

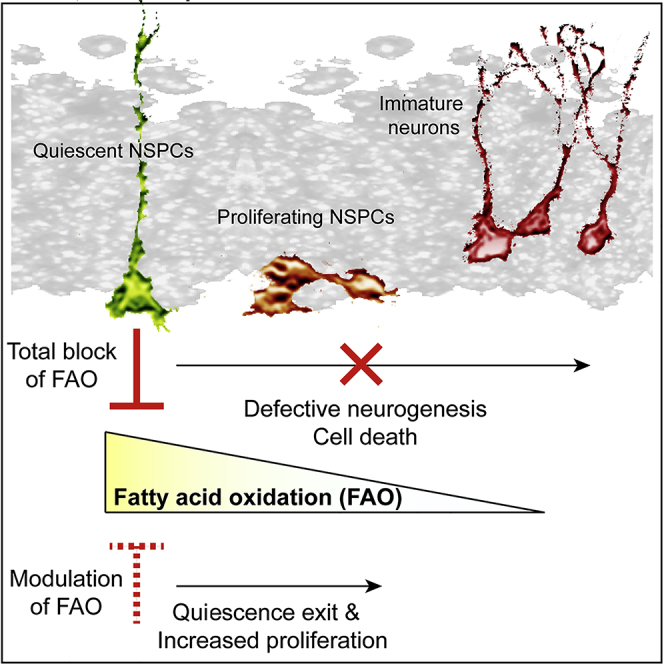
Summary
Hippocampal neurogenesis is important for certain forms of cognition, and failing neurogenesis has been implicated in neuropsychiatric diseases. The neurogenic capacity of hippocampal neural stem/progenitor cells (NSPCs) depends on a balance between quiescent and proliferative states. Here, we show that the rate of fatty acid oxidation (FAO) regulates the activity of NSPCs. Quiescent NSPCs show high levels of carnitine palmitoyltransferase 1a (Cpt1a)-dependent FAO, which is downregulated in proliferating NSPCs. Pharmacological inhibition and conditional deletion of Cpt1a in vitro and in vivo leads to altered NSPC behavior, showing that Cpt1a-dependent FAO is required for stem cell maintenance and proper neurogenesis. Strikingly, manipulation of malonyl-CoA, the metabolite that regulates levels of FAO, is sufficient to induce exit from quiescence and to enhance NSPC proliferation. Thus, the data presented here identify a shift in FAO metabolism that governs NSPC behavior and suggest an instructive role for fatty acid metabolism in regulating NSPC activity.
Keywords: neurogenesis, neural stem cell, hippocampus, beta-oxidation, metabolism, proliferation, quiescence
Graphical Abstract
Highlights
-
•
A metabolic shift defines NSPC quiescence versus proliferation
-
•
Quiescent NSPCs require high levels of FAO
-
•
Changing levels of a single metabolite is sufficient to induce NSPC proliferation
Controlled balance between proliferation and quiescence of neural stem/progenitor cells (NSPCs) is required for lifelong neurogenesis. Knobloch et al. identify a metabolic shift in fatty acid oxidation (FAO) that governs the proliferation of NSPCs. Further, their data suggest an instructive role for FAO in regulating NSPC activity. Thus, Knobloch et al. identify FAO as a key metabolic pathway to regulate NSPC activity.
Introduction
New neurons are generated throughout life in the mammalian hippocampus (Spalding et al., 2013, van Praag et al., 2002). This process, called adult neurogenesis, is critically involved in a variety of hippocampus-dependent forms of learning and memory (Clelland et al., 2009, Deng et al., 2010, Dupret et al., 2008, Gonçalves et al., 2016, Nakashiba et al., 2012, Sahay et al., 2011a, Sahay et al., 2011b). In addition, failing or altered neurogenesis has been associated with a number of neuropsychiatric diseases, such as major depression, epilepsy, and cognitive aging, suggesting adult hippocampal neurogenesis is relevant for human health and disease (Christian et al., 2014, Kempermann et al., 2008, Scharfman and Hen, 2007). Neural stem/progenitor cells (NSPCs) in the adult hippocampus reside in the subgranular zone (SGZ) of the dentate gyrus (DG), where they proliferate and generate new glutamatergic, excitatory granule cells that become integrated into pre-existing circuitries over the course of several weeks (Espósito et al., 2005, Ge et al., 2007, Lagace et al., 2007, Seri et al., 2001, Toni et al., 2008, Zhao et al., 2006). Previous reports have suggested a delicate balance between quiescent, radial glia-like NSPCs and more proliferative NSPCs controlled by key signaling pathways, such as Notch and BMP signaling, resembling molecular mechanisms identified in the developing brain (Ables et al., 2010, Ehm et al., 2010, Lugert et al., 2010, Ming and Song, 2011, Mira et al., 2010). In addition, accumulating evidence in NSPCs and other somatic stem cells, such as hematopoietic stem cells (HSCs), has suggested that cellular metabolism might govern the levels of activity of adult stem cells in vivo and during cellular reprogramming in vitro (Chorna et al., 2013, David, 2011, Folmes et al., 2011, Homem et al., 2015, Ito et al., 2012, Ito and Suda, 2014, Knobloch et al., 2013, Ryall et al., 2015). However, whether specific metabolic programs regulate the balance between NSPC quiescence and proliferation remains unknown. The brain is the organ with the highest glucose consumption rate (Mergenthaler et al., 2013), and neurons are mainly dependent on glucose and lactate for normal function. The role of lipids in brain metabolism has been much less studied, given the predominance of glucose consumption. Furthermore, the relatively small proportion of NSPCs compared to the cellular mass of the brain might have led to the overlooking of other metabolic pathways relevant for NSPCs. Indeed, we have previously identified an important role for lipid metabolism in NSPCs, showing that the build-up of lipids through de novo lipogenesis is crucial for proliferation (Knobloch et al., 2013). However, whether the metabolic counterpart, the breakdown of lipids called fatty acid oxidation (FAO), is important to regulate NSPC behavior remains poorly understood. We here characterized metabolic adaptations from a quiescent to an activated NSPC state and identified FAO as a key metabolic pathway to regulate NSPC quiescence.
Results
Quiescent NSPCs Have High Levels of FAO
To study metabolic adaptations during NSPC quiescence versus activation, we modified previously established in vitro protocols that are based on the induction of NSPC quiescence by bone morphogenic protein 4 (BMP4), leading to cellular quiescence over the course of three days (Figure 1A) (Martynoga et al., 2013, Mira et al., 2010). BMP4-induced quiescence was reversible with restored proliferation and differentiation potential after removal of quiescence cues, suggesting a reliable in vitro model of functional NSPC quiescence (Figures S1A–S1C). We first analyzed the whole proteome of proliferating compared to quiescent NSPCs and found proteins associated with FAO (the breakdown of fatty acids into acetyl-coenzyme A [CoA] in the mitochondria) to be highly enriched in quiescent NSPCs (Figures 1B and S1D–S1F; Tables S1 and S2). To test whether the high expression levels of proteins associated with FAO translate into functionally elevated levels of FAO, we used radioactive FAO measurements. A labeled fatty acid (3H-palmitic acid) was added to the medium. During oxidation of such labeled palmitic acid into eight acetyl-CoAs, measurable radioactive labeled water (3H2O) is produced, which serves as a readout of the rate of FAO. Strikingly, we found high levels of FAO in quiescent NSPCs that were substantially lower in proliferating NSPCs (Figure 1C).
Figure 1.

Quiescent NSPCs Have a High Rate of FAO
(A) Exposure of NSPCs to a BMP4-containing quiescence medium for three days leads to a massive decrease in proliferation. Shown are representative images of proliferating (prol) and quiescent (quie) NSPCs and quantification of the mitotic cell marker phospho histone 3 (pH3) (mean ± SEM).
(B) Mass spectrometric comparison of the proteome of proliferating and quiescent NSPCs reveals FAO to be enriched in quiescent NSPCs. The histogram shows the normalized abundance of proteins belonging to the GO term “Fatty acid oxidation” (gray = expression change of log2 < |1|, red = expression change of log2 ≤ −1, enriched in quiescent cells).
(C) Radioactive FAO measurements using 3H-labeled palmitic acid revealed a significant increase in the rate of FAO in quiescent NSPCs compared to proliferating NSPCs. Remarkably, this increase was reversed in quiescent NSPCs that had been re-exposed to proliferation conditions (ex quie), suggesting FAO is specifically upregulated upon quiescence entry (mean ± SD).
(D) mRNA levels of the key FAO enzyme Cpt1a are highly and reversibly upregulated in quiescent (quie) NSPCs compared to proliferating (prol) and formerly quiescent (ex quie) NSPCs (mean ± SEM).
(E) The increase in Cpt1a mRNA levels is also reflected on protein levels, as revealed by western blot analysis (mean ± SEM).
(F) mRNA levels of the previously described novel quiescence marker Spot14 are highly upregulated in quiescent (quie) NSPCs compared to proliferating (prol) NSPCs. This upregulation is reversible, as formerly quiescent (ex quie) NSPCs greatly reduce Spot14 mRNA levels. This suggests that the BMP4-induced in vitro quiescence system indeed reflects features of in vivo NSPC quiescence (mean ± SEM).
(G) The endogenous Cpt1a inhibitor malonyl-CoA is lowered in quiescent NSPCs compared to proliferating NSPCs, as measured by mass spectrometry analysis (mean ± SEM).
(H) Co-stainings against Cpt1a and a mitochondrial marker (Mitotracker) reveals the mitochondrial localization of Cpt1a in quiescent NSPCs. Shown is a representative confocal image of maximum projections of individual channels and a 3D reconstruction.
Scale bars represent 50 μm (A) and 20 μm (H). ∗∗∗p < 0.001; ∗∗p < 0.01.
See also Figure S1.
To characterize FAO in quiescent NSPCs, we analyzed the expression of carnitine palmitoyltransferase 1a (Cpt1a), a rate-limiting mitochondrial enzyme of FAO that mediates the transport of fatty acids into the mitochondria (Houten and Wanders, 2010). Corroborating the proteomics data, we found strong upregulation of Cpt1a using qRT-PCR and western blot analyses, showing a substantial increase in the expression of Cpt1a in quiescent compared to proliferating NSPCs (Figures 1D and 1E). In line with the radioactive FAO measurements, the increase in Cpt1a expression in quiescent NSPCs was reversible (Figure 1D). Cpt1a co-labeled with the mitochondrial dye Mitotracker in both quiescent and proliferating NSPCs (Figures 1H and S1H). In addition, Cpt1a was highly expressed in NSPCs compared to their neuronal progeny when directly isolated from the adult DG (3.5-fold upregulated in SOX2+ cells versus DCX+ cells), as described previously (Bracko et al., 2012, Shin et al., 2015). Collectively, these data indicate that compared to proliferating NSPCs that are highly lipogenic (Knobloch et al., 2013), quiescent NSPCs strongly express Cpt1a and show high levels of functional FAO.
Next, we aimed to understand the molecular mechanism underlying high levels of FAO in quiescent NSPCs. We have previously shown that Spot14 is selectively expressed in quiescent NSPCs in vivo (Knobloch et al., 2013, Knobloch et al., 2014). When we induced quiescence in vitro, we found a >30-fold upregulation of Spot14 mRNA using qRT-PCR (Figure 1F). Given that Spot14 negatively regulates malonyl-CoA levels (Colbert et al., 2010, Knobloch et al., 2013), we expected that high levels of Spot14 in quiescent NSPCs would lead to low levels of malonyl-CoA. Indeed, quiescent NSPCs showed a substantial decrease in malonyl-CoA, as measured with mass spectrometry (Figure 1G). The levels of acetyl-CoA, which are not affected by Spot14, were comparable between proliferating and quiescent NSPCs (Figure S1G). Because malonyl-CoA is an endogenous inhibitor of Cpt1a, its levels determine the rate of FAO (Folmes et al., 2013, Houten and Wanders, 2010). Thus, high levels of Spot14 accompanied by low levels of malonyl-CoA in quiescent NSPCs promote high FAO (see also summary scheme Figure 5E).
Figure 5.

Malonyl-CoA Levels Regulate NSPC Proliferation
(A–E) Manipulating the levels of the physiological, intrinsic Cpt1a inhibitor malonyl-CoA is sufficient to prevent NSPC quiescence and to induce proliferation under quiescence conditions.
(A) Schematic outline of the experimental setup.
(B) Addition of malonyl-CoA (100 or 200 μM) at the beginning of quiescence induction is sufficient to prevent quiescence entry in a dose-dependent manner, as revealed with the cell cycle marker Ki67 and the mitotic marker phospho Histone 3 (pH3). Shown are representative images of indicated doses and the quantification of cycling and proliferating cells after 3 days of quiescence induction (mean ± SEM).
(C) Schematic outline of the experimental setup.
(D) Replating NSPCs after fully established quiescence in quiescence medium containing malonyl-CoA (100 or 200 μM) is sufficient to trigger cell cycle re-entry. This suggests that malonyl-CoA levels can overrule the present quiescence cues. Shown are representative images of indicated doses and the quantification of cycling and proliferating cells after three days of quiescence induction (mean ± SEM).
(E) Summary scheme of the described findings.
Scale bars represent 50 μm. ∗∗∗p < 0.001; ∗∗p < 0.01, +p = 0.06.
See also Figure S5.
High Levels of FAO Are Required to Sustain Cellular Quiescence
To test for the functional relevance of FAO activity, we blocked FAO in quiescent NSPCs using the irreversible Cpt1 inhibitor Etomoxir. NSPCs were induced to quiescence and then exposed to various doses of Etomoxir. Cell survival was assessed using time-lapse imaging. Strikingly, such complete FAO inhibition in quiescent NSPCs led to massive cell death in a dose-dependent manner (Figure 2A). This finding indicates that FAO is critically involved in maintaining adult hippocampal NSPCs in a quiescent state and that absence of this pathway is detrimental. Complete blockage of FAO using Etomoxir also affected NSPCs kept under proliferating conditions by reducing their proliferation, as assessed by time-lapse imaging, flow cytometry analysis, and 5-ethynyl-2’-deoxyuridine (EdU)-pulse labeling (Figures S2A–S2C). These findings are in line with a recent report showing that proliferating NSPCs in the SVZ can oxidize fatty acids (Stoll et al., 2015). Given the detectable levels of FAO in proliferating NSPCs, (although much lower than in quiescent NSPCs; Figure 1C) and the effect of blocking FAO on proliferation, these data suggest that FAO is still to a certain extend relevant during proliferation. However, in contrast to quiescent NSPCs, abolishing FAO in proliferating NSPCs only mildly affected their cell survival (Figures S2A–S2C).
Figure 2.

High Levels of FAO Are Required to Sustain Cellular Quiescence
(A) Time-lapse analysis of quiescent NSPCs exposed to various doses of the irreversible Cpt1 inhibitor Etomoxir (50, 100, and 200 μM). Shown are a schematic outline of the experimental setup, the quantification of the area covered by quiescent NSPCs over time, and representative images. Complete block of FAO by Etomoxir during quiescence leads to a dramatic and dose-dependent decrease of area covered over time, caused by cell death upon FAO inhibition (mean ± SEM).
(B–E) Quiescent NSPCs use FAO for energy purposes and may use it as an alternative carbon source.
(B) Radioactive FAO measurements using 14C-labeled palmitic acid revealed a significant increase in 14CO2 in quiescent NSPCs compared to proliferating NSPCs, suggesting that at least part of the fatty acids are fully oxidized and might be used for energy purposes (mean ± SD).
(C) Energy charge measurements in quiescent NSPCs show that FAO indeed contributes to the amount of ATP generated, as treatment with various doses of Etomoxir (50, 100, and 200 μM) reduced the energy charge significantly (mean ± SEM).
(D) Scheme outlining the path of 13C-labeled palmitic acid upon FAO. The oxidation of fatty acids results in acetyl-CoA, which can be fed into the TCA. The metabolites measured with mass spectrometry in a 13C-incorporation assay are shown in bold.
(E) Quiescent NSPCs show an increase in 13C-incorporation in TCA intermediates and amino acids derived from TCA intermediates compared to proliferating NSPCs (mean ± SD). These 13C-incorporation assay results suggest that fatty acids might also serve as an alternative carbon source in quiescent NSPCs.
(F) High levels of FAO in quiescent NSPCs are at least partially regulated through the transcription factor PPARα. Treatment for 48 hr with 100 μM of the PPARα agonist WY14643 in proliferating NSPCs significantly reduced proliferation compared to control NSPCs, but proliferation was still far higher than in quiescent NSPCs, as assessed by EdU pulsing. Shown are representative images and the corresponding quantification of EdU-positive cells (mean ± SEM).
(G) WY14643 treatment led to an upregulation of PPARα and its target FAO genes in proliferating NSPCs compared to control NSPCs, however, to a far lesser extent than in quiescent NSPCs. Shown are the mRNA expression levels (mean fold change ± range) of PPARα, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), Cpt1a, medium-chain acyl-CoA dehydrogenase (Acadm), long-chain acyl-CoA dehydrogenase (Acadl), and very long-chain acyl-CoA dehydrogenase (Acadvl).
Scale bar represents 50 μm (A) and 20 μm (F). ∗∗∗p < 0.001; ∗∗p < 0.01.
Given the importance of FAO for NSPC behavior in vitro, we next investigated why quiescent NSPCs require FAO. We used two complementary approaches to analyze the fate of oxidized fatty acids in NSPCs. First, we used radioactively labeled 14C-palmitic acid to determine the complete oxidation of fatty acids, allowing for energy production. In contrast to the 3H-palmitic acid labeling, where radioactive labeled 3H2O is produced during the entire oxidation cycles into acetyl-CoAs, radioactively labeled 14C-palmitic acid yields measureable 14CO2 only if the resulting acetyl-CoAs are further oxidized in the tricarboxylic acid (TCA) cycle. We found significantly higher levels of 14CO2 in quiescent NSPCs compared to proliferating NSPCs, suggesting that quiescent NSPCs might use fatty acids as a fuel source (Figure 2B). To confirm that quiescent NSPCs use FAO for energy production, we determined their energy charge, as a readout for the amount of energy available in the form of adenosine triphosphate (ATP). We found that the energy charge significantly dropped following Etomoxir treatment, indicating that FAO is indeed contributing to the amount of ATP generated in quiescent NSPCs (Figure 2C), with the massive drop using 200 μm Etomoxir probably indicating reduced cell viability.
Next, we used 13C-labeled palmitic acid to trace the incorporation of labeled carbon atoms in quiescent versus proliferative NSPCs (Figure 2D). During complete oxidation of such labeled palmitic acid, labeled carbon atoms are cleaved off and transferred to TCA intermediates and amino acids derived from TCA intermediates. The amount of incorporation of labeled carbons can be measured using mass spectrometry. We found highly significant increases in the incorporation of 13C into TCA intermediates as well as into amino acids derived from TCA intermediates in quiescent NSPCs (Figure 2E), suggesting that the fatty acid-derived carbon atoms might be further used as an alternative carbon source.
To address whether the upregulation of FAO in quiescent NSPCs is transcriptionally regulated, we measured the expression levels of the Peroxisome proliferator-activated receptor alpha (PPARα), a known transcriptional regulator of genes involved in FAO by qRT-PCR (Leone et al., 1999). Indeed, expression of PPARα and its target genes was highly upregulated in quiescent NSPCs compared to proliferating NSPCs (Figures 2G and S2D), suggesting that upregulation of FAO upon quiescence is at least partially regulated on a transcriptional level. Given this transcriptional component of FAO regulation through PPARα, we next assessed whether FAO gene expression could be modulated in proliferating NSPCs to reach similar levels as in quiescent NSPCs and whether such an upregulation would render proliferating NSPCs more quiescent. Treatment with the PPARα agonist WY14643 indeed led to an upregulation of FAO genes in proliferating NSPCs compared to control NSPCs (Figure 2G and S2D), however, to a far lesser extent than in quiescent NSPCs. Similarly, although modulation of FAO genes by WY14643 significantly reduced proliferation compared to control NSPCs, proliferation was still far higher than in quiescent NSPCs (Figure 2F).
Cpt1a Is Expressed in Hippocampal NSPCs In Vivo
After identifying a role for FAO in NSPC quiescence in vitro, we next analyzed the expression of Cpt1a within the adult hippocampal neurogenic niche using a Cpt1a reporter mouse expressing GFP from the regulatory elements of the Cpt1a genomic locus (Genesat; hereafter called Cpt1a-GFP) (Gong et al., 2003). Besides GFP-positive classical astrocytes throughout all hippocampal subfields (Figures 3A and S3B), we found GFP expression to be highly enriched in the subgranular zone (SGZ) of the DG (Figures 3A and S3A). GFP-positive cells expressed Cpt1a, as confirmed with staining against endogenous Cpt1a (Figure S3C). Next, we phenotyped GFP-labeled cells in Cpt1a-GFP mice and found that GFP expression preferentially labeled non-mitotic, Ki67-negative hippocampal NSPCs and only a small subset of cycling NSPCs (Figure 3B). Virtually all Cpt1a-GFP-positive cells co-labeled with the radial and non-radial NSPC marker SOX2 (Figure 3C). Furthermore, the majority of radial processes positive for the radial NSPC marker Nestin were also GFP positive (83.7% ± 6.2%; Figure 3C). Cpt1a was virtually absent in doublecortin (DCX)-expressing neuronal progeny of hippocampal NSPCs (Figure 3D). Furthermore, Spot14 was co-expressed in Cpt1a-GFP-positive cells (Figures S3D and S3E), suggesting that FAO is indeed high in NSPCs and becomes downregulated with neuronal differentiation.
Figure 3.

Cpt1a Is Expressed in Adult NSPCs In Vivo
(A) Immunohistological analysis of a reporter mouse expressing GFP under the Cpt1a promoter reveals high GFP expression in the SGZ of the DG, where NSPCs reside. Shown is a representative image of a brain section from a 2-month-old animal.
(B) Co-stainings for Cpt1a-GFP and the proliferation marker Ki67 show that the majority of proliferating NSPCs do not express GFP, supporting the findings that FAO is specifically upregulated in quiescent NSPCs. Shown is a representative confocal image (maximum projection) from a 2-month-old Cpt1a-GFP reporter mouse. Arrows indicate Ki67+/GFP− cells; the arrowhead points to a Ki67+/GFP+ cell. The bar graph shows the percentage of GFP+ and GFP− NSPCs out of all Ki67+ cells in the SGZ of the DG (mean ± SEM).
(C) Cpt1a-GFP-positive NSPCs are almost all positive for the NSPC marker SOX2, and GFP+ processes co-stain with the NSPC marker Nestin. Shown is a representative confocal image (maximum projection) from a 2-month-old Cpt1a-GFP reporter mouse. Dotted lines show the outline of the granular zone of the DG, the boxed area is enlarged in the right panel. The bar graph shows the percentage of GFP+ and GFP− NSPCs out of SOX2+ cells (mean ± SEM).
(D) Cpt1a-GFP-positive NSPCs are almost all negative for the immature neuronal marker Doublecortin (DCX), suggesting that FAO is downregulated upon neuronal lineage commitment. Shown is a representative confocal image (maximum projection) from a 2-month-old Cpt1a-GFP reporter mouse. Dotted lines show the outline of the granular zone of the DG, the boxed area is enlarged in the right panel. The bar graph shows the percentage of GFP+ and GFP− NSPCs out of DCX+ cells (mean ± SEM).
Scale bars represent 200 μm (A), 50 μm (C and D, left), and 20 μm (B, C, and D, right).
See also Figure S3.
Cpt1a Is Required for Proper Neurogenesis In Vivo
To directly test for a role of Cpt1a-dependent FAO for NSPCs in vivo, we conditionally deleted Cpt1a specifically in adult quiescent NSPCs by crossing Cpt1a flox/flox mice (Cpt1a-conditional knockout [cKO]) (Schoors et al., 2015) with mice harboring tamoxifen (TAM)-inducible Spot14-driven Cre recombinase (S14iCre) (Knobloch et al., 2013) and yellow fluorescent protein (YFP) reporter alleles in the ROSA locus (R26YFP), inducing recombination at seven weeks of age. The low recombination efficiency of the S14iCre mouse line resulted in sparse labeling of cells, allowing the identification and classification of labeled progeny into potential clones according to their spatial clustering, similarly to previously published analyses referred to as clonal analysis (Bonaguidi et al., 2011). However, it needs to be noted that the classification of cellular clusters into clones according to spatial distance only assumes common lineage and does not ultimately prove the presence of cells of clonal origin as this would require an additional level of genetic lineage tracing (e.g., through genetic bookmarking as previously used in NSPCs; Fuentealba et al., 2015).
We first analyzed cell cluster size distribution 8 days after the first TAM administration using the sparse labeling method and found that size and composition of cellular clusters was not significantly altered between control and Cpt1a-cKO mice, although no larger clones were found in Cpt1a-cKO (Figures S4A and S4D). At this time point, slightly lower numbers of clusters were found in Cpt1a-cKO mice compared to control mice (17 clones in 5 mice versus 24 clones in 4 mice), but the number of YFP-positive cells per mouse was not significantly lower in the Cpt1a-cKO mice compared to control mice (7.2 ± 4.5 versus 11 ± 3.7; p > 0.1). Detailed cluster analysis did not reveal significant changes (Figures S4B, S4C, and S4F), although a small decrease in clones containing only R and a small, non-significant increase in active clones in Cpt1a-cKO might be suggestive of an initial activation upon Cpt1a-dependent knockout of FAO.
Next, we analyzed cell cluster composition and number 25 days after the first TAM administration. These analyses revealed a dramatic decrease in the number of YFP-positive cells per clone in Cpt1a-cKO mice compared to control littermates (Figures 4A, 4B, and S4E). The majority of clones in the Cpt1a-cKO mice contained only one to two cells, whereas more than half of the clones in control mice were composed of three to 12 or even more cells (Figures 4A, 4B, and S4E), suggesting a massive impairment of NSPC expansion upon FAO knockout. Furthermore, fewer clones were found in Cpt1a-cKO mice compared to control mice (34 clones in seven Cpt1a-cKO mice versus 62 clones in three control mice), and the number of YFP-positive cells per mouse was significantly lower in the Cpt1a-cKO mice compared to control mice (Cpt1a-cKO: 12.8 ± 5.8 versus 111 ± 65.6 in control mice; p < 0.05), implying that FAO knockout leads to cell death. This was also reflected by a decrease in the number of radial-glia like NSPCs in Cpt1a-cKO compared to controls (Cpt1a-cKO: 2.6 ± 1.5 versus 17 ± 10.4 in control mice; p = 0.06).
Figure 4.

Cpt1a Is Required for Proper Neurogenesis In Vivo
(A) Conditional Cpt1a knockout in adult quiescent NSPCs, by crossing Cpt1a flox/flox mice with Spot14-driven CreERT2 recombinase mice and ROSA YFP reporter mice, leads to disturbed neurogenesis. Shown are representative confocal images (maximum projection) and a composite image showing the reconstruction of individual clones from Cpt1a-cKO wild-type (WT)/WT mice (left) and Cpt1a-cKO flox/flox mice (right) 25 days after first TAM induction. The boxed area is enlarged in the reconstruction panel.
(B) Clonal analysis reveals marked reduction in clone size in the Cpt1a-cKO flox/flox mice compared to Cpt1a-cKO WT/WT littermates.
(C) Detailed clone composition analysis shows a marked increase in clones containing only one radial glia-like cell (R), a great reduction in clones containing an R and neural progeny (N), absence of clones containing an R, N, and astrocytes (A) and a slight increase in clones without an R in Cpt1a-cKO flox/flox compared to Cpt1a-cKO WT/WT.
(D) Active clones containing a radial glia-like cell (R) and any kind of progeny (X) are strongly reduced in Cpt1a-cKO flox/flox, suggesting that FAO is required to allow proper neurogenesis.
(E) Representative confocal images of different types of recombined, YFP-positive cells from Cpt1a cKO WT/WT mice 25 days after the first TAM injection. Radial glia-like cells (R) have a triangular-shaped soma (arrowhead) in the subgranular zone (SGZ) with a radial, arborized process into GCL (arrow) and are SOX2 positive. Neural progenitors (N) are non-radial cells with their soma in the SGZ (arrowhead), with one or more horizontal processes (arrows), sometimes still SOX2, some already DCX positive. Immature neurons (N) have a round soma (arrowheads) in the granular cell layer (GCL) with a long vertical process (arrow), and are usually DCX positive. Astrocytes (A) are multi-process-containing, star-shaped or bushy cells in the SGZ/Hilus or GCL.
Scale bars represent 50 μm (A) and 40 μm (E). nd, not detected. ∗∗∗p < 0.001; ∗∗p < 0.01, +p = 0.086.
See also Figure S4.
A detailed analysis of the clone compositions revealed an increase in clones containing only one radial glia-like cell (R), a reduction in clones containing an R and neural progeny (N), an absence of clones containing a R, N and astrocytes (A), and an increase in clones without a R in Cpt1a-cKO mice compared to controls. Further, there was a strong reduction in the number of active clones (containing a radial glia-like cell that generated progeny) (Figures 4C–4D). Thus, genetic inhibition of FAO in adult hippocampal NSPCs results in fewer recombined cells indicative of cell death and smaller as well as less active clones indicative of reduced cell proliferation, thus corroborating the in vitro results upon FAO inhibition. However, the detailed impact of FAO on the specific cellular stage (quiescence, proliferation, cell death, and survival) cannot be answered unambiguously with these snapshot data. Given that FAO is used by both proliferating and quiescent NSPCs in vitro (although the latter have much higher FAO levels and depend more on it), it is indeed likely that the strong decrease in progeny generation upon FAO ablation is due to an influence of FAO on several cellular stages.
Small hairpin RNA (shRNA)-mediated knockdown of Cpt1a using in utero electroporation in the developing mouse cortex at mid-neurogenesis (embryonic day 13 [E13]) corroborated the importance of FAO for proper neurogenesis in vivo that has been also previously shown by Xie et al. (2016) (Figures S4I and S4J). Although NSPCs are more proliferative at this stage compared to adulthood (Farkas and Huttner, 2008), staining against endogenous Cpt1a confirmed that Cpt1a is also highly enriched in NSPCs lining the ventricle during development (Figure S4H). 24 hours after electroporation (corresponding to E14) (Figure S4J), proliferation at the apical surface was significantly reduced upon Cpt1a knockdown, and general disorganization of mitoses in the ventricular zone was observed (Figures S4K and S4L). Furthermore, we found a massive increase in cell death upon Cpt1a knockdown, as measured by the apoptotic marker cleaved caspase-3 (Figure S4M), corroborating the in vitro results.
Malonyl-CoA Levels Regulate NSPC Proliferation
Given the important role of FAO for NSPCs in vitro and in vivo, determined by fully blocking this pathway with genetic and pharmacological means, we next aimed to test the significance of this regulatory pathway for NSPC activity in a more physiological manner. Metabolic pathways are finely tunable through substrate availability and intrinsic levels of metabolites have been shown to determine pathway activity. Such shifts in pathway activity are likely to reflect better the actual physiological situation than switching off a pathway by genetic deletion of its key players. For FAO, the metabolite malonyl-CoA functions as the endogenous inhibitor of Cpt1a and serves at the same time as a substrate for de novo lipogenesis, thus it has also been termed a “rheostat” regulating these two lipid metabolic pathways (Foster, 2012, McGarry and Brown, 1997). Thus, we reasoned that elevating levels of malonyl-CoA decreases Cpt1a-dependent FAO in a physiological way rather than completely blocking it and provides sufficient substrate to fuel FASN-dependent de novo lipogenesis that is required for NSPC proliferation (Knobloch et al., 2013). However, it is not known if malonyl-CoA can be taken up by cells when provided extracellularly. Thus, we first tested if exogenously provided malonyl-CoA can be metabolized and detected intracellularly. We isolated lipids of proliferating, highly lipogenic NSPCs (Knobloch et al., 2013) that were incubated with radioactively labeled malonyl-CoA (14C-malonyl-CoA) for 48 hr (Figure S5A). We detected incorporation of 14C-malonyl-CoA in polar lipids and triacylglycerides (TAGs, neutral lipids) (Figure S5B), clearly showing that exogenous malonyl-CoA can indeed be utilized by NSPCs. Next, we tested if exogenously applied malonyl-CoA could prevent BMP4-mediated induction of NSPC quiescence. Adult NSPCs were exposed to BMP4-containing medium in the presence of different concentrations of malonyl-CoA (Figure 5A). Indeed, elevated levels of malonyl-CoA dose-dependently prevented the induction of quiescence, as measured using the cell cycle markers Ki67 and phosphorylated histone H3 (pH3) (Figure 5B). These data show that manipulating FAO through malonyl-CoA levels is sufficient to override BMP4-induced quiescence and keeps NSPCs in a proliferating state.
Next, we induced BMP4-mediated quiescence over 3 days, after which proliferation is almost completely inhibited, followed by replating the cells in quiescence medium together with malonyl-CoA (Figure 5C). Strikingly, we found that malonyl-CoA triggered NSPCs to enter the cell cycle in a dose-dependent manner, despite a fully established quiescence state and the continuous presence of quiescence cues (Figure 5D). This increased proliferation is most likely mediated by an increase in FASN-dependent de novo lipogenesis, as the addition of the FASN inhibitor Orlistat (Kridel et al., 2004) into the BMP4-containing quiescence medium significantly reduced remaining proliferation and abolished the pro-proliferative effect of malonyl-CoA when applied together with malonyl-CoA (Figures S5C and S5D). These data reveal that differential metabolic states are not a mere consequence of a given stem cell state but that manipulating levels of FAO through malonyl-CoA is instructive to regulate the behavior of adult NSPCs (Figure 5E).
Discussion
Due to its significance for brain function, understanding the mechanisms regulating adult hippocampal neurogenesis is important to advance the current knowledge of brain plasticity in health and disease (Jessberger and Gage, 2014). In addition to candidate-based approaches testing the role of well-studied signaling pathways that had been implicated in regulating embryonic neurogenesis such as Notch- and WNT-signaling, a series of recent studies used unbiased approaches to characterize gene expression profiles during distinct developmental stages in the course of adult neurogenesis (Bracko et al., 2012, Llorens-Bobadilla et al., 2015, Shin et al., 2015). These studies all pointed toward an important role of cellular metabolism and, more specifically, lipid metabolism in the regulation of the neurogenic process in the adult brain. Supporting this, we have previously shown that NSPCs in the adult brain depend on FASN-dependent de novo lipogenesis for proper proliferation and have identified Spot14, which is selectively expressed in largely quiescent NSPCs, as the brake on de novo lipogenesis (Knobloch et al., 2013, Knobloch et al., 2014). However, it remained unclear if Spot14 merely suppresses de novo lipogenesis to keep NSPCs in a quiescence state or if NSPC quiescence requires a specialized metabolic state.
We here used several complementary approaches in vitro and in vivo to show that quiescent NSPCs rely on FAO and that blocking this pathway pharmacologically or genetically leads to massive disturbance of NSPC behavior. Using in vitro approaches, we find clear evidence that inhibition of FAO leads to enhanced cell death of quiescent cells but also reduced proliferation. The conditional deletion of the rate-limiting enzyme Cpt1a in vivo also results in fewer and smaller cellular clusters, suggesting that the role for FAO in vivo resembles the described function of cultured NSPCs in vitro. The conditional deletion of Cpt1a in quiescent Spot14-positive NSPCs using the Spot14-CreERT2 mouse line allowed us to study the role of FAO on a specific NSPC population, reducing possible side effects of altering FAO in astrocytes, as would be the case by using other Cre-lines, such as GLAST-CreERT2 or Nestin-CreERT2 (Lagace et al., 2007, Mori et al., 2006). However, the low recombination efficiency of the Spot14-CreERT2-line results in sparse labeling, enabling clonal analysis, but making assessments of proliferation or cell death with classical markers almost impossible. Thus, the detailed cellular effects (i.e., on exactly which stage FAO deletion exerts its main phenotype) remain to be elucidated and will require novel approaches to study neurogenesis within the endogenous niche such as chronic imaging.
Although glucose and lactate remain the main fuel sources for the brain (Lundgaard et al., 2015, Wyss et al., 2011), our data suggest that NSPCs might not only rely on these fuels. We provide mechanistic evidence that oxidized fatty acids are required for energy production and might serve as an alternative carbon source. Thus, our data reveal a metabolic shift in adult hippocampal NSPCs that defines the change from a quiescent to a proliferative state. Interestingly, many metabolic pathways are not only transcriptionally regulated but also highly dependent on substrate availability and inhibitory feedback loops of metabolic intermediates, providing a regulatory system than can be finely tuned rather than an “on-off” system (Lunt and Vander Heiden, 2011, Metallo and Vander Heiden, 2013). This is also the case for the breakdown and buildup of lipids (Houten et al., 2016, Menendez and Lupu, 2007). A central role in regulating the shift from FAO to lipogenesis appears to be mediated by the levels of malonyl-CoA. This physiological metabolite has been suggested to be a rheostat of stem cell fate (Folmes et al., 2013), being both an inhibitor of Cpt1a and a substrate for de novo lipogenesis (McGarry et al., 1983, Menendez and Lupu, 2007). Strikingly, we have previously shown that Spot14, which is highly expressed in quiescent NSPCs, indirectly reduces the levels of available malonyl-CoA and reduces lipid synthesis (Knobloch et al., 2013). BMP4-induced quiescent NSPCs recapitulate these features and show highly increased Spot14 levels and reduced malonyl-CoA levels, providing optimal conditions for FAO. Thus, our data support the hypothesis that malonyl-CoA acts as a rheostat with Spot14 taking a key role in this regulatory process (summarized in the graphical abstract). Our data corroborate previous findings by Stoll and colleagues who characterized the metabolic state of NSPCs in the subventricular zone (SVZ) and found that NSPCs in the adult SVZ depend on FAO for their proliferation as shown by infusing Etomoxir (thus pharmacologically inhibiting FAO) into the adult brain (Stoll et al., 2015). Interestingly, recent studies described a role for FAO in other stem cell systems: hematopoietic stem cells (HSCs) seem to require FAO to maintain their stem cell potential through a PPAR-delta mediated pathway and inhibition of FAO leads to HSC exhaustion (Ito et al., 2012). Similarly, quiescent muscle satellite cells are relying on FAO and pyruvate oxidation and undergo a metabolic shift toward glycolysis once they become activated (Ryall et al., 2015). Furthermore, a recent study has addressed the consequences of systemic inborn errors of FAO on brain development and showed that defects in FAO lead to enhanced progenitor generation and subsequently to a reduced embryonic NSPC pool (Xie et al., 2016). These studies emphasize the importance of FAO for proper stem cell behavior and suggest shared regulatory pathways involving FAO between different somatic stem cell types. Whether malonyl-CoA levels and Spot14 are the regulating entities in other somatic stem cells remains to be elucidated.
Excitingly, our data show that metabolic changes in the course of NSPC activation are not mere bystanders of other signaling pathways or transcriptional programs, but are effective to change NSPC behavior in vitro. Manipulating FAO through changing the levels of a single metabolite, its endogenous inhibitor malonyl-CoA, is sufficient to instruct quiescent NSPCs to enter cell cycle and to proliferate in vitro. Due to the short half-life of malonyl-CoA at 37°C (72 hr), in vivo experiments are currently not feasible. In contrast to the detrimental effects seen when completely switching off FAO genetically or pharmacologically, alteration of endogenous metabolite levels might better reflect the physiological relevance of metabolic shifts. Given the instructive role of metabolism on NSPC quiescence and proliferation behavior, manipulations of the metabolic state may thus represent a novel approach to achieve enhanced neurogenesis in the aging brain or in disease states where NSPC activity is reduced.
Experimental Procedures
Further details and an outline of the resources used in this work can be found in the Supplemental Experimental Procedures.
Mouse strains used were Cpt1a-EGFP reporter mice (STOCK Tg(Cpt1a-EGFP)IP41Gsat/Mmucd, MMRRC), Cpt1a cKO mice (Schoors et al., 2015), Spot14CreERT2 mice (S14iCre) Knobloch et al., 2013), ROSA26 YFP reporter mice (R26YFP), and C57/Bl6 mice (Janvier). All animal experiments were performed according to Swiss regulatory standards and approved by the Veterinary office of the Canton of Zurich. Cre-mediated recombination was induced by intraperitoneal injections of tamoxifen (180 mg/kg) at the age of 6 to 7 weeks. Lentiviral constructs were designed and viruses produced as previously described (Knobloch et al., 2013). Adult mouse DG NSPCs were cultured in DMEM with Ham’s F12, supplemented with N2 supplement plus epidermal growth factor (EGF), fibroblast growth factor, and heparin (Knobloch et al., 2013, Ray and Gage, 2006). Quiescence was induced over three days replacing EGF with BMP4, as previously described (Martynoga et al., 2013, Mira et al., 2010). For Cpt1a inhibition, Etomoxir or malonyl-CoA was added to the medium as outlined in the figures. Proteomic analysis was done according to previously established protocols (Wiśniewski et al., 2009). Radioactive FAO measurements were done using labeled 3H-palmitic acid and 14C-palmitic acid, and the amount of tritiated water or 14CO2 generated was assessed (Djouadi et al., 2003, Huynh et al., 2014). Quiescent and proliferating NSPCs were collected for RNA and protein isolation, and established protocols were used for subsequent processing and analysis (Knobloch et al., 2013). In utero electroporation of Cpt1a and control shRNA plasmid DNA into mouse embryos (embryonic day 13 [E13]) was carried out as described previously (Asami et al., 2011). For immunohistochemical analyses, brain tissues were sectioned, stained, and imaged using previously published methods (Bonaguidi et al., 2011, Knobloch et al., 2013). Mass spectrometry measurements to assess the amount of malonyl-CoA and to determine the energy charge were done with cellular extracts using liquid chromatography-tandem mass spectrometry LC-MS/MS (Knobloch et al., 2013, Schoors et al., 2015). To analyze C13 incorporation, quiescent and proliferating NSPCs were incubated 24 hr prior to collection/extraction with 13C-palmitic acid and analyzed with gas chromatography-mass spectrometry (GC-MS) as previously described (Schoors et al., 2015). Statistical analysis was performed using unpaired t tests, paired t tests, and one-way-ANOVA or two-way-ANOVA, followed by Holm-Sidak’s multiple comparisons tests. Significance levels are set at p < 0.05.
Author Contributions
M.K. co-developed the concept, performed the experiments, analyzed the data, and co-wrote the paper. G.-A.P. and M.K. performed the experiments in the embryonic brain. B.G. and M.K. performed the metabolite tracing experiments. T.W. performed the proteomics experiments. W.J.K. and M.K. performed the radioactive tracing experiments and the gene expression analyses. M.K. performed the in vivo experiments, with help from G.-A.P and D.L.M. D.L.M. contributed to in vivo Cpt1a expression analysis. M.H. and N.Z. performed malonyl-CoA measurements. P.C. contributed reagents and transgenic mice and provided critical conceptual input. All authors revised the manuscript. S.J. developed the concept and wrote the paper. The authors declare that patent applications concerning this work are pending.
Acknowledgments
We thank D.C. Lie for comments on the manuscript and the ZMB (UZH) and BIOP (EPFL) imaging facilities for help with imaging. We also thank J. Grossmann, P. Gehrig, and B. Roschitzki of the FGCZ (UZH and ETHZ) and the Zurich Neuroscience Center (ZNZ) of UZH and ETHZ. This study was supported by the Swiss National Science Foundation (BSCGI0_157859; 31003A_156943), the EMBO Young Investigator Program, the Théodore Ott Foundation, the Novartis Foundation, and the European Research Council (to S.J.). M.K. was supported by the Janggen-Pöhn Foundation, G.A.P. by an EMBO long-term fellowship, D.L.M. by a Human Frontier Science Program long-term fellowship, and T.W. by the Boehringer Ingelheim Fonds.
Published: August 29, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.08.029.
Contributor Information
Marlen Knobloch, Email: marlen.knobloch@unil.ch.
Sebastian Jessberger, Email: jessberger@hifo.uzh.ch.
Supplemental Information
References
- Ables J.L., Decarolis N.A., Johnson M.A., Rivera P.D., Gao Z., Cooper D.C., Radtke F., Hsieh J., Eisch A.J. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J. Neurosci. 2010;30:10484–10492. doi: 10.1523/JNEUROSCI.4721-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asami M., Pilz G.A., Ninkovic J., Godinho L., Schroeder T., Huttner W.B., Götz M. The role of Pax6 in regulating the orientation and mode of cell division of progenitors in the mouse cerebral cortex. Development. 2011;138:5067–5078. doi: 10.1242/dev.074591. [DOI] [PubMed] [Google Scholar]
- Bonaguidi M.A., Wheeler M.A., Shapiro J.S., Stadel R.P., Sun G.J., Ming G.L., Song H. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell. 2011;145:1142–1155. doi: 10.1016/j.cell.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracko O., Singer T., Aigner S., Knobloch M., Winner B., Ray J., Clemenson G.D., Jr., Suh H., Couillard-Despres S., Aigner L. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J. Neurosci. 2012;32:3376–3387. doi: 10.1523/JNEUROSCI.4248-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorna N.E., Santos-Soto I.J., Carballeira N.M., Morales J.L., de la Nuez J., Cátala-Valentin A., Chornyy A.P., Vázquez-Montes A., De Ortiz S.P. Fatty acid synthase as a factor required for exercise-induced cognitive enhancement and dentate gyrus cellular proliferation. PLoS ONE. 2013;8:e77845. doi: 10.1371/journal.pone.0077845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian K.M., Song H., Ming G.L. Functions and dysfunctions of adult hippocampal neurogenesis. Annu. Rev. Neurosci. 2014;37:243–262. doi: 10.1146/annurev-neuro-071013-014134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clelland C.D., Choi M., Romberg C., Clemenson G.D., Jr., Fragniere A., Tyers P., Jessberger S., Saksida L.M., Barker R.A., Gage F.H. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–213. doi: 10.1126/science.1173215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert C.L., Kim C.W., Moon Y.A., Henry L., Palnitkar M., McKean W.B., Fitzgerald K., Deisenhofer J., Horton J.D., Kwon H.J. Crystal structure of Spot 14, a modulator of fatty acid synthesis. Proc. Natl. Acad. Sci. USA. 2010;107:18820–18825. doi: 10.1073/pnas.1012736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David R. Stem cells: LKB1 maintains the balance. Nat. Rev. Mol. Cell Biol. 2011;12:4. doi: 10.1038/nrm3032. [DOI] [PubMed] [Google Scholar]
- Deng W., Aimone J.B., Gage F.H. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouadi F., Bonnefont J.P., Munnich A., Bastin J. Characterization of fatty acid oxidation in human muscle mitochondria and myoblasts. Mol. Genet. Metab. 2003;78:112–118. doi: 10.1016/s1096-7192(03)00017-9. [DOI] [PubMed] [Google Scholar]
- Dupret D., Revest J.M., Koehl M., Ichas F., De Giorgi F., Costet P., Abrous D.N., Piazza P.V. Spatial relational memory requires hippocampal adult neurogenesis. PLoS ONE. 2008;3:e1959. doi: 10.1371/journal.pone.0001959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehm O., Göritz C., Covic M., Schäffner I., Schwarz T.J., Karaca E., Kempkes B., Kremmer E., Pfrieger F.W., Espinosa L. RBPJkappa-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J. Neurosci. 2010;30:13794–13807. doi: 10.1523/JNEUROSCI.1567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espósito M.S., Piatti V.C., Laplagne D.A., Morgenstern N.A., Ferrari C.C., Pitossi F.J., Schinder A.F. Neuronal differentiation in the adult hippocampus recapitulates embryonic development. J. Neurosci. 2005;25:10074–10086. doi: 10.1523/JNEUROSCI.3114-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas L.M., Huttner W.B. The cell biology of neural stem and progenitor cells and its significance for their proliferation versus differentiation during mammalian brain development. Curr. Opin. Cell Biol. 2008;20:707–715. doi: 10.1016/j.ceb.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Folmes C.D., Nelson T.J., Martinez-Fernandez A., Arrell D.K., Lindor J.Z., Dzeja P.P., Ikeda Y., Perez-Terzic C., Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–271. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes C.D., Park S., Terzic A. Lipid metabolism greases the stem cell engine. Cell Metab. 2013;17:153–155. doi: 10.1016/j.cmet.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Foster D.W. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J. Clin. Invest. 2012;122:1958–1959. doi: 10.1172/JCI63967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba L.C., Rompani S.B., Parraguez J.I., Obernier K., Romero R., Cepko C.L., Alvarez-Buylla A. Embryonic origin of postnatal neural stem cells. Cell. 2015;161:1644–1655. doi: 10.1016/j.cell.2015.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S., Yang C.H., Hsu K.S., Ming G.L., Song H. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. 2007;54:559–566. doi: 10.1016/j.neuron.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves J.T., Schafer S.T., Gage F.H. Adult neurogenesis in the hippocampus: from stem cells to behavior. Cell. 2016;167:897–914. doi: 10.1016/j.cell.2016.10.021. [DOI] [PubMed] [Google Scholar]
- Gong S., Zheng C., Doughty M.L., Losos K., Didkovsky N., Schambra U.B., Nowak N.J., Joyner A., Leblanc G., Hatten M.E., Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Homem C.C., Repic M., Knoblich J.A. Proliferation control in neural stem and progenitor cells. Nature reviews. 2015;16:647–659. doi: 10.1038/nrn4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten S.M., Wanders R.J. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten S.M., Violante S., Ventura F.V., Wanders R.J. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu. Rev. Physiol. 2016;78:23–44. doi: 10.1146/annurev-physiol-021115-105045. [DOI] [PubMed] [Google Scholar]
- Huynh F.K., Green M.F., Koves T.R., Hirschey M.D. Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol. 2014;542:391–405. doi: 10.1016/B978-0-12-416618-9.00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Carracedo A., Weiss D., Arai F., Ala U., Avigan D.E., Schafer Z.T., Evans R.M., Suda T., Lee C.H., Pandolfi P.P. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012;18:1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S., Gage F.H. Adult neurogenesis: bridging the gap between mice and humans. Trends Cell Biol. 2014;24:558–563. doi: 10.1016/j.tcb.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Kempermann G., Krebs J., Fabel K. The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr. Opin. Psychiatry. 2008;21:290–295. doi: 10.1097/YCO.0b013e3282fad375. [DOI] [PubMed] [Google Scholar]
- Knobloch M., Braun S.M., Zurkirchen L., von Schoultz C., Zamboni N., Araúzo-Bravo M.J., Kovacs W.J., Karalay O., Suter U., Machado R.A. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493:226–230. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M., von Schoultz C., Zurkirchen L., Braun S.M., Vidmar M., Jessberger S. SPOT14-positive neural stem/progenitor cells in the hippocampus respond dynamically to neurogenic regulators. Stem Cell Reports. 2014;3:735–742. doi: 10.1016/j.stemcr.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kridel S.J., Axelrod F., Rozenkrantz N., Smith J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64:2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- Lagace D.C., Whitman M.C., Noonan M.A., Ables J.L., DeCarolis N.A., Arguello A.A., Donovan M.H., Fischer S.J., Farnbauch L.A., Beech R.D. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J. Neurosci. 2007;27:12623–12629. doi: 10.1523/JNEUROSCI.3812-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone T.C., Weinheimer C.J., Kelly D.P. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Bobadilla E., Zhao S., Baser A., Saiz-Castro G., Zwadlo K., Martin-Villalba A. Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell Stem Cell. 2015;17:329–340. doi: 10.1016/j.stem.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Lugert S., Basak O., Knuckles P., Haussler U., Fabel K., Götz M., Haas C.A., Kempermann G., Taylor V., Giachino C. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 2010;6:445–456. doi: 10.1016/j.stem.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Lundgaard I., Li B., Xie L., Kang H., Sanggaard S., Haswell J.D., Sun W., Goldman S., Blekot S., Nielsen M. Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 2015;6:6807. doi: 10.1038/ncomms7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Martynoga B., Mateo J.L., Zhou B., Andersen J., Achimastou A., Urbán N., van den Berg D., Georgopoulou D., Hadjur S., Wittbrodt J. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 2013;27:1769–1786. doi: 10.1101/gad.216804.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry J.D., Brown N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- McGarry J.D., Mills S.E., Long C.S., Foster D.W. Observations on the affinity for carnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem. J. 1983;214:21–28. doi: 10.1042/bj2140021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez J.A., Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P., Lindauer U., Dienel G.A., Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–597. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo C.M., Vander Heiden M.G. Understanding metabolic regulation and its influence on cell physiology. Mol. Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G.L., Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira H., Andreu Z., Suh H., Lie D.C., Jessberger S., Consiglio A., San Emeterio J., Hortigüela R., Marqués-Torrejón M.A., Nakashima K. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell. 2010;7:78–89. doi: 10.1016/j.stem.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Mori T., Tanaka K., Buffo A., Wurst W., Kühn R., Götz M. Inducible gene deletion in astroglia and radial glia--a valuable tool for functional and lineage analysis. Glia. 2006;54:21–34. doi: 10.1002/glia.20350. [DOI] [PubMed] [Google Scholar]
- Nakashiba T., Cushman J.D., Pelkey K.A., Renaudineau S., Buhl D.L., McHugh T.J., Rodriguez Barrera V., Chittajallu R., Iwamoto K.S., McBain C.J. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell. 2012;149:188–201. doi: 10.1016/j.cell.2012.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray J., Gage F.H. Differential properties of adult rat and mouse brain-derived neural stem/progenitor cells. Mol. Cell. Neurosci. 2006;31:560–573. doi: 10.1016/j.mcn.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Ryall J.G., Dell’Orso S., Derfoul A., Juan A., Zare H., Feng X., Clermont D., Koulnis M., Gutierrez-Cruz G., Fulco M., Sartorelli V. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–183. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A., Scobie K.N., Hill A.S., O’Carroll C.M., Kheirbek M.A., Burghardt N.S., Fenton A.A., Dranovsky A., Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A., Wilson D.A., Hen R. Pattern separation: a common function for new neurons in hippocampus and olfactory bulb. Neuron. 2011;70:582–588. doi: 10.1016/j.neuron.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman H.E., Hen R. Neuroscience. Is more neurogenesis always better? Science. 2007;315:336–338. doi: 10.1126/science.1138711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoors S., Bruning U., Missiaen R., Queiroz K.C., Borgers G., Elia I., Zecchin A., Cantelmo A.R., Christen S., Goveia J. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520:192–197. doi: 10.1038/nature14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seri B., García-Verdugo J.M., McEwen B.S., Alvarez-Buylla A. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J. Neurosci. 2001;21:7153–7160. doi: 10.1523/JNEUROSCI.21-18-07153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J., Berg D.A., Zhu Y., Shin J.Y., Song J., Bonaguidi M.A., Enikolopov G., Nauen D.W., Christian K.M., Ming G.L., Song H. Single-cell RNA-seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. 2015;17:360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding K.L., Bergmann O., Alkass K., Bernard S., Salehpour M., Huttner H.B., Boström E., Westerlund I., Vial C., Buchholz B.A. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153:1219–1227. doi: 10.1016/j.cell.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll E.A., Makin R., Sweet I.R., Trevelyan A.J., Miwa S., Horner P.J., Turnbull D.M. Neural stem cells in the adult subventricular zone oxidize fatty acids to produce energy and support neurogenic activity. Stem Cells. 2015;33:2306–2319. doi: 10.1002/stem.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N., Laplagne D.A., Zhao C., Lombardi G., Ribak C.E., Gage F.H., Schinder A.F. Neurons born in the adult dentate gyrus form functional synapses with target cells. Nat. Neurosci. 2008;11:901–907. doi: 10.1038/nn.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H., Schinder A.F., Christie B.R., Toni N., Palmer T.D., Gage F.H. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wyss M.T., Jolivet R., Buck A., Magistretti P.J., Weber B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 2011;31:7477–7485. doi: 10.1523/JNEUROSCI.0415-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Jones A., Deeney J.T., Hur S.K., Bankaitis V.A. Inborn errors of long-chain fatty acid β-oxidation link neural stem cell self-renewal to autism. Cell Rep. 2016;14:991–999. doi: 10.1016/j.celrep.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Teng E.M., Summers R.G., Jr., Ming G.L., Gage F.H. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci. 2006;26:3–11. doi: 10.1523/JNEUROSCI.3648-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.