

SUMMARY
Mutation of surface residues to charged amino acids increases resistance to aggregation and can enable reversible unfolding. We have developed a protocol using the Rosetta computational design package that “supercharges” proteins while considering the energetic implications of each mutation. Using a homology model, a single-chain variable fragment antibody was designed that has a markedly enhanced resistance to thermal inactivation and displays an unanticipated ≈30-fold improvement in affinity. Such supercharged antibodies should prove useful for assays in resource-limited settings and for developing reagents with improved shelf lives.
INTRODUCTION
Increasing the net charge of a polypeptide chain enhances solubility and causes intermolecular repulsion in the unfolded state, which in turn disfavors aggregation (Simeonov et al., 2011; Fink, 1998). Single-chain FV antibody fragments (scFVs) are useful reagents, but they tend to aggregate both in vivo and in vitro, particularly at increased temperatures. Thus, they must generally be refrigerated for storage, limiting their utility. David Liu’s research group (Lawrence et al., 2007) previously demonstrated that substitution of multiple surface residues with charged amino acids (“supercharging”) markedly increases resistance to thermally-induced aggregation in GFP, enabling reversible unfolding and thus the recovery of fluorescence after incubation at high temperatures. In this approach, residues were selected for substitution with positively- (Arg, Lys) or negatively-charged (Glu, Asp) amino acids based on solvent accessibility as determined by the AvNAPSA algorithm (average number of neighboring atoms per side-chain atom). A low AvNAPSA score indicates that a given residue has few nearby nonself atoms and is thus likely to be solvent-exposed and amenable to substitution (Lawrence et al., 2007).
However, when applied to a binding protein (streptavidin) and an enzyme (glutathione S-transferase) this approach conferred thermal resistance but diminished function (Lawrence et al., 2007). In the current work, we have developed a protocol using the Rosetta macromolecular modeling suite that considers the energetic consequences of such substitutions with the goal of predicting supercharging mutations in antibody fragments that avoided the introduction of unfavorable conformations or interactions, avoided the removal of existing, favorable interactions, and in many cases, introduced new, favorable interactions. Experimental validation of these predictions with the anti-MS2 scFV antibody fragment as a model revealed that substitution of up to 14 residues with arginine or lysine resulted in marked resistance to thermal denaturation while retaining or improving antigen-binding activity. The use of Rosetta protein design methods for the engineering of highly stable scFVs represents a generalizable approach to address the inherent propensity of single chain antibody fragments to aggregate, especially at elevated temperatures (Jung and Plückthun, 1997).
RESULTS AND DISCUSSION
Homology Modeling of an Anti-MS2 Antibody
“Ruggedizing” antibodies to remain functional over longer periods of time under a variety of environmental conditions has important implications for clinical and sensing applications. The bacteriophage MS2 is frequently used as a viral stimulant (McBride et al., 2003), and anti-MS2 coat protein antibodies are frequently used to prototype biosensor systems (Thomas et al., 2004). Unfortunately, no anti-MS2 antibody crystal structures exist to guide the placement of charged residues. Given that many antibodies lack structures, the success of homology modeling for supercharging would be particularly important for future design efforts.
We used the RosettaAntibody FV homology modeling server (Sircar et al., 2009) to create a series of structural models of the anti-MS2 scFV. This process began by finding antibody “pieces” of known structure that had high sequence homology to the anti-MS2 scFV. The most homologous VL and VH frameworks (from Protein Data Bank [PDB] 1QKZ and 2A77) and L1, L2, L3, H1, and H2 loops (1DLF, 1N4X, 1T66, 1H3P, and 1HIL) were culled from the RosettaAntibody database (Sivasubramanian et al., 2009). Sequence substitutions between the anti-MS2 antibody and the matching pieces were reconciled by modeling in the anti-MS2 antibody residues. The modeled loops were then grafted onto the modeled frameworks, and the overall homology model was further developed by identifying multiple, low energy conformations of the H3 loop. As loop conformations were explored, the VL-VH orientation and other complementarity determining region (CDR) conformations were permitted to change as needed. The 11 residue H3 loop of the anti-MS2 scFV is particularly important for ligand-binding; the nearest sequence in the database (2DBL) had 70% identity but a different loop length. Fortunately, loops in this size range have previously been modeled with RosettaAntibody; a typical heavy-atom root-mean-square deviation (rmsd) for this size loop is 2.4 Å (Sircar et al., 2009). After several cycles of modeling and refinement, gradient-based minimization was employed for final refinement. This process was performed 2,000 times to create a series of models, and the Rosetta full-atom scoring/energy function was used to select the best ten models (Figure 1). Outside of CDR H3, the ten selected models had a high degree of agreement at the atomic level (rmsd = 0.64 Å).
Figure 1.
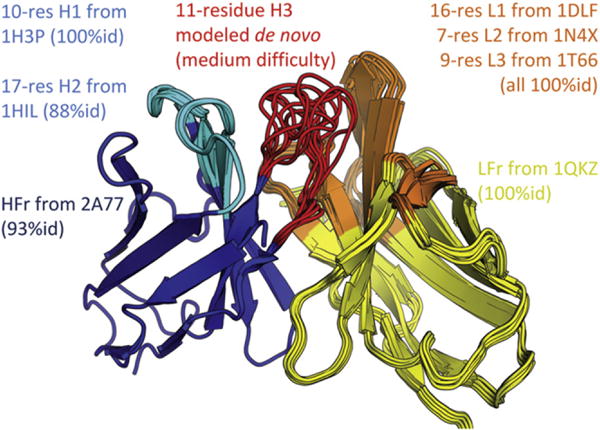
Ten Homology Models of the Anti-MS2 scFV Overlaid with Source PDBs Indicated for Grafted Regions See also Table S1.
Supercharging with Rosetta
We hypothesized that understanding the energetics of a mutation, rather than relying on the plasticity of the protein surface, would greatly facilitate the generation of functional, supercharged proteins. To identify residues that can be replaced by charged amino acids without a significant energetic cost, we employed the Rosetta suite of protein design tools (Dantas et al., 2003; Leaver-Fay et al., 2011), which can assess the detailed structural and stability effects of mutations. Rosetta’s energy function considers hydrogen bonding, salt-bridge interactions, rotamer preferences, solvation energies, van der Waals forces, and the intrinsic preferences of amino acids to participate in particular types of secondary structures (Figure 2A). Electrostatics in Rosetta are treated as a short-range phenomenon, which is suitable for proteins functional at roughly physiological pH values and ionic strengths. “Supercharging” at low ionic strengths would require the consideration of increased repulsive effects. We identified non-CDR positions that could be mutated without disturbing existing strong interactions while also potentially introducing additional stabilizing features such as hydrogen bonds or short-range electrostatic interactions. All ten homology models were used in Rosetta simulations, and a criterion for the selection of a mutation was that it be predicted for all models.
Figure 2.
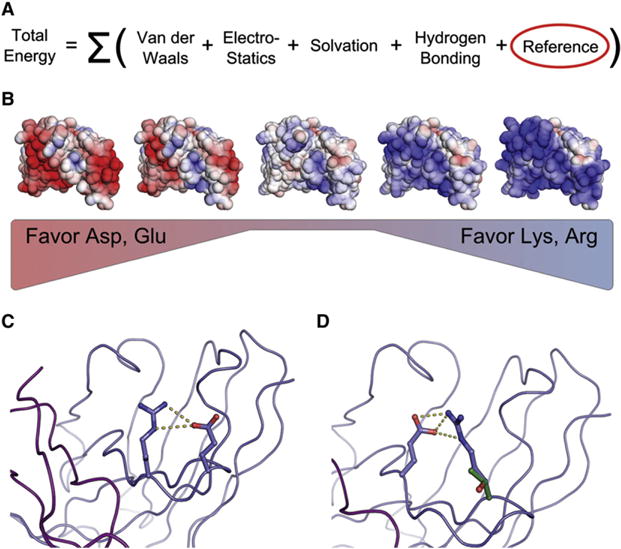
Supercharging with Rosetta (A and B) The major constituents of the Rosetta scoring function. The reference term (circled in red) biases the frequency of occurrence of a given amino acid, which was manipulated in a stepwise manner to produce (B), a series of designs with varying degrees of supercharging.
(C) A predicted salt bridge between R44 and E42 that would be removed by the mutation of E42 in positive-supercharging designs based solely on residue position due to the high solvent accessibility of E42. This interaction was preserved by Rosetta in the K-pos series as the energy of this interaction was insufficient to override the adjusted reference values.
(D) Consideration of structure while supercharging introduces a potential salt bridge through a T40 (green) to R (blue) mutation in the K-pos series.
See also Figure S4 and Table S2.
Because we were unsure at the outset what degree of supercharging would prove effective, we created a range of charge variants. These variants were generated by carrying out multiple runs of the algorithm in which we specified increasing energetic bonuses for mutations to charged residues (Figure 2B). For example, during the design of the supercharged variants an additional energetic bonus of from −0.40 to −1.40 Rosetta energy units per residue was granted to charged residues (arginine and lysine, or glutamate and aspartate), on top of the typical per-residue tally of −2 to −3 Rosetta energy units. Unlike the Av-NAPSA approach, no bonus was given for a charge-swap mutation. In this manner we were able to bias for the inclusion of charged residues while still considering the structure of the protein; for example, if placing a charged residue and thus obtaining an energetic bonus of −0.40 required removing a native hydrogen bond of energy −0.50, the deleterious effect would override the bonus, and that residue would not be chosen for substitution. Unsurprisingly, the majority of the predicted mutations were found at the surface, even though this was not an explicit requirement of the design process. Those few mutations that were predicted at less solvent accessible positions were considered high-risk upon visual inspection and were eliminated manually.
Ultimately, we generated five positively-supercharged designs with net charges varying from +15.4 to +29.5 (named K-pos-1–K-pos-5), and four negatively-supercharged designs with net charges varying from −0.5 to −19.5 (named K-neg-1–K-neg-4) (Table 1). The charge of the wild-type scFV is +7.5. Examination of the designs indicated that the energetic balance between the placement of charged residues and maintaining favorable interactions was achieved. As an example, H chain residue E43 was not mutated to R or K in any of the K-pos designs, despite being extremely solvent-exposed, as it was predicted to form a salt bridge with R44 (Figure 2C). Charged residues were also introduced into locations where they were predicted to form additional stabilizing interactions. For example, a T40R mutation in K-pos-1 is predicted to participate in a hydrogen bond with E46 (Figure 2D). Alignments of each variant sequence against wild-type are provided in Figure S1 available online and Supplemental File 1, and models of each design are included in Supplemental File 2.
Table 1.
Screening of Supercharged Single-Chain FV Variants
| Name | Charge | Rosetta Score(Δ Wild-Type) | Mutation Count | Expressiona | Binding | Thermal Resistance |
|---|---|---|---|---|---|---|
| K-neg-4 | −19.5 | −6.2 | 24 | + + | No | |
| K-neg-3 | −13.5 | −9.3 | 18 | + + | Yes (weak) | No |
| K-neg-2 | −8.5 | −8.6 | 14 | + + | Yes (strong) | No |
| K-neg-1 | −0.5 | −5.1 | 7 | + | No | |
| wt | 7.5 | 0 | 0 | + + + | Yes (strong) | Yes (trace) |
| K-pos-1 | 15.5 | −4.9 | 8 | + + | Yes (strong) | Yes (strong) |
| K-pos-2 | 18.5 | −8.9 | 11 | + | Yes (strong) | Yes (medium) |
| K-pos-3 | 22.5 | −10.1 | 14 | + | Yes (medium) | Yes (medium) |
| K-pos-4 | 29.5 | −11 | 21 | + | No | |
| K-pos-5 | 33.5 | −7.9 | 25 | (cytosol) | No |
wt, wild-type.
See also Figure S1 and Supplemental File 1.
Relative expression levels are indicated by +, ++, and +++ indicating low, medium, and high, respectively.
Experimental Validation
Genes encoding the codon-optimized anti-MS2 scFV and its supercharged variants were constructed by automated DNA synthesis and robotically-assisted assembly. The genes were cloned into the pAK400 vector (Krebber et al., 1997), in frame with the pelB signal peptide for secretion into the periplasm. Expressed proteins were initially harvested by osmotic shock. However, it was found that the use of lysozyme (pI ≈11) in the osmotic shock preparation led to aggregation of negatively-supercharged proteins, presumably due to a “molecular velcro” effect (Lawrence et al., 2007). Therefore, scFVs were purified from total cell lysates using IMAC chromatography followed by size-exclusion chromatography to remove additional impurities, dimers, and aggregates.
Additionally, we observed that positively-supercharged variants accumulated in the cytoplasm, rather than in the periplasmic space. Stretches of positively-charged amino acids are known to serve as a secretion block, preventing protein translocation across the cytosolic membrane (Wilson and Finlay, 1998). To circumvent this problem, we expressed highly-positive constructs in the cytosol using a vector (pET21a) that lacks the pelB signal peptide. Constructs were expressed in an Origami Escherichia coli strain whose cytoplasm is poised at a more oxidizing redox potential, thus favoring the formation of the two intramolecular disulfide bonds within the antibody fragment (Bessette et al., 1999).
Antigen-binding by the expressed and purified supercharged variants was first evaluated by ELISA with a dimer-forming mutant of the MS2 coat protein (Ni et al., 1995; Peabody and Ely, 1992) fused to a N-terminal FLAG tag (Figure 3A). K-neg-2 was the only negative variant to demonstrate appreciable binding to the antigen, whereas three positively-charged scFV variants (K-pos-1, K-pos-2, K-pos-3) displayed binding to MS2 (Table 1).
Figure 3. ELISA Screening and Characterization.
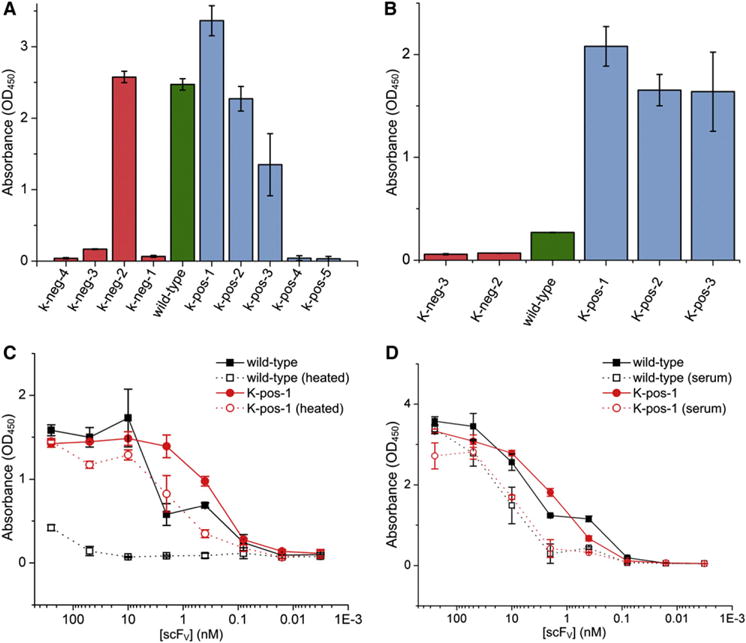
(A) The initial screen of the supercharged scFV variants for binding. ScFVs were added to the antigen-bound plate at a concentration of 5 μg/ml.
(B) Samples of constructs shown to bind in the initial screen were stored at 70°C for 1 hr, allowed to cool to room temperature, and assayed again to determine which constructs retained activity.
(C) A dilution-series ELISA comparing wild-type and K-pos-1 binding before and after incubation at 70°C for 1 hr.
(D) A dilution-series ELISA comparing the function of wild-type and K-pos-1 in 50% serum. Error bars represent SD calculated from the OD450 values of three identically-prepared wells.
See also Figures S2 and S3.
The four scFV variants that displayed significant antigen binding were evaluated for resistance to thermal inactivation following incubation at 70°C for 1 hr (Figure 3B). Although the wild-type anti-MS2 and K-neg-2 completely lost antigen-binding activity after heating, the positively-charged variants still bound, with K-pos-1 retaining ≈70% of its initial activity. The binding ability of K-pos-1 was further characterized as a function of antigen concentration (Figure 3C) and in 50% serum (Figure 3D). We also examined the binding kinetics of K-pos-1 via Biacore analysis (Figures S2A and S2B). Samples were held at 70°C for 1 hr and then flowed over the antigen-conjugated surface. Although the sensorgram of the wild-type parent showed no binding postheating (Figure S2C), the sensorgram of K-pos-1 again indicated that this variant retained binding activity (Figure S2D). The predominance of positively-charged thermoresistant variants was consistent with the observation that positively-charged amino acids generally make greater contributions to solubility (Shiraki et al., 2002). Also, because the antibody started at a native charge of +7.5, fewer substitutions were required for positive supercharging.
Not only was activity retained following incubation at high temperature, the protein was in general less prone to aggregation in typical storage conditions (4°C in phosphate-buffered saline). Dynamic light scattering (DLS) revealed that K-pos-1 was a highly-uniform solution of 2.6 nm particles with a polydispersity of 11.1%. In contrast, in the wild-type scFV 90% of the mass was attributed to a 3.4 nm population with 18.1% polydispersity whereas 10% of the mass was represented by highly-polydisperse larger particles (Figure S3A). In line with the DLS data, preparations of K-pos-1 also had fewer dimers and higher-order multimers, as indicated by size exclusion chromatography (Figure S3B). Interestingly, K-pos-1 was not only thermoresistant, but also more thermostable. The mid-point for thermal denaturation (Tm) of K-pos-1 determined by differential scanning calorimetry was slightly higher than that of the parental scFV, 69.1°C and 67.2°C, respectively (Figure S3C).
Given that CDR loop positions were not mutated during supercharging, Biacore analysis surprisingly revealed that K-pos-1 displayed an improved on-rate (ka = 2.5 × 106 ± 7.6 × 105 M−1s−1 versus the wild-type 3.77 × 105 ± 1.0 × 104 M−1s−1) and a slower off-rate (kd = 2.66 × 10−3 ± 9.9 × 10−4 s−1 versus 1.34 × 10−2 ± 8.6 × 10−5 s−1), resulting in a 30-fold lower equilibrium dissociation binding constant (Kd = 1 ± 0.7 nM and 36± 1.2 nM for K-pos-1 and the wild-type anti-MS2 scFV,, respectively). No binding to BSA was detected, suggesting that the increased affinity of K-pos-1 was not due to nonspecific binding or bulk electrostatic attraction (because BSA has a small net negative charge) (Kiel et al., 2004). The additional charge could have played a role in stabilizing the bound form (thus decreasing the off-rate) because the charged residues were peripheral to the CDR loops and they might not have experienced unfavorable desolvation upon binding (Sheinerman et al., 2000).
For comparison, we created a series of supercharged antibody designs based on AvNAPSA values, taking into account identity and position rather than energetics (Supplemental File 1). None of these antibodies proved to be thermoresistant (Table S1). Of the six most highly-exposed positions chosen to be mutated in all AvNAPSA-designed antibodies, only two of those positions were mutated by Rosetta. This is likely because Rosetta preserves energetically favorable interactions. As previously mentioned, H-chain residue E42 was preserved in the K-pos series of designs as it was predicted to participate in a salt bridge. This residue would be mutated in AvNAPSA-based positive-supercharging designs due to its high solvent-accessibility (and thus low AvNAPSA score). The finding that designs made using the AvNAPSA approach were not functional after a thermal challenge, even with the same mutational load or net charge, indicates that structure-based computational design was more effective in producing functional charged antibodies than simple consideration of solvent exposure.
EXPERIMENTAL PROCEDURES
Rosetta Design of Supercharged FV Antibody Fragments
The Rosetta molecular modeling program (version 3.1) (Leaver-Fay et al., 2011) was used to computationally supercharge the single-chain FV antibody fragment. Simulations were performed with Rosetta’s “fixbb” protocol. This protocol holds the protein backbone fixed and uses simulated annealing to perform rotamer-based optimization of the sequence. All residues outside of the CDR loops (light chain residues 24–34, 50–56, 89–97; heavy chain residues 26–35, 50–56, 95–102) were allowed to vary during the design simulations. During simulations to create positively-supercharged variants, allowed amino acids were arginine, lysine, and the native amino acid. For negatively-super charged variants, allowed amino acids were aspartate, glutamate, and the native amino acid. Separate simulations were run with incrementing reference energies for arginine and lysine, or aspartate and glutamate, to achieve a variety of net charges (see Table S2 for actual reference values). These amino acid restrictions were implemented using a residue input file, or “resfile” (Supplemental File 3 for positive and Supplemental File 4 for negative supercharging).
The effect of manipulating the reference energies was relatively subtle. The inclusion of the desired residues yielded a moderately favorable effect on the reference energy term (as would be expected) and favorable short-range electrostatic interactions were found by placing charged residues near oppositely-charged partners, yielding additional favorable changes in the energy terms. The primary detrimental effect seen as a result of manipulating the potential function was the inclusion of less-favorable side-chain poses with less favorable rotamer scores. A chart of these energies can be found in Supplemental File 5.
For each of the 10 homology models, 15 design trajectories were run, and only the sequence of best scoring model of the 15 was considered. The set of 150 design simulations was repeated for each reference energy increment. The simulations were performed using Dunbrack’s backbone dependent rotamer library (Dunbrack and Cohen, 1997), with additional sampling of χ 1 and χ 2 angles. Following rotamer-based optimization, side chain torsion angles were further optimized with gradient-based minimization. The full command line used for the simulations is provided in Figure S4.
A mutation was accepted if it occurred in all ten backbone models. The intention of this criterion was to ensure robustness of mutation given possible inaccuracies in the modeled backbone and side chain conformations. For experimental testing, five positively-supercharged variants and four negatively-supercharged variants were selected from these simulations.
Gene Synthesis and Cloning
Genes encoding the supercharged scFVs were synthesized from oligonucleotides using methods previously published by Cox et al. (2007) with modifications (vide infra). In brief, long DNA oligonucleotides (60 < n < 120) were first assembled into primary fragments of length (200 < n < 600) using inside-out PCR. These primary fragments were combined without purification in a secondary overlap-extension reaction that formed the final desired product. Custom software aided in the design of the assembly schemes and the efficient re-use of oligonucleotides across multiple related sequences.
In the case of the genes encoding the wild-type and supercharged scFVs, oligonucleotides with an average length of 77 base pairs were synthesized and used in two thermodynamically-balanced inside-out PCRs to assemble primary fragments of length 435 and 423. We updated the software to apply melting-temperature normalization across oligonucleotide overlaps to increase assembly robustness at this stage. The two primary reactions were combined with flanking primers to form the final, 834-base-pair product consisting of the scFV gene with flanking, incompatible SfiI sites.
The pAK400 vector (Krebber et al., 1997) was used for expression into the periplasm in Jude-1 cells (Griswold et al., 2005). The SfiI sites on the synthesized genes were designed to facilitate cloning into pAK400, where it is inserted between an N-terminal PelB leader and a C-terminal hexahistidine tag. The pET21a vector (Novagen) was used to express the scFVs in the Origami DE3 cells (Novagen), and the synthesized genes were re-amplified with primers to replace the SfiI sites with NdeI and XhoI sites suitable for insertion into the vector prior to the C-terminal hexahistidine tag. Inserts were verified by dye-terminator sequencing.
Purification of scFV Constructs from the Periplasm via Sonication
Anti-MS2 scFV constructs in the pAK400 vector were transformed into electrocompetent Jude-1 cells and plated on LB agar plates. All culture steps were performed in the presence of 34 μg/ml chloramphenicol to maintain the pAK400 plasmid. Individual colonies were picked, inoculated into 5 ml LB, and grown overnight at 37°C. Overnight cultures were diluted 1:1,000 into 1 l LB media and grown for 18 hr at 30°C in a shaking incubator. Cells were isolated by centrifugation at 4,000 × g for 20 min at 4°C, and resuspended in 1 l Superior Broth (Athena ES). These cultures were placed in a shaking incubator at 18°C for 1 hr prior to induction by the addition of IPTG (Cellgro) to a final concentration of 1 mM. Induction was allowed to proceed for 18 hr at 18°C, after which cells were harvested by centrifugation at 4,000 × g for 20 min. at 4°C and resuspended in 50 ml IMAC Buffer (20 mM MOPS, 500 mM NaCl, 20 mM imidazole, pH 7.4). One thousand units of benzonase (Novagen) were added and the resuspended cells were lysed by sonication at 40% amplitude for 4 min with a 50% duty cycle (Model 500 Sonic Dismembrator, Fisher Scientific). The lysate was cleared by centrifugation at 40,000 × g for 30 min at 4°C. The cleared lysate was filtered through a 0.45 μm Supor filter and applied to a 1 ml column of nickel-charged chelating Sepharose resin (GE) that had been equilibrated with IMAC buffer. The column was washed with 10 ml of IMAC buffer and 5 ml of IMAC buffer supplemented to 100 mM imidazole. Bound scFVs were then eluted with 5 ml of IMAC buffer supplemented to 400 mM imidazole. Expression and subsequent purification were verified by SDS-PAGE (Novex NuPAGE 4%–12% Bis-Tris gels, Invitrogen). The eluates were further purified by size exclusion chromatography on an S75 HiLoad 26/60 column (GE) in 10 mM sodium phosphate, 150 mM NaCl, pH 7.4. Fractions corresponding the monomer scFV were pooled and concentrated using Amicon Ultra-15 centrifugal spin columns with a 10 kDa cutoff (Millipore).
Purification of scFV Constructs from the Cytosol
Anti-MS2 scFV constructs in the pET21a vector were transformed into chemically competent Origami DE3 cells (Novagen). All culture steps were performed in the presence of 100 μg/ml ampicillin to maintain the pET21a plasmid, and 15 μg/ml kanamycin and 12.5 μg/ml tetracycline to maintain the redox features of the Origami DE3 cells. Individual colonies were inoculated into 5 ml LB, and the overnight cultures were diluted 1:1000 into 1 l Superior Broth at 37°C in a shaking incubator. When the culture reached an OD600 of 2.0, the culture was chilled to 18°C, expression was induced by the addition of IPTG to a final concentration of 1 mM, and induction was allowed to proceed with shaking at 18°C for 18 hr. Cells were harvested by centrifugation at 4,000 × g for 20 min at 4°C. Sonic disruption and purification were performed as described previously (vide supra).
Purification of the Dimer-Forming MS2 Coat Protein Variant
The gene encoding a mutant of the MS2 coat protein that favors dimer formation (Ni et al., 1995; Peabody and Ely, 1992) was synthesized in the same manner as the scFV constructs. The gene was designed with a leading NdeI site, an N-terminal FLAG tag (DYKDDDDK), and a C-terminal stop codon immediately before an XhoI site. This insert was cloned into pET21a via the aforementioned restriction sites. A single colony was inoculated into 5 ml LB plus 100 μg/ml ampicillin, grown overnight, diluted 1:1,000 into 1 l Power Broth (AthenaES) and allowed to grow at 37°C in a shaking incubator. At an OD600 of 2.0, the cultures were chilled to 18°C, IPTG was added to 1 mM, and induction proceeded for 18 hr at 18°C. Cells were harvested by centrifugation at 4,000 × g for 20 min at 4°C. Pellets were resuspended into 50 ml TBS (50 mM Tris, 500 mM NaCl, pH 7.4) with 1,000 units of benzonase (Novagen). Cells were lysed by sonication at 40% amplitude for 4 min with a 50% duty cycle, and the lysate was cleared by centrifugation at 40,000 × g for 30 min at 4°C. The cleared lysate was filtered through a 0.45 μm Supor filter and applied to a 2.5 ml column of anti-FLAG M2 IgG1 resin (Sigma), equilibrated with TBS. The flowthrough was reapplied to the column four times to increase binding efficiency. The column was washed with 50 ml of TBS, followed by five 2.5-ml elutions with 100 μg/ml FLAG peptide (Sigma) in TBS. The eluates were further purified by size exclusion chromatography on an S75 HiLoad 26/60 column (GE) in 10 mM sodium phosphate, 150 mM NaCl, pH 7.4. The MS2 dimer fractions were pooled and concentrated using Amicon Ultra-15 centrifugal spin columns with a 10 kDa cutoff (Millipore).
ELISA
Costar 96-well ELISA plates (Corning) were coated with 100 μl of 4 μg/ml FLAG-tagged MS2 antigen in ELISA buffer (10 mM sodium phosphate, 150 mM NaCl, pH 7.4). Wells used as background controls were coated with 2% (w/v) skim milk in ELISA buffer. The coated plates were covered and incubated overnight at 4°C, then decanted and blocked with 2% skim milk in ELISA buffer for 1 hr at room temperature. The blocking agent was decanted, the plates were washed three times with ELISA buffer, and the respective scFVs were applied to the plate.
Supercharged variants or the wild-type anti-MS2 scFV were serially-diluted from a 1 μM stock into ELISA buffer or ELISA buffer with 50% (v/v) human blood plasma (Gulf Coast Regional Blood Center) to generate a 1:5 dilution series from 250 nM to 3.2 fM. For the thermostability assays, 1 μM stock solutions of the scFVs were subjected to a heat shock of 70°C for 1 hr followed by a 5 min re-equilibration to room temperature, and a 1:5 serial dilution was again generated.
Antigen-antibody binding reactions proceeded for 1 hr at room temperature with gentle mixing. Plates were decanted, thoroughly air-dried, and washed three times with 200 μl of PBS plus 0.05% Tween-20 (PBS+T). The plate was probed with an HRP-conjugated antipolyhistidine antibody (Sigma, clone HIS-1; diluted 1:5,000 in PBS+T) for 1 hr at room temperature with gentle mixing. The plate was washed three times with PBS+T as before, briefly airdried and developed for 5 min using the 1-step Ultra TMB substrate (Thermo-Scientific). Reactions were terminated with 1 M H2SO4 and absorbances were measured at a wavelength of 450 nm.
DLS
For DLS analysis, five 20-μl aliquots of each sample were pipetted into wells of a quartz 384-well plate and centrifuged for 2 min at 1500 rpm in an Eppendorf Model 5430R centrifuge to remove trapped air bubbles. Mineral oil (Sigma) was pipetted on top of each sample to prevent desiccation, and measurements were performed using a DynaPro temperature-controlled DLS plate reader (Wyatt). Each aliquot was scanned ten times for 5 s each at 25°C, and averaged to determine the polydispersity for the sample using the Wyatt Technology Dynamics software.
Differential Scanning Calorimetry
For differential scanning calorimetry (DSC) experiments, samples were diluted to 0.5 mg/ml and dialyzed overnight in PBS. Samples were then degassed for 5 min before analysis and were added to the sample cell of a MicroCal VP-DSC. The dialysis buffer was added to the reference cell of the calorimeter, and a buffer scan was used as the baseline for all experiments. The samples were scanned from 15°C to 100°C at a rate of 60°C/hr in duplicate. The transition midpoint (Tm) of the protein was determined by analysis of the data using the Origin 7.0 software.
Surface Plasmon Resonance Assays
All Biacore analysis was performed on a Biacore 3000 instrument (GE) at room temperature. Prior to the kinetic analysis of the scFVs on the instrument, the Biacore 3000 was calibrated and primed using three cycles of (deionized and degassed) water followed by three cycles of degassed HBS+P (10 mM HEPES pH 7.4, 150 mM NaCl, 3.4 mM EDTA, 0.025% (v/v) surfactant P20). A new CM5 sensor chip (GE) was attached and primed using three cycles of the HBS+P buffer. The chip was activated using two sequential injection regimes of 50 mM NaOH, 10 mM HCl, and water. Following chip activation, a pH series of the FLAG-tagged MS2 coat protein dimer was generated in 1 M sodium acetate ranging from pH 6.5 to 8 was applied; pH 7.1 was found to be optimal for antigen absorption. The antigen (10 μg/ml in 1 M sodium acetate, pH 7.1) was then immobilized at a flow rate of 5 μl/min (immobilization level of 300 RU) using an EDC (0.4 M of 1-ethyl-3-(3-dimethylpropyl)-carbodiimide in water) and NHS (0.1 M N-hydroxysuccinimide in water) driven amine-coupling reaction. Unreacted groups were quenched with 1 M ethanolamine HCl, pH 8.5. On an adjacent channel, BSA in 1 M sodium acetate, pH 5.3 was immobilized.
Following the preparation of the chip, the scFVs were prepared in HBS+P buffer at concentrations of 2 μg/ml, 1.5 μg/ml, 1 μg/ml, 0.75 μg/ml, 0.5 μg/ml, 0.375 μg/ml, 0.25 μg/ml, 0.125 μg/ml, and 0.0625 μg/ml, each made from the same 1 μM stock. For the thermoresistance analysis, stocks (1 μM) were incubated at a temperature of 70°C for 1 hr, followed by a 5 min re-equilibration to room temperature. All samples were injected at a rate of 50 μl/min, and sensorgrams were analyzed using the Biacore kinetic software. Following every antigen-antibody binding run, the chip was regenerated using 50 mM phosphoric acid injected at a rate of 12 μl/min. All samples were normalized using background and BSA-channel subtraction. Unwanted areas on the sensorgrams, including the regeneration pulse, were deleted and data were fit using a standard 1:1 Langmuir fit model. For kinetic analysis, binding curves were fit (yielding kon and koff values) and binding constants (Kd) were derived using the BIAevaluation software (GE).
Supplementary Material
SIGNIFICANCE.
Here we outline a protein design approach that capitalizes on structure-based design to identify well-tolerated charged amino acid substitutions, which in turn lead to enhanced thermoresistance. These results are of particular note as the predictions were made based on a computationally-derived homology model of an antibody for which no high-resolution structural information existed. Our strategy should generally enable the optimization of highly stable antibody fragments that resist aggregation at physiological or supraphysiological temperatures, and that therefore have long shelf lives and avoid cold storage requirements. Supercharged antibody fragments such as K-pos-1 may now serve as a scaffold for the creation of synthetic libraries for the selection of thermoresistant scFvs with different specificities. Finally, given that positively-supercharged GFP can penetrate into mammalian cells (McNaughton et al., 2009), our results suggest that it may be possible to design self-internalizing antibodies that can disrupt intracellular functions.
Acknowledgments
The authors wish to acknowledge Arti Pothukuchy and Carolyn Hargrave for technical support. This work was funded by the DARPA Antibody Technology Program (HR-0011-10-1-0052).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures, two tables, Supplemental Experimental Procedures, and five Supplemental Files and can be found with this article online at doi:10.1016/j.chembiol.2012.01.018.
References
- Bessette PH, Åslund F, Beckwith J, Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA. 1999;96:13703–13708. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JC, Lape J, Sayed MA, Hellinga HW. Protein fabrication automation. Protein Sci. 2007;16:379–390. doi: 10.1110/ps.062591607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas G, Kuhlman B, Callender D, Wong M, Baker D. A large scale test of computational protein design: folding and stability of nine completely redesigned globular proteins. J Mol Biol. 2003;332:449–460. doi: 10.1016/s0022-2836(03)00888-x. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL, Jr, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- Griswold KE, Kawarasaki Y, Ghoneim N, Benkovic SJ, Iverson BL, Georgiou G. Evolution of highly active enzymes by homology-independent recombination. Proc Natl Acad Sci USA. 2005;102:10082–10087. doi: 10.1073/pnas.0504556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Plückthun A. Improving in vivo folding and stability of a single-chain Fv antibody fragment by loop grafting. Protein Eng. 1997;10:959–966. doi: 10.1093/protein/10.8.959. [DOI] [PubMed] [Google Scholar]
- Kiel C, Selzer T, Shaul Y, Schreiber G, Herrmann C. Electrostatically optimized Ras-binding Ral guanine dissociation stimulator mutants increase the rate of association by stabilizing the encounter complex. Proc Natl Acad Sci USA. 2004;101:9223–9228. doi: 10.1073/pnas.0401160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebber A, Bornhauser S, Burmester J, Honegger A, Willuda J, Bosshard HR, Plückthun A. Reliable cloning of functional anti-body variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J Immunol Methods. 1997;201:35–55. doi: 10.1016/s0022-1759(96)00208-6. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MT, Gammon S, Pitesky M, O’Brien TW, Smith T, Aldrich J, Langlois RG, Colston B, Venkateswaran KS. Multiplexed liquid arrays for simultaneous detection of simulants of biological warfare agents. Anal Chem. 2003;75:1924–1930. doi: 10.1021/ac026379k. [DOI] [PubMed] [Google Scholar]
- McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proc Natl Acad Sci USA. 2009;106:6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni CZ, Syed R, Kodandapani R, Wickersham J, Peabody DS, Ely KR. Crystal structure of the MS2 coat protein dimer: implications for RNA binding and virus assembly. Structure. 1995;3:255–263. doi: 10.1016/S0969-2126(01)00156-3. [DOI] [PubMed] [Google Scholar]
- Peabody DS, Ely KR. Control of translational repression by protein-protein interactions. Nucleic Acids Res. 1992;20:1649–1655. doi: 10.1093/nar/20.7.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein-protein interactions. Curr Opin Struct Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Shiraki K, Kudou M, Fujiwara S, Imanaka T, Takagi M. Biophysical effect of amino acids on the prevention of protein aggregation. J Biochem. 2002;132:591–595. doi: 10.1093/oxfordjournals.jbchem.a003261. [DOI] [PubMed] [Google Scholar]
- Simeonov P, Berger-Hoffmann R, Hoffmann R, Sträter N, Zuchner T. Surface supercharged human enteropeptidase light chain shows improved solubility and refolding yield. Protein Eng Des Sel. 2011;24:261–268. doi: 10.1093/protein/gzq104. [DOI] [PubMed] [Google Scholar]
- Sircar A, Kim ET, Gray JJ. RosettaAntibody: antibody variable region homology modeling server. Nucleic Acids Res. 2009;37:W474–9. doi: 10.1093/nar/gkp387. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivasubramanian A, Sircar A, Chaudhury S, Gray JJ. Toward high resolution homology modeling of antibody Fv regions and application to antibody–antigen docking. Proteins. 2009;74:497–514. doi: 10.1002/prot.22309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH, Kim SK, Hesketh PJ, Halsall HB, Heineman WR. Bead-based electrochemical immunoassay for bacteriophage MS2. Anal Chem. 2004;76:2700–2707. doi: 10.1021/ac035503c. [DOI] [PubMed] [Google Scholar]
- Wilson DR, Finlay BB. Phage display: applications, innovations, and issues in phage and host biology. Can J Microbiol. 1998;44:313–329. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.