

Abstract
Autism spectrum disorder (ASD) occurs in 1 in 68 births, preferentially affecting males. It encompasses a group of neurodevelopmental abnormalities characterized by impaired social interaction and communication, stereotypic behaviors and motor dysfunction. Although recent advances implicate maternal brain-reactive antibodies in a causative role in ASD, a definitive assessment of their pathogenic potential requires cloning of such antibodies. Here, we describe the isolation and characterization of monoclonal brain-reactive antibodies from blood of women with brain-reactive serology and a child with ASD. We further demonstrate that male but not female mice exposed in utero to the C6 monoclonal antibody, binding to contactin-associated protein-like 2 (Caspr2), display abnormal cortical development, decreased dendritic complexity of excitatory neurons and reduced numbers of inhibitory neurons in the hippocampus, as well as impairments in sociability, flexible learning and repetitive behavior. Anti-Caspr2 antibodies are frequent in women with brain-reactive serology and a child with ASD. Together these studies provide a methodology for obtaining monclonal brain-reactive antibodies from blood B cells, demonstrate that ASD can result from in utero exposure to maternal brain-reactive antibodies of single specificity and point toward the exciting possibility of prognostic and protective strategies.
INTRODUCTION
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder characterized by impaired communication and social skills, repetitive/stereotypic and inflexible behavior.1 The incidence of ASD has been increasing sharply worldwide over the last decade and it is now estimated to affect 1 of every 68 children in United States.2 The etiology of ASD is complex, and includes both genetic and environmental factors.
Several studies suggest that the maternal in utero environment can contribute to ASD in offspring. Maternal half-sibs of an ASD-affected individual are more likely to develop ASD than paternal half-sibs.3 Maternal immune activation (for example, infection) has been implicated in ASD in human and murine offspring.4 In murine models, elevated maternal production of interleukin-65 or interleukin-176 can alter brain development and lead to an ASD-like phenotype in the offspring. In epidemiologic studies, maternal influenza infection was associated with increased risk of ASD.7 Moreover, genetic and environmental risk factors cooperate in determining ASD severity; children who both possess large chromosomal copy number variations (CNVs) and experience in utero exposure to maternal infection exhibit more severe symptoms than children who are exposed in utero to maternal infection, or have CNVs.8
We and others have shown that significantly more mothers of children with ASD have brain-reactive antibodies than unselected women of child-bearing age or mothers of a developmentally normal child.9–13 These antibodies can enter the fetal brain14 and disrupt development. When serum or polyclonal antibodies from these mothers were administered to pregnant mice or monkeys, the offspring exhibited neurodevelopmental and behavioral alterations.13,15,16
Most recently, potential antigens recognized by serum of mothers of a child with ASD were identified.15,17 The availability of monoclonal antibodies derived from these mothers permits a clear identification of the fine specificities that impair brain development. In this paper we demonstrate methodology to obtain brain-reactive monoclonal antibodies cloned from B cells from mothers with brain-reactive serology and a child with ASD. We further demonstrate that the presence of a single monoclonal antibody targeting contactin-associated protein-like 2 (Caspr2) in the serum of a healthy pregnant female mouse can cause neurodevelopmental abnormalities in the offspring.
Caspr2, encoded by the gene CNTNAP2, has been linked to ASD.18 It is a molecule of the neurexin family, initially described to stabilize voltage-gated potassium channels on the myelinated axons.19 Later studies have indicated a role for Caspr2 much earlier in development. Mice lacking Caspr2 show neuronal migration abnormalities, reduced GABAergic neurons, and ASD-like behavior.20 Caspr2 is expressed in adult brain in the cerebral cortex and hippocampus. During development, it is highly expressed in proliferating zones, consistent with its reported role in early neuronal development.20
MATERIALS AND METHODS
Research subjects
Plasma from mothers with an ASD child was obtained from the Simons Simplex Collection (http://sfari.org/resources/simons-simplex-collection).21 Control plasma from women of child-bearing age were obtained from the Northwell Health (previously North Shore-LIJ Health System) clinical laboratory and participants in a registry at the Feinstein Institute for Medical Research (http://www.gapregistry.org). Both cohorts were described previously.9
Plasma of mothers of a typically developing child (determined by the mother report) were obtained from the Genotype and Phenotype registry (http://www.gapregistry.org) at the Feinstein Institute for Medical Research. The age of the mothers at the time the plasma was drawn matched the previous cohorts9 (mean = 36.6, s.d. = 7.4, range = 22–50).
All individuals provided informed consent through the appropriate institutional review boards.
Sample collection
Blood was collected into heparinized tubes from consenting mothers enrolled in the Simons Simplex Collection, previously identified as having brain-reactive antibodies.9 The protocol was approved by the Simons Simplex Collection as well as by the Feinstein Institute for Medical Research Institute Review Board.
Single-cell sorting
Isolation of single human memory antigenic-specific B cells was performed as previously described22 with several modifications. B cells were purified from fresh mononuclear cells by negative selection using a B cell kit (StemCell Technology, Vancouver, BC, Canada). They were then incubated for 30 min at room temperature with human fetal brain lysate (3 μg ml−1, Nouvus, Littleton, CO, USA) labeled with biotin using the EZ-Link Sulfo-NHS-Biotin labeling kit (Life Technologies, Carlsbad, CA, USA). Cells bound by biotinylated brain antigens were isolated with a biotin selection kit (StemCell Technologies) and stained with fluoroscein isothiocyanate-conjugated anti-human CD19, phycoerythrin-conjugated anti-human CD27 and allophycocyanin streptavidin to allow the separation of CD19+, CD27+, brain lysate+ memory B cells. As a control, the fraction that initially was identified as nonbrain-reactive was incubated with biotinylated brain antigen and stained as described above. No allophycocyanin-positive cells were detected in this fraction. Finally, CD19+, CD27+, allophycocyanin+ single cells were isolated on a BD FACSAria as described in ref. 23.
Complementary DNA synthesis and reverse transcriptase-PCR
Complementary DNA synthesis of individual immunoglobulin H (γ only) and immunoglobulin L chain (Kor λ) was performed as previously described.22,23 Heavy- and light-chain variable region genes were ligated into immunoglobulin G (IgG)1 or κ constant region containing plasmids (a gift from M Nussenzweig, Rockefeller University, New York, NY, USA).
Antibody production
Antibodies were expressed in vitro as described previously23 (See also Supplementary methods). Purified antibody was dialyzed extensively against PBS; integrity was determined by nonreducing sodium dodecyl sulphate gels stained with Coomassie blue and concentration was measured by both anti-human IgG ELISA22,23 and Nanodrop.
Binding assays using transfected HEK-293T cells
Plasma and the human monoclonal antibodies, C6 and B1,24 were analyzed for binding to Caspr2 using a live cell-based immunofluorescence assay as previously described.25 See Supplementary Methods.
Absorption of Caspr2 antibodies from plasma with cells expressing Caspr2
Absorption using live cell-based immunofluorescence assay was described previously.26 HEK-239T Cells were transiently transfected with turbo-green florescent protein (tGFP)-Caspr2 or tGFP only in 96-well plate as described.25 Seventy-two hours after transfection, 50 μl fractions of plasma were incubated with the cells for one hour at 4 °C. The incubation was repeated seven times. The plasma was centrifuged for 15 min at 12 000 g. Immunoglobulin G concentration in the plasma was determined by ELISA and the presence or absence of Caspr2 antibodies were determined by immunofluorescence using the cell-based assay described above.
Caspr2 protein expression
Caspr2 RNA expression
Antibody administration to pregnant dams
C57BL/6 mice (6–8 weeks old) were obtained from the Jackson Laboratory. Animal use was in accordance with institutional guidelines of the Feinstein Institute for Medical Research. For timed pregnancy, two females and one male were housed together for 14 h. The time when the male mouse was removed from the cage was designated embryonic (E) day 0.5. Pregnant females were randomly chosen to be injected with C6 antibody or B1 control antibody (200 μ g),24 tGFP-Caspr2-absorbed plasma, or tGFP absorbed plasma or unmanipulated plasma (200 μl containing 300 μg of IgG). IgG or plasma fractions were administered by retro-orbital injection to time-pregnant mice under light anesthesia at E13.5. Embryos were harvested at E15.5 and processed for sex identification (described in ref. 27) and fetal brain pathology. Additional pregnancies were allowed to reach full-term.
Immunohistology of fetal brains
Staining of Phospho-Histone H3 (PH3) and Nestin
E15.5 brains were fixed in paraformaldehyde (4%) overnight at 4 °C followed by sucrose solution (30%) for 48 h at 4 °C and then frozen in OCT compound (VWK, Randor, PA, USA) on dry ice and stored at −80 °C. Sagittal sections were cut (12 μm thick) on a Cryostat (Leica, Billerica, MA, USA) and mounted on gelatin-coated slides and stored at −80 °C. Prior to staining, sections were thawed to room temperature, rinsed twice with PBS and blocked for 1 h with PBS (5%) with bovine serum albumin in Triton X100 (0.1%) at room temperature. Anti PH3 antibody (1:100, Millipore 06–570) or antinestin antibody (1:200, Millipore MAB353, Wetzlar, Germany) and DAPI (1 μg per ml, Life Technologies) were added overnight at 4 °C. After washing in PBS/0.1%Tween, antibody binding was detected using Alexa 488 goat anti-rabbit or anti-mouse IgG (Life Technologies) and visualized with an Axio-Imager (Z-1, Axio-Vision 4.7, Zeiss, Peabody, MA, USA). PH3+ cell quantification was performed as described in ref. 24. Cortical plate and cortical width measurements were obtained from multiple sections of each animal as described.24 In all studies, the investigator was blinded to mouse treatment.
Staining of C6 and B1
Immunohistology of adult brains
Brain sections were prepared by anesthetizing mice with isoflurane prior to perfusion. They were perfused with paraformaldehyde (4%), following replacement of blood with heparinized preperfusion buffer. In all studies of mice exposed in utero to C6 or B1, the investigator was blinded to group. Immunostaining for brain-reactive antibodies from plasma or cell supernatant was performed as described9 on non-manipulated C57BL/6 (Jackson Laboratories, Bar Harbor, ME, USA) or CNTNAP2 −/− mice (a gift from Dr Brett S Abrahams, Albert Einstein College of Medicine, Bronx, NY, USA).
Nearest neighbor analysis
For this analysis, we randomly chose 16–20-week-old mice that had undergone behavioral assessment. Brains were sectioned by microtome (40 μm thick) and every fourth section was collected and mounted as before.24 Sections were stained with anti-NeuN antibody (Millipore, Mab 337). An assessment of the nearest neighbor (MBF, Williston, VT, USA) generated information about the distribution of neurons within the layers examined. The analysis is described in Supplementary methods.
Golgi staining and analysis
Mice exposed in utero to antibody were randomly chosen and studied at 2 weeks or 16–20 weeks of age (after the completion of behavioral assessment). Preparation of brains and Golgi staining were done by FD Rapid GolgiStain Kit (Ellicott City, MD, USA), according to the manufacturer’s protocol. Details of the analysis are described in Supplementary Methods.
Parvalbumin staining
Staining was performed similarly to NeuN staining described above on the same set of 16–20-week-old mice. Anti-parvalbumin antibody (Abcam, ab11427, Cambridge, UK) was used. For quantification details see Supplementary Methods.
Behavioral assessment
A primary screen was performed on mice that were exposed in utero to C6 or B1 at 5–7 weeks of age, followed by behavioral tests at 10–14 weeks of age
They were maintained on a reverse schedule of darkness (0900 to 2100) and light (2100 to 0900), with ad libitum access to food and water. Mice undergoing behavioral assessments were analyzed according to their cage number, assigned randomly. Cage numbers did not indicate the antibody the mice were exposed to in utero and therefore the testing was performed in a blinded fashion. Behavioral assays are described in Supplementary Methods.
Statistical analysis
We performed analysis of variance as well as Student’s t-test for data sets that were normally distributed (and with samples larger than 10). For smaller data sets, we performed the Mann-Whitney test. To analyze categorical data, a χ2-test for independence was used. The nonparametric Kolmogorov-Smirnov test was used for large data sets that were not normally distributed. All tests were performed with the statistical toolbox of Origin (versions 9 and 11), and are indicated in the text. Values were considered significant for P < 0.05. Data are presented as mean and error bars represent standard error. All tests were performed two-sided tailed.
RESULTS
Isolating brain-reactive monoclonal antibodies from a mother of an ASD child
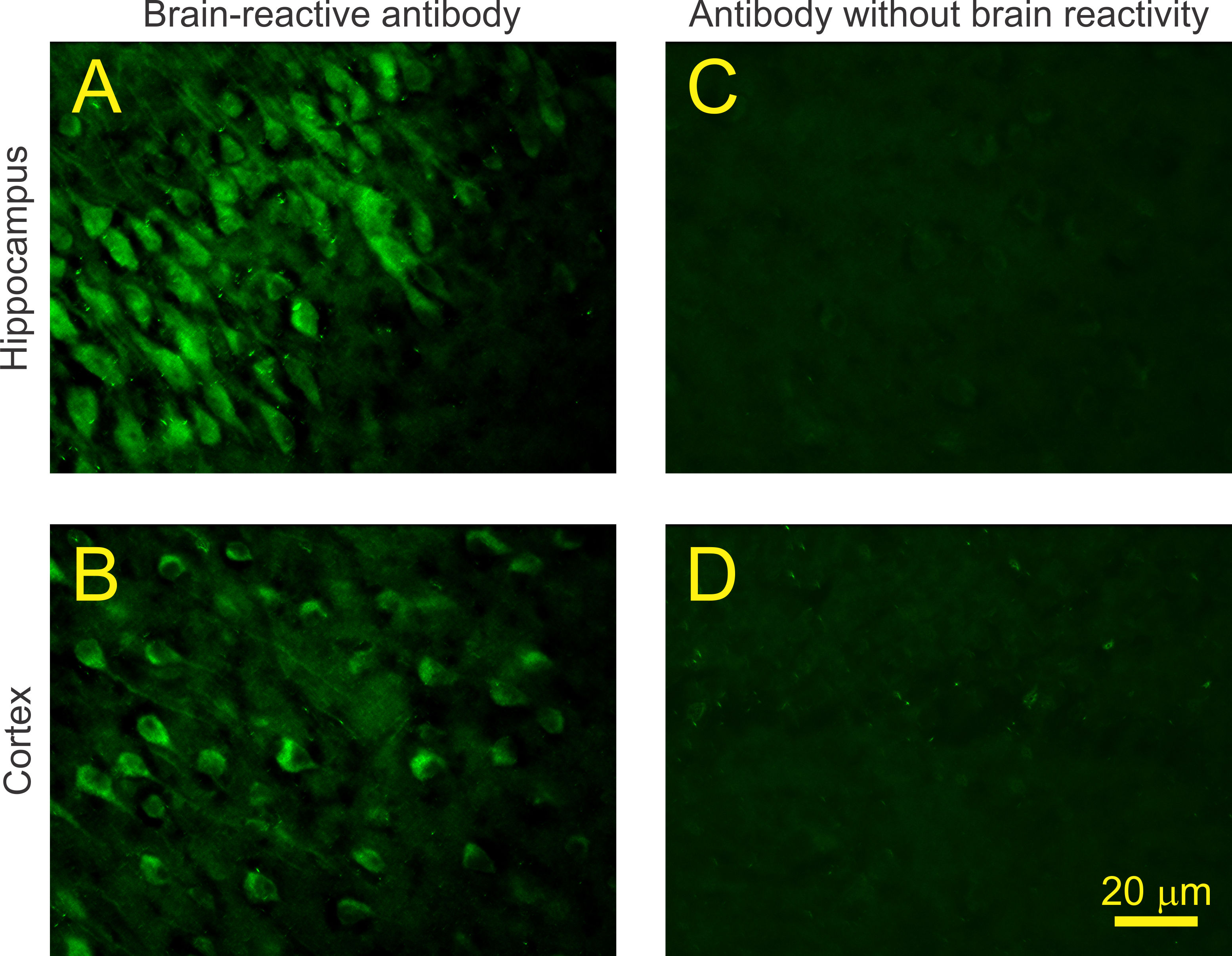
As a full appreciation of the pathology induced by brain-reactive antibodies requires cloning the pathogenic antibodies we established a protocol to identify antigen-specific B cells from mothers with brain-reactive serology and a child with ASD. We biotinylated human fetal brain lysate to tag brain-reactive B cells, and then performed single-cell cloning and expression of IgG. Because blood was obtained number of years after the birth of the affected child, we isolated memory (CD27+) B cells with reactivity to the fetal brain lysate.
Immunofluorescence revealed that the derived monoclonal antibodies stained mouse brain (Supplementary Figure 1). Here, we focus on the C6 monoclonal antibody that was found to bind the extracellular domain of Caspr2 by assessing its reactivity to ASD candidate antigens (Figure 1a). Caspr2 is a transmembrane protein in the soma, axon, dendrites and spines of neurons.19,28,29 It stabilizes surface expression of voltage-gated potassium channels along axons,30 glutamate receptors on dendrites29 and the formation of new synaptic spines.31
Figure 1.
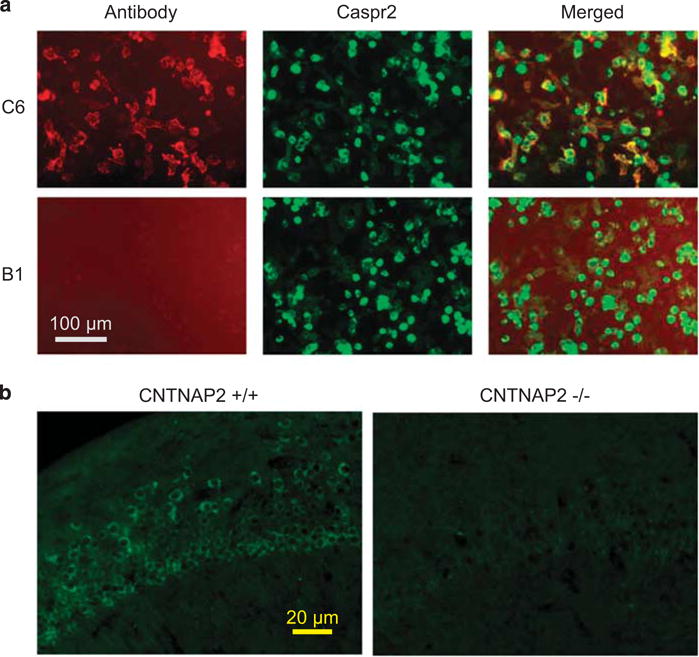
Brain-reactive monoclonal antibody C6 binds to Caspr2. (a) C6 (top panels), but not control B1 (bottom panels) antibody co-localize with Caspr2 on HEK-293T cells, expressing tGFP-Caspr2. No staining was seen on cells expressing only tGFP or non-transfected cells (data not shown). (b) C6 antibodies show reduced staining to the CA1 region in the hippocampus of CNTNAP2−/− (right) compared with wild-type (left) mice. Caspr2, contactin-associated protein-like 2; tGFP, turbo-green florescent protein.
We chose to focus on an anti-Caspr2 antibody as deletion of Caspr2 has been shown to lead to neurodevelopmental abnormalities,20 and Caspr2 mutations in human pedigrees associate with neurologic abnormalities including ASD.32–34 Thus, our choice permitted a proof-of-principle study of the potential impact of a monoclonal brain-reactive antibody on fetal brain development.
Immunohistochemical analysis revealed diminished binding of C6 to brains of mice lacking Caspr2 compared with brains of wildtype mice (Figure 1b), confirming its specificity for Caspr2. Importantly, plasma from the woman from whom C6 was derived also displayed diminished binding to Caspr2-deficient mouse brain (Supplementary Figure 2A–B). Caspr2 expression in the brain begins during fetal development, and is similar between females and males (Supplementary Figure 3). At E14, Caspr2 mRNA expression was localized to the developing cortex and to proliferating zones including the ventricular zone where excitatory projection neurons arise.19,20 Immunohistochemistry of fetal brain (E15.5) with C6 demonstrated staining in the cortex and the ventricular zone (Supplementary Figure 2C–D).
In utero exposure to C6 affects cortex integrity
To assess the pathogenic potential of C6, we intravenously administered either non-brain-reactive control antibody, B124 or C6 to timed-pregnant mice on day E13.5. Maternal antibody begins to cross the placenta and enter the fetal circulation at the beginning of the second trimester of pregnancy.35 As the blood-brain barrier is not fully formed in the developing fetus, maternal antibody present in fetal circulation can penetrate brain parenchyma starting at embryonic day E12.5. By E16.5–E17.5 the blood-brain barrier will normally exclude IgG.14
Strikingly, male, but not female, fetuses from dams killed 2 days after C6 administration exhibited a thinned cortical plate (Figures 2a and b). C6-exposed male, but not female, fetuses showed a decrease in proliferating cells in the developing cortex (Figures 2c and d). To confirm that the anti-Caspr2 antibodies were responsible for the alterations in the developing cortex, we performed additional experiments using plasma of the mother from whom C6 was isolated. For these studies we used: (i) unmanipulated plasma, (ii) plasma preabsorbed with cells expressing tGFP or (iii) plasma preabsorbed with cells expressing tGFP-Caspr2 (Supplementary Figure 4). Each of these preparations was administered intravenously to timed-pregnant mice on day E13.5 and the developing fetuses were analyzed 2 days later. Male fetuses exposed in utero to either unmanipulated plasma or plasma preabsorbed with cells expressing tGFP alone displayed a thinner cortex and a decreased number of proliferating cells (Supplementary Figure 4) than fetuses exposed to Caspr2-absorbed plasma, confirming the effects of anti-Caspr2 antibodies on brain development.
Figure 2.
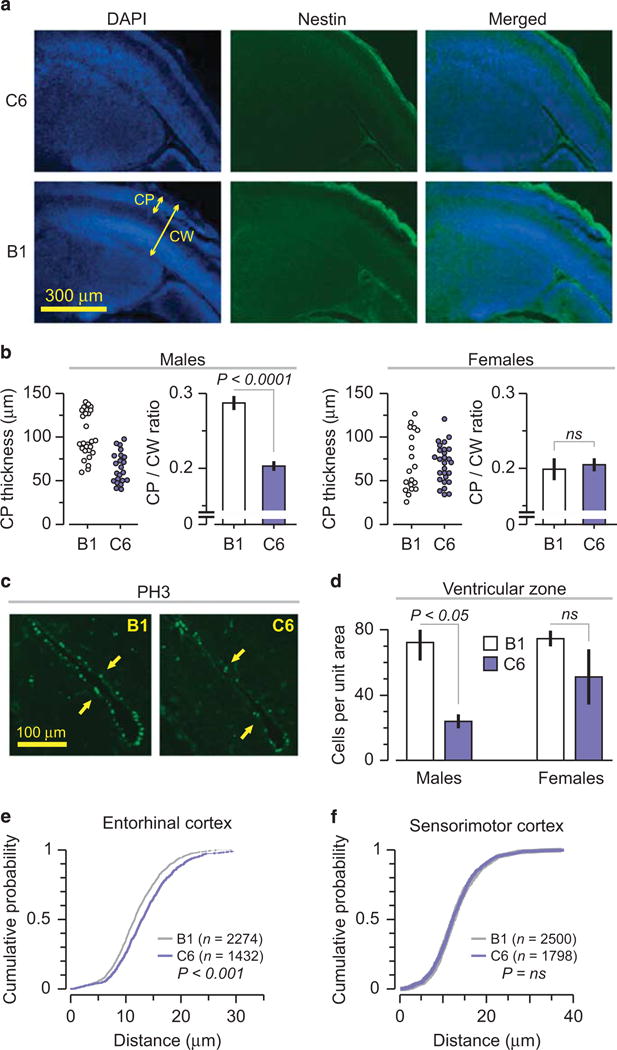
Male mice exposed in utero to C6 manifest cortical abnormalities. (a–d) Number of mice included in the analysis: Male: B1 = 5, C6 = 4; Female: B1 = 5, C6 = 5. Mice were derived from two litters for each antibody. (a) The cortical plate is abnormally thin in C6-exposed (top) relative to control B1-exposed (bottom) male brains as revealed by DAPI (left) and nestin (middle) staining; CP, cortical plate; CW, cortical width. (b) (Left) Quantification of cortical plate thickness. Dots correspond to number of measurements performed (Right). The ratio of cortical plate to cortical width in male and female fetal brain; P < 0.0001, t = 7.15, t-test. (c) C6-exposed male brains (right) display fewer mitotic cells than B1-exposed male brains (left) as revealed by PH3 staining. Arrows identify regions of neurogenesis in the ventricular zone (VZ). (d) Quantification of mitotic cells (PH3+) in the VZ, Intermediate zone and sub-plate for both males and females, as indicated. Unit area = 62.5 mm2; P < 0.05, Z = 2.34, Mann-Whitney test. (e–f) Cumulative probability based on nearest neighbor analysis in five adult B1- and four adult C6-exposed mice (two litters each). Dots represent the number of cells. (e) Entorhinal cortex (Cell number: B1, n = 2274, C6, n = 1432), P < 0.001, KS. (f) Somatosensory cortex (Cell number: B1, n = 2500, C6, n = 1798), P = N.S.
In adulthood, male offspring exposed in utero to C6 showed focal cortical changes. Based on nearest neighbor analysis the distance between neurons was significantly different in the entorhinal cortex between C6- and B1-exposed mice (Figure 2e). In contrast, in the somatosensory cortex there was no apparent differences (Figure 2f).
In utero exposure to C6 affects hippocampal neurons
As the entorhinal cortex represents a major input and output structure for the hippocampus, we assessed pyramidal hippocampal neurons and observed fewer dendritic arbors and dendritic spines, and less-dendritic branching in the neurons of C6-exposed male mice compared to B1-exposed male mice (Figures 3a–e). We also examined CA1 neurons in 2-week-old male mice before full development of these cells has occurred36 to determine whether the abnormalities observed in the adult mice represented a failure in maturation, or a late consequence of in utero exposure to antibody (Figures 3a–d). We found the 2-week-old C6-exposed mice exhibited reduced apical dendritic branches when compared with age-matched B1-exposed mice (Figures 3b–d), demonstrating a maturational defect.
Figure 3.
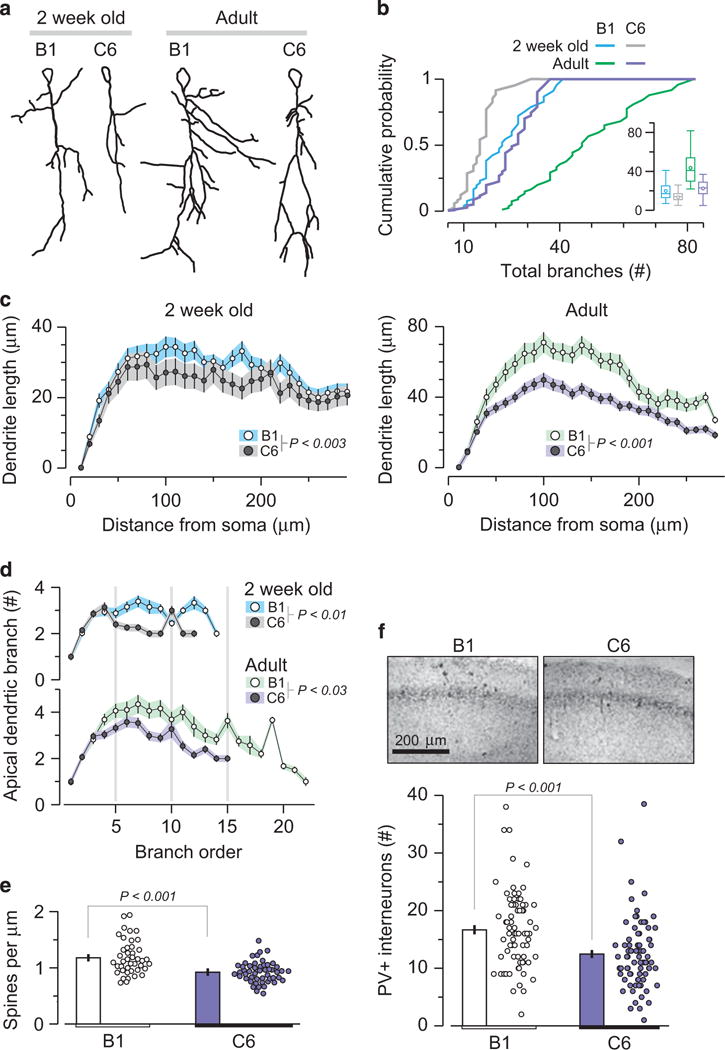
C6-exposed male offspring display significantly reduced hippocampal dendritic complexity. CAI pyramidal neurons of C6-exposed male mice show reduced dendritic length and spine density. Analysis includes: 2-week-old mice (n=4 per group); neurons: B1 = 45, C6 = 49 and adult animals (n = 4 per group); neurons: B1 = 70, C6 = 83. Each group includes animals from two litters. (a) Traced drawings of representative Golgi-impregnated CA1 pyramidal neurons from 2-week-old (left) and adult (16–20-week old) (right) B1- and C6-exposed mice. (b) Cumulative probability of total number of branches. 2-week-old B1 vs C6, P < 0.003, D = 0.35, Kolmogorov-Smirnov (KS) test; adult, P < 0.001, D = 0.64, KS test. Insert, box plot, represents total number of branches, with data presented as mean and quartiles. (c) Scholl analysis depicts dendritic length as a function of distance from the soma. Left: 2-week-old mice, P < 0.005, D = 0.44, KS. Right: adult mice, P < 0.001, D = 0.97, KS test. (d) Number of dendritic branches as a function of branch order, centrifugally defined to start at the origin of the tree and continue out towards the termination, and as the number of segments traversed from the origin. The C6- and B1-exposed mice differ significantly at both ages: 2-week-old, P < 0.01, D = 0.6, KS test; adult, P < 0.03, D = 0.45, KS test. (e) Reduced density of synaptic dendritic spines in CA1 neurons in C6-exposed male mice. Dots represent individual dendrites. B1 = 1.18 ± 0.04 spines μm−1; c6 = 0.92 ± 0.02 spines μm−1; P < 0.001, t = 4.78, t-test. (f) Top, representative photomicrographs of the CA1 field showing labeled PV+ interneurons. Bottom, quantification of PV+ neurons in adult B1 (n = 1251) and C6 (n = 998) groups, P < 0.001, Z = 4.25, Mann-Whitney, four animals per group, two litters.
Finally, we stained for GABAergic parvalbumin positive (PV+) neurons in adult male mice exposed in utero to C6 or B1, as mice with genetic deletion of Caspr2 display fewer GABAergic neurons. Fewer PV+ neurons were present in the hippocampus in C6-exposed male mice (Figure 3f), consistent with the data from Caspr2-deficient mice, which exhibit a 20% reduction in PV+ hippocampal interneurons.20
Behavioral abnormalities in male mice exposed to C6 in utero
To examine C6-mediated effects on behavior, adult offspring were studied. In total, 5–7-week-old C6-exposed mice of either sex were indistinguishable from B1-exposed of the same sex and age (B1 and C6 male: n = 16, five litters each;female: B1 n = 16, C6 = 11, five litters each), in body weight, coat, grip strength, body tone, reflexes and sickness behavior as described in ref. 37 (Supplementary Table 1). Moreover, in the open-field test, mobility and time spent in the center of the arena (suggestive of anxietylike behavior) were not different (t-test, P> 0.4; analysis of variance center/periphery × group, P>0.8, respectively).
When subjected to tasks that focus on core ASD symptoms,38 10–14-week-old C6-exposed male mice displayed behavioral abnormalities.
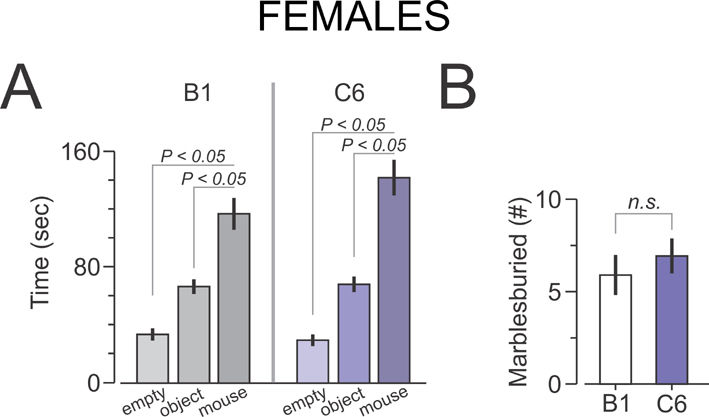
C6-exposed mice showed abnormal sociability; they spent equal time in proximity to the unfamiliar mouse and an unfamiliar object in the social approach test (Figure 4a). In contrast, B1-exposed mice spent more time in proximity to the unfamiliar mouse than the unfamiliar object (Figure 4a).
Figure 4.
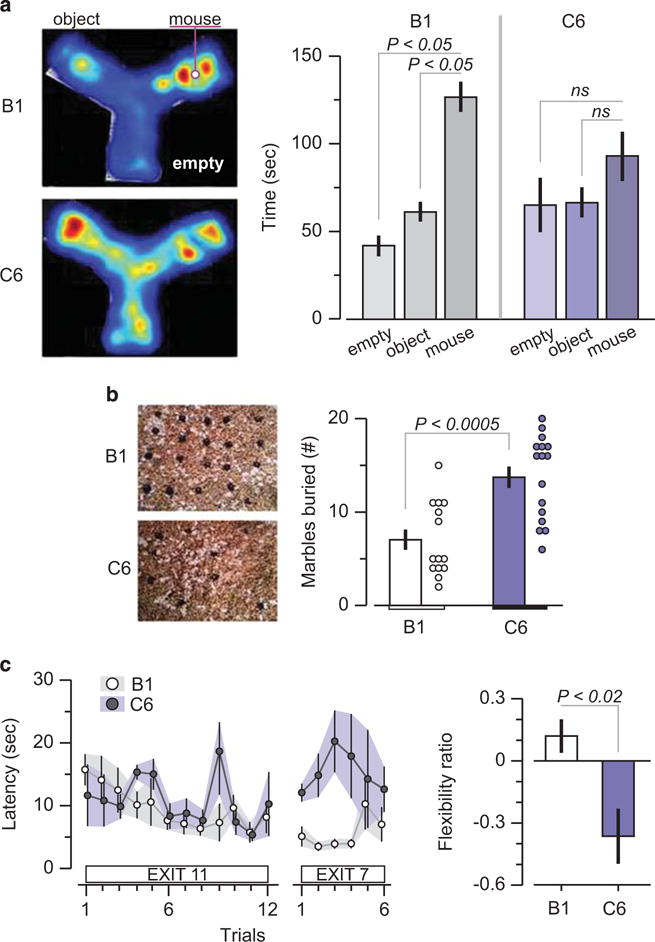
Impaired performance of C6-exposed male offspring in tasks that resemble core autism spectrum disorder symptoms. Maternal antibody-exposed male mice were subjected to behavioral assessment at adulthood (10–14 weeks). (a) Social approach task. (Left) representative heat map showing social patterns in B1- and C6-exposed mice. (Right) B1-exposed male mice (n = 15) displayed normal sociability, defined as spending significantly more time with the novel mouse compared with the novel object, whereas C6-exposed male mice (n = 14) spent a similar amount of time near the novel object and the novel mouse. Five litters per group. ANOVA, followed by Bonferroni test, P < 0.05. Two C6 and one B1 mouse were excluded due to a technical failure (b) Marble-burying task. (Left) representative examples of burying patterns in the two groups. (Right) C6-exposed mice (n = 16) display enhanced stereotypic behavior, that is, they bury more marbles than the B1-exposed mice (n = 14); P < 0.0005, t = 4.2, t-test; five litters per group. One B1 mouse was excluded because it scored more than three s.d.s above the group mean (c) Clock maze task. (Left) the graph shows the latency to escape from the center of the maze to a peripherally located exit. C6-exposed male mice (n = 6) perform similarly to B1-exposed male mice (n = 11) in the initial phase (Exit flexibility ratio (defined in Methods) shows that C6-exposed mice are significantly less likely to switch from a familiar exit to a novel one, showing impaired flexible learning; P < 0.02, Z = 2.36, Mann-Whitney test; 2–3 litters per group.
Previous ASD models defined impaired sociability as the lack of difference between time spent with the unfamiliar mouse and the unfamiliar object.38,39 Time spent in the non-occupied area is not included in the definition of impaired sociability; C6-exposed mice spent equal time with the unfamiliar object and in the empty space. Although this might be interrupted as general motivational impairment, that is, lack of interest regardless of the stimulus, it is important to note that C6-exposed male mice spent as much time as B1-exposed male mice near the unfamiliar object (Figure 4a). This suggests that the diminished interest of C6-exposed mice in the unfamiliar mouse was specific to that stimulus and less likely to reflect a general motivational problem.
It is possible that C6-exposed mice are impaired in olfaction and therefore less likely to initiate social behavior. Although this hypothesis has not been formally tested in our study, it should be noted the C6-exposed mice did initiate sniffing in proximity to the unfamiliar mouse (data not shown).
C6-exposed mice buried significantly more marbles than B1-exposed mice in a test that measures stereotypic and/or compulsive behavior40,41 (Figure 4b). Although they learned the initial exit in the clock maze test with similar efficiency as B1-exposed mice, they were impaired in learning a new exit, demonstrating a specific defect inflexible learning (Figure 4c). C6- and B1-exposed female offspring performed equivalently in these tasks, confirming the absence of an effect of C6 on the developing female fetal brain (Supplementary Figure 5).
Mothers with anti-brain antibodies and with an ASD child are more likely to harbor anti-Caspr2 antibodies
We next examined how frequently antibodies with this specificity are present in women with an ASD child. We analyzed plasma of mothers of a child with ASD that had brain-reactive serology9 using indirect immunofluorescence on non-permeabilized Caspr2-transfected HEK-293T cells. We found that 37% (20 of 53) displayed robust Caspr2 membrane staining, compared with 12% (8 of 63) of plasma from mothers of an ASD child lacking brain-reactive antibodies, 12% (6 of 51) of plasma from unselected women of child-bearing age and 7.6% (4 of 52) of plasma from mothers of a typically developing child bound Caspr2 (χ2-test, P < 0.001).
DISCUSSION
In this study we have report a protocol for cloning brain-reactive antibody from mothers with an ASD child. Using this methodology we have demonstrated that a single monoclonal antibody can elicit profound structural brain abnormalities and lead to core ASD behavioral impairments in offspring. There is a period of time when the fetal blood-brain barrier is porous to antibodies;14 thus potentially pathogenic autoantibodies present in the mother’s blood can affect the developing brain even in the absence of detectable disease in the mother. The antibody we studied has specificity for Caspr2, a protein critical to normal brain development. We found that antibodies with Caspr2 specificity are significantly more frequent in the plasma of mothers with anti-brain antibodies and a child with ASD.
This paradigm, in which maternal antibodies affect fetal brain development with long-term cognitive effects, has already been reported in systemic lupus.24,27,42 In utero exposure at day E13.5 to a subset of lupus anti-DNA monoclonal antibody that cross-react with the GluN2 A/B subunit of the N-methyl-D-aspartate cause long-term and selective cognitive impairments in male offspring24 and death of female fetuses.27 In the course of these studies, it was confirmed that maternal IgG penetrates equally male and female fetal brain parenchyma,27 thereby setting the foundation for the current study.
The pathogenic antibody, C6, targets Caspr2. In humans, a rare variant of CNTNAP2, the gene that encodes Caspr2, is associated with ASD.32 Families with mutant CNTNAP2 display epilepsy,18 obsessive compulsive behaviors34 and variable, gender-dependent structural abnormalities in the brain.43 Similarly, mice with a deletion of CNTNAP2 display disrupted neuronal migration, a loss of inhibitory GABAergic neurons and behavioral characteristics that are considered to represent an ASD-like phenotype in mice, such as hyperactivity, stereotypic behaviors, reduced flexible learning in the Morris water maze and social behavior abnormalities.20 In vitro downregulation of Caspr2 leads to a decrease in dendritic processes28 and spine density.31 In these studies no data are provided on whether male and female mice are equally or differentially affected. In our model exposure to C6 led to gender effects as in the human pedigrees and to histopathologic and behavioral similarities to Caspr2 deficiency in vivo20 and in vitro,28,31 including loss of inhibitory GABAergic neurons, reduced dendrites and spines arborization and an ASD-like phenotype.
Although the parallels between the CNTNAP2−/− mice and our study are striking, there are also some differences. The mechanisms leading to neurodevelopmental abnormalities in either Caspr2-deficient mice or in mice exposed in utero to anti-Caspr2 antibody are not fully understood. It interesting to speculate that C6 may create a functional hypomorph of Caspr2 at a critical moment in brain development. This hypothesis requires further studies.
Changes in cortical thickness as well as decreased neuronal packing44 have been observed in ASD. It is possible that cortical thinning may reflect aberrant migration of developing neurons that is mediated by transient axonal glycoprotein-1, which colocalizes with Caspr230 and has a role in the migration of proliferating neurons in the cortex.45,46 Alternatively, Caspr2 may regulate neuronal migration through its effects on glutamate receptor density,28,29 as the N-methyl-D-aspartate receptor is known to regulate neuronal migration during fetal brain development.
Both neuroanatomical and behavioral alterations in C6-exposed mice involved the cortico-hippocampal circuits.40,47 Particularly strong expression of Caspr2 has been documented in the cortex and hippocampus.48 Further studies should examine whether increased expression explains the preferentially effect on those area, or whether particular neurons are preferentially affected by the antibody.
It is of interest that only male mice exposed in utero to C6 antibody exhibited neurodevelopmental abnormalities given the strong male bias in ASD.2 Antibody penetration to the placenta and to the fetal brain is not sex dependent.14,24,27 We also did not observe differences in the expression of Caspr2 between male and female fetuses, or at any time postpartum, and we are not aware of published data on the phenotype of CNTNAP2 −/− female mice. Male gender is a factor in the linkage association between CNTNAP2 and impaired language in a human genealogy.32 The basis for gender preference requires further exploration; however a recent paper showed that estrogen agonist can reverse the behavioral Phenotype in Zebrafish CNTNAP2 Mutants.49 As by day E13.5, there are hormonal differences in male and female fetuses, estrogen may extract a protective effect in C6-exposed female fetuses.
Our data suggest that anti-Caspr2 antibodies are more frequent in mothers with anti-brain antibodies and a child with ASD. It remains to be determined whether these antibodies target the same epitope on Caspr2 as the C6 antibody, and whether epitope specificity is critical to pathogenicity. It is documented that patients with encephalitis, neuromyotonia and Morvan’s syndrome have antibodies against Caspr2.50,51 It is not known how these antibodies penetrate the adult blood-brain barrier, or whether they might lead to fetal brain abnormalities.
The method for deriving human monoclonal brain-reactive antibodies from mothers with an ASD child, and the demonstration that a monclonal maternal anti-brain antibody can mediate structural brain abnormalities and behavioral changes provides the technical and conceptual basis to explore a model of ASD pathogenesis with significant diagnostic and therapeutic implications.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the Simons Foundation and Elena Kowalsky at the Feinstein Institute for help obtaining blood samples. We also thank Dr Kevin J Tracey for helpful comments. The research was funded by the Department of Defense (AR130137), NIH (R43 MH106195), and The Nancy Lurie Marks Foundation and The Simons Foundation. LB is a recipient of a Brain and Behavior NARSAD Young Investigator Foundation Grant. We thank Dr. Czeslawa Kowal for her assistance in perfusing embryos.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Diagnostic and Statistical Manual of Mental Disorders. 5th. American Psychiatric Publishing; Arlington, VA, USA: 2013. [Google Scholar]
- 2.Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorder among children aged 8 years – autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63:1–21. [PubMed] [Google Scholar]
- 3.Risch N, Hoffmann TJ, Anderson M, Croen LA, Grether JK, Windham GC. Familial recurrence of autism spectrum disorder: evaluating genetic and environmental contributions. Am J Psychiatry. 2014;171:1206–1213. doi: 10.1176/appi.ajp.2014.13101359. [DOI] [PubMed] [Google Scholar]
- 4.Patterson PH. Maternal infection and autism. Brain Behav Immun. 2012;26:393. doi: 10.1016/j.bbi.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–939. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atladottir HO, Henriksen TB, Schendel DE, Parner ET. Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics. 2012;130:e1447–e1454. doi: 10.1542/peds.2012-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazina V, Gerdts J, Trinh S, Ankenman K, Ward T, Dennis MY, et al. Epigenetics of autism-related impairment: copy number variation and maternal infection. J Dev Behav Pediatr. 2015;36:61–67. doi: 10.1097/DBP.0000000000000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brimberg L, Sadiq A, Gregersen PK, Diamond B. Brain-reactive IgG correlates with autoimmunity in mothers of a child with an autism spectrum disorder. Mol Psychiatry. 2013;18:1171–1177. doi: 10.1038/mp.2013.101. [DOI] [PubMed] [Google Scholar]
- 10.Croen LA, Braunschweig D, Haapanen L, Yoshida CK, Fireman B, Grether JK, et al. Maternal mid-pregnancy autoantibodies to fetal brain protein: the early markers for autism study. Biol Psychiatry. 2008;64:583–588. doi: 10.1016/j.biopsych.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braunschweig D, Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Croen LA, et al. Autism: maternally derived antibodies specific for fetal brain proteins. Neurotoxicology. 2008;29:226–231. doi: 10.1016/j.neuro.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singer HS, Morris CM, Gause CD, Gillin PK, Crawford S, Zimmerman AW. Antibodies against fetal brain in sera of mothers with autistic children. J Neuroimmunol. 2008;194:165–172. doi: 10.1016/j.jneuroim.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Dalton P, Deacon R, Blamire A, Pike M, McKinlay I, Stein J, et al. Maternal neuronal antibodies associated with autism and a language disorder. Ann Neurol. 2003;53:533–537. doi: 10.1002/ana.10557. [DOI] [PubMed] [Google Scholar]
- 14.Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6:263ra158. doi: 10.1126/scitranslmed.3009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauman MD, Iosif AM, Ashwood P, Braunschweig D, Lee A, Schumann CM, et al. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl Psychiatry. 2013;3:e278. doi: 10.1038/tp.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singer HS, Morris C, Gause C, Pollard M, Zimmerman AW, Pletnikov M. Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: a pregnant dam mouse model. J Neuroimmunol. 2009;211:39–48. doi: 10.1016/j.jneuroim.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P, et al. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry. 2013;3:e277. doi: 10.1038/tp.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 19.Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24:1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- 20.Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Jacobi AM, Mackay M, Aranow C, Wang T, Chinnasamy P, et al. Identification of DNA-reactive B cells in patients with systemic lupus erythematosus. J Immunol Methods. 2008;338:79–84. doi: 10.1016/j.jim.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329:112–124. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JY, Huerta PT, Zhang J, Kowal C, Bertini E, Volpe BT, et al. Neurotoxic autoantibodies mediate congenital cortical impairment of offspring in maternal lupus. Nat Med. 2009;15:91–96. doi: 10.1038/nm.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mader S, Lutterotti A, Di Pauli F, Kuenz B, Schanda K, Aboul-Enein F, et al. Patterns of antibody binding to aquaporin-4 isoforms in neuromyelitis optica. PLoS One. 2010;5:e10455. doi: 10.1371/journal.pone.0010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Zhou D, Lee J, Niu H, Faust TW, Frattini S, et al. Female mouse fetal loss mediated by maternal autoantibody. The J Exp Med. 2012;209:1083–1089. doi: 10.1084/jem.20111986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, Sudhof TC. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci USA. 2012;109:18120–18125. doi: 10.1073/pnas.1216398109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varea O, Martin-de-Saavedra MD, Kopeikina KJ, Schurmann B, Fleming HJ, Fawcett-Patel JM, et al. Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc Natl Acad Sci USA. 2015;112:6176–6181. doi: 10.1073/pnas.1423205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gdalyahu A, Lazaro M, Penagarikano O, Golshani P, Trachtenberg JT, Geschwind DH. The autism related protein contactin-associated protein-like 2 (CNTNAP2) stabilizes new spines: an in vivo mouse study. PLoS One. 2015;10:e0125633. doi: 10.1371/journal.pone.0125633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falivelli G, De Jaco A, Favaloro FL, Kim H, Wilson J, Dubi N, et al. Inherited genetic variants in autism-related CNTNAP2 show perturbed trafficking and ATF6 activation. Hum Mol Genet. 2012;21:4761–4773. doi: 10.1093/hmg/dds320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics. 2003;82:1–9. doi: 10.1016/s0888-7543(03)00097-1. [DOI] [PubMed] [Google Scholar]
- 35.Simister NE. Placental transport of immunoglobulin G. Vaccine. 2003;21:3365–3369. doi: 10.1016/s0264-410x(03)00334-7. [DOI] [PubMed] [Google Scholar]
- 36.Casanova JR, Nishimura M, Swann JW. The effects of early-life seizures on hippocampal dendrite development and later-life learning and memory. Brain Res Bull. 2014;103:39–48. doi: 10.1016/j.brainresbull.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, et al. HMGB1 mediates cognitive impairment in sepsis survivors. Mol Med. 2012;18:930–937. doi: 10.2119/molmed.2012.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFarlane HG, Kusek GK, Yang M, Phoenix JL, Bolivar VJ, Crawley JN. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. 2008;7:152–163. doi: 10.1111/j.1601-183X.2007.00330.x. [DOI] [PubMed] [Google Scholar]
- 40.Deacon RM. Digging and marble burying in mice: simple methods for in vivo identification of biological impacts. Nat Protoc. 2006;1:122–124. doi: 10.1038/nprot.2006.20. [DOI] [PubMed] [Google Scholar]
- 41.Thomas A, Burant A, Bui N, Graham D, Yuva-Paylor LA, Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology (Berl) 2009;204:361–373. doi: 10.1007/s00213-009-1466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang EH, Volpe BT, Mackay M, Aranow C, Watson P, Kowal C, et al. Selective impairment of spatial cognition caused by autoantibodies to the N-methyl-d-aspartate receptor. EBioMed. 2015;2:755–764. doi: 10.1016/j.ebiom.2015.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan GC, Doke TF, Ashburner J, Wood NW, Frackowiak RS. Normal variation in fronto-occipital circuitry and cerebellar structure with an autism-associated polymorphism of CNTNAP2. Neuroimage. 2010;53:1030–1042. doi: 10.1016/j.neuroimage.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simms ML, Kemper TL, Timbie CM, Bauman ML, Blatt GJ. The anterior cingulate cortex in autism: heterogeneity of qualitative and quantitative cytoarchitectonic features suggests possible subgroups. Acta Neuropathol. 2009;118:673–684. doi: 10.1007/s00401-009-0568-2. [DOI] [PubMed] [Google Scholar]
- 45.Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D. The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development. 2001;128:4635–4644. doi: 10.1242/dev.128.22.4635. [DOI] [PubMed] [Google Scholar]
- 46.Furley AJ, Morton SB, Manalo D, Karagogeos D, Dodd J, Jessell TM. The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell. 1990;61:157–170. doi: 10.1016/0092-8674(90)90223-2. [DOI] [PubMed] [Google Scholar]
- 47.Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56. doi: 10.1002/(SICI)1098-1063(2000)10:1<47::AID-HIPO5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 48.Gordon A, Salomon D, Barak N, Pen Y, Tsoory M, Kimchi T, et al. Expression of Cntnap2 (Caspr2) in multiple levels of sensory systems. Mol Cell Neurosci. 2016;70:42–53. doi: 10.1016/j.mcn.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 49.Hoffman EJ, Turner KJ, Fernandez JM, Cifuentes D, Ghosh M, Ijaz S, et al. Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron. 2016;89:725–733. doi: 10.1016/j.neuron.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol. 2011;69:303–311. doi: 10.1002/ana.22297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010;133:2734–2748. doi: 10.1093/brain/awq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.