. coli, placed behind a strong ribosome binding sequence (Life Technologies, Grand Island, NY), and inserted upstream of the λ Red recombinase genes on plasmid pKD46 using standard methods (33), resulting in plasmid pKD46-I-SceI. I-SceI enzyme functionality was tested in an overnight culture grown with 1 mM arabinose to induce I-SceI expression. Cells were harvested, washed, and frozen at −20°C before lysis in I-SceI buffer (10 mM Tris-HCl, 10 mM MgCl2, 1 mM dithiothreitol [DTT], 100 mg/ml bovine serum albumin, pH 8.8). The cell extract was incubated with plasmid pGPS2 (New England BioLabs, Waltham, MA) at 37°C for 1 h. Samples were taken every 15 min and analyzed by gel electrophoresis to confirm I-SceI digestion activity (see Fig. S1B in the supplemental material).
Plasmids pJM180 and pJM182, expressing trans-activating clustered regularly interspaced short palindromic repeat (CRISPR) RNAs (tracrRNAs) targeting the pcaH RBS, were generated through inverse PCR of pTarget using primers pTarget FWD and pTarget REV (34). The appropriate oligonucleotides were then assembled into the pTarget backbone using HiFi master mix and transformed into 10-β E. coli (NEB, Waltham, MA). Plasmid pJM179, targeting flu, and plasmid pJM160, targeting elfC for integration of praI, were constructed in a similar fashion.
Strain construction.
The PCA 3,4-degradation pathway was inserted into the chromosome of E. coli BW25113 using a “Landing Pad” method (35). A chloramphenicol (Cm) resistance gene cassette flanked by I-SceI restriction sites and “landing pad” regions of homology for λ Red recombination (1, 5′-TACGGCCCCAAGGTCCAAACGGTGA-3′; 2, 5′-GATGGCGCCTCATCCCTGAAGCCAA-3′) was PCR amplified from pUC57-LP with primers ompT-LP-up/ompT-LP-down or primers pflB-LP-up/pflB-LP-down to add flanking 50-bp homology regions upstream and downstream of either ompT or pflB. The ompT PCR product was transformed into E. coli BW25113 containing pKD46 and expressing the λ Red recombinase system. Chloramphenicol-resistant colonies were screened for gene replacement by PCR at ompT (ompT-FWD/ompT-REV). Correct strains were cotransformed with pKD46-I-SceI and either the pET30a-LP-control or pET30a-LP-pcaHGBDC plasmid and selected using kanamycin and carbenicillin. Plasmid transformation was confirmed by restriction digestion, and strains containing both plasmids were grown in LB plus Kan plus Carb at 30°C for 3 h, harvested, resuspended in LB plus Carb with 1 mM arabinose to induce the λ Red recombinase system and I-SceI expression, and grown at 30°C for 1 h. The culture (10 μl) was streaked on LB plates and incubated at 37°C overnight to cure the pKD46-I-SceI plasmid. Colonies were patched to LB and LB plus Cm at 37°C, and all Cm-sensitive colonies were PCR screened at ompT. Correct strains for each integration (ΔompT and ompT::pcaHGBDC) were streaked on LB and patched on LB, LB plus Kan, and LB plus Carb at 30°C to verify the expected phenotypes, and all Kan- and Carb-sensitive colonies were again PCR screened at ompT to confirm integration. The process was repeated in the ΔompT and ΔompT::pcaHGBDC strains for ΔpflB or replacement with pcaIJFK, resulting in strains ΔompT ΔpflB (AG977) and ΔompT::pcaHGBDC ΔpflB::pcaIJFK (AG978). Strains correct by PCR were sequence verified at both the ompT and pflB loci.
Growth measurements.
Cultures were grown to saturation in LB plus 1 mM IPTG, diluted 100-fold into fresh M9 plus 1 mM IPTG plus substrate, and grown as triplicate 100-μl cultures in a Bioscreen C plate reader (Oy Growth Curves Ab Ltd., Helsinki, Finland). Growth was monitored using optical density at 600 nm (OD600). Growth rates were calculated during exponential growth using CurveFitter software (36). OD600 is linearly proportional to the number of live cells, as calculated by counting CFU per milliliter, over this range.
Experimental evolution.
The parental strain, AG978, was streaked to single colonies. Three separate colonies were grown to saturation in M9 plus 1 mM IPTG plus 0.2% glucose and were then diluted 128-fold into M9 plus 1 mM IPTG plus 1 g/liter PCA. When the cultures reached saturation, initially after 48 h but later after 24 h, they were diluted a further 128-fold into fresh M9 plus 1 mM IPTG plus 1 g/liter PCA. Culture aliquots were periodically frozen at −80°C for later analysis. After 500 generations, each culture was streaked to single colonies. Eight colonies from each plate were picked and analyzed for growth in M9 plus 1 mM IPTG plus 1 g/liter PCA.
Genome resequencing and reconstruction.
Genomic DNA was prepared using a DNeasy blood and tissue kit (Qiagen, Valencia, CA) according to the manufacturer's directions. Purified DNA was quantified using a Qubit fluorimeter (Thermo Fisher, Waltham, MA), resequenced by the Joint Genome Institute using a MiSeq system (Illumina, San Diego, CA) to approximately 150× coverage, and analyzed using Geneious (Biomatters, Auckland, NZ). Targeted mutations were introduced into the parental strain on single-stranded DNA (ssDNA) oligonucleotides or double-stranded DNA (dsDNA) gBlocks using λ-RED in combination with CRISPR-Cas (34). Mutations were verified by Sanger sequencing of PCR amplicons (Eurofins Operon, Louisville, KY). The gene cassette for elfC::praI was synthesized by Gen9 (Cambridge, MA) and integrated into the appropriate chromosomal locus following the same process as for the targeted mutations. The promoter and terminator for praI expression were chosen from previously characterized genetic parts (37, 38).
RBS design and analysis.
The parental and mutant ribosome binding sites were analyzed using RBS Calculator with Free Energy model v2.0 (39). Mutant ribosome binding sites were designed using RBS Calculator (25).
qPCR.
Gene copy numbers were determined by quantitative PCR using Phusion DNA polymerase (NEB) and EvaGreen (Biotium, Fremont, CA) on a CFX96 Touch thermocycler (Bio-Rad, Hercules, CA). Whole cells from triplicate cultures were used as the DNA templates. pcaH was measured using primers pcaH_SeqF (5′-CGCTCACAATTCCACAACG-3′) and pcaH_SeqR (5′-CTTTTGGGTGCCAATTTCTATC-3′). Copy numbers were normalized to 16S rRNA gene copy numbers determined using primers 515F (5′GTGCCAGCMGCCGCGGTAA-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′).
Proteomics.
Parent, evolved, and engineered E. coli strains were grown in triplicate to an OD600 of 0.3 in 5 ml of M9 plus 1 mM IPTG plus 1 g/liter PCA, pelleted by centrifugation, and frozen in liquid nitrogen until processing. Cells were resuspended in 500 μl of 4% sodium deoxycholate (SDC)–100 mM ammonium bicarbonate (ABC) (pH 7.8) and lysed via pulse sonication (Branson Sonifier) (amplitude, 10%; 2 s on, 2 s off; 1 min total). Crude lysates were precleared by centrifugation at 21,000 × g, adjusted to 10 mM dithiothreitol to reduce disulfide bonds, and incubated at 95°C for 5 min. Cysteine residues were then alkylated with iodoacetamide (30 mM) by 20 min of incubation at room temperature in the dark. Samples were transferred to a 10,000-molecular-weight-cutoff (MWCO) spin filter (Vivaspin 2; Sartorius) and centrifuged at 4,500 × g to remove supernatant and to collect denatured and reduced proteins atop the filter membrane. Proteins were washed with ABC (2× the initial sample volume; 1,000 μl) followed by centrifugation and were finally resuspended in a 1× volume of ABC (500 μl) prior to proteolytic digestion. Protein samples, now roughly 2% SDC, were digested in situ using proteomics-grade porcine trypsin (Sigma) at a 1:50 (wt/wt) ratio and incubated at 37°C overnight. Samples were diluted with a 1× volume of 100 mM ABC (500 μl), supplied with another 1:50 (wt/wt) aliquot of trypsin, and incubated at 37°C for 3 h. Appropriately sized tryptic peptides were then collected by centrifugation and transferred to a 2-ml microcentrifuge tube, where they were acidified to 0.5% formic acid to precipitate the residual SDC. The detergent precipitate was removed from the peptide solution by addition of 1 ml water-saturated ethyl acetate as previously described (40). Ethyl acetate cleanup was repeated twice. The aqueous phase was transferred to a new microcentrifuge tube and concentrated via the use of a SpeedVac to ∼100 μl. Peptide concentrations were measured by bicinchoninic acid (BCA) assay (Pierce).
Peptide samples were diluted 1:1 with solvent A (95% liquid chromatography-mass spectrometry [LC-MS]-grade H2O, 5% LC-MS-grade acetonitrile, 0.1% formic acid) and placed into autosampler vials for automated two-dimensional LC-tandem MS (LC-MS/MS) analysis using a Vanquish ultra-high-performance LC (UHPLC) instrument directly in line with a Q Exactive Plus mass spectrometer (QE+; Thermo Scientific) outfitted with an in-house-pulled nanospray emitter (75-μm-inner-diameter [ID] fused silica) packed with 15 cm of 1.7-μm-particle-size Kinetex C18 reversed-phase (RP) resin (Phenomenex). For each sample, 5 μg of peptides was loaded onto a triphasic 100-μm-ID fritted precolumn (constructed in-house) packed with 10 cm of 5-μm-particle-size Luna strong-cation exchange resin (SCX; Phenomenex) sandwiched between two 5-cm lengths of 5-μm-particle-size Kinetex C18 RP resins. Peptides bound to the first RP resin were effectively desalted by this 30-min loading step (2 μl/min direct flow of 100% solvent A) and portioned to the SCX “mid-phase” by a 30-min, split-flow (300 nl/min postsplit) gradient to 100% solvent B (30% LC-MS-grade H2O, 70% LC-MS-grade acetonitrile, 0.1% formic acid).
Peptides bound to the SCX mid-phase were then portioned to the latter RP resin over two successive salt cuts of ammonium acetate (50 mM and 500 mM, autosampled, 2 μl/min direct flow), each followed by a 90-min, split-flow (300 nl/min) organic gradient (0% to 3% solvent B over 1 min; 3% to 25% solvent B over 80 min; 25% to 50% solvent B over 5 min; 50% to 0% solvent B over 4 min) to separate peptides along both the latter RP resin of the precolumn and, subsequently, the RP-packed nanospray emitter. Eluting peptides were measured and sequenced by the use of a QE+ with the following parameters: data-dependent acquisition; mass range = 300 to 1,500 m/z; MS/MS loop count = 15; MS and MS/MS resolution = 70K and 15K, respectively; isolation window = 1.6 m/z; charge exclusion = unassigned, 1, 6 to 8; exclude isotopes = on; peptide match = preferred; dynamic exclusion window and duration = 10 ppm and 45 s, respectively.
MS/MS spectra were searched against the E. coli K-12 proteome concatenated with exogenous PCA pathway protein sequences and common protein contaminants using MyriMatch v.2.2 (41) with the following settings: mass tolerances of 5 and 10 ppm were used for monoisotopic precursor and fragment ion scoring, respectively; peptide spectrum matches (PSM) were required to be fully tryptic with any number of missed cleavages; a static modification of 57.0214 Da was used for cysteine (carbamidomethylated) while a dynamic modification of 15.9949 Da was used for methionine (oxidized) residues. PSMs were filtered using IDPicker v.3.0 (42) with an experiment-wide false-discovery rate (assessed by PSMs to decoy sequences) initially controlled at <1% at the peptide level. Peptide intensities were assessed by chromatographic determination of the area under the curve using IDPicker's embed spectrum/label-free quantification option. Proteotypic peptides were downselected using Perseus (43) to remove stochastic peptide identifications and the resulting peptide abundance distributions normalized in InfernoRDN (44), as previously described (45). Peptide abundance trends were then used to derive overall protein abundance values via RRollup and missing values imputed to simulate the MS instrument's limit of detection. Proteomic data are available in the MassIVE proteome database as data set MSV000081122.
Supplementary Material
ACKNOWLEDGMENTS
Genome resequencing and analysis were performed by Christa Pennacchio, Natasha Brown, Anna Lipzen, and Wendy Schackwitz at the Joint Genome Institute.
The work conducted by the U.S. Department of Energy Joint Genome Institute, a Department of Energy (DOE) Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the DOE under contract no. DE-AC05-00OR22725. This work was supported by the BioEnergy Science Center, a U.S. Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science, and by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle, LLC, for the U.S. Department of Energy. The funders had no involvement in the design, analysis, or interpretation of these experiments.
S.M.C., D.M.K., J.G.E., and A.M.G. designed, constructed, and tested strains AG977 and AG978. R.J.G. performed proteomic measurements. J.K.M. designed and performed experiments for evolutionary optimization, characterization, and pathway extension. S.M.C., A.M.G., and J.K.M. wrote the paper with input from all authors.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01313-17.
REFERENCES
- 1.U.S. Department of Energy. 2016. 2016 billion-ton report: advancing domestic resources for a thriving bioeconomy, volume 1: economic availability of feedstocks. U.S. Department of Energy, Oak Ridge, TN. [Google Scholar]
- 2.Ragauskas AJ, Beckham GT, Biddy MJ, Chandra R, Chen F, Davis MF, Davison BH, Dixon RA, Gilna P, Keller M, Langan P, Naskar AK, Saddler JN, Tschaplinski TJ, Tuskan GA, Wyman CE. 2014. Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843. doi: 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- 3.Sannigrahi P, Ragauskas AJ, Tuskan GA. 2010. Poplar as a feedstock for biofuels: a review of compositional characteristics. Biofuels Bioprod Biorefining 4:209–226. doi: 10.1002/bbb.206. [DOI] [Google Scholar]
- 4.Samuel R, Pu Y, Raman B, Ragauskas AJ. 2010. Structural characterization and comparison of switchgrass ball-milled lignin before and after dilute acid pretreatment. Appl Biochem Biotechnol 162:62–74. doi: 10.1007/s12010-009-8749-y. [DOI] [PubMed] [Google Scholar]
- 5.Trajano HL, Engle NL, Foston M, Ragauskas AJ, Tschaplinski TJ, Wyman CE. 2013. The fate of lignin during hydrothermal pretreatment. Biotechnol Biofuels 6:110. doi: 10.1186/1754-6834-6-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beckham GT, Johnson CW, Karp EM, Salvachúa D, Vardon DR. 2016. Opportunities and challenges in biological lignin valorization. Curr Opin Biotechnol 42:40–53. doi: 10.1016/j.copbio.2016.02.030. [DOI] [PubMed] [Google Scholar]
- 7.Bugg TDH, Ahmad M, Hardiman EM, Rahmanpour R. 2011. Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep 28:1883. doi: 10.1039/c1np00042j. [DOI] [PubMed] [Google Scholar]
- 8.Salvachúa D, Karp EM, Nimlos CT, Vardon DR, Beckham GT. 2015. Towards lignin consolidated bioprocessing: simultaneous lignin depolymerization and product generation by bacteria. Green Chem 17:4951–4967. doi: 10.1039/C5GC01165E. [DOI] [Google Scholar]
- 9.Linger JG, Vardon DR, Guarnieri MT, Karp EM, Hunsinger GB, Franden MA, Johnson CW, Chupka G, Strathmann TJ, Pienkos PT, Beckham GT. 2014. Lignin valorization through integrated biological funneling and chemical catalysis. Proc Natl Acad Sci U S A 111:12013–12018. doi: 10.1073/pnas.1410657111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrández A, Garciá JL, Díaz E. 1997. Genetic characterization and expression in heterologous hosts of the 3-(3-hydroxyphenyl)propionate catabolic pathway of Escherichia coli K-12. J Bacteriol 179:2573–2581. doi: 10.1128/jb.179.8.2573-2581.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Díaz E, Ferrández A, García JL. 1998. Characterization of the hca cluster encoding the dioxygenolytic pathway for initial catabolism of 3-phenylpropionic acid in Escherichia coli K-12. J Bacteriol 180:2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burlingame R, Chapman PJ. 1983. Catabolism of phenylpropionic acid and its 3-hydroxy derivative by Escherichia coli. J Bacteriol 155:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harwood CS, Parales RE. 1996. The β-ketoadipate pathway and the biology of self-identity. Annu Rev Microbiol 50:553–590. doi: 10.1146/annurev.micro.50.1.553. [DOI] [PubMed] [Google Scholar]
- 14.Masai E, Katayama Y, Fukuda M. 2007. Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. Biosci Biotechnol Biochem 71:1–15. doi: 10.1271/bbb.60437. [DOI] [PubMed] [Google Scholar]
- 15.Maruyama K, Shibayama T, Ichikawa A, Sakou Y, Yamada S, Sugisaki H. 2004. Cloning and characterization of the genes encoding enzymes for the protocatechuate meta-degradation pathway of Pseudomonas ochraceae NGJ1. Biosci Biotechnol Biochem 68:1434–1441. doi: 10.1271/bbb.68.1434. [DOI] [PubMed] [Google Scholar]
- 16.Kasai D, Fujinami T, Abe T, Mase K, Katayama Y, Fukuda M, Masai E. 2009. Uncovering the protocatechuate 2,3-cleavage pathway genes. J Bacteriol 191:6758–6768. doi: 10.1128/JB.00840-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ornston LN, Stanier RY. 1964. Mechanism of beta-ketoadipate formation by bacteria. Nature 204:1279–1283. doi: 10.1038/2041279a0. [DOI] [PubMed] [Google Scholar]
- 18.Doten RC, Ngai KL, Mitchell DJ, Ornston LN. 1987. Cloning and genetic organization of the pca gene cluster from Acinetobacter calcoaceticus. J Bacteriol 169:3168–3174. doi: 10.1128/jb.169.7.3168-3174.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong K-K, Vongsangnak W, Vemuri GN, Nielsen J. 2011. Unravelling evolutionary strategies of yeast for improving galactose utilization through integrated systems level analysis. Proc Natl Acad Sci U S A 108:12179–12184. doi: 10.1073/pnas.1103219108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herring CD, Raghunathan A, Honisch C, Patel T, Applebee MK, Joyce AR, Albert TJ, Blattner FR, van den Boom D, Cantor CR, Palsson BØ. 2006. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet 38:1406–1412. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- 21.Michener JK, Camargo Neves AA, Vuilleumier S, Bringel F, Marx CJ. 2014. Effective use of a horizontally-transferred pathway for dichloromethane catabolism requires post-transfer refinement. Elife 3:e04279. doi: 10.7554/eLife.04279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou H-H, Chiu H-C, Delaney NF, Segrè D, Marx CJ. 2011. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332:1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark IC, Melnyk RA, Youngblut MD, Carlson HK, Iavarone AT, Coates JD. 2015. Synthetic and evolutionary construction of a chlorate-reducing Shewanella oneidensis MR-1. mBio 6:e00282-15. doi: 10.1128/mBio.00282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nichols NN, Harwood CS. 1997. PcaK, a high-affinity permease for the aromatic compounds 4-hydroxybenzoate and protocatechuate from Pseudomonas putida. J Bacteriol 179:5056–5061. doi: 10.1128/jb.179.16.5056-5061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salis HM, Mirsky EA, Voigt CA. 2009. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnetz K, Rak B. 1992. IS5: a mobile enhancer of transcription in Escherichia coli. Proc Natl Acad Sci U S A 89:1244–1248. doi: 10.1073/pnas.89.4.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kershner JP, McLoughlin SY, Kim J, Morgenthaler A, Ebmeier CC, Old WM, Copley SD. 2016. A synonymous mutation upstream of the gene encoding a weak-link enzyme causes an ultrasensitive response in growth rate. J Bacteriol 198:2853–2863. doi: 10.1128/JB.00262-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harden MM, He A, Creamer K, Clark MW, Hamdallah I, Martinez KA, Kresslein RL, Bush SP, Slonczewski JL. 2015. Acid-adapted strains of Escherichia coli K-12 obtained by experimental evolution. Appl Environ Microbiol 81:1932–1941. doi: 10.1128/AEM.03494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diderichsen B. 1980. flu, a metastable gene controlling surface properties of Escherichia coli. J Bacteriol 141:858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, Pilpel Y, Dahan O. 2012. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci U S A 109:21010–21015. doi: 10.1073/pnas.1211150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blount ZD, Barrick JE, Davidson CJ, Lenski RE. 2012. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature 489:513–518. doi: 10.1038/nature11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ornston LN, Stanier RY. 1966. The conversion of catechol and protocatechuate to beta-ketoadipate by Pseudomonas putida. J Biol Chem 241:3776–3786. [PubMed] [Google Scholar]
- 33.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. doi: 10.1128/AEM.04023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhlman TE, Cox EC. 2010. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res 38:e92. doi: 10.1093/nar/gkp1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delaney NF, Kaczmarek ME, Ward LM, Swanson PK, Lee M-C, Marx CJ. 2013. Development of an optimized medium, strain and high-throughput culturing methods for Methylobacterium extorquens. PLoS One 8:e62957. doi: 10.1371/journal.pone.0062957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosuri S, Goodman DB, Cambray G, Mutalik VK, Gao Y, Arkin AP, Endy D, Church GM. 2013. Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc Natl Acad Sci U S A 110:14024–14029. doi: 10.1073/pnas.1301301110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y-J, Liu P, Nielsen AAK, Brophy JAN, Clancy K, Peterson T, Voigt CA. 2013. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat Methods 10:659–664. doi: 10.1038/nmeth.2515. [DOI] [PubMed] [Google Scholar]
- 39.Espah Borujeni A, Channarasappa AS, Salis HM. 2014. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res 42:2646–2659. doi: 10.1093/nar/gkt1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masuda T, Tomita M, Ishihama Y. 2008. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J Proteome Res 7:731–740. doi: 10.1021/pr700658q. [DOI] [PubMed] [Google Scholar]
- 41.Tabb DL, Fernando CG, Chambers MC. 2007. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res 6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma Z-Q, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, Halvey PJ, Schilling B, Drake PM, Gibson BW, Tabb DL. 2009. IDPicker 2.0: improved protein assembly with high discrimination peptide identification filtering. J Proteome Res 8:3872–3881. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. 2016. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 44.Taverner T, Karpievitch YV, Polpitiya AD, Brown JN, Dabney AR, Anderson GA, Smith RD. 2012. DanteR: an extensible R-based tool for quantitative analysis of -omics data. Bioinformatics 28:2404–2406. doi: 10.1093/bioinformatics/bts449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Q, Resch MG, Podkaminer K, Yang S, Baker JO, Donohoe BS, Wilson C, Klingeman DM, Olson DG, Decker SR, Giannone RJ, Hettich RL, Brown SD, Lynd LR, Bayer EA, Himmel ME, Bomble YJ. 2016. Dramatic performance of Clostridium thermocellum explained by its wide range of cellulase modalities. Sci Adv 2:e1501254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.