

Abstract
A mitotically stable linear extra chromosome obtained in a Leishmania donovani strain rendered mycophenolic acid-resistant has been physically mapped. This 290-kb chromosome has an inverted duplicated structure around a central inversion region, and is derived from a conservative amplification event of a ∼140-kb subtelomeric end of chromosome 19. Large-sized targeted deletions of the central region were performed through homologous recombination using three specific transfection vectors. The size of the extra chromosome was thus successfully reduced from 290 to 260, 200 and 120 kb respectively. The mitotic stability of these chromosomes was then analysed in drug-free cultures over >140 days. Results differed according to the deletion created. By contrast with the smallest deletion the two largest deletions altered mitotic stability, leading to progressive loss of the size-reduced chromosomes with similar kinetics in both mutants. The 30-kb region common to both deletions may therefore be considered as involved in mitotic stability. A 44-kb contig covering this region could be assembled and sequenced. The analysis of this sequence did not reveal any sequence elements typical of centromeric DNA. By contrast, its enrichment in homopolymer tracts suggests that this region might contain an origin of replication.
INTRODUCTION
Leishmania is a protozoan parasite that belongs to the family of Trypanosomatids and is responsible for a group of diseases whose symptoms range from mild cutaneous lesions to fatal visceral involvement. The population at risk is ∼350 million, and 1–2 million cases are estimated to occur annually (1). The species complex Leishmania donovani is the agent of human visceral leishmaniasis, also known as kala-azar, and is present in four continents. In the last few years, important efforts have been made toward understanding the genome and biology of this parasite and characterizing new chemotherapeutic and vaccine targets. Leishmania is thought to be ‘essentially’ diploid (2,3). The haploid genome is 35 Mb in size, and it comprises 36 heterologous chromosomes (4). The complete sequence of the smallest chromosome (chromosome 1) of the reference ‘Friedlin’ strain has been obtained recently by Myler et al. (5). The genome is remarkably stable among evolutionarily distant Leishmania species (4,6–8), but paradoxically it exhibits a high degree of inter- and intraspecific plasticity (4,9) essentially due to the instability of the subtelomeric ends of the chromosomes (10,11). This genomic plasticity is also observed by the frequent amplification of extrachromosomal DNAs, occurring either spontaneously (12,13) or in response to drug selection (14,15).
After selection in increasing concentrations of mycophenolic acid (MPA), a potent inhibitor of inosine monophosphate dehydrogenase (IMPDH), the penultimate enzyme in guanylate nucleotide synthesis, Wilson et al. (16,17) obtained an L.donovani cell line that had amplified its IMPDH gene copy number in the form of a linear extrachromosomal DNA (then sized as 280 kb). In contrast to other reported linear extrachromosomal DNAs in drug-resistant Leishmania (14,15,18–20), this extra chromosome and the drug resistance phenotype were shown to be stable in the absence of selective pressure for at least 2 years (17; P.Dubessay, unpublished data). This extra chromosome (XC), which exhibits all the stability features of a constitutive chromosome, was chosen as a model for the present study. Our objective is to characterize DNA regions involved in mitotic chromosomal stability, and ultimately to begin to elucidate the molecular mechanisms giving rise to stable drug resistance in Leishmania. The DNA sequence elements participating in the replication and segregation functions are largely unknown in Trypanosomatids. The analysis of the complete nucleotide sequence of several chromosomes, in particular Leishmania major Fiedlin chromosome 1 (5), has not revealed any homologies to known or putative centromeric DNA of yeasts (21) or Plasmodium falciparum (22,23). As regards origins of replication, although some authors have isolated putative autonomously replicating elements in plasmids in Trypanosoma brucei, no sequence data are available (see Discussion). In Leishmania, the only candidate so far is a 1.6-kb ‘switch’ region on chromosome 1 where the polarity of the two large gene clusters (made of 50 and 29 coding DNA sequences respectively) changes (5,24). In this context, we started by constructing a physical restriction map of the stable XC cited above, using 27 specific DNA markers, which showed a ‘mirror-like’ inverted repeat structure. The mitotic stability of this XC was then evaluated following large targeted deletions (up to 170 kb) of its central region. We thus show that chromosomal knock-outs >30 kb can substantially modify, but do not abolish, the mitotic stability of this XC, and that stability localizes to a 30 kb region within the XC. At this stage, we cannot yet infer whether this region supports a replication or a segregation function.
MATERIALS AND METHODS
Cell lines, culture and transfection conditions
The L.donovani clone DI700 and its MPA-resistant cloned derivative MPA100 were generated from strain 1S (MHOM/SD/83/1S) (16). The cell lines were cultivated in RPMI medium supplemented with 10% foetal calf serum (FCS), essential and non-essential MEM amino acids, sodium pyruvate, glutamine and antibiotics. In transfection and cellular cloning experiments, a 20% FCS concentration was used. In this study, MPA100 was maintained without MPA. All cultures were grown at 26°C in a humidified 5% CO2 incubator. Cell growth curves were established as follows: cultures were seeded with 2 × 105 cells ml–1 at day 0 (D0), and promastigotes were counted daily on a haemocytometer from day 2 (D2) to day 7 (D7). For the study of chromosomal stability, the percentage of drug-resistant cells in a given line was estimated by cultivating this line in the presence and absence of antibiotics and comparing the cell concentrations in both cultures at D2 and D3 (mid-log phase).
For transfection experiments, MPA100 cells grown to mid-log phase were resuspended at a density of 108 ml–1 and incubated for 10 min in chilled electroporation cuvettes either with 10 µg of linearised vector DNA or without DNA as the controls. Electroporation was performed in an Easyject Plus (Equibio) electroporator using the following conditions: 2000 V cm–1, 2310 Ω, 25 µF. Each cuvette product was divided into four samples, and cells were transferred to 1 ml of drug-free medium for 24 h. At D1, the selective antibiotic hygromycin B (Sigma) was added. Stable transfectants were selected at a concentration of 50 µg/ml, a concentration at which no survivors were observed in the mock-transfected controls. At D4, all cultures were divided into hygromycin-supplemented medium. After 2 weeks, transfectant derivatives were harvested and DNA prepared for pulsed field gel electrophoresis (PFGE). Cultures displaying a size-reduced XC were cloned by limiting dilution. For each deletion (termed Δ-658, Δ-kin and Δ-impdh), about 10 resultant clones were analysed by PFGE. Three clones exhibiting the size-reduced XC were then selected for further analysis and cultivated in the presence of hygromycin until the study of chromosomal stability was started.
DNA preparation and analysis, and DNA probes
Leishmania DNA purification, PFGE conditions and restriction analysis on either total DNA or purified chromosomes were performed as described previously (11). Southern blot and hybridisation conditions were as reported (6). The IMPDH probe was as described previously (16). The Leishmania mexicana cysteine protease a (LmCPA) gene (25) probe was kindly provided by J. C. Mottram (Glasgow University, UK). The ST515 probe for the 81-bp minisatellite sequence LiSTIR1 has been reported (26). The probe for the minisatellite type Lmet2 sequence (27) was generously provided by Michael Miles (London School of Hygiene and Tropical Medicine, UK). Anonymous DNA markers (termed As) were selected from a shotgun library of the XC (see below), sequenced and used for mapping. Two coding DNA sequences (CDSs) were identified at the beginning of this work and were used as DNA probes for chromosome mapping; the first one exhibited high homology to genes encoding members of the AAA ATPase family (GenBank accession no. AF319040), and the second exhibited high homology to the mitogen activated protein (MAP)-kinase family (termed LmMAPK1, GenBank accession no. AF176312).
Shotgun DNA library and sequencing
PFGE-purified DNA from the 290-kb XC from clone MPA100 was sheared by sonication. The resulting fragments were then subjected to mung bean nuclease digestion and T4 polynucleotide kinase and size-selected in an agarose gel. Fragments of 0.5–3 kb were then cloned into the EcoRV site of pBluescript. Individual plasmid clones were used as probes to screen Southern blots of PFGE gels and selected for their ability to recognise the XC. The selected clones (termed As) were then sequenced using a kit from Epicenter. Nucleotide sequences were determined in both directions from double-stranded DNA using dye-primer technology and Licor and Vistra (Amersham) automated sequencers. Raw sequences were assembled using the Sequencher software (Gene Codes). The contig sequence was examined for putative protein-coding ORFs by using the GCG program TESTCODE. The predicted amino acid sequences from each putative gene were subjected to BLASTP and TBLASTN searches using the GenBank databases.
Construction of vector pVV
The vector pVV was designed for systematic knock-outs in the Leishmania genome. The vector construct was built from the pGEM3Zf plasmid (Promega). In order to drive the expression of the drug resistance gene, 5′ and 3′ DNA sequences were first cloned simultaneously into pGEM3Zf in which the BamHI site had been removed. The flanking DNAs were from the 5′ and 3′ untranslated regions of the L.major dihydrofolate reductase-thymidilate synthase (DHFRTS) gene (28) (GenBank accession no. U59231). A 953-bp fragment of the 5′ DHFRTS region was PCR-amplified from L.major DNA using the primers 5′-CTTTCCGGGTCGACGGGG-3′ and 5′-TTTTCATCACTAGTGCTCGG-3′ containing SalI and SpeI sites respectively (underlined). A 1738-bp fragment of the 3′ DHFRTS region was amplified with the primers 5′-ACTAGTCGGGATCCAGTAGATGCCGAC-3′ and 5′-TCGCGGCCGTCGACGTCTTC-3′ with SpeI and BamHI sites present in the first one, and a SalI site in the second one. Both fragments were gel-purified, and after digestion with SalI and SpeI were cloned simultaneously into the SalI site of pGEM3Zf. The accurate construction of this new plasmid (termed pVI) was assessed by PCR amplification of the junctions. A selectable gene marker was then introduced into pVI: the hygromycin phosphotransferase (HYG) gene (29), which confers resistance to hygromycin B, was obtained by PCR amplification of the template vector pMAT2 (generously provided by J. C. Mottram) using primers 5′-CGAGCACTAGTGATGAAAAAGCC-3′ and 5′-GCGGATCCCTATTCCTTTGCCCT-3′ containing SpeI and BamHI sites respectively. Following digestion with SpeI and BamHI, the HYG gene was inserted into pVI between the DHFRTS 5′ and DHFRTS 3′ regions: the resulting construct was termed pVI′. Correct HYG gene expression in pVI′ was assessed by transfection of the plasmid.
Polylinker cloning sites CS1 and CS2 were then added to pVI′ by replacement of the DHFRTS 5′ and DHFRTS 3′ regions with identical flanking sequences containing different restriction sites. The DHFRTS 5′ region was amplified by PCR from pVI′ and the primers 5′-CCCAAGCTTCTGAAGGTTAACGGCAATTGGTCGACGGGGTGATGGAGAG-3′ containing HindIII, Eco57I (bold), HpaI and MfeI sites, and 5′-TTTTCATCACTAGTGCTCGG-3′ containing the SpeI site. The HindIII + SpeI-digested fragment was inserted in place of the DHFRTS 5′ region in pVI′, taking advantage of the HindIII site present in the vector. Similarly, the DHFRTS 3′ region was amplified using the primers 5′-ACTAGTCGGGATCCAGTAGATGCCGAC-3′ containing the BamHI site and 5′-GGGAATTCCTGAAGGGTACCGGTCTAGAGTCGACGTCTTCTTCTTGGCGC-3′ containing EcoRI, Eco57I, KpnI and XbaI sites. The EcoRI + BamHI-digested fragment was then inserted in the location of the DHFRTS 3′ region in the EcoRI site of pVI′. The resulting transfection vector was termed pVV.
Homologous DNA sequences can be introduced into pVV within the HpaI/MfeI and XbaI/KpnI cloning sites in both possible orientations. Moreover, since these sites are rare-cutters in the GC-rich Leishmania genome, they offer an extensive choice for insertion of homologous DNA sequences. In this work, for each construct, target sequences for homologous recombination were identical on both sides of the resistance gene but with inverted orientations. Two IIB-type Eco57I restriction sites (CTGAAGN16/GACTTCN14) in the construct allowed for release of the targeting fragment by excision within the homologous sequences. The sequence of vector pVV was submitted to GenBank under the accession number AF315645.
Construction of pVV-derived transfection vectors
The target DNA sequences employed in this study (termed impdh for IMPDH, kin for MAPK1 and 658 for As658) were PCR-amplified from total Leishmania DNA using 12 primer pairs. Three fragments, impdh (1014 bp), kin (328 bp) and 658 (330 bp) were amplified using oligonucleotides 5′-ATGGCGACCAACAACGCG-3′ (A) and 5′-AGGACGACCGCAAGCTAG-3′ (B), 5′-AATGGCTACAGAAGAGGCAA-3′ (C) and 5′-AGTGCACCGTCAGTGGACG-3′ (D), 5′-CGAGCAAGAGATCTGAGCTG-3′ (E) and 5′-AAACTCGTGTAAGGAGCGTAC-3′ (F) respectively. A 5′ tail was added to each oligonucleotide, containing one of four restriction sites: HpaI, 5′-GGGGTTAAC-3′ (H); MfeI, 5′-GGGCAATTG-3′ (M); XbaI, 5′-GGGTCTAGA-3′ (X); or KpnI, 5′-GGGGGTACC-3′ (K). Twenty-four primers were thus designed, and the combinations used for the PCR amplification of the homologous sequences to be inserted in cloning sites CS1 and CS2 are listed in Table 1. These PCR fragments were double-digested with either HpaI and MfeI or KpnI and XbaI and then introduced into pVV in both possible orientations for each vector. Six different targeting vectors were ultimately obtained, and these were designated pVVa-imp, pVVb-imp, pVVa-kin, pVVb-kin, pVVa-658 and pVVb-658 (see Fig. 2A).
Table 1. Combinations of primer pairs used for the PCR amplification of homologous sequences for the construction of the six specific pVV vectors used in this study.
| Vectora |
CS1 (HpaI–MfeI)b |
CS2 (XbaI–KpnI)c |
| PVVa-imp |
HB and MA |
XA and KB |
| PVVb-imp |
HA and MB |
XB and KA |
| PVVa-kin |
HD and MC |
XC and KD |
| PVVb-kin |
HC and MD |
XD and KC |
| PVVa-658 |
HF and ME |
XE and KF |
| PVVb-658 | HE and MF | XF and KE |
aThe names of the vectors and homologous sequences are explained in Materials and Methods.
bCS1, cloning site 1 (see text).
cCS2, cloning site 2 (see text).
The primers A, B, C, D, E, F, H, K, M and X are described in Materials and Methods.
Figure 2.

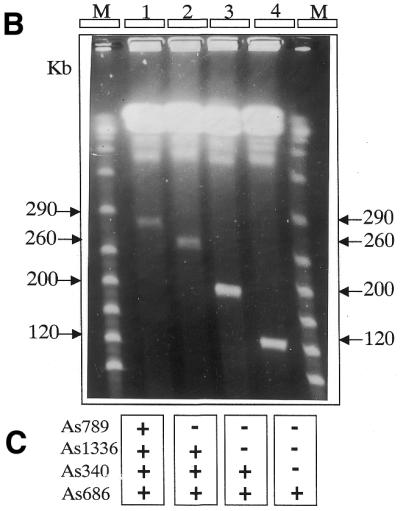

Large-sized targeted deletions on XC-290. (A) Schematic representation of the targeted deletions and homologous sequences. The location and size of the targeted deletions are shown (double-headed arrows) above the restriction map of XC-290. The restriction sites of the map are detailed in Figure 1. The locations of the targeted homologous sequences, i.e. IMPDH (impdh), MAPK1 (kin) and As658, are shown below the map. The orientation of these sequences in the six different pVV constructs is shown by arrows. The region sequenced and analysed in Figure 5 is indicated by shaded boxes. (B) PFGE analysis of chromosomal DNA from the parental cell line MPA100 (1), and the transfectant clones Δ-658 (2), Δ-kin (3) and Δ-impdh (4). The size of each XC is indicated on each side of the gel in kilobases. The molecular weight markers (M) are polymers of bacteriophage lambda DNA. The PFGE gel was run at 7.5 V cm–1 on a home-made apparatus with a 30 s pulse time for 48 h as described (4,11). (C) Summary of the Southern analysis of the gel shown in (B). The locations of the markers (termed As), external or internal to the targeted deletion, are shown in Figure 1. (+) and (–) refer to positive or negative hybridisation with XC-290 (lane 1 in B) and its down-sized derivatives (lanes 2, 3 and 4 in B). The HYG probe hybridized to the three size-reduced chromosomes (not shown). (D) Schematic representation of the predicted recombination events between XC-290 and the targeting construct pVV bearing HYG (H). The hatched box represents the targeted central region of the extra chromosome bound by both homologous sequences. The arrows show the orientation of the homologous sequences. Tel, telomeric repeats. In the targeting event depicted by arrow 1, the construct harboured both homologous sequences in the correct ‘head-to-head’ orientation and the generated clones are analysed in (B). In contrast, no recombination of the type shown by arrow 2 between two possible homologs of XC-290 was observed when the homologous sequences were present in inverted ‘tail-to-tail’ orientation.
RESULTS
The mitotically stable extra chromosome has an inverted repeat structure
Wilson et al. (16,17) demonstrated that the linear extrachromosomal DNA present in the MPA-resistant L.donovani clone MPA100 was the result of an amplification bearing the IMPDH gene and originating from a 700-kb chromosome: a conservative amplification event was suggested as the possible mechanism. This hypothesis was confirmed by our mapping of 27 selected DNA markers (see Materials and Methods for description), including the leishmanial IMPDH and LmCPA genes and two novel CDSs, that localized both to the source chromosome and to the XC. The source chromosome was identified as chromosome 19 according to the nomenclature adopted by the Leishmania Genome Network (3,4). The amplification event was further investigated by the construction of a physical map of the XC (hereafter termed XC-290) (Fig. 1). The mapping of these 27 DNA probes on the pulsed field gel-excised and purified XC showed their symmetric distribution on both sides of one central fragment of 20 kb. This inverted duplicated structure was inferred from three sets of results. (i) Restriction experiments on the purified XC using the enzymes AseI, DraI, HpaI, SfiNI, SpeI and SspI yielded a small number of fragments with variable ethidium bromide-staining intensities (data not shown). The sum of the sizes of these fragments was always between 155 and 190 kb, less than that of the XC. However, considering the 2-fold increase in staining intensity as the result of the presence of two co-migrating fragments, a total size of 290 kb could be calculated for the XC. (ii) Hybridisation of these restriction fragments with a telomeric probe [5′-AGGG(TTAGGG)16-3′] (11) revealed a single band in every digest, with a size varying from 15 kb for DraI to 130 kb for HpaI (data not shown). These data suggest a structural identity for both XC ends up to the HpaI site (130 kb). (iii) Accordingly, the 27 marker probes also revealed a single-sized fragment in all the restriction digests of the purified XC (data not shown). Taken together, the most plausible explanation for these three sets of experiments is that the XC has a ‘mirror-like’ inverted repeat structure (Fig. 1), at least from the telomere up to the most proximal SspI site (135 kb). Two subtelomeric minisatellite repeats, LiSTIR1 and Lmet2, previously shown to be present on chromosomes 1, 5, 19 and 22 in L.donovani (11,26) were also used as hybridisation probes. Both mapped to the same telomeric DraI fragment of the XC (data not shown), strongly implying that the XC originated from one subtelomeric end of the source chromosome. Finally, partial shotgun sequencing of XC-290 (see below) allowed the identification of a subtelomeric tandemly repeated DNA, of which the repeat unit has a consensus sequence of 276 bp (GenBank accession nos AF393161–AF393182). This sequence co-localises with LiSTIR and Lmet2 in the terminal DraI fragment of XC-290 and its derivatives (not shown, see Fig. 1) and corresponds (with 78% homology) to the 272-bp subtelomeric repeats identified in L.major Friedlin chromosome 1 (5).
Figure 1.

Middle-range restriction map of the extra chromosome XC-290. The restriction enzymes used were AseI (A), DraI (D), SfiNI (F), HpaI (H), SpeI (P) and SspI (S). Numbers below the map indicate the size of the restriction fragments (the scale is not proportional). The locations of the 27 markers used as probes (boxes) on the chromosome are shown by the arrows. One copy of each marker is present on each side of a central region bound by the two HpaI sites, except for the most central markers, As757 and As789, for which this information is not known. LmCPA, L.major cystein protease a; MAPK1, LmMAPK1; AAA-p, AAA ATPase gene (see Materials and Methods). Tel, telomeric repeat sequences. The region sequenced and analysed in Figure 5 is indicated by shaded boxes above the map. The 44-kb contig nucleotide sequence confirmed the positions of the restriction sites around and in the segment MAPK1–As658, except for two more DraI sites distant by ∼1 kb and located in the HpaI–DraI 20-kb fragment at 6 kb from the DraI site.
Comparative physical mapping between the XC and chromosome 19 was carried out by restriction analysis. Total DNA from MPA100 containing both chromosomes 19 and XC-290 was digested with the same enzymes as above (AseI, DraI, HpaI, SfiNI, SpeI and SspI) and hybridised with the 27 marker probes; each of these labelled a single-sized band in each digest (data not shown). These data confirm that the XC is the result of a conservative inverted duplication of one subtelomeric end from chromosome 19.
Large deletions of central regions of the extra chromosome XC-290
In order to identify DNA sequences potentially involved in chromosomal stability, different-sized targeted deletions of the central region of the XC were made by targeted DNA replacements in MPA100 cells. The three DNA sequences targeted for replacements were segments of the IMPDH gene, MAPK1 gene and the anonymous sequence As658 (see Materials and Methods). From the physical map shown in Figure 1, each of these DNA sequences should be present as two inverted copies along the XC. Since the orientation of these three target sequences within the XC was not known, they were introduced into the pVV vector in both possible orientations (see Materials and Methods). The three target sequences targeted for homologous recombination and the different deletions expected with the six vector constructs are shown in Figure 2A. The predicted deletions between both copies of As658, kin or impdh span 30, 90 or 170 kb of the XC respectively.
Of the six constructs, only pVVa-658, pVVa-kin and pVVa-imp (those with the homologous sequences in ‘head-to-head’ orientation) yielded the expected deletions (termed Δ-658, Δ-kin and Δ-impdh respectively). For each of the three targeted deletions, cell clones were obtained in which XC-290 had disappeared and been replaced by a size-reduced copy (Fig. 2B). Only one clone exhibited the original XC-290 together with a size-reduced copy (see below). The sizes of these mutant XCs were determined by PFGE gels and found to be, as expected, 260, 200 and 120 kb in clones Δ-658, Δ-kin and Δ-impd respectively. The identities of the size-reduced chromosomes, as well as the correct location of the different deletions, were then verified by Southern analysis using several DNA marker probes (Fig. 2C) as well as a HYG probe (not shown). The identity of the mutant XCs was also confirmed by restriction analysis with AseI, SfiNI and SpeI, followed by Southern blotting and hybridization with probes As686, As340, As1336, As789 and IMPDH (data not shown). All these data demonstrated that the recombination events had occurred exactly as expected. No other recombination event was detected in these clones. The fact that the recombination events occurred exactly as expected further confirmed the inverted duplicated structure of XC-290. Finally, it shows that the transcription orientation of both genes, IMPDH and MAPK1, on XC-290 is from the centre toward the telomeres.
Conversely, the three pVV constructs with the homologous sequences in a ‘tail-to-tail’ orientation, pVVb-imp, pVVb-kin and pVVb-658, gave rise to different and complex recombination events involving chromosome 19 (data not shown). These cell lines were not analysed further. Nevertheless, it is noteworthy that we never observed a recombination event between two possible homologs of XC-290, which would have generated a larger XC with a duplication of the central region adjoined by both homologous sequences (Fig. 2D).
Analysis of the mitotic stability of the deleted chromosomes
In order to examine whether the different large-sized deletions affected the mitotic stability of the XC, three sibling clones (nos 1, 2 and 3) of each line harbouring a truncated XC were incubated in the absence of hygromycin and the mitotic stability of the size-reduced chromosome assessed. In each case, the three sibling clones yielded comparable results. Each clone was cultivated in the absence of drug pressure for >140 days, and cells were analysed at various time intervals. The percentage of cells exhibiting both the hygromycin-resistant phenotype and the size-reduced XC was assessed by three methods. First, aliquots of each of these samples were transferred into both hygromycin-containing and drug-free medium, and growth was measured and compared as described in Materials and Methods. Secondly, 20–30 subclones were isolated by limiting dilution from one or two of the three sibling clones (i.e. nos 1, 2 or 3), and the hygromycin-resistant phenotype and presence of the truncated XC were assessed. Thirdly, a PFGE analysis of the whole culture at the time of the sampling was also performed.
Cell clones harbouring Δ-658 and incubated without hygromycin maintained the hygromycin-resistance phenotype and their truncated XC for the entire 140 days of the study (Fig. 3). The resistance phenotype was maintained in 100% of the cells; all subclones isolated during two subcloning experiments were drug-resistant; and the 260-kb size-reduced XC persisted up to 140 days with the same ethidium bromide-staining intensity in pulsed field gels (Figs 3 and 4A). By contrast, clones Δ-kin and Δ-impdh exhibited a progressive decrease in the percentage of hygromycin-resistant cells (Fig. 3), as well as a progressive loss of intensity of the size-reduced chromosomes in pulsed field gels (Fig. 4A and B). Moreover, in one Δ-kin clone (Δ-kin 2), which contained both the full-length XC-290 and the truncated XC, XC-290 was maintained while the smaller XC disappeared in the absence of selective pressure (Fig. 4A). It is noteworthy that for both Δ-kin and Δ-impdh, the diminution curves were very similar, i.e. after 25 days of cultivation in drug-free medium ∼55% of the cells did not exhibit a resistant phenotype, and this percentage increased to 90% after 80 days (Fig. 3). Likewise, the subcloning experiments described above showed a decrease in the proportion of cells possessing the hygromycin-resistance phenotype and the size-reduced XC in the culture (closed symbols in Fig. 3). For the three deletions (shown for Δ-impdh in Fig. 4C), there was a strict correlation between the resistance phenotype and the presence of the truncated XC in all subclones. This whole body of data clearly demonstrates that deletions of the 30-kb region bound by MAPK1 and As658 durably affect the mitotic stability of XC-290.
Figure 3.

Mitotic stability of the size-reduced XCs. Mitotic stability was measured as the percentage of hygromycin-resistant cells in different samples taken at various time intervals from the transfectant cultures in drug-free medium. First (curves and open symbols), these percentages were assessed by the cultivation of the different samples in parallel both in the presence or absence of the drug and the comparison of growth curves (as described in Materials and Methods). Open triangles, squares and circles show the results obtained with clones Δ-658, Δ-kin and Δ-impdh respectively. The smallest deletion of 30 kb (Δ-658) yielded a chromosome stable for >140 days in vitro after removing the drug selection, while the larger deletions (Δ-kin and Δ-impdh) yielded mitotically unstable chromosomes. Secondly (closed symbols), subcloning of the same cultures was carried out, the resultant subclones were tested for their resistance to the antibiotic and the percentage of hygromycin-resistant subclones was calculated. Closed triangles, squares and circles show the results obtained with clones Δ-658, Δ-kin and Δ-impdh respectively. Both sets of results are correlated, although lower figures were obtained with subcloning than with direct assessment.
Figure 4.

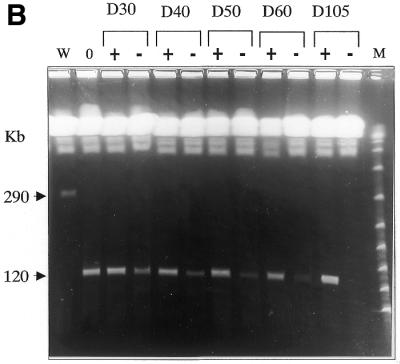
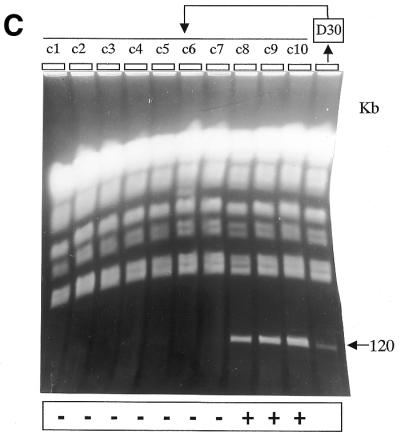
PFGE analysis of the mitotic stability of the deleted chromosomes. Chromosomal DNA was prepared from clones Δ-658, Δ-kin and Δ-impdh and cultivated in the presence or absence of hygromycin for the number of days indicated above the gels, i.e. 0, 20, 40, or 105 days. (A) W, MPA100 wild-type cells; M, lambda polymer. The size of the XCs is indicated on both sides in kilobases. In clone Δ-kin1, the proportion of cells containing the size-reduced XC decreased with time to finally render the XC not visible. In clone Δ-kin2, the mitotic stability was conserved for XC-290, while the size-reduced copy was progressively lost with time. The two gels depicted were run independently using identical parameters (see Fig. 2B) in different apparatuses. (B) The same experiment is shown for Δ-impdh parasites. The control cultures maintained in the presence of hygromycin are indicated as (+) and the cultures in drug-free medium as (–). (C) The figure shows the chromosomal analysis of 10 clones (c1–c10) isolated from the Δ-impdh culture maintained 30 days without drug selection (D30); c1–c7 are hygromycin-sensitive clones; c8–c10 are hygromycin-resistant clones. Hygromycin resistance was always associated to the size-reduced XC.
Moreover, the possibility that some cells from the Δ-kin and Δ-impdh clones propagated without hygromycin had retained their truncated XC as a consequence of some epigenetic effect that permitted recovery of the stability of the size-reduced XC (30) was tested. Two subclones issued from Δ-kin and Δ-impdh at D60 and D70 respectively, and exhibiting both the hygromycin-resistance phenotype and the size-reduced chromosome, were then cultured for an additional 100–110 days in the absence of selective pressure. In these two subclones, a progressive loss of the resistance phenotype and the size-reduced XC was observed with the same kinetics shown in Figure 3 for the initial cultures (data not shown). Thus, cells maintaining the XC for long periods of time in the absence of drug had not experienced a modification that conferred a ‘neo-stability’ to the XC.
Sequencing of the 30-kb MAPK1–As658 region
The shotgun cloning and sequencing of the whole PFG-purified XC-290 has been started in the laboratory. The complete nucleotide sequence of XC-290 is not yet available. However, a 44273-bp contig covering the 30-kb region bound by MAPK1 and As658 could be assembled (GenBank accession no. AY028171). The analysis of this sequence revealed the presence of 11 CDSs, including the known genes LmCPA and MAPK1, all located on the same strand with a transcription orientation toward the telomere (Fig. 5). Two interspersed repeats, named LIR1 and LIR2, were also identified: their consensus sequences are 582 and 720 bp in size and they are present in four and two copies respectively (Fig. 5). One of the LIR1 repeats shows an inverted orientation as compared with the other ones. Analysis of the databases revealed that these repeats are frequently found in the Leishmania genome and present on several chromosomes. The most remarkable feature of the whole sequence is a large (∼10 kb) segment devoid of any CDS (located between CDS7 and CDS8 in Fig. 5). Although this segment does not contain any other repetitive element than one LIR1 and one LIR2 copy, it is comparatively rich in poly(dA), (dC), (dG) and (dT) tracts: as an example, poly(dA)n with n ≥ 6 are indicated in Figure 5. Similarly, homo-oligomers with n ≥ 3 are twice as frequent in this segment as in the remaining non-coding regions of the 44 kb. Apart from those homopolymer tracts, no segment particularly rich in (typically di- and trinucleotide) microsatellite sequences was found on the whole contig (not shown). The contig exhibited good sequence homology with the cosmid clones L6754 (GenBank accession no. AL358632) and L2185 (GenBank accession no. AL358712) from chromosome 19 of L.major Friedlin; in particular the distribution of the 11 CDSs and of the repeat elements was almost identical between the two species.
Figure 5.

Gene map of the entire 44273-bp contig sequence. The region bounded by As658 and the MAPK1 gene is indicated. The CDSs are shown as black boxes; CDS7, LmCPA; CDS10, MAPK1. The location and orientation of the interspersed repeats LIR1 and LIR2 are shown by arrows. Each poly(dA)n with n ≥ 6 found in the whole contig tract is shown as a vertical line. Poly(dC)n, (dG)n and (dT)n tracts are present with a similar distribution but are not shown for the sake of clarity.
DISCUSSION
Targeted DNA deletions have been exploited to initiate a dissection of chromosome stability DNA elements in L.donovani. Three different large-sized deletions of 30, 90 and 170 kb were generated on the mitotically stable supernumerary chromosome XC-290 of MPA-selected cells. Two of these deletions induced the progressive loss of the mutant chromosome in culture, indicating a partial loss of mitotic stability of this XC. Thus, a 30-kb candidate region involved in mitotic stability has been mapped and sequenced for the first time in this protozoan.
Origin and ploidy of XC-290
Numerous extrachromosomal DNA amplifications have been described in Leishmania and they can exhibit either a circular or linear topology (reviewed in 20). Physical mapping of the best characterised linear amplicons LD1s (31) revealed their inverted repeat structure, probably originating from the duplication of a portion of the subtelomeric region (32,33) of chromosome 35 (4,12,13). The extrachromosomal linear DNA XC-290 found in MPA-selected L.donovani showed no homology to the LD1 family nor to any other known amplicon (17). Nevertheless, XC-290 also exhibited an inverted repeat structure and originated from the mirror duplication of ∼140 kb from one subtelomeric end of chromosome 19. This was proposed based upon the extensive colinearity of DNA markers and restriction sites mapped on both halves of the XC and on the source chromosome. These results confirm the hypothesis of a conservative duplication event as the mechanism for the formation of this XC.
The results of the targeted deletion experiments show that, apart from one exception, the creation of the deleted chromosomes is always accompanied by the disappearance of XC-290 (see Figs 2B, 4A and B), suggesting that the latter is monosomic. However, we cannot completely rule out the hypothesis that XC-290 is disomic, at least occasionally (as for clone Δ-kin2, Fig. 4A). Aneuploidy may indeed be more frequent than previously thought in Leishmania, mainly as monosomic or trisomic chromosomes (3,34–36, P.Dubessay, unpublished data), and seems to be well tolerated in this organism.
Mitotic stability of the size-reduced chromosomes
The extra chromosome XC-290 is a particularly pertinent model for dissecting the molecular determinants ensuring chromosomal mitotic stability. Since all the genes and markers characterised on XC-290 are also found on chromosome 19, its presence should not confer any growth advantage or disadvantage in vitro. Indeed, no significant differences in the growth rate of DI700, MPA100 or the mutant clones Δ-658, Δ-kin and Δ-impdh were observed (data not shown).
Two major functions are necessary for chromosomal stability during mitosis: replication and correct segregation. Here, both functions may play a role in the persistent or partial loss of mitotic stability of our size-reduced XCs. The deletion of ∼30 kb of the central region of XC-290 did not affect its mitotic stability. Therefore, the deleted region does not contain, at least exclusively, sequences necessary for either chromosomal replication or segregation. The two deletions of 90 and 170 kb of the central region of XC-290 in the Δ-kin and Δ-impdh mutants induced a progressive loss of the deleted chromosome in the absence of drug pressure with similar kinetics. These results indicate that the region common to both deletions, bound by MAPK1 and As658 (shaded in Figs 1 and 2A), is involved in the loss of stability of the XC. However, this loss of stability is only partial. Indeed, a complete loss of the replication function on a chromosome should rapidly lead to the dilution of the latter in the cultivated population [(1/2)n after n generations]. Similarly, a random chromosome segregation would leave only (3/4)n cells possessing the supernumerary chromosome after n generations e.g. <5% after 10 generations (in the hypothesis of a random chromosomal segregation, each mitotic division will lead to the following distribution of genotypes: absence of the chromosome = 1/4; presence of 2-fold more of the chromosome = 1/4; genotype identical to the initial one = 2/4). In the Δ-kin and Δ-impdh mutants, this percentage is only attained after more than 110 generations (Fig. 3). This indicates that the truncated XCs analysed in this study can be replicated and/or segregated but not as faithfully as XC-290 or the constitutive chromosomes. Several alternative hypotheses (detailed below) may explain these results.
Is chromosome size involved in mitotic stability?
The chromosome size might play a role in mitotic stability. In yeast, truncated versions of the 530-kb chromosome III, all containing the centromeric DNA, were created by chromosomal fragmentation (37). Size reductions down to 100 kb weakly affected chromosomal segregation. By contrast, below 100 kb, small changes in size resulted in sharp decreases in stability, e.g. ∼2 and 8% loss per division for 40 and 29 kb. The faithful separation of sister chromatids during mitosis depends upon their prior cohesion and condensation (38). A drastic size reduction of such chromatids might disrupt these structural changes and induce segregation disorders (37,39). In T.brucei, variously-sized linear ‘artificial mini-chromosomes’ were produced by transfection with 10- and 13-kb plasmidic constructs containing telomeres and subtelomeric sequences at each end (40). They all had integrated unidentified genomic sequences. Two of them were analysed for mitotic stability in the absence of drug pressure: one of ∼40 kb was lost from 80% of the population at 32 generations, while the other one, ∼145 kb in size, was stable during 60 generations. In the absence of complete knowledge on the nature of the newly integrated DNA sequences, it was postulated that mitotic stability of linear chromosomes is proportional to chromosome length in this protozoan. Nevertheless, several points argue against this ‘size’ hypothesis applied to our data. Trypanosoma brucei contains constitutive mini-chromosomes of size ∼30–150 kb which are stable in mitosis (41). Moreover, one of these mini-chromosomes of size ∼60 kb and bearing rDNA promoter sequences was tagged with either the NEOr gene (42) or the HYG gene (40) and found to be mitotically stable after 130 generations by the first group, but unstable by the second group. In a recent study on L.major, we created a linear ‘artificial mini-chromosome’ of 155 kb in size that was stable during >120 generations in the absence of drug pressure (P.Dubessay, C.Ravel, P.Bastien, K.Stuart, J.-P.Dedet, C.Blaineau and M.Pagès, manuscript in preparation). Finally, in the present study, the loss of stability of the Δ-kin and Δ-impdh derivatives (200 and 120 kb in size respectively) was quantitatively similar, ∼3% per division. All this strongly suggests that the partial loss of stability of the XCs of our study is not correlated with the size reduction. Rather, one may consider the presence on the MAPK1–As658 region of sequence elements participating to replication or to centromeric function.
Origins of replication?
The region MAPK1–As658 may first contain one or several origins of replication. In this respect, in Trypanosomatids, the only precise data concern the identification of replication origins on the mitochondrial DNA minicircles and maxicircles that have been inferred from nucleotide sequences in various genera, including Leishmania (43) and Trypanosoma (44): the most serious candidate being a highly conserved 12mer sequence. Accordingly, plasmids containing 1-kb fragments of mitochondrial DNA minicircle of T.brucei were found to replicate under selective pressure (45). The nuclear chromosome replication issue was addressed by several authors working on artificial constructs in T.brucei. Patnaik et al. (46,47) isolated a panel of putative autonomously replicating sequence (ARS) elements by introduction into plasmids; and artificial mini-chromosomes were constructed in T.brucei that were stable under drug selection, suggesting they contained ARS elements (40,48). Unfortunately, we could not find any further analysis of the nucleotide sequences involved in these experiments. In Saccharomyces cerevisiae, the deletion of a functional ARS can be complemented by a neighbouring sequence (49). This model may be operative in our study, i.e. the dependence upon secondary replication origins of lower efficiency would not enable the cyclical and regular replication of the chromosome in an absolute faithful manner, and would therefore lead to its progressive loss in culture. The nucleotide sequence analysis of the region involved in mitotic stability does not allow the identification of origins of replication that are known as highly heterogeneous in yeasts as well as metazoa (50,51). Nevertheless, a 10-kb segment of this region appears devoid of CDSs and is particularly rich in homopolymer tracts. One of the general features of origins of replication is their enrichment in homopolymers and short (A+T)-rich tracts (52,53). Also, in Schizosaccharomyces pombe, ARS activity does not depend on an ARS consensus sequence like in budding yeast but rather on multiple poly(dA) or (dT) stretches with n ≥ 3 (54). It has been shown in prokaryotes and yeast that the nucleotide sequence may directly intervene in the formation of ARSs by influencing the higher-order DNA structure (53,55). In particular, the periodicity of A3 and T3 tracts can give rise to anti-bent DNA structures (53,56), which appear as essential components of functional ARS elements (55). A similar analysis of chromosome 1 of L.major Friedlin also reveals that poly(dA) and (dT) stretches with n ≥ 6 are more concentrated in two ‘hot spots’, one encompassing the 1.6-kb ‘switch’ region between the two strand-specific gene clusters, at 76–84 kb from the ‘left’ end of the sequence (GenBank accession no. NC001905), and the other one in another intergenic region at ∼10 kb from the ‘right’ end of the same sequence (P.Dubessay, unpublished data). A functional analysis of these regions is presently being carried out in our laboratory.
Sequence elements involved in segregation?
Alternatively, the loss of stability of our size-reduced XCs may be explained by the presence in the region MAPK1–As658 of sequence elements participating in segregation. Centromeres are totally unknown in Leishmania and Trypanosoma. In particular, in T.brucei, the artificial mini-chromosomes constructed in the 1990s that were stable under drug selection were more or less rapidly lost from the population on the removal of selective pressure—the only condition allowing to distinguish between forced maintenance and true segregation (46–48). Here, as the loss of stability was only partial, the presumed segregation elements would not be as in S.cerevisiae, where the absence of the <150 bp point centromere induces a complete loss of the segregation function (21). Rather, they would be like the complex regional centromeres present in S.pombe and higher eukaryotes. In S.pombe, the centromeric sequences comprise various repeated and single-copy elements distributed along large (40–100 kb) genomic regions (21). However, the sequence analysis of the region MAPK1–As658 shows that it is covered by 11 CDSs and does not comprise any repetitive elements that might recall centromeric DNA from other organisms. Moreover, recent data from our group suggest that the centromeric DNA might be located elsewhere on this XC (P.Dubessay, C.Ravel, P.Bastien, K.Stuart, J.-P.Dedet, C.Blaineau and M.Pagès, manuscript in preparation): in this work, the fragmentation of L.major chromosome 1 created a mini-chromosome of 155 kb that only comprised plasmidic sequences, a 272-bp repeat satellite DNA and telomeres; this XC has been mitotically stable during 120 generations without drug selection, suggesting a centromeric role for this subtelomeric satellite DNA. A 276-bp satellite homologous to the latter is present in L.donovani both on chromosome 19 and on XC290 and its size-reduced derivatives (not shown).
If this sequence acts as a centromere on these XCs, then the hypothesis cited above of a replication origin in the region MAPK1–As658 would clearly be favoured against that of centromeric sequences. The experimental identification of candidate sequences for replication origins by direct mapping (57,58) is one strategy envisioned to confirm this hypothesis. In any case, large-sized deletions created through homologous recombination, developed in Leishmania initially on the α-tubulin gene cluster (59) and recently on chromosome 5 and 23 by telomere-mediated chromosome breakage (36), appear to be a powerful tool to analyse DNA elements involved in mitotic stability.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Professor Jean-Pierre Dedet who provided continuous support for carrying out this study in his department. We gratefully acknowledge the technical help of Michèle Lefebvre and Lucien Crobu. We are also indebted to Dr Jeremy Mottram (Glasgow University, UK) who provided us with the LmCPA probe and the template vector pMAT2. This study received financial support from the Centre National de la Recherche Scientifique (CNRS), from the GDR 1077 of the CNRS/DGA-DSP, from the European Community INCO-DC programme (Contract no. 970256), and from the NIH (Grant no. AI23682). P.D. was a recipient of a PhD grant from the Ministère de l’Enseignement Supérieur et de la Recherche.
DDBJ/EMBL/GenBank accession nos+ To whom correspondence should be addressed. Tel: +33 4 67 63 55 13; Fax: +33 4 67 63 00 49; Email: genpara@sc.univ-montp1.fr AF315645, AF176312, AF319040, AY028171, AF393161–AF393182
References
- 1.Desjeux P. (1999) Global control and Leishmania HIV co-infection. Clin. Dermatol.,17, 317–325 [DOI] [PubMed]
- 2.Bastien P., Blaineau,C. and Pagès,M. (1992) Leishmania: Sex, lies and karyotype. Parasitol. Today, 8, 174–177. [DOI] [PubMed] [Google Scholar]
- 3.Ravel C., Dubessay,P., Blackwell,J.M., Ivens,A.C., Bastien,P. and the Leishmania Genome Network (1998) The complete chromosomal organization of the reference strain of the Leishmania Genome Project, L.major ‘Friedlin’. Parasitol. Today, 14, 301–303. [DOI] [PubMed] [Google Scholar]
- 4.Wincker P., Ravel,C., Blaineau,C., Pagès,M., Jauffret,Y., Dedet,J.P. and Bastien,P. (1996) The Leishmania genome comprises 36 chromosomes conserved across widely divergent human pathogenic species. Nucleic Acids Res., 24, 1688–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myler P.J., Audelman,L., De Vos,T., Hixson,G., Kiser,P., Lemley,G., Magness,C., Rickel,E., Sisk,E., Sunkin,S., Swartzell,S., Westlake,T., Bastien,P., Fu,G., Ivens,A. and Stuart,K. (1999) Leishmania major Friedlin chromosome I has an unusual distribution of protein-coding genes. Proc. Natl Acad. Sci. USA, 96, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ravel C., Macari,F., Bastien,P., Pagès,M. and Blaineau,C. (1995) Conservation among Old World Leishmania species of six physical linkage groups defined in Leishmania infantum small chromosomes. Mol. Biochem. Parasitol., 69, 1–8. [DOI] [PubMed] [Google Scholar]
- 7.Britto C., Ravel,C., Bastien,P., Blaineau,C., Pagès,M., Dedet,J.P. and Wincker,P. (1998) Conserved linkage groups associated with large-scale chromosomal rearrangements between Old World and New World Leishmania genomes. Gene, 222, 107–117. [DOI] [PubMed] [Google Scholar]
- 8.Ravel C., Dubessay,P., Britto,C., Blaineau,C., Bastien,P. and Pages,M. (1999) High conservation of the fine-scale organisation of chromosome 5 between two pathogenic Leishmania species. Nucleic Acids Res., 27, 2473–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bastien P., Blaineau,C. and Pagès,M. (1992) Molecular karyotype analysis in Leishmania. In Avila,J.L. and Harris,R. (eds), Intracellular Parasites Subcellular Biochemistry. Plenum Press, New York, NY, Vol. 18, pp. 131–187. [DOI] [PubMed]
- 10.Blaineau C., Bastien,P., Rioux,J.A., Roizès,G. and Pagès,M. (1991) Long-range restriction maps of size-variable homologous chromosomes in Leishmania infantum. Mol. Biochem. Parasitol., 46, 293–302. [DOI] [PubMed] [Google Scholar]
- 11.Ravel C., Wincker,P., Blaineau,C., Britto,C., Bastien,P. and Pagès,M. (1996) Medium range restriction maps of five chromosomes of Leishmania infantum and localization of size-variable regions. Genomics, 35, 509–516. [DOI] [PubMed] [Google Scholar]
- 12.Liu J.H., Gajendran,N., Muthui,D., Muyldermans,S., Dujardin,J.C., De Doncker,S., Jacquet,D., Le Ray,D., Mathieu-Daudé,F. and Hamers,R. (1991) Chromosome rearrangement in Leishmania mexicana M379. Mol. Biochem. Parasitol., 46, 53–60. [DOI] [PubMed] [Google Scholar]
- 13.Tripp C.A., Myler,P.J. and Stuart,K. (1991) A DNA sequence (LD1) which occurs in several genomic organizations in Leishmania. Mol. Biochem. Parasitol., 47, 151–161. [DOI] [PubMed] [Google Scholar]
- 14.Beverley S.M. (1991) Gene amplification in Leishmania. Annu. Rev. Microbiol., 45, 417–444. [DOI] [PubMed] [Google Scholar]
- 15.Ouellette M. and Papadopoulou,B. (1993) Mechanisms of drug resistance in Leishmania. Parasitol. Today, 9, 150–153. [DOI] [PubMed] [Google Scholar]
- 16.Wilson K., Collard,F.R., Huberman,E., Stringer,J.R. and Ullman,B. (1991) Amplification and molecular cloning of the IMP Dehydrogenase gene of Leishmania donovani. J. Biol. Chem., 266, 1665–1671. [PubMed] [Google Scholar]
- 17.Wilson K., Beverley,S.M. and Ullman,B. (1992) Stable amplification of a linear extrachromosomal DNA in mycophenolic acid-resistant Leishmania donovani. Mol. Biochem. Parasitol., 55, 197–206. [DOI] [PubMed] [Google Scholar]
- 18.Hanson S., Beverley,S.M., Wagner,W. and Ullman,B. (1992) Unstable amplification of two extrachromosomal elements in alpha-difluoromethylornithine resistant Leishmania donovani. Mol. Cell. Biol., 12, 5499–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papadopoulou B., Roy,G. and Ouellette,M. (1993) Frequent amplification of a short chain dehydrogenase gene as part of circular and linear amplicons in methotrexate resistant Leishmania. Nucleic Acids Res., 21, 4305–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olmo A., Arrebola,R., Bernier,V., Gonzalez-Pacanowska,D. and Ruiz-Pérez,L.M. (1995) Co-existence of circular and multiple linear amplicons in methotrexate-resistant Leishmania. Nucleic Acids Res., 23, 2856–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiens G.R. and Sorger,P. (1998) Centromeric chromatin and epigenetic effects in kinetochore assembly. Cell, 93, 313–316. [DOI] [PubMed] [Google Scholar]
- 22.Gardner M.J., Tettelin,H., Carucci,D.J., Cummings,L.M., Aravind,L., Koonin,E.V., Shallom,S., Mason,T., Yu,K., Fujii,C. et al. (1998) Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science, 282, 1126–1132. [DOI] [PubMed] [Google Scholar]
- 23.Bowman S., Lawson,D., Basham,D., Brown,D., Chillingworth,T., Churcher,C.M., Craig,A., Davies,R.M., Devlin,K., Feltwell,T. et al. (1999) The complete nucleotide sequence of chromosome 3 of Plasmodium falciparum. Nature, 400, 532–538. [DOI] [PubMed] [Google Scholar]
- 24.McDonagh P.D., Myler,P.J. and Stuart,K. (2000) The unusual gene organization of Leishmania major chromosome 1 may reflect novel transcription processes. Nucleic Acids Res., 28, 2800–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mottram J.C., Robertson,C.D., Coombs,G.H. and Barry,J.D. (1992) A developmentally regulated cysteine proteinase gene of Leishmania mexicana. Mol. Microbiol., 6, 1925–1932. [DOI] [PubMed] [Google Scholar]
- 26.Ravel C., Wincker,P., Bastien,P., Blaineau,C. and Pagès,M. (1995) A polymorphic minisatellite sequence in the subtelomeric regions of chromosome I and V in Leishmania infantum. Mol. Biochem. Parasitol., 74, 31–34. [DOI] [PubMed] [Google Scholar]
- 27.Howard M.K., Kelly,J.M., Lane,R.P. and Miles,M.A. (1991) A sensitive repetitive DNA probe that is specific to the Leishmania donovani complex and its use as an epidemiological and diagnostic reagent. Mol. Biochem. Parasitol., 44, 63–72. [DOI] [PubMed] [Google Scholar]
- 28.Ryan K.A., Dasgupta,S. and Beverley,S.M. (1993) Shuttle cosmid vectors for the trypanosomatid parasite Leishmania. Gene, 131, 145–150. [DOI] [PubMed] [Google Scholar]
- 29.Cruz A., Coburn,C.M. and Beverley,S.M. (1991) Double targeted gene replacement for creating null mutants. Proc. Natl Acad. Sci. USA, 88, 7170–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steiner N.C. and Clarke,L. (1994) A novel epigenetic effect can alter centromere function in fission yeast. Cell, 79, 865–874. [DOI] [PubMed] [Google Scholar]
- 31.Segovia M. and Ortiz,G. (1997) LD1 Amplifications in Leishmania. Parasitol. Today, 13, 342–348. [DOI] [PubMed] [Google Scholar]
- 32.Navarro M., Liu,J., Muthui,D., Ortiz,G., Segovia,M. and Hamers,R. (1994) Inverted repeat structure and homologous sequences in LD1 amplicons of Leishmania spp. Mol. Biochem. Parasitol., 68, 69–80. [DOI] [PubMed] [Google Scholar]
- 33.Ortiz G., Navarro,M. and Segovia,M. (1995) Location in the source chromosome of the 180-kb mini-chromosome of Leishmania major and characterization of the novel junction. Mol. Biochem. Parasitol., 71, 153–161. [DOI] [PubMed] [Google Scholar]
- 34.Cruz A.K., Titus,R. and Beverley,S.M. (1993) Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc. Natl Acad. Sci. USA, 90, 1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunkin S.M., Kiser,P., Myler,P.J. and Stuart,K. (2000) The size difference between Leishmania major Friedlin chromosome one homologues is localized to sub-telomeric repeats at one chromosomal end. Mol. Biochem. Parasitol., 109, 1–15. [DOI] [PubMed] [Google Scholar]
- 36.Tamar S. and Papadopoulou,B. (2001) A telomere-mediated chromosome fragmentation approach to assess mitotic stability and ploidy alterations of Leishmania chromosomes. J. Biol. Chem., 276, 11662–11673. [DOI] [PubMed] [Google Scholar]
- 37.Surosky R.T., Newlon,C.S. and Tye,B.K. (1986) The mitotic stability of deletion derivatives of chromosome III in yeast. Proc. Natl Acad. Sci. USA, 83, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirano T. (2000) Chromosome cohesion, condensation and separation. Annu. Rev. Biochem., 69, 115–144. [DOI] [PubMed] [Google Scholar]
- 39.Murray A.W., Schultes,N.P. and Szostak,J.W. (1986) Chromosome length controls mitotic chromosome segregation in yeast. Cell, 45, 529–536. [DOI] [PubMed] [Google Scholar]
- 40.Lee M.G., E,Y. and Axelrod,N. (1995) Construction of trypanosome artificial mini-chromosomes. Nucleic Acids Res., 23, 4893–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alsford S., Wickstead,B., Ersfeld,K. and Gull,K. (2001) Diversity and dynamics of the minichromosomal karyotype in Trypanosoma brucei. Mol. Biochem. Parasitol., 113, 79–88. [DOI] [PubMed] [Google Scholar]
- 42.Zomerdjik J.C.B.M., Kieft,R. and Borst,P. (1992) A ribosomal RNA gene promoter at the telomere of a mini-chromosome in Trypanosoma brucei. Nucleic Acids Res., 20, 2725–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheline C. and Ray,D.S. (1989) Specific discontinuities in Leishmania tarentolae minicircles map within universally conserved sequence blocks. Mol. Biochem. Parasitol., 37, 151–157. [DOI] [PubMed] [Google Scholar]
- 44.Myler P.J., Glick,D., Feagin,J.E., Morales,T.H. and Stuart,K.D. (1993) Structural organization of the maxicircle variable region of Trypanosoma brucei: identification of potential replication origins and topoisomerase II binding sites. Nucleic Acids Res., 21, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Metzenberg S. and Agabian,N. (1994) Mitochondrial minicircle DNA supports plasmid replication and maintenance in nuclei of Trypanosoma brucei. Proc. Natl Acad. Sci. USA, 91, 5962–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patnaik P.K., Kulkarni,S.K. and Cross,G.A.M. (1993) Autonomously replicating single-copy episomes in Trypanosoma brucei show unusual stability. EMBO J., 12, 2529–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patnaik P.K., Fang,X. and Cross,G.A.M. (1994) The region encompassing the procyclic acidic repetitive protein (PARP) gene promoter plays a role in plasmidic DNA replication in Trypanosoma brucei. Nucleic Acids Res., 22, 4111–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patnaik P.K., Axelrod,N., Van der Ploeg,L.H.T. and Cross,G.A.M. (1996) Artificial linear mini-chromosomes for Trypanosoma brucei. Nucleic Acids Res., 24, 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vujcic M., Miller,C.A. and Kowalski,D. (1999) Activation of silent replication origins at autonomously replicating sequence elements near HML locus in budding yeast. Mol. Cell. Biol., 19, 6098–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilbert D.M. (1998) Replication origins in yeast versus metazoa: separation of the haves and the have nots. Curr. Opin. Genet. Dev., 8, 194–199. [DOI] [PubMed] [Google Scholar]
- 51.Bogan J.A., Natale,D.A. and Depamphilis,M.L. (2000) Initiation of eukaryotic replication: conservative or liberal? J. Cell Physiol., 184, 139–150. [DOI] [PubMed] [Google Scholar]
- 52.Boulikas T. (1996) Common structural features of replication origins in all life forms. J. Cell. Biochem., 60, 297–316. [DOI] [PubMed] [Google Scholar]
- 53.Eckdall T.T. and Anderson,J.N. (1990) Conserved DNA structures in origins of replication. Nucleic Acids Res., 18, 1609–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okuno Y., Satoh,H., Sekiguchi,M. and Masukata,H. (1999) Clustered adenine/thymine stretches are essential for function of a fission yeast replication origin. Mol. Cell. Biol., 19, 6699–6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marilley M. (2000) Structure-function relationships in replication origins of the yeast Saccharomyces cerevisiae: higher-order structural organization of DNA in regions flanking the ARS consensus sequence. Mol. Gen. Genet., 263, 854–866. [DOI] [PubMed] [Google Scholar]
- 56.Nelson H.C., Finch,J.T., Luisi,B.F. and Klug,A. (1987) The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature, 330, 221–226. [DOI] [PubMed] [Google Scholar]
- 57.Dijkwel P.A. and Hamlin,J.L. (1997) Mapping replication origins by neutral/neutral two dimensional gel electrophoresis. Methods, 13, 235–245. [DOI] [PubMed] [Google Scholar]
- 58.Giaccia M., Pelizon,C. and Falaschi,A. (1997) Mapping replication origins by quantifying relative abundance of nascent DNA strands using competitive Polymerase Chain Reaction. Methods, 13, 301–312. [DOI] [PubMed] [Google Scholar]
- 59.Curotto de Lafaille M.A. and Wirth,D.F. (1992) Creation of Null/+ mutants of the α-tubulin gene in Leishmania enrietti by gene cluster deletion. J. Biol. Chem., 267, 23839–23846. [PubMed] [Google Scholar]