

Abstract
Aims/hypothesis
Obesity triggers complex inflammatory networks within the innate immune system. During pregnancy, the placenta amplifies the low-grade inflammation through activation of Toll-like receptor 4 (TLR4) signalling pathways. The purpose of this study was to investigate the impact of obesity on placental TLR4 expression and inflammatory signals. The secondary aim was to analyse the placental cell type responsible for TLR4 activation.
Methods
Thirty-nine women recruited at term-scheduled Caesarean section were grouped according to their pre-gravid BMI (< 25 kg/m2 and > 30 kg/m2). Placenta, venous maternal and cord blood were obtained at delivery for analysis. Data were analysed with linear regression and Spearman’s rank correlation coefficient analysis.
Results
TLR4, IL6 and IL8 expression was increased three- to ninefold (p < 0.001) in the placenta of obese vs lean women. There was a positive correlation between placental TLR4 and maternal systemic and placental IL6 and IL8 concentrations. Placental TLR4 expression was correlated with maternal pre-gravid BMI, insulin resistance index, plasma insulin and C-reactive protein (r = 0.57, 0.31, 0.35, 0.53, respectively; p < 0.001) but not with plasma glucose, maternal age, gestational age and gestational weight gain (r < 0.2; p > 0.1). TLR4 was located in both trophoblast and macrovascular endothelial cells lining fetal vasculature. Lipopolysaccharide-induced TLR4 activation was more robust in trophoblasts than in endothelial vascular cells (100-fold vs tenfold; p < 0.001).
Conclusions/interpretation
Trophoblastic TLR4 is implicated in the propagation of placental inflammation. Placental inflammation is related to maternal metabolic conditions such as pre-gravid BMI, whilst gestational weight gain or gestational age are not. These results implicate the pre-gravid condition as a significant contributor to metabolic inflammation in late pregnancy.
Keywords: Gestational weight gain, Inflammation, LPS, Macrovascular endothelial cells, Obesity, Placenta, Pregnancy, Toll-like receptor 4, Trophoblast
Introduction
Obesity is emerging as a trans-generational disease that propagates through an adverse programming effect in the progeny [1]. Infants of obese women develop short- and long-term health consequences [2]. Of particular concern is the increased risk to the offspring for excess neonatal adiposity and metabolic disease in adolescence and adulthood [3, 4]. Other obesity-related traits, including insulin resistance, are associated with fetal macrosomia [5–8]. However, the causal nature of the factors that result in higher fetal body adiposity remain to be elucidated. Likewise, the mechanisms underlying the in utero programming effects are not well defined.
Human and animal studies have clearly established that obesity is associated with a state of chronic low-grade inflammation [9]. During pregnancy, obese women exhibit metabolic (low-grade) inflammation characterised by elevated levels of pro-inflammatory cytokines in the systemic circulation and adipose tissue compared with lean women [10–12]. Low-grade inflammation encompasses the placenta [13], suggesting that maternal obesity exposes the developing fetus to an inflammatory environment. In this context, early-life exposure to metabolic inflammation may represent a mechanism by which metabolic dysfunction will manifest later in life. One current challenge of developmental programming is the gathering of data with which to dissect out the obesogenic stimuli that drive the increased in utero inflammation in obese women, particularly at the maternal–fetal interface.
Current literature supports a functional connection between triggers of metabolic inflammation and the innate immune system [14]. Toll-like receptor 4 (TLR4) is one isoform of pattern-recognition receptors, which trigger immune responses in response to various infectious agents and metabolic stressors [9, 15, 16]. TLR4 binds a wide spectrum of exogenous and endogenous ligands including bacterial lipopolysaccharide (LPS), its primary ligand. The sequence of events generated upon TLR4 activation leads to the release of various effectors of innate immunity, cytokines and pro-inflammatory mediators through the nuclear translocation of NFκB [17]. TLR4 is found in virtually all metabolic tissue and cells. It is overexpressed in adipose tissue of individuals who are obese or have type 2 diabetes, as well as in obese pregnant women [18]. TLR4 is expressed in the human placenta, suggesting that crosstalk may arise between immune and endocrine functions at the maternal–fetal interface. Against this background, the objective of this study was to investigate the impact of maternal obesity-related traits on placental TLR4 expression and inflammatory signals. The secondary aim was to analyse the placental cell type responsible for the propagation of TLR4-induced inflammation.
Methods
Study participants
Women with uncomplicated pregnancies were recruited at term-scheduled Caesarean delivery. Those with multiple gestation, fetal anomalies, intrauterine growth restriction and diabetes (pre-existing and gestational) were excluded. Obesity was defined as pre-gravid BMI > 30 kg/m2 and lean body type as BMI < 25 kg/m2. Pre-conception weight was obtained from charts as self-reported weight. Late-pregnancy weight was obtained from charts as measured at the last prenatal visit. Total gestational weight gain (GWG) was calculated as the difference between last prenatal visit and pre-conception maternal weight. Excess GWG was estimated by applying 2009 Institute Of Medicine (IOM) guidelines for each BMI category [19].
All participants provided informed consent; they were fully aware of the study procedures and made an informed decision on their participation. Our investigations were approved by the MetroHealth Medical Center institutional review board (IRB) for collection of maternal blood, placenta and venous cord blood according to the protocol (IRB05-288). Maternal pre-gravid BMI was obtained from medical records. Placentas were weighed after the umbilical cord and fetal membranes had been trimmed by research staff. Anthropometric variables of the study cohort are presented in Table 1.
Table 1.
Anthropometrics of study cohort
| Variable | Lean (n=20) | Obese (n=19) | p value |
|---|---|---|---|
| Maternal age (years) | 28.0±6.4 | 29.7±7.4 | 0.225a |
| Gestational age (weeks) | 38.9±0.6 | 39.1±0.4 | 0.114a |
| Pre-gravid BMI (kg/m2) | 21.1±2.4 | 40.3±7.7 | 0.000a |
| BMI at delivery (kg/m2) | 27.4±2.5 | 46.7±7.1 | 0.000a |
| GWG (kg) | 16.8±5.5 | 17.3±9.5 | 0.421a |
| Excess GWG (%) | 65.0 | 73.7 | 0.557b |
| Race (%) | |||
| White | 70 | 47.4 | 0.151b |
| African-American | 30 | 42.1 | 0.431b |
| Hispanic | 0 | 10.5 | 0.136b |
| Smoke (%) | 25.0 | 10.5 | 0.239b |
| Neonatal sex (%) | |||
| Female | 55 | 26 | 0.069b |
| Male | 45 | 74 | |
| Plasma NEFA (mmol/l) | |||
| Maternal | 0.7±0.2 | 0.7±0.1 | 0.292a |
| Cord | 0.2±0.0 | 0.3±0.1 | 0.195a |
Data are presented as means±SD or as percentage
Student’s t test
χ2 test
Biological specimen collection
Blood and placental tissue were obtained from 39 women at term pregnancy scheduled for elective Caesarean section. Maternal venous blood samples were collected in EDTA before placement of intravenous lines for hydration. Placental and cord blood samples were obtained immediately after double-clamping of the umbilical cord. Fragments of placental villous tissue were collected for either paraffin embedding for immunohistochemistry or molecular analysis, and stored at −80°C. Adjacent placental fragments (total 50 g) were immediately processed for isolation of trophoblast cells. Placental macrovascular veins were dissected within15 min for isolation of endothelial cells.
Plasma assays
Plasma glucose was assessed by the glucose oxidase method, using YSI 2300 STAT Plus Glucose & Lactate Analyzer (YSI Incorporated, Yellow Springs, OH, USA) [11]. Plasma insulin was measured using ELISA (EMD Millipore Corporation, Billerica, MA, USA) with an intra-assay CV of 3.0–8.2%. Leptin, IL8 and IL6 were measured using ELISA kits (R&D Systems, Minneapolis, MN, USA) with intra-assay CVs of 0.11–10.7%, 0.4–4.7% and 0.1–7.2%, respectively. All plasma samples were assayed in duplicate. Plasma C-reactive protein (CRP) concentration was measured by ELISA (Alpha Diagnostics International, San Antonio, TX, USA). Insulin resistance was estimated using HOMA-IR, as published previously [20]. Plasma NEFA levels were measured by a HR series NEFA kit (Wako Diagnostics, Richmond, VA, USA).
Immunofluorescence
Paraffin blocks of villous tissue were sectioned (5 μm thick) and collected on Excell Slides (Fisher Scientific, Pittsburgh, PA, USA). The slides were deparaffinised, followed by 30 min incubation in 10 mmol/l sodium citrate with 0.05% (vol./vol.) Tween solution (pH 6.0) in a 100°C water bath for antigen retrieval. The tissue sections were blocked for non-specific staining with PBS containing 0.3% (vol./vol.) Triton X-100 (Sigma-Aldridge, St Louis, MO, USA), 5% (vol./vol.) Normal Donkey Serum, 1% (vol./vol.) IgG-free BSA (Jackson Immunoresearch, West Grove, PA, USA) for 30 min. The slides with isolated cells or villous tissue were incubated overnight at 4°C with the following antibodies: TLR4 (H-80, 1:50; Santa Cruz, Dallas, TX, USA) and CD34 (ab8536, 1:400; Abcam, Cambridge, MA, USA). For visualisation, Alexa Fluor 488 and 594 secondary antibodies (1:1000; Jackson Immunoresearch, West Grove, PA, USA) were used. The nucleus was labelled with To-Pro 3 (Sigma-Aldridge). The slides were imaged with Leica Microsystem SP2 Confocal Microscope (Leica Microsystems, Buffalo Grove, IL, USA). All antibodies were diluted in PBS and validated by negative control.
Placental cell isolation and cell culture
Trophoblast cells from obese women were isolated by sequential trypsin and DNase digestion followed by gradient centrifugation [21]. Flow cytometry analysis of isolated placental cells indicated that 86.7 ± 6.7% cells stained positive for cytokeratine 7, 1.6 ± 0.8% for placental alkaline phosphatase and 4.8 ± 1.7% for CD68 (data not shown). Cells were placed into 12-well plates at a density of 1.5×106 cells/well and cultured overnight in Iscoves’s modified DMEM culture medium supplemented with 10% (vol./vol.) FBS (Gibco, Grand island, NY, USA) and 1% (vol./vol.) penicillin/streptomycin at 37°C under 5% CO2. The medium was replaced with 1 ml DMEM with 1% (vol./vol.) FBS 2 h before addition of 100 ng/ml LPS (Sigma-Aldridge) and plates were left for 24 h. Medium with PBS alone was used as control.
For placental macrovascular endothelial cell (PVEC) isolation, the amnion was removed and macrovascular veins from the placental chorionic plate of obese women were cut out in lengths of 3–5 cm and washed with Hank’s Balanced Salt Solution (HBSS; 2.5% (vol./vol.) penicillin/streptomycin). A cannula was inserted into the vessel and fixed with sterilised surgical thread. Following insertion of a syringe containing 0.05% (wt/vol.) collagenase/dispase solution (Sigma Aldrich) in HBSS pre-warmed to 37°C, 20 ml was slowly rinsed through the vessel by using a syringe pump (syringe diameter of 30 ml, 27.6 mm, flow rate, 2.5 ml/min). The released cells were collected in a 50 ml Falcon centrifuge tube (Corning, Corning, NY, USA) containing 5 ml FBS. The cell suspension was centrifuged at 900 rpm (200 g) for 5 min at 4°C, then resuspended in complete media (500ml Endothelial Basal Medium [EBM; Clonetics, Lonza, Walkersville, MD, USA] + one EGM-MV BulletKit [Clonetics, Lonza] + 10% (vol./vol.) FBS + 1% (vol./vol.) penicillin/streptomycin) and cultured in 12-well plates that had been pre-coated with 1% (vol./vol.) gelatin (Sigma Aldrich) in HBSS for 1 h at 37°C. After reaching 80% confluence at 21% oxygen and 37°C, cells were transferred to 25 cm2 flasks and expanded for identification purposes and determination of purity [22–24]. The endothelial identity of the cells was confirmed by immunohistochemical staining, with the endothelial markers anti-von Willebrand factor (A0082, 1:2000; Dako, Carpinteria, CA, USA), Vimentin (M0725, 1:100; Dako) and Desmin (M0760, 1:200; Dako). Lack of immunoreactivity with a marker for smooth muscle cells (smooth muscle actin, M0851, 1:100; Dako) confirmed cell purity (data not shown). Cells were placed into 12-well plates in 5% (vol./vol.) FBS + EBM + EGM-MV BulletKit culture medium at 37°C under 5% CO2. After reaching 90% confluence, the medium was replaced with EBM + EGM-MV BulletKit 2 h before adding 50 ng/ml LPS and plates were left for 24 h. PBS was used as control.
IL6, IL8 and TNF-α concentrations in culture medium were measured by ELISA (HS600B (S6050), HS800 (S8000C) and, HSTA00D (STA00C), respectively; R&D System, Minneapolis, MN, USA).
RNA preparation and quantitative real-time PCR
Total RNA was extracted from 30 mg frozen placental tissue or trophoblast cells and endothelial cells (cultured on 12-well plates) using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) or TRIzol (Invitrogen, Carlsbad, CA, USA). RNA was reversed-transcribed using a Superscript II RNase H-Reverse Transcriptase system (Invitrogen). Gene expression was measured in duplicates by quantitative real-time PCR (qPCR) using a Roche thermal cycler (Roche Applied Science, Indianapolis, IN, USA) with Lightcycler Fast-start DNA Sybr Green 1 master mix and primers (Integrated DNA Technologies, Coralville, IA, USA). Specific primers are shown in electronic supplementary material (ESM) Table 1. Results were normalised for β-actin and performed by the comparative Ct method. Data were quantified as copy number/(ng RNA) calculated against the standard curve of human term placental cDNA or expressed as fold change of treated vs control groups in cultured trophoblast cells and endothelial cells.
TLR4 protein detection
Total protein was extracted from 25 mg frozen placenta tissue, 1.5 × 106 cultured trophoblast cells or 1.5 × 106 endothelial cells. The protein lysates were prepared by homogenisation in buffer (10 mmol/l Tris pH 8, 130 mmol/l NaCl, 1% [vol./vol.] Triton X-100, 10 mmol/l sodium fluoride, 10 mmol/l sodium phosphate and 10 mmol/l sodium pyrophosphate) with protease inhibitors (P8340; Sigma-Aldridge, St Louis, MO, USA). Lysates were centrifuged at 20,000 g for 10 min at 4°C. Protein concentrations were measured with a BCA protein assay kit (Pierce, Carlsbad, CA, USA). Proteins were electrophoresed on a 7.5% SDS gel (Bio-Rad, Hercules, CA, USA), loaded with 100 μg total protein per well, and then transferred to a nitrocellulose filter (Invitrogen, Carlsbad, CA, USA). The membrane was blocked with 5% (wt/vol.) non-fat milk for 1 h, incubated with rabbit polyclonal TLR4 (H-80, 1:200; Santa Cruz) and β-actin (1:2000; Abcam, Cambridge, MA, USA) overnight then secondary antibodies (1:2000 and 1:6000) for 1 h. Amersham ECL Plus Western blotting Reagents (GE Healthcare, Aurora, OH, USA) was used for detection. All antibodies were diluted in PBS and validated by Precision Plus Protein Kaleidoscope Standards (BIO-RAD, Hercules, CA, USA). Densitometric data from autoradiograms were quantified by Image J (https://imagej.nih.gov/ij/download.html).
Statistical analysis
Values in Tables 1 and 2 are presented as means ± SD. Differences among dependent variables were analysed by Student’s t test and χ2 test. Associations between placental TLR4 mRNA expression (copy number/ng RNA) and maternal variables were analysed by Spearman’s rank correlation coefficient and linear regression analysis. Statistical mean differences for qPCR data were calculated by Kruskal–Wallis test for non-parametric data. Statistical significance was set at p < 0.05.
Table 2.
Metabolic and inflammatory variables
| Maternal variable at delivery | Lean (n=20) | Obese (n=19) | p value |
|---|---|---|---|
| HOMA-IR | 2.2±1.1 | 4.1±1.0 | 0.000 |
| Plasma glucose (mmol/l) | 4.1±0.5 | 4.4±0.4 | 0.026 |
| Plasma insulin (pmol/l) | 80.6±8.3 | 161.8±73.6 | 0.000 |
| Plasma leptin (ng/ml) | 23.6±4.4 | 81.8±4.7 | 0.000 |
| Plasma IL6 (pg/ml) | 2.1±1.0 | 5.1±2.7 | 0.007 |
| Plasma IL8 (pg/ml) | 1.9±0.9 | 3.2±1.5 | 0.011 |
| Plasma CRP (nmol/l) | 49.9±34.6 | 122.3±64.1 | 0.001 |
Data are presented as means±SD
Results
Metabolic inflammation at the maternal–fetal interface
Metabolic variables were characterised in obese and lean women at term normal delivery. As expected, obese women were hyperinsulinaemic and hyperleptinaemic and had a higher insulin resistance index compared with their lean counterparts (Table 2). They also displayed low-grade systemic inflammation, with an increased plasma concentration of CRP, IL6 and IL8 (p < 0.05). Placentas of obese women displayed higher mRNA levels of TLR4, IL6 and IL8 (Fig. 1). Sex of offspring had no effect on the pattern of placental TLR4 expression (data not shown). Placental TLR4 mRNA levels were strongly correlated with the levels of IL6 and IL8 in maternal systemic circulation and expression of IL6 and IL8 in the placenta (Fig. 2).
Fig. 1.

Inflammatory cytokine and TLR4 expression in placenta of lean and obese women. (a–c) IL6 (a), IL8 (b) and TLR4 (c) mRNA expression in placental tissue from lean (n=20) and obese (n=19) women was measured by qPCR. Results were normalised to β-actin and presented as fold change of obese vs lean group. (d) TLR4 protein in placental tissue was measured by western blot. Results were normalised to β-actin, n=8. AU, arbitrary units. Data are shown as means±SEM; *p<0.05 obese vs lean
Fig. 2.

Correlation between placental TLR4 expression and systemic or placental inflammation. The correlation between placental TLR4 and pre-gravid maternal plasma IL6 (a), IL8 (b) and CRP (c) levels orplacental IL6 (d) and IL8 (e) expression was analysed by simple linear regression analysis. Statistical significance was set at p<0.05
Association between placental TLR4 and maternal metabolic variables
Multiple correlation analyses (Table 3) indicated that placental TLR4 exhibited a strong positive correlation with maternal pre-gravid BMI and BMI at delivery (r = 0.57 and 0.67, respectively; p <0.0001) as well as with plasma insulin, leptin and CRP (r = 0.35, 0.73 and 0.53, respectively; p < 0.01). HOMA-IR estimated at delivery showed a weaker correlation with TLR4 and this did not quite reach statistical significance (p < 0.06). In contrast, maternal plasma glucose, gestational age and total GWG were not significantly associated with placental TLR4. Therefore, in our population, maternal pre-gravid obesity and not GWG appears to be the more important factor associated with placental innate immune response.
Table 3.
Correlation between placental TLR4 mRNA and body composition or metabolic status in pregnant women
| Variable | Correlation with placental TLR4 mRNA (copies/ng RNA) | |
|---|---|---|
| Spearman r | p value | |
| Maternal age | 0.007 | 0.967 |
| Gestational age | 0.080 | 0.630 |
| Pre-gravid BMI | 0.570 | 0.000 |
| BMI at delivery | 0.667 | 0.000 |
| Total GWG | 0.178 | 0.278 |
| HOMA-IR | 0.309 | 0.062 |
| Plasma glucose | 0.007 | 0.969 |
| Plasma insulin | 0.350 | 0.034 |
| Plasma leptin | 0.729 | 0.000 |
| Plasma CRP | 0.532 | 0.004 |
Regulation of placental TLR4 expression
TLR4 staining was detected by immunohistochemistry on syncytiotrophoblasts and the perivascular layer of the placental villous, including fetal vascular endothelium (Fig. 3). The presence of TLR4 was confirmed by western blot analysis on cells isolated from the same placentas (Fig. 3c). The signalling mechanisms of TLR4 activation were characterised in primary cultures of term trophoblast cells and PVECs from obese women (Fig. 4). LPS, the primary ligand of TLR4, was used to evaluate TLR4-stimulated cytokine release. The amount of IL6 and IL8 released by trophoblast cells was increased by 80-fold and 100-fold, respectively (p < 0.01) vs control upon 24 h treatment with 100 ng/ml LPS. Under the same stimulation conditions, the release of IL6 and IL8 was increased 10-fold and 20-fold, respectively, in PVECs, p < 0.05. The release of TNF-α was stimulated six- to ninefold (p < 0.001) in trophoblast cells whereas there was no detectable release by PVECs. IL6 and IL8 levels in culture media of LPS-treated trophoblast cells were four- to fivefold higher than those in PVEC media.
Fig. 3.
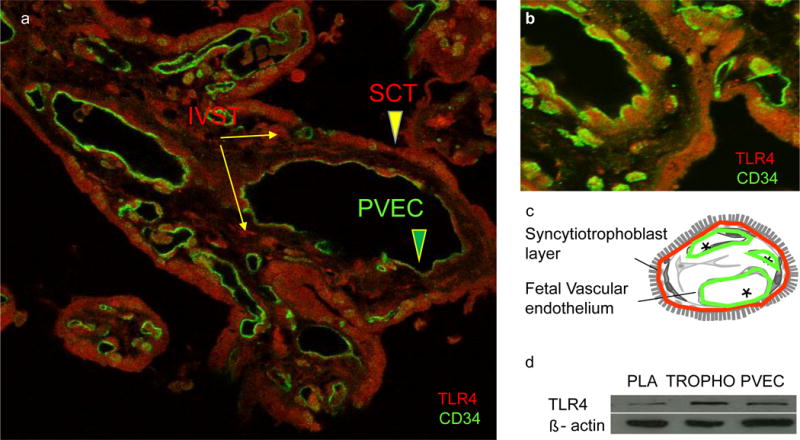
TLR4 localisation in human term placenta. (a, b) Immunofluorescence analysis showing TLR4 (red) localised in the syncytiotrophoblast (SCT) layer (a) and perivascular layer (b) with an endothelial cell marker CD34 (green). (c) TLR4 protein was examined by western blot in placental tissue (PLA), term human trophoblast cells (TROPHO) and PVECS, n=3. IVST, intravillous stromal cells
Fig. 4.
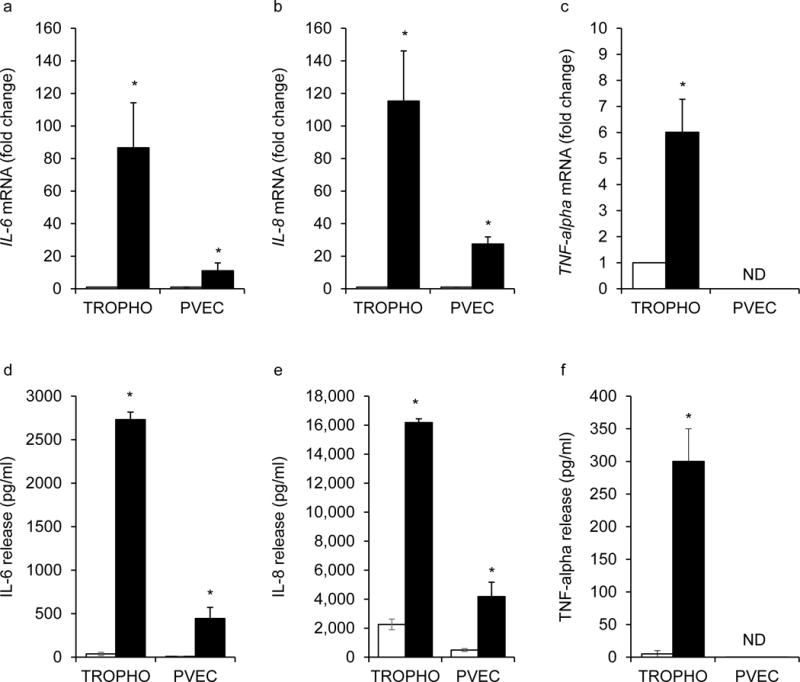
Regulation of inflammatory cytokines in cultured human trophoblast cells and placental venous endothelial cells. Primary human trophoblast cells (TROPHO) and PVECs from obese placentas were treated with LPS. IL6, IL8 and TNF-α (a–c) mRNA was detected by qPCR and (d–f) protein levels by ELISA. TNF-α expression in endothelial cells was below the detection threshold. qPCR results were normalised to β-actin and expressed as fold change vs control (no LPS). Data are shown as means±SEM for 3–6 independent experiments; *p<0.05 vs control. White bars, control; black bars, LPS. CTL, control; ND, non-detectable
Discussion
In this prospective cohort study of healthy pregnant women at term, we observed a significant induction of TLR4-induced inflammatory signals in the placenta of obese compared with lean women. These findings suggest that factors related to the obesogenic environment of the pregnant women had triggered the propagation of an inflammatory cascade through activation of TLR4-induced pathways. The molecular mechanisms of TLR4 activation have been well described in adipose tissue, particularly in rodent models of obesity [25]. LPS and saturated fatty acids both have the ability to activate TLR4 immune responses leading to activation of NFκB and several inflammatory genes in adipocytes of non-pregnant individuals [17, 26, 27] and pregnant women [18]. Likewise, we have recently reported that saturated fatty acids promote the activation of TLR4 in primary human trophoblast cells [28]. The intense TLR4 staining detected in trophoblast cells extends the previous observation made by Powell et al, of TLR4 expression in term placenta [29]. It further supports our original hypothesis that ligand binding sites of placental TLR4 are readily accessible to factors derived from the maternal systemic circulation. An important finding is the localisation of TLR4 in the immediate perivascular region of placental–fetal vessels, which is in accordance with TLR4 expression in umbilical endothelial cells [30]. The human placental structure allows direct contact between the fetal vascular endothelial cells and fetal blood. Hence, the vascular TLR4 may be directly targeted by factors derived from the fetal circulation. This may contribute to regulation of the local cytokine environment and convey to the placenta immune signals originating from the fetus [31]. Beside LPS, glucose, NEFA and LDL are additional metabolic stimulators of TLR4-induced innate immune response [32–34]. In the context of this study, obese women did not exhibit overt hyperglycaemia or glucose intolerance (data not shown); this does not point towards a role for maternal plasma glucose in placental TLR4 activation. However, in late pregnancy the decreased ability of obese women to respond to the anti-lipolytic effect of insulin results in an increased circulating NEFA concentration under insulin-stimulated condition [35–37]. Furthermore, the placentas of obese women, compared with their lean counterparts, have an enhanced capacity for lipid storage [38]. It has been suggested that triacylglycerols and phospholipid species stored in the placenta are made available for use as fatty acids through the activation of endogenous lipases [39–41]. We have previously reported that in cultured trophoblast cells long-chain saturated fatty acids such as palmitate induce a TLR4 inflammatory cascade and cytokine release through NFκB activation [28]. Together these data suggest that in obese women the lipid overload at the maternal–fetal interface may trigger TLR4-induced placental inflammation. There is currently no data relating to the regulation of trophoblast TLR4 by other maternal lipid species such as LDL or HDL. As regards to fetal lipid substrates, HDL/ApoE has been proposed as a regulator of gene expression in human placental endothelial cells [42]. However, it is unclear whether or not specific LDL species are present in the placental perivascular milieu in proximity to the fetal circulation. If they are, this could represent a mechanism for regulation of placental TLR4 through fetal-derived stimuli and needs further examination.
In addition to nutritional adaptations, increased fat mass and insulin resistance are the hallmarks of obesity whose mechanisms have been best defined [43, 44]. In this cohort of women at term pregnancy, we observed a strong association between maternal pre-gravid BMI and placental TLR4 expression and a weaker association with maternal insulin resistance estimated in late pregnancy. Current knowledge favours metabolic inflammation as a trigger of chronic insulin resistance, rather than the reverse situation. Together, these observations suggest that it is placental inflammation that enhances insulin resistance in obese women through cytokine release rather than pre-existing maternal insulin resistance enhancing placenta TLR4 activation.
We report that maternal pre-conception and late BMI had the strongest association with placental TLR4 expression. In contrast, maternal GWG was not significantly correlated with TLR4. Importantly, the timing of weight gain rather than total GWG may have greater impact on fetal body composition [45]. Hence, our analysis in which we included total GWG leaves open the possibility that weight gain in early pregnancy may positively associate with placental inflammation.
Our findings suggest that in obese women, the maternal pre-gravid metabolic condition has a stronger impact on the regulation of feto–placental adaptations than gestational changes such as GWG [5]. Our observations are consistent with those made in many studies [46–48], although not all [49], relating to the importance of pre-pregnancy obesity as a risk factor for neonatal adiposity.
In conclusion, TLR4 is strongly implicated in the propagation of placental inflammation. It is not merely the increased mass of adipose and other maternal tissues accrued during gestation that is related to placental TLR4 activation but rather an excess maternal BMI at the start of pregnancy. The distribution of TLR4 in placental cell types may have important functional implications in favouring access to maternal rather than fetal obesogenic stimuli. Therefore, we propose that trophoblastic TLR4 represents an accessible target for nutritional intervention in pregnant women. Overall, these findings contribute to our understanding of the programming mechanisms of obesity and provide compelling pre-clinical evidence for a potential strategy for improving pregnancy metabolic outcomes.
Supplementary Material
Acknowledgments
P. Glazebrook and J. Minium (Center for Reproductive Health, MetroHealth Medical Center, OH, USA) were associated with the study; P. Glazebrook contributed to imaging and J. Minium recruited patients and obtained biological samples.
Funding
This work was supported by a grant(NIH HD 22965-19).
Abbreviations
- CRP
C-reactive protein
- GWG
Gestational weight gain
- HBSS
Hanks’ balanced salt solution
- LPS
Lipopolysaccharide
- PVEC
Placental macrovascular endothelial cell
- TLR4
Toll-like receptor 4
- qPCR
quantitative real-time PCR
Footnotes
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
XY and MH contributed to study design, acquisition and analysis of data and manuscript drafting. ML performed statistical analysis and manuscript drafting. PMC, PO’T-G and SHM engaged in interpretation of data and critical revision of the manuscript. All authors gave their approval to the final version of the manuscript. SHM was responsible for the integrity of the work as a whole.
References
- 1.Nicholas LM, Morrison JL, Rattanatray L, Zhang S, Ozanne SE, McMillen IC. The early origins of obesity and insulin resistance: timing, programming and mechanisms. Int J Obes (Lond) 2016;40:229–238. doi: 10.1038/ijo.2015.178. [DOI] [PubMed] [Google Scholar]
- 2.Catalano PM, Ehrenberg HM. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG. 2006;113:1126–1133. doi: 10.1111/j.1471-0528.2006.00989.x. [DOI] [PubMed] [Google Scholar]
- 3.Catalano PM. Obesity and pregnancy–the propagation of a viscous cycle? J Clin Endocrinol Metab. 2003;88:3505–3506. doi: 10.1210/jc.2003-031046. [DOI] [PubMed] [Google Scholar]
- 4.Ozanne EM, Shieh Y, Barnes J, Bouzan C, Hwang ES, Esserman LJ. Characterizing the impact of 25 years of DCIS treatment. Breast Cancer Res Treat. 2011;129:165–173. doi: 10.1007/s10549-011-1430-5. [DOI] [PubMed] [Google Scholar]
- 5.Catalano PM, Kirwan JP. Maternal factors that determine neonatal size and body fat. Curr Diab Rep. 2001;1:71–77. doi: 10.1007/s11892-001-0013-y. [DOI] [PubMed] [Google Scholar]
- 6.Lepercq J, Hauguel-De Mouzon S, Timsit J, Catalano PM. Fetal macrosomia and maternal weight gain during pregnancy. Diabetes Metab. 2002;28:323–328. [PubMed] [Google Scholar]
- 7.Crume TL, Shapiro AL, Brinton JT, et al. Maternal fuels and metabolic measures during pregnancy and neonatal body composition: the healthy start study. J Clin Endocrinol Metab. 2015;100:1672–1680. doi: 10.1210/jc.2014-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berggren EK, Groh-Wargo S, Presley L, Hauguel-de Mouzon S, Catalano PM. Maternal fat, but not lean, mass is increased among overweight/obese women with excess gestational weight gain. Am J Obstet Gynecol. 2015 doi: 10.1016/j.ajog.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 10.Haghiac M, Basu S, Presley L, Serre D, Catalano PM, Hauguel-de Mouzon S. Patterns of adiponectin expression in term pregnancy: impact of obesity. J Clin Endocrinol Metab. 2014;99:3427–3434. doi: 10.1210/jc.2013-4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basu S, Haghiac M, Surace P, et al. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring) 2011;19:476–482. doi: 10.1038/oby.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jarvie E, Hauguel-de-Mouzon S, Nelson SM, Sattar N, Catalano PM, Freeman DJ. Lipotoxicity in obese pregnancy and its potential role in adverse pregnancy outcome and obesity in the offspring. Clin Sci (Lond) 2010;119:123–129. doi: 10.1042/CS20090640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29:274–281. doi: 10.1016/j.placenta.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. 2016;12:15–28. doi: 10.1038/nrendo.2015.189. [DOI] [PubMed] [Google Scholar]
- 15.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Kim JK. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab. 2006;4:417–419. doi: 10.1016/j.cmet.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr Opin Lipidol. 2009;20:379–385. doi: 10.1097/MOL.0b013e32832fa5c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haghiac M, Yang XH, Presley L, et al. Dietary Omega-3 Fatty Acid Supplementation Reduces Inflammation in Obese Pregnant Women: A Randomized Double-Blind Controlled Clinical Trial. PLoS One. 2015;10:e0137309. doi: 10.1371/journal.pone.0137309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasmussen KM, Abrams B, Bodnar LM, Butte NF, Catalano PM, Maria Siega-Riz A. Recommendations for weight gain during pregnancy in the context of the obesity epidemic. Obstet Gynecol. 2010;116:1191–1195. doi: 10.1097/AOG.0b013e3181f60da7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 21.Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF., 3rd Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118:1567–1582. doi: 10.1210/endo-118-4-1567. [DOI] [PubMed] [Google Scholar]
- 22.Lang I, Schweizer A, Hiden U, et al. Human fetal placental endothelial cells have a mature arterial and a juvenile venous phenotype with adipogenic and osteogenic differentiation potential. Differentiation. 2008;76:1031–1043. doi: 10.1111/j.1432-0436.2008.00302.x. [DOI] [PubMed] [Google Scholar]
- 23.Jones S, Bischof H, Lang I, et al. Dysregulated flow-mediated vasodilatation in the human placenta in fetal growth restriction. J Physiol. 2015;593:3077–3092. doi: 10.1113/JP270495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang I, Pabst MA, Hiden U, et al. Heterogeneity of microvascular endothelial cells isolated from human term placenta and macrovascular umbilical vein endothelial cells. Eur J Cell Biol. 2003;82:163–173. doi: 10.1078/0171-9335-00306. [DOI] [PubMed] [Google Scholar]
- 25.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vitseva OI, Tanriverdi K, Tchkonia TT, et al. Inducible Toll-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obesity (Silver Spring) 2008;16:932–937. doi: 10.1038/oby.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 28.Yang X, Haghiac M, Glazebrook P, Minium J, Catalano PM, Hauguel-de Mouzon S. Saturated fatty acids enhance TLR4 immune pathways in human trophoblasts. Hum Reprod. 2015;30:2152–2159. doi: 10.1093/humrep/dev173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beijar EC, Mallard C, Powell TL. Expression and subcellular localization of TLR-4 in term and first trimester human placenta. Placenta. 2006;27:322–326. doi: 10.1016/j.placenta.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 30.Ma Y, Kadner SS, Guller S. Differential effects of lipopolysaccharide and thrombin on interleukin-8 expression in syncytiotrophoblasts and endothelial cells: implications for fetal survival. Ann N Y Acad Sci. 2004;1034:236–244. doi: 10.1196/annals.1335.025. [DOI] [PubMed] [Google Scholar]
- 31.Krishnaswamy G, Kelley J, Yerra L, Smith JK, Chi DS. Human endothelium as a source of multifunctional cytokines: molecular regulation and possible role in human disease. J Interferon Cytokine Res. 1999;19:91–104. doi: 10.1089/107999099314234. [DOI] [PubMed] [Google Scholar]
- 32.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 33.Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes. 2008;57:3090–3098. doi: 10.2337/db08-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- 35.Metzger JD, Sebesta DK. Role of Endogenous Growth Regulators in Seed Dormancy of Avena fatua: I. Short Chain Fatty Acids. Plant Physiol. 1982;70:1480–1485. doi: 10.1104/pp.70.5.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boden G, Chen X, Rosner J, Barton M. Effects of a 48-h fat infusion on insulin secretion and glucose utilization. Diabetes. 1995;44:1239–1242. doi: 10.2337/diab.44.10.1239. [DOI] [PubMed] [Google Scholar]
- 37.Vidakovic AJ, Jaddoe VW, Gishti O, et al. Body mass index, gestational weight gain and fatty acid concentrations during pregnancy: the Generation R Study. Eur J Epidemiol. 2015;30:1175–1185. doi: 10.1007/s10654-015-0106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pathmaperuma AN, Mana P, Cheung SN, et al. Fatty acids alter glycerolipid metabolism and induce lipid droplet formation, syncytialisation and cytokine production in human trophoblasts with minimal glucose effect or interaction. Placenta. 2010;31:230–239. doi: 10.1016/j.placenta.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 39.Varastehpour A, Radaelli T, Minium J, et al. Activation of phospholipase A2 is associated with generation of placental lipid signals and fetal obesity. J Clin Endocrinol Metab. 2006;91:248–255. doi: 10.1210/jc.2005-0873. [DOI] [PubMed] [Google Scholar]
- 40.Qiao L, Guo Z, Bosco C, et al. Maternal High-Fat Feeding Increases Placental Lipoprotein Lipase Activity by Reducing SIRT1 Expression in Mice. Diabetes. 2015;64:3111–3120. doi: 10.2337/db14-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gauster M, Hiden U, van Poppel M, et al. Dysregulation of placental endothelial lipase in obese women with gestational diabetes mellitus. Diabetes. 2011;60:2457–2464. doi: 10.2337/db10-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Augsten M, Hackl H, Ebner B, et al. Fetal HDL/apoE: a novel regulator of gene expression in human placental endothelial cells. Physiol Genomics. 2011;43:1255–1262. doi: 10.1152/physiolgenomics.00109.2011. [DOI] [PubMed] [Google Scholar]
- 43.Catalano PM, Huston L, Amini SB, Kalhan SC. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am J Obstet Gynecol. 1999;180:903–916. doi: 10.1016/s0002-9378(99)70662-9. [DOI] [PubMed] [Google Scholar]
- 44.Catalano PM, Farrell K, Thomas A, et al. Perinatal risk factors for childhood obesity and metabolic dysregulation. Am J Clin Nutr. 2009;90:1303–1313. doi: 10.3945/ajcn.2008.27416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Widen EM, Whyatt RM, Hoepner LA, et al. Excessive gestational weight gain is associated with long-term body fat and weight retention at 7 y postpartum in African American and Dominican mothers with underweight, normal, and overweight prepregnancy BMI. Am J Clin Nutr. 2015;102:1460–1467. doi: 10.3945/ajcn.115.116939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abrams BF, Laros RK., Jr Prepregnancy weight, weight gain, and birth weight. Am J Obstet Gynecol. 1986;154:503–509. doi: 10.1016/0002-9378(86)90591-0. [DOI] [PubMed] [Google Scholar]
- 47.Waters TP, Huston-Presley L, Catalano PM. Neonatal body composition according to the revised institute of medicine recommendations for maternal weight gain. J Clin Endocrinol Metab. 2012;97:3648–3654. doi: 10.1210/jc.2012-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heerman WJ, Bian A, Shintani A, Barkin SL. Interaction between maternal prepregnancy body mass index and gestational weight gain shapes infant growth. Acad Pediatr. 2014;14:463–470. doi: 10.1016/j.acap.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starling AP, Brinton JT, Glueck DH, et al. Associations of maternal BMI and gestational weight gain with neonatal adiposity in the Healthy Start study. Am J Clin Nutr. 2015;101:302–309. doi: 10.3945/ajcn.114.094946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.