

Abstract
Common autoimmune diseases are relatively heterogeneous with both genetic and environmental factors influencing disease susceptibility and progression. As the populations in developed countries age, these chronic diseases will become an increasing burden in human suffering and health care costs. By contrast, rare immune diseases that are severe and develop early in childhood are frequently monogenic and fully penetrant, often with a Mendelian inheritance pattern. Although these may be incompatible with survival or cured by hematopoietic stem cell transplantation, we will argue that they constitute a rich source of genetic insights into immunological diseases. Here, we discuss five examples of well-studied Mendelian disease-causing genes and their known or predicted roles in conferring susceptibility to common, polygenic diseases of autoimmunity. Mendelian disease mutations, as experiments of nature, reveal human loci that are indispensable for immune regulation and, therefore, most promising as therapeutic targets.
Introduction
The genetic basis for autoimmunity, defined as genomic DNA sequence variants conferring disease, has been intensively studied in common autoimmune disorders including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), type 1 diabetes (T1D), Crohn's disease, celiac disease, and others with the intention of finding new causative pathways and potential therapeutic targets. Identification of specific MHC alleles as important for disease susceptibility was a significant initial success1. Since that time, genome-wide association (GWA) studies have correlated an abundance of single nucleotide polymorphisms (SNPs) with autoimmune disease susceptibility2,3. However, even individuals harboring large numbers of susceptibility SNPs did not necessarily develop disease and those who developed disease did not necessarily have all, or even most, of the susceptibility SNPs, making the genetic argument for causality tenuous. The wide availability of whole exome (WES) or whole genome (WGS) DNA sequencing has offered a more comprehensive view into genome sequence variation related to immunological disease. It allows, far more efficiently than ever before, a means to associate large numbers of genomic differences, especially rare and private variants, with disease susceptibility. It also provides the most effective means yet to solve the molecular basis of rare, monogenic immune disorders. This allows gradual building of a catalog of autoimmune disease genes and pathways. Here, we discuss integrating findings in Mendelian genomics research on rare diseases with GWA studies in common autoimmune diseases to elucidate key pathways and potential therapeutic targets.
An important distinction between GWA studies and gene discovery in Mendelian disorders is that they detect fundamentally different types of gene variation. GWA studies query SNPs to find a statistical enrichment in diseased individuals compared to controls4. Typically, associations were based on hybridization chips that interrogate common SNPs, with 1-5% allele frequencies, which, because they have been selected to have sufficient fitness to be inherited through generations broadly within the population, have reduced disease impact. Because most of the genome is not protein-coding, many such SNPs were in unclassified regions of the genome rather than the exome2. Moreover, disease-associated SNPs generally have quantitatively small susceptibility effects, and few are sufficiently well understood to be diagnostically or therapeutically actionable5. By contrast, Mendelian genomics identifies deleterious and highly penetrant nucleotide variants and copy number variations (CNVs) that mostly affect protein-coding exome sequences in rare individuals with severe immune disorders. Less severe alleles of these same genes allow sufficient reproductive fitness that they can be associated with common autoimmune diseases. Already, several genes (AIRE, FOXP3, CTLA4) detected in both gene discovery approaches, are clear causes of autoimmunity. Also, both approaches are converging on genomic DNA sequencing which is rapidly shifting from WES to WGS. The latter provides a far superior computational exome and enables significant advances in understanding risk loci that map to non-coding regions of the genome or in genes with unknown function. Identifying the regions of the genome and pathways underlying common and rare autoimmune disorders will facilitate translation of these findings to clinical benefit.
Gene discovery in rare Mendelian immune disorders
Common autoimmune disorders are not monogenic or Mendelian in their inheritance patterns. Instead, these disorders, which affect millions of patients, are caused by a constellation of susceptibility genes and environmental exposures that are insufficiently defined at present. The first attempts at gene associations came from human twin studies in which disease concordance between two identical twins compared to fraternal twins or the normal population gave a quantitative estimate of heritability6. Similarly, the sibling recurrence risk in non-twin siblings was higher for most common autoimmune diseases7. Though generally involving small numbers of patients, such studies give a clear indication of genetic susceptibility to autoimmune disease but no actual disease gene identification. Linkage dysequilibrium (LD) studies in both humans and mice were successful in identifying gene variation in autoimmune diseases but these investigations were laborious and yielded genes with small effects on disease7. The process of associating genetic variation with autoimmune disease was accelerated by GWA studies that correlated SNPs with disease for many autoimmune conditions4. All of these methods pointed toward a mode of inheritance in which susceptibility alleles were inherited in complex combinations with incomplete penetrance perhaps due to environmental factors7. One challenge was the fact that all of the genetic variation identified through LD or GWA studies seemed to fall short of the apparent heritability of disease raising the problem of “missing heritability.” Understanding how genetic variation could contribute to disease pathogenesis and how it could be targeted therapeutically remained elusive. New approaches were needed to answer these questions more effectively.
The emergence of inexpensive, high-throughput, next-generation (nextgen) DNA sequencing (NDS) technology enabling complete exome and genome interrogation offered a complementary approach to GWA and LD studies5. These technologies had a profound impact on our conception of the genome by revealing millions of rare and private variants in each sequenced sample, far more variation than had been previously conceived5. NDS was a powerful new approach to identifying causative genes in rare and severe/early onset autoimmune disorders that are inherited in a Mendelian fashion and, therefore, are likely to be largely monogenic. The lens provided by these rare diseases allowed us to catch glimpses of genes and pathways that, when altered in more subtle ways, likely contribute to susceptibility to the more common autoimmune diseases. These findings can help guide interventions and therapies. As shown in Figure 1, the more common diseases with greater allele frequency can be analyzed using GWA studies or, more comprehensively, WES/WGS to identify genetic variants associated with disease phenotype. We argue that, despite their rarity, the Mendelian immune diseases with high penetrance are valuable to discover gene mutations that can be examined mechanistically to provide direct evidence in humans for the importance of particular genes or pathways in immune health. Indeed, we discuss below several examples of DNA sequence variants in genes that are implicated in both rare Mendelian immune disorders and GWA studies of common autoimmune diseases. As genetic research on autoimmune diseases progresses and shifts from WES to WGS, we predict that genetic defects in previously poorly understood regions of the genome, including intergenic and intronic regions, will shed more light on existing GWA data and population studies that implicate non-coding or unknown genes as risk loci.
Figure 1.
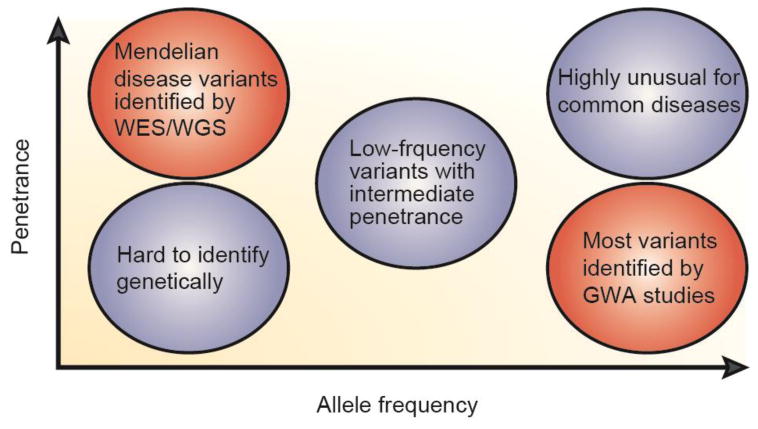
Genetic research has identified common variants in common diseases through GWA studies (bottom right) and rare and private disease-causing mutations in uncommon diseases through WES/WGS (top left). (Adapted from Nature Reviews Genetics; May 2008; vol 9:356.)
In this essay, we have chosen to highlight 5 monogenic diseases with autoimmune features that already have or are likely to be linked to common, polygenic autoimmune disorders. This is not meant to be an exhaustive list of genetic autoimmune disorders, which would be beyond the scope of this essay. Instead, we hope to describe some classic Mendelian autoimmune diseases that have been well-studied and some recently discovered disorders that we have investigated to underscore the synergy between WES/WGS analysis in Mendelian diseases and GWA studies in common diseases. Many other important autoimmune gene mutations (e.g., those causing interferonopathies, complement deficiency, STAT3 and STAT1 gain-of-function, CD25 deficiency, IL-10 deficiency, STING gain-of-function, PLCG2 gain-of-function) have been described and similarly offer important insights. The genes we will discuss as examples are listed in Table 1.
Table 1.
The genes discussed here are listed together with the Mendelian and common diseases in which they have been implicated. APECED: autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; IPEX: immunodysregulation polyendocrinopathy enteropathy X-linked syndrome; ALPS: autoimmune lymphoproliferative syndrome; PASLI: p110δ-activation with senescent T cells, lymphadenopathy, and immunodeficiency; APDS: activated p110δ syndrome; CHAI: CTLA4 haploinsufficiency with autoimmune infiltration.
| GENE | Basic Function | Mendelian Disease with Causative Mutation | Common Disease with Susceptibility Association |
|---|---|---|---|
| AIRE | Transcriptional regulator for tolerance to self antigens | APECED | Rheumatoid arthritis |
| FOXP3 | Transcription factor for regulatory T cells | IPEX | Vitiligo |
|
FAS (and related) |
Pro-apoptotic | ALPS | Systemic lupus erythrematosis |
|
PI3K (several genes) |
Cell growth, proliferation, and survival | PASLI/APDS | PI3K inhibition ameliorates RA, SLE |
|
CTLA4 (and related) |
T cell inhibitory molecule | CHAI | Myasthenia gravis, alopecia areata |
The AIRE gene and APECED disease
A clinical condition known as APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy) was identified as an autosomal recessive, fully penetrant Mendelian disease by clinicians as early as the 1960s8. Diagnosis of APECED is made when a patient presents with two of the following three diagnostic criteria: chronic mucocutaneous candidiasis, chronic hypoparathyroidism, and autoimmune adrenal insufficiency. APECED was found in 1997 to be caused by deleterious mutations in the AIRE (autoimmune regulator) gene9,10, thereby initiating intense interest in this gene product as a critical molecule preventing autoimmunity in healthy individuals. Mice with genetically engineered Aire gene deficiency also suffered autoimmunity11,12. This work defined Aire as a transcriptional regulator that promotes ectopic expression of tissue-specific antigens in thymic epithelial cells to promote negative selection of autoreactive T cells during development. As would be predicted for a key mediator of immunological tolerance, subsequent GWA studies identified an association between variants in the AIRE gene and susceptibility to autoimmune disease, including RA 13,14. Although not currently feasible to therapeutically modulate a transcriptional regulator such as AIRE, the discovery of APECED/AIRE defects highlights the concept that uncovering the genetic basis for rare, severe Mendelian immune disorders can point toward genes of interest to evaluate variants in more common and complex diseases.
The FOXP3 gene and IPEX disease
Another rare autoimmune disease called IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked syndrome) was described in the 1982 and genetically linked in 2000 to the recessive mutations in the FOXP3 gene15,16. Patients with IPEX often present with early-onset insulin-dependent diabetes mellitus, diarrhea, and dermatitis. Foxp3-deficient mice exhibit a similar phenotype, which was attributed to severe autoimmune infiltration of lymphocytes into peripheral tissues due to loss of regulatory T cells (Tregs). A large number of publications have now defined the role of FOXP3 as a master transcription factor in Treg development and function. Thus, IPEX patients, combined with consequent genetic studies in mice, provided important insights into the mechanisms of immunological tolerance and autoimmunity. Genetic variants in FOXP3 have now been identified through GWA studies in humans with autoimmunity and underscore the wider importance of Tregs in healthy immune homeostasis17,18. Therapies for autoimmune diseases that are focused on increasing the number and function of Tregs are being intensely pursued and may offer promise as a means of therapeutic suppression of autoimmunity.
The FAS pathway and ALPS disease
The autoimmune lymphoproliferative syndrome (ALPS) is characterized by benign lymphadenopathy and splenomegaly, autoimmune cytopenias (hemolytic anemia, thrombocytopenia), accumulation of lymphocytes, particularly mature peripheral alpha-beta T cells that do not express CD4 or CD8 (double-negative T cells), and a marked propensity to develop B cell lymphomas. Most ALPS patients have heterozygous dominant-interfering mutations in the gene encoding the tumor necrosis factor receptor superfamily cell death inducing receptor, FAS. Examination of larger cohorts of patients revealed that ALPS is also caused by mutations in the FASL, CASP10, and NRAS genes19-26. A large body of work in human patients and mice defined FAS as a critical regulator of lymphocyte homeostasis. This pathway is important for controlling mature lymphocyte cell fate through the activation of a caspase cascade to mediated the extrinsic pathway of immune cell apoptosis. The autoimmunity observed in ALPS patients seems to result from failure to properly cull superfluous T cells. From the genetic elucidation of this pathway, lymphocyte cell death and homeostasis emerged as an additional layer of peripheral immune tolerance (beyond AIRE-mediated thymic deletion and FOXP3-mediated Treg suppression). Interestingly, the penetrance of clinical disease in individuals harboring dominant-interfering Fas mutations is only 60% indicating that other genes or environmental factors are critical27. Again, GWA studies identified polymorphisms in the FAS gene that confer susceptibility to autoimmunity28-31.
The PI3K pathway and PASLI/APDS disease
An immunodeficiency disease with lymphoproliferation and predisposition to lymphoma and autoimmunity was recently found to be caused by gain-of-function, heterozygous mutations in PIK3CD32,33 and PIK3R134,35. This disorder has been called p110δ-activation with senescent T cells, lymphadenopathy, and immunodeficiency (PASLI) disease/activated p110δ syndrome (APDS). Multiple groups have reported additional families and expanded the spectrum of clinical findings36-38. The PI3K signaling pathway promotes cell growth, survival, and proliferation. PASLI/APDS patients have revealed that hyperactive PI3K signaling in immune cells disrupts immune homeostasis, increasing susceptibility to autoimmune cytopenias and other forms of autoimmunity (e.g., arthritis). Although there is little evidence for clear direct associations between variants in PI3K genes and autoimmune diseases, there is strong evidence that inhibition of the leukocyte-enriched PI3K molecules ameliorates disease in several autoimmune conditions39,40. The activation of the mTOR kinase due to PI3K suggest that rapamycin or direct p110δ inhibitors may be effective in select autoimmune conditions.
The CTLA4 gene and CHAI disease
The CTLA4 gene has been the subject of much research since its discovery as an inhibitory receptor that binds B7 ligands and suppresses T cell activation by dampening signals through the costimulatory CD28 receptor41,42. Mice genetically deficient in the ctla4 gene exhibit excessive T cell proliferation and infiltration leading to broad tissue destruction and death in the perinatal period 43. Subsequent GWA studies of patients with organ-destructive autoimmunity identified associations with human CTLA4 variants44-46. More recently, a Mendelian human autoimmune disorder caused by heterozygous mutation in CTLA4 has been delineated and termed CTLA4 haploinsufficiency with autoimmune infiltration (CHAI) disease47. These patients suffer from T lymphocyte infiltration in the lung, gut, and brain, and their disease points to an important difference between humans and mice since, unlike the human patients, mice with one mutant ctla4 allele show no overt phenotype. These findings emphasize that the expression level of CTLA4 in humans must be tightly regulated to ensure immune tolerance. Indeed, another Mendelian immune disease in humans has now been linked to reduced CTLA4 expression. Homozygous, loss-of-function mutations in the gene encoding LRBA48 have been found to result in reduced CTLA4 protein expression due to excessive protein degradation in lysosomes. For this reason, patients with LRBA deficiency clinically phenocopy CHAI disease with autoimmune tissue infiltration, and we have termed this disorder LATAIE (LRBA deficiency with autoantibodies, Treg defects, autoimmune infiltration, and enteropathy) 49. Both CTLA4 haploinsufficiency (CHAI disease) and recessive loss of LRBA (LATAIE disease) result in lymphadenopathy/splenomegaly, defective immunoglobulin production, autoantibodies, Treg defects, and tissue infiltration. Disease in CHAI patients has only 50% penetrance, and it remains to be determined what the penetrance will be in patients with LRBA deficiency50. Thus, CTLA4 is a substantiated target for modulation in autoimmune diseases, and therapies to mimic inhibitory activity of CTLA4 using a CTLA4-Ig fusion protein have recently been FDA-approved.
Conclusions
Despite the success of GWA studies in identifying many associations between common DNA sequence variants and common autoimmune disorders, tangible translational advances in the form of clinical treatments or reliable biomarkers of disease prognosis or progression have not yet been widely achieved. Similarly, the field of Mendelian genomics research has been fruitful in identifying causative gene mutations that are sometimes not actionable for therapy or are considered fairly narrowly in the context of rare disease. However, each approach has the capability of surmounting the shortcomings of the other to achieve a useful genetic understanding of autoimmunity. We propose that the next wave of genomics research will integrate more broadly the lessons learned from GWA and Mendelian genomics research to prioritize the gene candidates that contribute to autoimmunity in both types of studies. Cell signaling and immune effector functions are likely complex adaptive systems in which a web of a large number of self-organizing molecular interactions create unexpected robustness punctuated by tipping points to severe dysfunction in response to gene variation51. To understand this, systems approaches combining genomic sequencing with gene expression, proteomics, and modeling data will be instrumental in defining the cellular signaling network regulating autoimmunity, both broadly and within more narrowly defined disease phenotypes52.
Highlights.
Mendelian genomics research identifies key autoimmunity genes in humans
Study of rare Mendelian disorders is complementary to GWA studies of common diseases
Immune tolerance is critically regulated by AIRE, FOXP3, FAS, PI3K, and CTLA
Acknowledgments
This work was supported by the Intramural Research Program of NIH, NIAID. C.L.L. was supported by the Postdoctoral Research Associate (PRAT) Fellowship, NIGMS, NIH. The authors thank Drs. Helen Su and Yu Zhang for critically reading the manuscript.
References
- 1.Svejgaard A, et al. HLA and diabetes. Progress in clinical and biological research. 1982;103 Pt B:55–64. [PubMed] [Google Scholar]
- 2*.Fahr KK, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. The authors assess genomic DNA variants in 21 autoimmune disorders and find that ∼90% of causal variants are non-coding with many mapping to immune-cell enhancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 4.Wellcome Trust Case Control, C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogdanos DP, et al. Twin studies in autoimmune disease: genetics, gender and environment. Journal of autoimmunity. 2012;38:J156–169. doi: 10.1016/j.jaut.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Broadley SA, Deans J, Sawcer SJ, Clayton D, Compston DA. Autoimmune disease in first-degree relatives of patients with multiple sclerosis. A UK survey. Brain : a journal of neurology. 2000;123(Pt 6):1102–1111. doi: 10.1093/brain/123.6.1102. [DOI] [PubMed] [Google Scholar]
- 8.Blizzard RM, Kyle M. Studies of the Adrenal Antigens and Antibodies in Addison's Disease. The Journal of clinical investigation. 1963;42:1653–1660. doi: 10.1172/JCI104851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finnish-German AC. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nature genetics. 1997;17:399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 10.Nagamine K, et al. Positional cloning of the APECED gene. Nature genetics. 1997;17:393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 11.Anderson MS, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 12.Ramsey C, et al. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Human molecular genetics. 2002;11:397–409. doi: 10.1093/hmg/11.4.397. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Lozano JR, et al. Association of the AIRE gene with susceptibility to rheumatoid arthritis in a European population: a case control study. Arthritis research & therapy. 2013;15:R11. doi: 10.1186/ar4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terao C, et al. The human AIRE gene at chromosome 21q22 is a genetic determinant for the predisposition to rheumatoid arthritis in Japanese population. Human molecular genetics. 2011;20:2680–2685. doi: 10.1093/hmg/ddr161. [DOI] [PubMed] [Google Scholar]
- 15.Bennett CL, et al. X-Linked syndrome of polyendocrinopathy, immune dysfunction, and diarrhea maps to Xp11.23-Xq13.3. American journal of human genetics. 2000;66:461–468. doi: 10.1086/302761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. The Journal of pediatrics. 1982;100:731–737. doi: 10.1016/s0022-3476(82)80573-8. [DOI] [PubMed] [Google Scholar]
- 17.Birlea SA, et al. Comprehensive association analysis of candidate genes for generalized vitiligo supports XBP1, FOXP3, and TSLP. The Journal of investigative dermatology. 2011;131:371–381. doi: 10.1038/jid.2010.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang D, et al. Accounting for eXentricities: analysis of the X chromosome in GWAS reveals X-linked genes implicated in autoimmune diseases. PloS one. 2014;9:e113684. doi: 10.1371/journal.pone.0113684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bi LL, et al. Dominant inhibition of Fas ligand-mediated apoptosis due to a heterozygous mutation associated with autoimmune lymphoproliferative syndrome (ALPS) Type Ib. BMC medical genetics. 2007;8:41. doi: 10.1186/1471-2350-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher GH, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 21.Oliveira JB, et al. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8953–8958. doi: 10.1073/pnas.0702975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rieux-Laucat F, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 23.Sneller MC, et al. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. The Journal of clinical investigation. 1992;90:334–341. doi: 10.1172/JCI115867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sneller MC, et al. Clincal, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–1348. [PubMed] [Google Scholar]
- 25.Straus SE, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. doi: 10.1182/blood.v98.1.194. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, et al. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell. 1999;98:47–58. doi: 10.1016/S0092-8674(00)80605-4. [DOI] [PubMed] [Google Scholar]
- 27.Price S, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123:1989–1999. doi: 10.1182/blood-2013-10-535393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horiuchi T, et al. Association of Fas/APO-1 gene polymorphism with systemic lupus erythematosus in Japanese. Rheumatology. 1999;38:516–520. doi: 10.1093/rheumatology/38.6.516. [DOI] [PubMed] [Google Scholar]
- 29.Huang QR, Danis V, Lassere M, Edmonds J, Manolios N. Evaluation of a new Apo-1/Fas promoter polymorphism in rheumatoid arthritis and systemic lupus erythematosus patients. Rheumatology. 1999;38:645–651. doi: 10.1093/rheumatology/38.7.645. [DOI] [PubMed] [Google Scholar]
- 30.Huang QR, Morris D, Manolios N. Identification and characterization of polymorphisms in the promoter region of the human Apo-1/Fas (CD95) gene. Molecular immunology. 1997;34:577–582. doi: 10.1016/s0161-5890(97)00081-3. [DOI] [PubMed] [Google Scholar]
- 31.Kanemitsu S, et al. A functional polymorphism in fas (CD95/APO-1) gene promoter associated with systemic lupus erythematosus. The Journal of rheumatology. 2002;29:1183–1188. [PubMed] [Google Scholar]
- 32**.Angulo I, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342:866–871. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33**.Lucas CL, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nature immunology. 2014;15:88–97. doi: 10.1038/ni.2771. These two manuscripts were the first to describe heterozygous, gain-of-function mutations in the gene encoding p110δ that cause immunodeficiency with lymphoproliferative disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deau MC, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. The Journal of clinical investigation. 2014;124:3923–3928. doi: 10.1172/JCI75746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucas CL, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. The Journal of experimental medicine. 2014;211:2537–2547. doi: 10.1084/jem.20141759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crank MC, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. Journal of clinical immunology. 2014;34:272–276. doi: 10.1007/s10875-014-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartman HN, et al. Gain of Function Mutations of PIK3CD as a Cause of Primary Sclerosing Cholangitis. Journal of clinical immunology. 2014 doi: 10.1007/s10875-014-0109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kracker S, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase delta syndrome. The Journal of allergy and clinical immunology. 2014;134:233–236. doi: 10.1016/j.jaci.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banham-Hall E, Clatworthy MR, Okkenhaug K. The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases. The open rheumatology journal. 2012;6:245–258. doi: 10.2174/1874312901206010245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle DL, Kim HR, Topolewski K, Bartok B, Firestein GS. Novel phosphoinositide 3-kinase delta,gamma inhibitor: potent anti-inflammatory effects and joint protection in models of rheumatoid arthritis. The Journal of pharmacology and experimental therapeutics. 2014;348:271–280. doi: 10.1124/jpet.113.205955. [DOI] [PubMed] [Google Scholar]
- 41.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. The Journal of experimental medicine. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linsley PS, et al. CTLA-4 is a second receptor for the B cell activation antigen B7. The Journal of experimental medicine. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tivol EA, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 44.Gough SC, Walker LS, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunological reviews. 2005;204:102–115. doi: 10.1111/j.0105-2896.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- 45.Petukhova L, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113–117. doi: 10.1038/nature09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renton AE, et al. A genome-wide association study of myasthenia gravis. JAMA neurology. 2015;72:396–404. doi: 10.1001/jamaneurol.2014.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47**.Kuehn HS, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345:1623–1627. doi: 10.1126/science.1255904. This manuscript describes autoimmune patients with lymphocytic tissue infiltration caused by inherited, heterozygous mutations in the CTLA4 gene. Unlike mice, one normal CTLA4 allele is insufficient to prevent disease in humans (i.e., CTLA4 haploinsufficiency is disease-causing) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez-Herrera G, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. American journal of human genetics. 2012;90:986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49*.Lo B, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349:436–440. doi: 10.1126/science.aaa1663. Deficiency in the vesicle trafficking-associated molecule LRBA in humans is shown to cause reduction in CTLA4 protein stability, resulting in a phenocopy of CTLA4 haploinsufficient patients with autoimmunity and tissue infiltration. [DOI] [PubMed] [Google Scholar]
- 50.Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nature medicine. 2014;20:1410–1416. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holland JH. Studying Complex Adaptive Systems. J Systems Science & Complexity. 2006;19:8. [Google Scholar]
- 52.Gottschalk RA, et al. Recent progress using systems biology approaches to better understand molecular mechanisms of immunity. Seminars in immunology. 2013;25:201–208. doi: 10.1016/j.smim.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]