


Abstract
We describe a format for production of protein arrays termed ‘protein in situ array’ (PISA). A PISA is rapidly generated in one step directly from PCR-generated DNA fragments by cell-free protein expression and in situ immobilisation at a surface. The template for expression is DNA encoding individual proteins or domains, which is produced by PCR using primers designed from information in DNA databases. Coupled transcription and translation is carried out on a surface to which the tagged protein adheres as soon as it is synthesised. Because proteins generated by cell-free synthesis are usually soluble and functional, this method can overcome problems of insolubility or degradation associated with bacterial expression of recombinant proteins. Moreover, the use of PCR-generated DNA enables rapid production of proteins or domains based on genome information alone and will be particularly useful where cloned material is not available. Here we show that human single-chain antibody fragments (three domain, VH/K form) and an enzyme (luciferase) can be functionally arrayed by the PISA method.
INTRODUCTION
With the completion of the genome sequences of many organisms, it is now seen as a priority to use high-throughput approaches to analyse protein function, in order to gain a comprehensive understanding of complex cellular processes and pathways (1). Array technologies allow large-scale analysis of the interactions and functions of biological molecules in parallel and simultaneous quantitative comparison of individual assay values (2). They are well established for global analysis of nucleic acids, i.e. oligonucleotide and cDNA microarrays, and are widely used for the analysis of gene expression and polymorphisms (3). For application to proteomics, the array approach has also been adapted for high-throughput functional display and analysis of peptides and proteins (4). Protein arrays have been generated by immobilisation of purified or clonally expressed proteins onto suitable solid surfaces, such as treated glass slides, microtitre plates, polyacrylamide gel pads or absorptive membranes such as nitrocellulose or polyvinylidene fluoride (PVDF) (4). In order to provide the necessary material, methods for genome-wide production of recombinant proteins from bacteria and yeast have been described (5–7). In some cases, the proteins were expressed as constructs fused to an affinity tag, such as hexahistidine or glutathione-S-transferase and used either as crude lysates or after affinity purification. Antibody arrays have been prepared by displaying recombinant Escherichia coli colonies on filters and allowing expressed proteins to adhere directly to a PVDF membrane for large-scale screening by antigen binding (8).
A significant bottleneck in the production of proteins for microarray technologies is the comprehensive production of functional recombinant proteins. This is due both to poor expression, aggregation and degradation of many eukaryotic proteins in bacterial systems, as well as time-consuming DNA cloning and gene identification procedures which are required before protein expression can occur (9). Furthermore, where denaturants are used in the extraction, proteins will often be rendered non-functional. To overcome those problems, we describe a format termed ‘protein in situ array’ (PISA) in which protein arrays are rapidly generated in one step, directly from DNA templates, by cell-free protein expression and simultaneous in situ immobilisation at a surface (Fig. 1). Individual genes or gene fragments are produced by PCR or RT–PCR, depending on the source of genetic material, using primers designed from information in databases. The PISA is generated by cell-free protein synthesis using coupled transcription and translation to produce a double (His)6-tagged protein, the reaction being carried out on a Ni–NTA-coated surface to which the protein adheres as soon as it is synthesised. Advantages of the method are that proteins generated by cell-free synthesis are usually soluble and functional (10), while the use of PCR-generated DNA enables rapid production of proteins or domains based only on genome information, particularly where cloned material is not available. Here, we show that human single-chain antibody fragments and an enzyme (luciferase) can be functionally arrayed by the PISA method.
Figure 1.
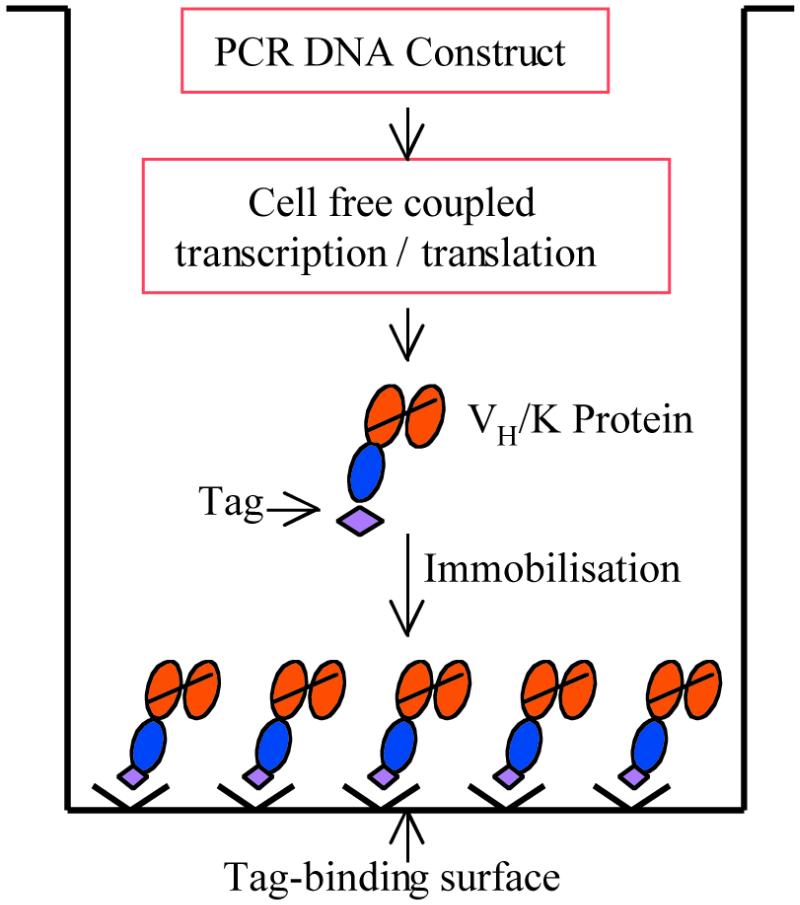
The PISA procedure. The process of cell-free protein expression and in situ immobilisation occurs in a single well, starting with a PCR-generated DNA construct. Expression and immobilisation of a VH/K protein are illustrated.
MATERIALS AND METHODS
Single chain antibody VH/K fragments and luciferase
VH/K fragments comprise a VH domain linked to the complete κ light chain, as described (11). Here we have used cloned human anti-progesterone VH/K fragments previously obtained by ribosome display selection from a transgenic mouse library (12). The T7 control plasmid containing luciferase was obtained from Promega.
Design of PCR construct for in vitro expression of tagged proteins
Figure 2 outlines a general PCR strategy for construction of DNA suitable for in vitro protein synthesis by the PISA method. The construct contains a T7 promoter for transcription by T7 RNA polymerase and a Kozak sequence for translation initiation in cell-free eukaryotic systems (13). To increase the efficiency of protein immobilisation on a Ni–NTA-coated surface and to allow the re-use of the protein arrays, a double (His)6-tag domain was designed. A flexible 19 residue linker (14) is placed between the protein to be arrayed and the His-tag domain, in order to reduce any possible interference of the tag sequence on folding of the attached protein. A poly(A)28 tail and a transcription terminator are incorporated at the 3′ end of the DNA to increase transcription efficiency (13). To ensure translation termination and release of the nascent polypeptide from the ribosome complex, two stop codons (TAATAA) are included following the double (His)6-tag sequence.
Figure 2.

PCR strategy for construction of DNA suitable for PISA, illustrated with a VH/K antibody fragment. The primers are: (1) T7Ab/back; (2) Ab-linker/for; (3) Linker-tag/back; (4) His-tag/for. The broken line indicates the linker sequence.
DNA constructs in which either antibody VH/K fragments or luciferase were linked to the double (His)6-tag domain (VH/K-His) were produced by PCR with the primers described below.
Primers for PCR generation of VH/K-linker fragments
T7Ab/back, 5′-GCAGCTAATACGACTCACTATAGGAACAGACCACCATG(C/G)AGGT(G/C)CA(G/C)CTCGAG(C/G)AGTCTGG-3′ provides the T7 promoter and Kozak signal (underlined) and a degenerate sequence complementary to the 5′ region of the antibody heavy chain (italics). The initiation codon is indicated in bold. Ab-linker/for, 5′-GCCACCGCCTCTAGAGCGGCTCAGCGTCAGGGTGCTGCT-3′ provides a sequence complementary to the 3′ region of the human κ constant domain (Cκ) and a sequence (underlined) overlapping a linker-tag/back primer (see below) used for generation of the double (His)6-tag domain.
Primers for PCR generation of luciferase-linker fragment
T7Luci/back, 5′-GCAGCTAATACGACTCACTATAGGAACAGACCACCATGGAAGACGCCAAAAAC-3′. The T7 promoter and Kozak signal are underlined. Italics indicate the sequence complementary to the 5′ region of luciferase. ATG is the initiation codon. Luci-linker/for, 5′-GCCACCGCCTCTAGAGCGCAATTTGGACTTTCCGCC-3′. The underlined sequence overlaps a linker-tag/back primer (see below) used for generation of the double (His)6-tag domain.
Primers for PCR generation of the His-tag domain
Linker-tag/back, 5′-GCTCTAGAGGCGGTGGCTCTGGTGGCGGTTCTGGCGGTGGCACCGGTGGCGGTTCTGGCGGTDDCAAACGGGCTGATGCTGCA-3′ provides a sequence (underlined) overlapping the primers Ab-linker/for and Luci-linker/for, used for VH/K-linker or luciferase construction (above) and the linker sequence for PCR generation of the double (His)6-tag domain (below). His-tag/for, 5′-TCCGGATATAGTTCCTCC-3′
Plasmid pTA-His encoding a double (His)6-tag domain
The plasmid pTA-His contains a fragment encoding (in order) a flexible linker and a double (His)6-tag, followed by two stop codons, a poly(A) tail and a transcription termination region. The sequence of this fragment was: 5′-GCTCTAGAggcggtggctctggtggcggttctggcggtggcaccggtggcggttctggcggtggcAAACGGGCTGATGCTGCACATCACCATCACCATCACTCTAGAGCTTGGCGTCACCCGCAGTTCGGTGGTCACCACCACCACCACCACTAATAA(A)28CCGCTGAGCAATAACTAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTGCTGAAAGGAGGAACTATATCCGGA-3′. The linker encoding a 19 amino acid sequence is shown in lower case (14), the double (His)6 tag is underlined, stop codons are shown in bold and (A)28 indicates a poly(A) region comprising 28× A. The transcription terminator is shown in italics.
Construction of PCR fragments
In general, standard PCR was carried out for 30 cycles to obtain the VH/K-linker, luciferase-linker and double (His)6-tag domain DNA fragments in separate reactions using Taq polymerase (Qiagen, UK) according to the manufacturer’s instructions. The resulting fragments were analysed and eluted from a 1% agarose gel using a gel-extraction kit (Qiagen, UK). For assembly, double (His)6-tag domain was mixed with either the VH/K-linker or luciferase-linker in equimolar ratios (total DNA 10–50 ng) and added to a PCR mixture containing 2.5 µl 10× PCR buffer (supplied with Taq DNA polymerase), 1 µl 2.5 mM dNTPs, 1 U Taq DNA polymerase and water to a final volume of 25 µl. After thermal cycling for eight cycles (94°C for 30 s, 54°C for 1 min and 72°C for 1 min), 2 µl product was subjected to a second PCR in a final volume of 50 µl for 30 cycles using primers T7Ab/back and His-tag/for to generate VH/K-His (Fig. 2) or T7 Luci/back and His-tag/for to generate the Luci–His construct.
Generation of protein in situ array
Two surfaces for protein immobilisation were used, namely Ni–NTA-coated microtiter plates or Ni–NTA magnetic agarose beads. A standard 50 µl coupled transcription/translation reaction was prepared by adding 0.5–1 µg PCR DNA (purified or unpurified), together with 1 µl 1 mM methionine and 0.5 µl 100 mM magnesium acetate, into 40 µl rabbit reticulocyte ‘TNT T7 Quick for PCR DNA’ kit (Promega, UK). Thereafter, for the microtiter plate method, 25 µl mixture was added to each well of a Ni–NTA HisSorb strip or plate (Qiagen, UK). After incubation at 30°C for 2 h with gentle shaking, the mixture was removed and the well washed three times with 100 µl wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole pH 8.0), followed by a final wash with 100 µl phospate buffered saline (PBS). For protein immobilisation on beads, 5 µl Ni–NTA-coated magnetic agarose beads (Qiagen, UK) were washed once with 50 µl PBS in a 0.5 ml Eppendorf tube and 25 µl TNT mixture was added. Incubation and washing procedures were as described for plates.
Detection and functionality of immobilised VH/K-His fragments
For detection of human single chain VH/K-His fragments immobilised on Ni–NTA coated wells, 25 µl horseradish peroxidase (HRP)-linked rabbit anti-human κ antibody (DAKO, Denmark) diluted 1:500 in Superblock buffer (Pierce, UK) was added to each well and incubated at room temperature for 1 h with vibration. HRP-linked rat monoclonal anti-mouse κ (Zymed, San Francisco), 1:1000 dilution, was used as a control. After washing three times with the wash buffer as above and once with PBS, HRP activity was developed using tetramethyl benzidine substrate (Sigma, UK) and H2O2 in acetate buffer pH 6 (11). Colour was developed as either blue (no addition of acid) or yellow with addition of 5 µl of 2 M H2SO4.
The antigens progesterone-11α-hemisuccinyl-bovine serum albumin (P-BSA) and BSA were from Sigma. Antigen biotinylation was carried out as described (15) using biotin from Pierce. For detection of the functional binding activity of immobilised VH/K-His fragments, 25 µl biotinylated antigen (3 µg/ml in Superblock buffer) was added to each well and incubated at room temperature for 1 h with vibration. After three washes with the wash buffer and one with PBS, 25 µl of a 1:500 dilution of HRP-linked streptavidin (Amersham Pharmacia Biotech, UK) was added per well and incubated at room temperature for 1 h, followed by washing and development of HRP activity as above.
Functional assay of free and immobilised luciferase
Luciferase Assay Reagent (Promega, UK) was used to measure luciferase activity. For free luciferase, 2 µl TNT mixture after translation was added into 100 µl Luciferase Assay Reagent. After quickly mixing, light intensity was measured immediately on a BioOrbit luminometer. The reading was recorded twice in a 10 s interval. For PISA-immobilised luciferase, 100 µl Luciferase Assay Reagent was added to the luciferase-immobilised beads and quickly mixed. Light intensity was measured as above.
SDS–PAGE and western blot analysis
Procedures for SDS–PAGE, protein transfer to PVDF membranes and detection with HRP-linked antibodies were as described (11). For sample preparation, proteins produced in the rabbit reticulocyte TNT mixture (above) were treated with equal volumes of Laemmli sample buffer (Bio-Rad, USA). Proteins immobilised on Ni–NTA-coated wells were eluted by incubation with 40 µl Laemmli sample buffer diluted 1:1 with TE buffer (10 mM Tris–HCl, 1 mM EDTA pH 8.0) at room temperature for 10 min. His-tagged proteins were detected by HRP-linked anti-polyhistidine antibody (Sigma, UK), at a dilution of 1:2000. Antibody κ chains were detected using HRP-linked anti-human κ antibody (DAKO, Denmark), at a dilution of 1:1000. HRP activity was developed using the Supersignal kit (Pierce, UK), following the manufacturer’s instructions. As standards for quantitation, P5-17 VH/K-His was expressed in E.coli as described (12) and purified by absorption and elution from Ni–NTA agarose magnetic beads (Qiagen, UK). Protein concentration was determined by measuring optical density at 280 nm. Blotted and stained proteins were scanned and quantitated by densitometry using a Joyce Loebel Chromoscan 3.
Stripping and re-use of PISAs
After exposure to developing reagents, the array wells were washed three times with 100 µl PBS, 0.05% Tween, followed by 50 µl freshly prepared stripping buffer [1 M (NH4)2SO4, 1 M urea] (16) at room temperature for 2 h, followed by three further washes with PBS–Tween and one with PBS. Arrays were re-exposed to antigen or antibodies as above.
RESULTS
Coupled expression and immobilisation of a human single-chain anti-progesterone antibody fragment by PISA on microtiter plates
A human single-chain anti-progesterone VH/K fragment (P5-17) (12) was chosen to test the PISA procedure. Bacterially expressed P5-17 binds specifically to P-BSA but not to BSA (12). For PISA, the P5-17 VH/K DNA fragment was extended into P5-17 VH/K-His and used to generate 16 coated wells by cell-free transcription/translation on a HisSorb plate (Fig. 3). As a control, eight wells were also incubated with a DNA-free TNT mixture. Expression and immobilisation of P5-17 VH/K-His protein was demonstrated using HRP-linked anti-human κ chain antibody: a positive reaction (blue) was obtained from P5-17 VH/K-His arrayed wells, but not from control wells or with anti-mouse κ on P5-17 VH/K-His wells. Specific antigen (progesterone) binding of the arrayed P5-17 VH/K-His was demonstrated using biotinylated P-BSA followed by HRP-streptavidin. P-BSA, but not BSA, produced positive colour development on the P5-17 VH/K-His wells, consistent with the known specificity of P5-17, while the control wells were non-reactive. In addition, it was shown that P5-17 VH/K-His immobilisation required the C-terminal double (His)6-tag domain, as no protein was detected on Ni–NTA-coated wells using the construct without a double (His)6-tag (not shown).
Figure 3.

PISA of a human single chain anti-progesterone VH/K-His fragment (P5-17). Four wells were analysed with each reagent. Wells in the upper two rows were probed with biotinylated antigens, followed by HRP-linked streptavidin. The lower two rows were probed with HRP-linked anti-human or anti-mouse κ chain reagents. Con, TNT mixture control lacking PCR DNA.
Western blot analysis of expression and immobilisation of P5-17 VH/K-His
To confirm and quantitate the cell-free expression and immobilisation of P5-17 VH/K-His, western blot analysis was carried out, with detection either by HRP-linked anti-human κ or anti-polyhistidine antibody (Fig. 4A and B). For standardisation, the P5-17 VH/K-His construct was expressed in E.coli and protein purified by Ni–NTA magnetic agarose beads (not shown). The bacterially expressed VH/K-His, in the range of 4–32 ng, was used as a standard for comparison in the same western blot with in vitro synthesised VH/K-His (Fig. 4C). The blot indicated that a total (T) of 120 ng of VH/K-His was produced from 25 µl TNT cell-free system, while ∼50 ng was detected as the unbound fraction (U) after immobilisation on a Ni–NTA-coated well. After washing the well with wash buffer and PBS, treatment with SDS sample buffer (without heating) led to recovery of 25 ng VH/K-His protein (P) (Fig. 4B).
Figure 4.
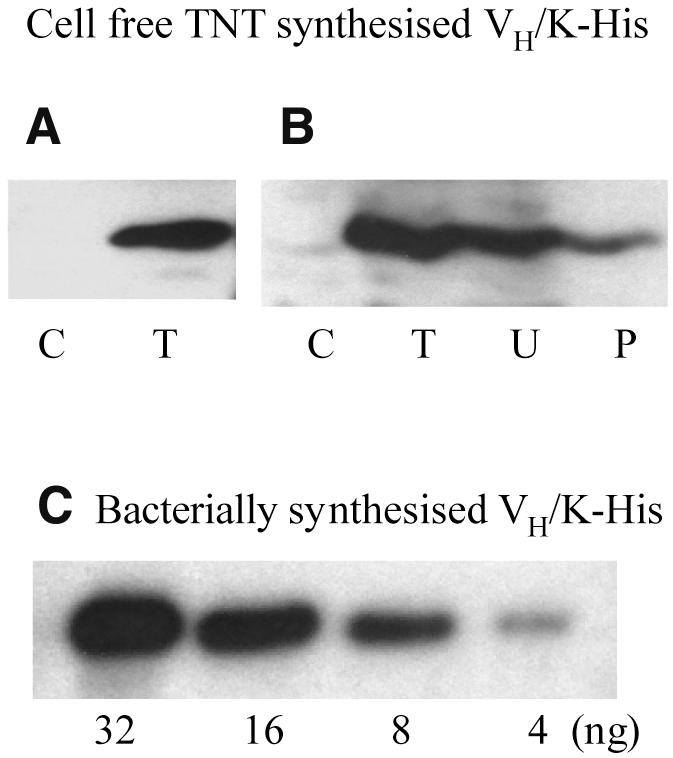
Western blot analysis of VH/K-His expression and immobilisation. (A) Cell-free TNT-synthesised P5-17 VH/K-His, detected by HRP-linked anti-human κ chain. (B) Cell-free TNT-synthesised P5-17 VH/K-His, detected by HRP-linked anti-polyhistidine. C, TNT mixture control without PCR DNA; T, TNT translation of P5-17 VH/K-His without immobilisation (5 µl loaded from total 30 µl); U, unbound fraction (10 µl loaded from total 25 µl) after immobilisation of total translation mixture on a Ni–NTA-coated well; P, P5-17 VH/K-His eluted from Ni–NTA coated well by SDS sample buffer (10 µl loaded out of total 40 µl). (C) Bacterially synthesised P5-17 VH/K-His as standard, detected with HRP-linked anti-polyhistidine; loading amounts of P5-17 VH/K-His are indicated.
Array screening of human VH/K fragments from a cloned library
To apply the PISA method for the screening of antibody fragments, an array was generated comprising nine cloned human antibody VH/K fragments from a transgenic mouse library (12). Five non-antigen binding VH/K clones and four progesterone-binding VH/K clones (P5-8, P5-10, P5-16, P5-17), the latter obtained by ribosome display (12), were arrayed. It was previously shown by ELISA screening of E.coli extracts that the soluble VH/K fragments 1–5 had no P-BSA binding activity, while the antigen-selected fragments bound specifically to P-BSA (12). After conversion into VH/K-His format, PCR-generated DNA fragments from each clone were used to generate a protein array by PISA (Fig. 5). The array was first screened using biotinylated P-BSA; the wells were then treated with stripping buffer (16) and re-screened with anti-human κ to confirm protein expression and immobilisation. Screening by P-BSA (Fig. 5A) showed that all the antigen-selected fragments had binding activity (yellow), whereas fragments before selection showed no detectable interaction. Anti-human κ confirmed that all fragments were expressed and immobilised on wells and mostly at a similar level (Fig. 5B). As this result agrees with E.coli screening (12), it demonstrates the feasibility of using PISA to identify antigen-binding fragments from a library.
Figure 5.

PISA of nine human VH/K-His fragments from a transgenic mouse library (12). (A) Analysis with biotinylated P-BSA, followed by HRP-linked streptavidin. (B) The same array after treatment with stripping buffer and re-analysis with HRP-linked anti-human κ chain. Wells 1–5, non-antigen binding VH/K-His fragments from clones of the transgenic mouse library; P5-8, -10, -16, -17, specific anti-progesterone VH/K-His fragments from the same library; Con, TNT mixture control lacking PCR DNA.
Coupled expression and immobilisation of luciferase on magnetic beads
Luciferase was chosen for the generation of a functional enzyme immobilised on the solid surface of magnetic beads using PISA. A construct encoding luciferase with a C-terminal double (His)6-tag (Luci–His) was produced by PCR; as a control, luciferase DNA lacking the double (His)6-tag domain (Luci) was also generated. After cell-free expression in the presence of Ni–NTA-coated magnetic beads, the latter were separated from the translation mixture and washed. Luciferase activity both free in the translation mixture supernatant and immobilised on the beads was measured using a luminometer (Fig. 6). While the Luci construct lacking the double (His)6-tag domain only produced activity in the supernatant, the Luci–His construct generated activity both in the supernatant and on the beads, demonstrating the successful immobilisation of functional luciferase through the double (His)6-tag domain.
Figure 6.
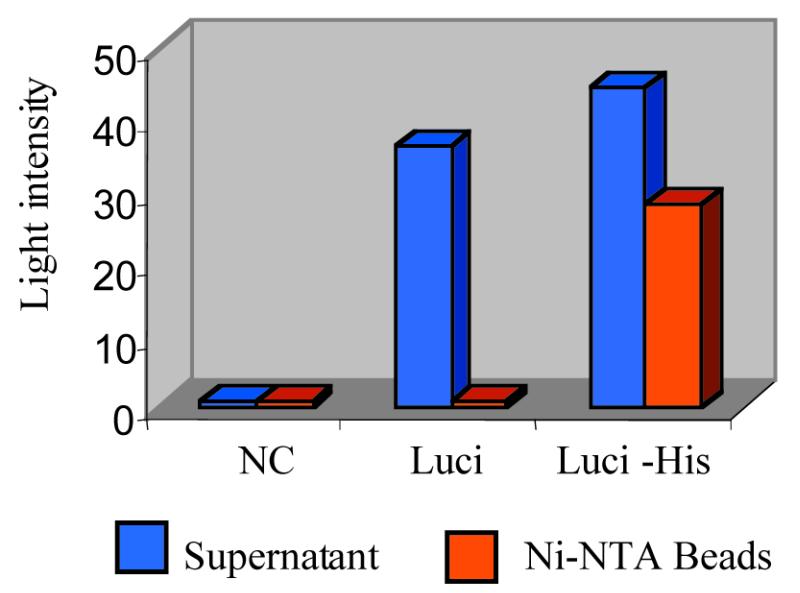
Functional assay of free and PISA-immobilised luciferase by luminometry. Supernatant, TNT mixture after incubation with Ni–NTA magnetic agarose beads, followed by magnetic removal of the beads; NC, TNT mixture negative control lacking PCR DNA; Luci, TNT mixture containing the PCR construct encoding luciferase without double (His)6-tag domain; Luci-His, TNT mixture containing the PCR construct encoding luciferase fused with double (His)6-tag domain.
DISCUSSION
Here we report a rapid procedure, PISA, designed to generate protein arrays directly from PCR-generated DNA through cell-free synthesis of a tagged protein and simultaneous immobilisation on a suitably treated surface. The method offers a number of advantages for protein array production. With the rapid expansion and annotation of genome databases, individual genes or gene fragments for cell-free expression can be easily obtained from diverse genetic materials (DNA, mRNA or cloned plasmids) by PCR or RT–PCR using database-designed specific primers. Protein synthesis in cell-free systems has advantages over cell-based gene expression by reliably generating soluble, functional proteins, including toxic proteins (10). Moreover, eukaryotic cell-free systems such as rabbit reticulocyte permit processing of a variety of post-translational modifications and are also capable of assembly of functional multi-subunit protein complexes (www.promega.com/techserv/tntbib.html contains a bibliography of published uses of the TNT system) making it possible to generate a wide range of arrays.
We have demonstrated the functional immobilisation of antibody fragments by PISA. As a model, a human anti-progesterone antibody fragment of known specificity was shown to be expressed and immobilised, retaining its original antigen-binding specificity (Figs 3 and 4). Cloned antibody fragments from a library were also arrayed and screened with labelled antigen, distinguishing binders from non-binders in a reproducible way (Fig. 5). The reverse process, of screening antibodies on a PISA antigen array, can also be envisaged. Uses of antibody arrays include proteomics applications (1), such as protein expression profiling and diagnostics. Luciferase was also functionally immobilised in situ on Ni–NTA- linked beads, supporting the general applicability of the PISA procedure.
Quantitation by western blotting showed that in our standard reaction volume of 25 µl, ∼120 ng VH/K protein was obtained, of which ∼50% became immobilised in a microtitre well. Immobilisation efficiency may be enhanced by raising protein expression yield; recently, cell-free technologies have been improved to increase protein production level, with yields of hundreds of µg/ml in a coupled E.coli S30 extract (17) and up to 4 mg/ml in a modified wheatgerm system (18). To monitor variations in the expression and immobilisation of individual proteins, cell-free labelling methods could be used for standardisation.
In order for the PCR strategy to have general applicability, a double (His)6-tag domain, comprising a flexible linker, double (His)6-tag, stop codons, poly(A) tail and transcription terminator, was designed and cloned into a plasmid (Fig. 2). The functional generation of antibody arrays and of luciferase on beads suggests that the C-terminal tag domain had no deleterious effect. The designed double (His)6 sequence was highly efficient in immobilising protein onto Ni–NTA surfaces, to the extent that it allowed re-use of the arrays after washing with stripping buffer. The PISA-immobilised proteins could be re-assayed at least three times without significant loss of binding reaction and stored at 4°C for at least 2 weeks.
In combination with sensitive detection and readout methods, the PISA procedure, including the PCR DNA construction strategy (Fig. 2), would be amenable to high-throughput applications. It could be particularly useful for the generation of protein arrays where cloned genes are not available or proteins cannot be functionally produced in heterologous expression systems. It would also enable the rapid arraying of individual protein domains for detailed analysis directly from sequence information without cloning, e.g. for mapping the localisation of functional sites for interactions with ligands. The power of the method could be further enhanced by combination with in vitro display technologies such as ribosome display (19,20) or phage display (21) for high-throughput identification of protein–protein interactions or genome-wide screening of very large libraries. Finally, since transcription/translation can occur on immobilised DNA (22), PISA may allow the possibility of converting a DNA array into a protein array by cell-free protein synthesis.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Hong Liu for excellent technical assistance. Research at the Babraham Institute is supported by the BBSRC.
References
- 1.Pandey A. and Mann,M. (2000) Proteomics to study genes and genomes. Nature, 405, 837–846. [DOI] [PubMed] [Google Scholar]
- 2.Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science, 270, 467–470. [DOI] [PubMed] [Google Scholar]
- 3.Young R.A. (2000) Biomedical discovery with DNA arrays. Cell, 102, 9–15. [DOI] [PubMed] [Google Scholar]
- 4. Zhu,H. and Snyder,M. (2001) Protein arrays and microarrays. Curr. Opin. Chem. Biol., 5, 40–45. [DOI] [PubMed]
- 5.Martzen M.R., McCraith,S.M., Spinelli,S.L., Torres,F.M., Fields,S., Grayhack,E.J. and Phizicky,E.M. (1999) A biochemical genomics approach for identifying genes by the activity of their products. Science, 286, 1153–1155. [DOI] [PubMed] [Google Scholar]
- 6.Bussow K., Cahill,D., Nietfeld,W., Bancroft,D., Scherzinger,E., Lehrach,H. and Walter,G. (1998) A method for global protein expression and antibody screening on high-density filters of an arrayed cDNA library. Nucleic Acids Res., 26, 5007–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uetz P., Giot,L., Cagney,G., Mansfield,T.A., Judson,R.S., Knight,J.R., Lockshon,D., Narayan,V., Srinivasan,M., Pochart,P. et al. (2000) A comprehensive analysis of protein–protein interations in Saccharomyces cerevisiae. Nature, 403, 623–627. [DOI] [PubMed] [Google Scholar]
- 8.Holt L.J., Bussow,K., Walter, G and Tomlinson,I.M. (2000) Bypassing selection: direct screening for antibody–antigen interactions using protein arrays. Nucleic Acids Res., 28, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevens R.C. (2000) Design of high-throughput methods of protein production for structural biology. Structure Fold. Des., 8, R177–R185. [DOI] [PubMed] [Google Scholar]
- 10.Nakano H. and Yamane,T. (1998) Cell-free protein synthesis systems. Biotechnology Advances, 16, 367–384. [DOI] [PubMed] [Google Scholar]
- 11.He M., Kang,A.S., Hamon,M., Humphreys,A.S., Gani,M. and Taussig,M.J. (1995) Characterization of a progesterone-binding, three-domain antibody fragment (VH/K) expressed in Escherichia coli. Immunology, 84, 662–668. [PMC free article] [PubMed] [Google Scholar]
- 12.He M., Menges,M., Groves,M.A., Corps,E., Liu,H., Brüggemann,M. and Taussig,M.J. (1999) Selection of a human anti-progesterone antibody fragment from a transgenic mouse library by ARM ribosome display. J. Immunol. Methods, 231, 105–117. [DOI] [PubMed] [Google Scholar]
- 13.Sachs A.B. and Varani,G. (2000) Eukaryotic translation initiation: there are (at least) two sides to every story. Nature Struct. Biol., 7, 356–361. [DOI] [PubMed] [Google Scholar]
- 14.Robinson C.R. and Sauer,R.T. (1998) Optimizing the stability of single-chain proteins by linker length and composition mutagenesis. Proc. Natl Acad. Sci. USA, 95, 5929–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taussig M.J., Groves,M.A., Menges,M., Liu,H. and He,M. (2000) ARM complexes for in vitro display and evolution of antibody combining sites. In Shepherd,P. and Deans,C. (eds), Monoclonal Antibodies: A Practical Approach. Oxford University Press, Oxford, UK, pp. 91–109.
- 16.Ge H. (2000) UPA, a universal protein array system for quantitative detection of protein–protein, protein–DNA, protein–RNA and protein–ligand interactions. Nucleic Acids Res., 28, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chekulayeva M.N., Kurnasov,O.V., Shirokov,V.A. and Spirin,A.S. (2001) Continuous-exchange cell-free protein-synthesizing system: synthesis of HIV-1 antigen Nef. Biochem. Biophys. Res. Commun., 280, 914–917. [DOI] [PubMed] [Google Scholar]
- 18.Madin K., Sawasaki,T., Ogasawara,T. and Endo,Y. (2000) A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos: plants apparently contain a suicide system directed at ribosomes. Proc. Natl Acad. Sci. USA, 97, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He M. and Taussig,M.J. (1997) Antibody-ribosome-mRNA (ARM) complexes as efficient selection particles for in vitro display and evolution of antibody combining sites. Nucleic Acids Res., 25, 5132–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanes J. and Plückthun,A. (1997) In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl Acad. Sci. USA, 94, 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCafferty J., Griffiths,A.D., Winter,G. and Chiswell,D.J. (1990) Phage antibodies: filamentous phage displaying antibody variable domains. Nature, 348, 552–554. [DOI] [PubMed] [Google Scholar]
- 22.Andreadis J.D. and Chrisey,L.A. (2000) Use of immobilized PCR primers to generate covalently immobilized DNAs for in vitro transcription/translation reactions. Nucleic Acids Res., 28, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]