

Abstract
Sialic acids play many important roles in several physiological and pathological processes, including cancers, infection and blood diseases. Sialic acids are fragile and prone to fragmentation under electrospray ionization and matrix-assisted laser desorption/ionization. It is crucial to modify sialic acids for qualitative and quantitative identification of their change in abundance in complex biological samples. Permethylation is a method of choice for sialic acid stabilization, but the harsh conditions during permethylation may lead to the decomposition of O-acetyl groups. Esterification or amidation in solution effectively protects sialic acids, yet, it is not trivial to purify glycans from their reagents. Quantitative analysis of glycans can be achieved by labeling their reducing end using fluorescent tags. Loss of sialic acids during labeling is a major concern. In this study, we demonstrated the utility of sialic acids modification for the analysis sialyl oligosaccharides and glycopeptides. Without modification, sialic acids are partially or completely lost during sample preparation, leading to the presence of false glycans or glycopeptides in the sample. The stabilized sialic acids not only result in accurate identification of sialylated glycans, but they also improve the characterization of intact glycopeptides. The modification of sialic acids on the solid support facilitates analysis of glycans and their intact glycoproteins.
Keywords: Sialic Acid, Chemoenzymatic, Glycoproteomics, Mass Spectrometry
Graphical Abstract
INTRODUCTION
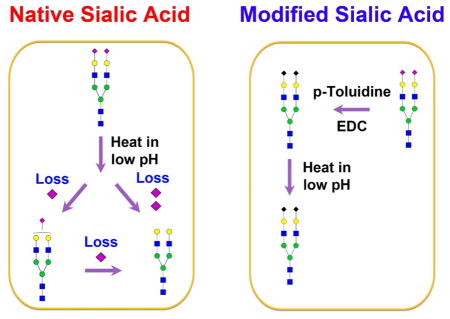
Sialic acids have a nine-carbon backbone and are the most diverse sugars found on glycan chains of mammalian cell surfaces 1,2. The diversity of sialic acids arises from their various linkages to the underlying sugar chain on the 2-position and different types of substitutions at position 4, 5, 7, 8, and 9. However, the sialic acids are fragile in acidic solution and can also be easily lost in matrix-assisted laser desorption/ionization (MALDI) or in positive-ion mode electrospray ionization (ESI)-mass spectrometry (MS) 3,4. In addition, acid-catalyzed hydrolysis can cause severe loss of sialic acids 5. Therefore, it is crucial to protect sialic acids to prevent their loss during sample preparation and MALDI or ESI ionization.
Chemical derivatization methods are commonly used for the stabilization of sialic acids. Methyl esterification converts carboxylic acid to methyl ester, producing strong positive-ion signals and stabilizing the sialic acid moieties under MALDI conditions 6,7. Perbenzolylation replaces carboxylic acids and hydroxides with benzoyl groups 8–10. However, the perbenzolylation is performed at 110°C under aerobic conditions, which is unfavorable for the modification of sialylated glycopeptides. Permethylation has been widely used for substituting carboxylic acids, amines and hydroxides with methyl groups in recent decades 11. Yet, the harsh conditions used for permethylation may impact the stability of O-acetyl groups 4. In addition, proteins or peptides are severely degraded under strong basic conditions. Recently, an amidation approach by carbodiimide coupling has been developed to convert the carboxylic acids to amide in the presence of both an amine constituent and a condensing agent 3. Amidation is performed in water solution at room temperature in the presence of a condensation reagent such as 1-ethyl-3-(3-dimentylaminopropyl) carbodiimide (EDC) 4. This chemical derivatization is an effective method for sialic acid modification. It is difficult to remove reagents such as EDC from the products when the reaction is performed in solution because the chemical compound has a similar hydrophobicity as the reacted product. As a result, it is challenging to get rid of excess reagents by chromatography-based purification 12,13.
Fluorescent labeling on glycan reducing-end has been widely used for the analysis of glycans without sialic acid stabilization 14–17. When N-glycans are released by PNGase F from their glycoproteins (on asparagine), they contain an active alditol on their reducing-end that can be tagged with a primary amine. For example, 2-aminobenzamide (2-AB) or 2-aminobenzoic acid (2-AA) has been used for tagging N-glycans by a reductive amination reaction 14,18. The tagged N-glycans are hydrophobic and photoactive so that they can be purified or separated by high performance liquid chromatography (HPLC) 19,20. To determine the structure of the labeled N-glycans, the same tag can be used to generate a dextran ladder as an internal standard for the assignment of glucose units (GU) to unknown glycans 19. The labeling reaction involving a two-step process, Schiff’s base formation and reduction, is usually conducted in 30% acetic acid (HOAc) and 70% dimethyl sulfoxide (DMSO) at 65°C for 2–4 h. Given the pKa of HOAc at 4.76, the pH for 30% acetic acid is approximately 2.02 (SI Table S1). It has been shown that sialic acids may be lost when they are subjected to high temperature (> 28°C) or extreme pH (such as low pH) (Ludger product guide: glycan labeling kit). A slightly lower temperature (60°C) is reported to reduce the acid-catalyzed loss of sialic acids within 2 h of labeling 21. Loss of sialic acids is indeed a concern during fluorescent labeling, possibly resulting in the formation of false glycans in the sample. However, the systematic characterization of sialic acids in low pH solution has not yet been conducted.
Solid-phase methods are an alternative approach for the analysis of glycoproteins via chemical immobilization 22 or affinity enrichment 23. Chemoenzymatic methods offer excellent specificity since they enable the permanent conjugation of amino acids 24,25 or glycans 22 of glycoproteins on the solid support for versatile chemical and enzymatic reactions. To study glycosite-containing peptides, glycans on glycoproteins are oxidized for specific enrichment of N-linked glycopeptides via hydrazide chemistry 22. To analyze glycans and intact glycopeptides, lysines or N-termini of glycoproteins are conjugated to amine-reactive solid supports via reductive amination 24–26. Several advantages have been demonstrated for the analysis of glycans using chemoenzymatic methods. For example, polymers (optimal cutting temperature medium (OCT)) for fixing tissues are successfully removed by washing while proteins are immobilized on the solid support 27; sialic acids are effectively protected using glycoprotein immobilization for glycan extraction (GIG) 24,28; and intact glycopeptides are directly analyzed by a solid-phase platform 17,26. These techniques have great potential for the quantitative analysis of protein glycosylation.
Questions are raised as to why the chemoenzymatic solid-phase method is necessary in the analysis of protein glycosylation in comparison with conventional approaches. As discussed previously, solid-phase methods such as GIG allow for the convenient modification of sialic acids, while the excess chemical reagents are removed by washing steps 26. Without protection of sialic acids in advance, it is highly probable that sialic acids will be partially or completely lost during sample preparation, especially in high temperature and low pH solution such as fluorescent labeling. In this study, we demonstrate that stabilization of sialic acids using GIG can mitigate the loss of sialic acids, preventing the occurrence of false glycans. Moreover, we investigate the possible loss of sialic acids in low pH solutions such as 0.1% and 1% Trifluoroacetic acid (TFA). The loss of sialic acids can be prevented by carbodiimide coupling and our method can improve sialic acid identification and quantification. The intact sialylated glycopeptides are simultaneously stabilized for accurate analysis.
EXPERIMENTAL PROCEDURES
Sialic Acid in DMSO-HOAc
Glycoproteins on resin were subjected to 30% HOAc at different temperatures (SI Figure S1a). To compare whether sialic acids are hydrolyzed in 30% acetic acid (typical conditions for glycan fluorescent labeling), one set of samples was incubated in DMSO-HOAc prior to sialic acid protection, while the second set was modified by pT in the presence of EDC. The first set was further modified by pT-EDC for protection of the remaining sialic acids; the second set was then incubated in the DMSO-HOAc solution. N-glycans were cleaved off by PNGase F digestion (37°C, overnight). The purified N-glycans were dried in Speed-Vac for QUANTITY (Quaternary Amine Containing Isobaric Tag for Glycan) labeling (65°C, 4 h) 29. Ten μL of 100 mM QUANTITY was mixed with N-glycans in the presence of 1 M NaCNBH3. The QUANTITY-tagged N-glycans were purified by either Carbograph or C18 solid-phase-extraction (SPE) column. Materials, chemical reagents, glycoprotein immobilization, TFA concentration effect on sialic acids, acid-catalyzed hydrolysis of intact glycoprotein, and mass spectrometry is described in Supporting Information.
RESULTS AND DISCUSSION
Acid-catalyzed Sialic Acid Hydrolysis
The goal of this set of experiments is to demonstrate whether sialic acids are affected by a high concentration of acids (30% HOAc) at different temperatures. Fluorescent labeling of glycans is typically conducted at 65°C in 30% HOAc and 70% DMSO 14,21. To inspect the dynamics of sialic acid hydrolysis, SGP (sialylglycopeptide) was incubated for 4 h at 4°C, 23°C (room temperature), 37°C, and 65°C respectively. One set of samples was directly treated by HOAc-DMSO prior to protection by pT-EDC; while another set was protected by pT-EDC before incubation in HOAc-DMSO solution (Figure 1a). This is a unique feature of the chemoenzymatic technique since samples can be conveniently treated in solution with glycoproteins immobilized on the solid support. According to the manufacturer’s specifications (Fushimi; Japan), SGP contains N2H2S2 (dominant), N2H2S, and H2HS (least) (where N = HexNAc, H = Hexose, and S = Neu5Ac; N-glycan core is excluded (N2H3)). When one sialic acid of N2H2S2 is lost, the dominant peak becomes N2H2S in MS; if both sialic acids of N2H2S2 are lost, it yields N2H2. Overall, we expected a decreased amount of N2H2S2 at higher temperatures, whereas N2H2 would increase significantly with increased temperature. The results in Figure 1 revealed such prediction: without protection by pT-EDC first, we observed a decreased amount of N2H2S2 due to the hydrolysis of sialic acids, or a significantly decreased amount of N2H2S2 at 65°C (Figure 1a). With protection by pT-EDC first, the hydrolysis of sialic acids was negligible (Figure 1b). More importantly, a gradually increased amount of N2H2S or N2H2 was observed from 4°C to 65°C, in which one or two sialic acids were lost (Figure 1c&e); while the N2H2S remained at the same level (Figure 1d). These results indicate that acid-catalyzed hydrolysis does occur during reducing-end labeling. The loss of sialic acids, however, can be effectively prevented if they are protected on the solid support first.
Figure 1. Acid-catalyzed hydrolysis of sialic acids in dimethyl sulfide (DMSO)-acetic acid (AA).

(a) Hydrolysis of native sialic acid (N2H2S2) (Sialylglycopeptide; SGP) in DMSO-AA at 4°C, 23°C, 37°C, and 65°C; (b) p-Toluidine stabilized sialic acid (N2H2S2) in DMSO-AA at 4°C, 23°C, 37°C, and 65°C; (c) Increase of N2H2S from hydrolysis of N2H2S2 in DMSO-AA; (d) No change on N2H2S due to the stabilized N2H2S2 with p-Toluidine; (e) Increase of N2H2 due to hydrolysis of N2H2S and N2H2S2 in DMSO-AA at different temperature; (f) Increase of N2H2 due to hydrolysis of N2H2S and N2H2S2 incubated 0.1% TFA from 2h to 24h (23°C). Triplicate experiments were conducted.
Sample preparation is often conducted at room temperature. To determine whether the prolonged incubation of samples in 0.1% TFA could lead to increased acid-catalyzed hydrolysis of sialic acids, a time course study was performed from 1 h to 24 h. We chose 0.1% TFA since it is commonly used for sample clean up (e.g., C18) or buffer for LC-MS. Figure 1f illustrates the increased amount of N2H2 glycan with the extended incubation in 0.1% TFA (all tested at room temperature). The data indicates that loss of sialic acids (from N2H2S2) occurs within one hour incubation in 0.1% TFA. The pKa of TFA is approximately 0.23 at room temperature, thus the pH of 0.1% TFA equals 2.09 (SI Table S1). The loss of sialic acids become fast with a longer incubation. This will cause the inaccurate characterization of intact sialylated glycopeptides if samples are stored in 0.1% TFA for a longer period of time.
Temperature effect on hydrolysis of sialic acids
To test the degree of hydrolysis of sialic acids in low pH at different temperature, we dissolved 10 μg of SGP in 0.1% TFA and incubated for 4 h at 4°C, 23°C, 37°C, and 65°C. Figure 2a demonstrates the relative abundance of each N-glycan at different temperatures. Several observations were made: (1) the amount of native sialylated N-glycans (N2H2S2) decreases when incubated in high temperature; (2) the intermediate glycans (N2H2S or NHS) also decrease with increased temperature; (3) however, glycans that lost both sialic acids (N2H2) are highly abundant at an elevated temperature. These results indicate that loss of sialic acids is a severe issue in the analysis of sialylated glycans since samples are often treated in 0.1% TFA at room temperature or 37°C.
Figure 2. Effect of 0.1% TFA on the hydrolysis of Sialylglycopeptide (SGP) at different temperatures.

(a) Without hydrolysis, SGP consists of N2H2S2 only. Incubated in 0.1% TFA, it gradually hydrolyzed at low temperatures while most sialic acids were lost at an elevated temperature (65°C). SGP glycan was converted to asialo N2H2. The relative abundance was N2H2S2 (4°C) > N2H2S2 (23°C) > N2H2S2 (37°C) > N2H2S2 (65°C), while N2H2 (4°C) < N2H2 (23°C) < N2H2 (37°C) < N2H2S2 (65°C); (b) Increased asialo N-glycans in bovine Fetuin; (c) pT-stabilized sialic acids remained constant at different temperatures, while without pT, the sialic acid content was significantly reduced at an elevated temperature.
We then used bovine fetuin to illustrate the loss of sialic acids and the resulting increase of asialo N-glycans. Bovine fetuin has five major N-glycans: N2H2S, N2H2S2, N3H3S2, N3H3S3, and N3H3S4. If sialic acids are completely removed, the two remaining asialo N-glycans are N2H2 and N3H3. We compared the change of two asialo N-glycans and their sialylated N-glycans (N2H2S2 ⇒ N2H2 and N3H3S3 ⇒ N3H3). Figure 2b shows the relative abundance of N2H2 and N3H3 incubated at different temperatures. At low temperature (4°C or 23°C), there are few N2H2 detectable, while their abundance significantly increased at 37°C up to 65°C. Compared to the amount of N2H2 at 4°C, it increases by 10 fold at 37°C and ~200 fold at 65°C. Similar observations were made for N3H3. Its relative abundance increased by ~3 fold at 37°C and ~10 fold at 65°C. On the other hand, the reverse change is observed for sialylated N-glycans of N2H2S2 and N3H3S3 (Figure 2c). Without pT modification, both N2H2S2 and N3H3S3 have decreased abundance versus the elevated temperature, indicating their enhanced hydrolysis. As a comparison, the modified N2H2S2 and N3H3S3 have been well preserved even though they are incubated in 0.1% TFA at 65°C for longer than 4 h. These results reveal that it is important and necessary to protect sialic acids in order to prevent acid-catalyzed hydrolysis; the pT stabilization is effective and can prevent hydrolysis at an elevated temperature.
Perspectives of Glycan and Intact Glycopeptides
Stabilization of sialic acids is an effective approach for accurate characterization of protein glycosylation. Without sialic acid modification, the hydrolysis of sialic acids can complicate the analysis and generate false species which do not exist endogenously in the sample. To this end, we plotted a schematic diagram to illustrate how a sialylated glycan or glycopeptide can produce additional glycans after the hydrolysis of sialic acids (SI Figure S2). Using N2H2S2 as an example, it can change to N2H2S and N2H2 if one or two sialic acids are lost. Samples can contain three N-glycans, including N2H2S2, N2H2S, and N2H2 (SI Figure S2a). For intact glycopeptides, they can generate quite a few additional intact glycopeptides by the partial or complete loss of sialic acids. For example, as shown in SI Figure S2 and SI Table S4, an intact glycopeptide with N3H3S4 can form four additional intact glycopeptides, including peptides linked to N3H3S3, N3H3S2, N3H3S, and N3H3. In addition, the second glycopeptide that may have the same or different peptide sequence has asparagine conjugated with N3H3S3; after partial hydrolysis, it generates N3H3S2, N3H3S, and N3H3. These additional glycopeptides will undoubtedly change the relative abundance of each intact glycopeptide, causing inaccurate or even incorrect characterization of glycopeptides from the sample. Therefore, it is essential to stabilize sialylated glycopeptides in order to identify the actual abundance of each glycopeptide in the complex biological sample.
Our studies show that sialic acids can be affected by acid-catalyzed hydrolysis in acidic buffer at room or elevated temperature. Sialylated glycans are often reacted in acid solution without stabilization. For example, N-glycans are labeled with fluorescent compounds such as 2-AA, 2-AB, 2-AP (2-Aminopyridine), via Schiff’s base reaction. The labeling is typically conducted in 30% acetic acid and 70% DMSO, whose pH is approximately 2.02 (SI Table S1). Samples are also heated up to 65°C for 2–4 h for completion. As shown in Figure 1, over 50% of sialic acids could be lost during the reaction (65°C for 4 h) without sialic acid modification, while the sialylated glycans remain stable after modification. Therefore, it is likely that some of the sialylated glycans have been de-sialylated by acid-catalyzed hydrolysis. A lower temperature may be applied for labeling; however, it requires a much longer incubation time to be completed. The longer the incubation, the more extensive the hydrolysis (Figure 1d).
Acid has been widely used in sample preparation. One example is to use 0.1% TFA for peptide cleanup. As calculated in SI Table S1, the pH of 0.1% TFA is approximately 2.09. For C18 cartridges, the cleanup procedure is usually performed at room temperature. In 0.1% TFA, sialic acid is partially lost after 4 h incubation (Figure 2a), resulting in the formation of false glycans after C18 cleanup. Our data on Fetuin N-glycans in 0.1% TFA demonstrated that asialo N-glycan (N3H3) at 23°C is increased by 2-fold in comparison to its abundance at 4°C (Figure 2b). The sialylated N-glycans such as N3H3S3 were slightly decreased, as expected (Figure 2c). The second example is to use 1% TFA in 80% ACN as the washing buffer for intact glycopeptide enrichment with a hydrophilic interaction liquid chromatography (HILIC) column 30 or 1% TFA used in mobile phases for increased glycopeptide enrichment efficiency 31. When using 1% TFA, we observed a significant loss of sialic acids in fetuin N-glycans at room temperature (SI Figure S3). It is thus possible that we lost some of the sialic acids during HILIC enrichment and separation.
Several strategies can be applied for the stabilization of sialic acids. In addition to our method, sialic acids can be modified by esterification, such as methyl esterification 7,32 and ethyl esterification 33. Non-reduced N-glycans are directly reacted with methanol or ethanol in the presence of equal molar of 1-Hydroxybenzotriazole hydrate and EDC. Intact glycopeptides can also be modified by esterification 34, providing an approach for the stabilization of sialylated glycopeptides. The chemical compound, 4-(4,6-dimethoxy-1,3,5-triazin-2yl)-4-methlmorpholinium chloride (DMT-MM) has been synthesized for the modification of sialic acids 3; methyl ester can also be achieved by dissolving glycans in methanol and treating with DMT-MM 35, enabling concomitant differentiation of α2,3 and α2,6. Amidation is another effective method for stabilizing sialic acids. Acetohydrazide can modify sialylated glycans in the presence of EDC at room temperature 4,36. Methylamidation is performed in the presence of methylamine and (7-azabenzotriazol-1-yloxy) trispyrrolidinophosphonium hexafluorophosphate (PyAOP) 37. These methods are very effective for stabilizing sialic acids. Adapting them to the solid-phase can further simplify the modification of sialylated glycans and glycopeptides.
CONCLUSION
Sialic acids are one of the important forms of protein glycosylation that regulates diverse biological functions. Accurate characterization of protein sialylation can provide insight into how sialylation is associated with physiology and pathology. Loss of sialic acids has been a major concern in sample preparation. Samples are often processed in acidic buffer at room or elevated temperature, causing significant hydrolysis of sialic acids.
In this study, we characterized the dynamics of the acid-catalyzed hydrolysis of sialic acids. This characterization was conducted on the solid support, in which glycoproteins are immobilized on Aminolink resin. We compared N-glycans with and without p-Toluidine modification, showing that sialic acids are hydrolyzed even at room temperature and over 50% are lost at 65°C. On the other hand, sialic acids modified by pT are very stable in low pH even at elevated temperatures. Without modification, sialic acids on sialylated glycopeptides are also partially or completely lost during sample preparation. This led to the increased number of false glycopeptides, consequently impacting the analysis of glycopeptides. It is thus significant to stabilize sialylated glycans for the accurate characterization of both glycans and their glycopeptides (SI Table S2).
This study demonstrates that it is a prerequisite to characterize protein sialylation by first stabilizing sialic acids. We confirm the loss of sialic acids as a result of fluorescent labeling and HILIC cleanup. Carbodiimide coupling on the solid support, together with other methods for sialic acid modification, should be incorporated for glycan and glycopeptide analysis.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health, National Cancer Institute, the Early Detection Research Network (EDRN, U01CA152813), the Clinical Proteomic Tumor Analysis Consortium (CPTAC, U24CA160036), National Heart Lung and Blood Institute, Programs of Excellence in Glycosciences (PEG, P01HL107153), and the National Institute of Allergy and Infectious Diseases (R21AI122382), by Maryland Innovation Initiative (MII), and by The Patrick C. Walsh Prostate Cancer Research Fund.
Abbreviation
- GIG
glycoprotein immobilization for glycan extraction
- LC
liquid chromatography
- MS
mass spectrometry
- QUANTITY
Quaternary Amine Containing Isobaric Tag for Glycan
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have declared no conflict of interest.
SUPPORTING INFORMATION AVAILABLE
The Supporting Information is available free of charge via the Internet at http//pubs.acs.org.
References
- 1.Varki NM, Varki A. Lab Invest. 2007;87:851–857. doi: 10.1038/labinvest.3700656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Varki A. Trends Mol Med. 2008;14:351–360. doi: 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sekiya S, Wada Y, Tanaka K. Anal Chem. 2005;77:4962–4968. doi: 10.1021/ac050287o. [DOI] [PubMed] [Google Scholar]
- 4.Toyoda M, Ito H, Matsuno Y-k, Narimatsu H, Kameyama A. Anal Chem. 2008;80:5211–5218. doi: 10.1021/ac800457a. [DOI] [PubMed] [Google Scholar]
- 5.Varki A, Diaz S. Anal Biochem. 1984;137:236–247. doi: 10.1016/0003-2697(84)90377-4. [DOI] [PubMed] [Google Scholar]
- 6.Neises B, Steglich W. Angew Chem Int Ed. 1978;17:522–524. [Google Scholar]
- 7.Powell AK, Harvey DJ. Rapid Commun Mass Spectrom. 1996;10:1027–1032. doi: 10.1002/(SICI)1097-0231(19960715)10:9<1027::AID-RCM634>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 8.Daniel PF. Methos Enzymol. 1987;138:94–116. doi: 10.1016/0076-6879(87)38009-7. [DOI] [PubMed] [Google Scholar]
- 9.Chen P, Werner-Zwanziger U, Wiesler D, Pagel M, Novotny MV. Anal Chem. 1999;71:4969–4973. doi: 10.1021/ac990674w. [DOI] [PubMed] [Google Scholar]
- 10.Perreault H, Costello CE. Can J Chem. 1996;74:1682–1695. [Google Scholar]
- 11.Juhasz P, Costello CE. J Am Soc Mass Spectrom. 1992;3:785–796. doi: 10.1016/1044-0305(92)80001-2. [DOI] [PubMed] [Google Scholar]
- 12.Yang S, Zhang H. Curr Protoc Chem Biol. 2014;6:191–208. doi: 10.1002/9780470559277.ch140085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang S, Rubin A, Eshghi ST, Zhang H. Proteomics. 2016;16:241–256. doi: 10.1002/pmic.201500266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bigge J, Patel T, Bruce J, Goulding P, Charles S, Parekh R. Anal Biochem. 1995;230:229–238. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- 15.Harvey DJ. J Am Soc Mass Spectrom. 2000;11:900–915. doi: 10.1016/S1044-0305(00)00156-2. [DOI] [PubMed] [Google Scholar]
- 16.Evangelista RA, Liu MS, Chen FTA. Anal Chem. 1995;67:2239–2245. [Google Scholar]
- 17.Suzuki H, Müller O, Guttman A, Karger BL. Anal Chem. 1997;69:4554–4559. doi: 10.1021/ac970090z. [DOI] [PubMed] [Google Scholar]
- 18.Prien JM, Prater BD, Qin Q, Cockrill SL. Anal Chem. 2010;82:1498–1508. doi: 10.1021/ac902617t. [DOI] [PubMed] [Google Scholar]
- 19.Guile GR, Rudd PM, Wing DR, Prime SB, Dwek RA. Anal Biochem. 1996;240:210–226. doi: 10.1006/abio.1996.0351. [DOI] [PubMed] [Google Scholar]
- 20.Ruhaak LR, Deelder AM, Wuhrer M. Anal Bioanal Chem. 2009;394:163–174. doi: 10.1007/s00216-009-2664-5. [DOI] [PubMed] [Google Scholar]
- 21.Ruhaak L, Zauner G, Huhn C, Bruggink C, Deelder A, Wuhrer M. Anal Bioanal Chem. 2010;397:3457–3481. doi: 10.1007/s00216-010-3532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Li X-j, Martin DB, Aebersold R. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 23.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K-i, Takahashi N, Isobe T. Nat Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- 24.Yang S, Li Y, Shah P, Zhang H. Anal Chem. 2013;85:5555–5561. doi: 10.1021/ac400761e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun S, Shah P, Eshghi ST, Yang W, Trikannad N, Yang S, Chen L, Aiyetan P, Höti N, Zhang Z. Nat Biotechnol. 2015;34:84–88. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S, Mishra S, Chen L, Zhou J-y, Chan DW, Chatterjee S, Zhang H. Anal Chem. 2015;87:9671–9678. doi: 10.1021/acs.analchem.5b01663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah P, Zhang B, Choi C, Yang S, Zhou J, Harlan R, Tian Y, Zhang Z, Chan DW, Zhang H. Anal Biochem. 2015;469:27–33. doi: 10.1016/j.ab.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah P, Yang S, Sun S, Aiyetan P, Yarema KJ, Zhang H. Anal Chem. 2013;85:3606–3613. doi: 10.1021/ac3033867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S, Wang M, Chen L, Yin B, Song G, Turko IV, Phinney KW, Betenbaugh MJ, Zhang H, Li S. Sci Rep. 2015;5:17585–17593. doi: 10.1038/srep17585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmisano G, Lendal SE, Engholm-Keller K, Leth-Larsen R, Parker BL, Larsen MR. Nat Protoc. 2010;5:1974–1982. doi: 10.1038/nprot.2010.167. [DOI] [PubMed] [Google Scholar]
- 31.Mysling S, Palmisano G, Højrup P, Thaysen-Andersen M. Anal Chem. 2010;82:5598–5609. doi: 10.1021/ac100530w. [DOI] [PubMed] [Google Scholar]
- 32.Karkas JD, Chargaff E. J Biol chem. 1964;239:949–957. [PubMed] [Google Scholar]
- 33.Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M. Anal Chem. 2014;86:5784–5793. doi: 10.1021/ac500335t. [DOI] [PubMed] [Google Scholar]
- 34.Gomes de Oliveira AG, Roy R, Raymond C, Bodnar ED, Tayi VS, Butler M, Durocher Y, Perreault H. Rapid Commun Mass Spectrom. 2015;29:1817–1826. doi: 10.1002/rcm.7287. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler SF, Domann P, Harvey DJ. Rapid Commun Mass Spectrom. 2009;23:303–312. doi: 10.1002/rcm.3867. [DOI] [PubMed] [Google Scholar]
- 36.Gil GC, Iliff B, Cerny R, Velander WH, Cott KEV. Anal Chem. 2010;82:6613–6620. doi: 10.1021/ac1011377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Qiu H, Lee RK, Chen W, Li J. Anal Chem. 2010;82:8300–8306. doi: 10.1021/ac101831t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.