

Abstract
Background
Phomopsis longicolla T. W. Hobbs (syn. Diaporthe longicolla) is a seed-borne fungus causing Phomopsis seed decay in soybean. This disease is one of the most devastating diseases reducing soybean seed quality worldwide. To facilitate investigation of the genomic basis of pathogenicity and to understand the mechanism of the disease development, the genome of an isolate, MSPL10–6, from Mississippi, USA was sequenced, de novo assembled, and analyzed.
Results
The genome of MSPL 10–6 was estimated to be approximately 62 Mb in size with an overall G + C content of 48.6%. Of 16,597 predicted genes, 9866 genes (59.45%) had significant matches to genes in the NCBI nr database, while 18.01% of them did not link to any gene ontology classification, and 9.64% of genes did not significantly match any known genes. Analysis of the 1221 putative genes that encoded carbohydrate-activated enzymes (CAZys) indicated that 715 genes belong to three classes of CAZy that have a direct role in degrading plant cell walls. A novel fungal ulvan lyase (PL24; EC 4.2.2.-) was identified. Approximately 12.7% of the P. longicolla genome consists of repetitive elements. A total of 510 potentially horizontally transferred genes were identified. They appeared to originate from 22 other fungi, 26 eubacteria and 5 archaebacteria.
Conclusions
The genome of the P. longicolla isolate MSPL10–6 represented the first reported genome sequence in the fungal Diaporthe-Phomopsis complex causing soybean diseases. The genome contained a number of Pfams not described previously. Information obtained from this study enhances our knowledge about this seed-borne pathogen and will facilitate further research on the genomic basis and pathogenicity mechanism of P. longicolla and aids in development of improved strategies for efficient management of Phomopsis seed decay in soybean.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4075-x) contains supplementary material, which is available to authorized users.
Keywords: Genome, Phomopsis longicolla, Phomopsis seed decay, Soybean
Background
Phomopsis longicolla T. W. Hobbs (syn. Diaporthe longicolla) is a seed-borne fungus primarily causing Phomopsis seed decay (PSD) in soybean, Glycine max (L.) Merrill [1–4]. This disease decreases seed quality and has been found in most soybean production areas, worldwide [2, 4, 5]. The common symptoms of PSD include discolored seed that are both shriveled and elongated or cracked seed coats and are chalky-white in appearance. Soybean seed infected by P. longicolla often lack of visible symptoms or signs at harvest [6]. It has been reported that soybean seed infected by P. longicolla, whether symptomless or having symptoms, could have very low seed germination, reduced seedling vigor, and poor stands [4, 7]. Poor seed quality of soybean could be due to the alteration of seed composition, reduction of oil quality, or moldy and/or split seed caused by P. longicolla [8]. PSD is one of the most economically important diseases of soybean. This disease has caused significant soybean yield loss. [9, 10]. If the environment is warm and humid during the late growing season from pod fill through harvest, it favors pathogen growth and PSD development [11].
Management of PSD is very challenging. Inconsistent reductions of PSD have been reported when common agronomic practices were used. Practices included crop rotation with non-host or non-legume crops, conventional tillage to reduce pathogen inoculum, and prompt harvest when soybeans matured to avoid late season wet weather [2]. In addition, fungicide treatments could be used as an option to reduce PSD and other soybean diseases. However, they were not always effective in controlling PSD [12–14]. Planting cultivars with resistance to PSD is a long-term strategy to manage PSD. In past decades, most research conducted on the host resistance, such as identifying resistance sources by screening soybean germplasms, commercial cultivars, and breeding lines [5, 15–17], breeding for resistant lines and cultivars [18], and investigating inheritance of resistance to PSD [19–21]. In addition, genetic mapping of resistance to PSD was reported [22]. However, information about the genomic features and mechanisms underlying the pathogenicity of P. longicolla on soybean were lacking. It is well-known that plant cell walls are the primary barrier against pathogen invasions. In order to infect plants, a plant pathogen should have the ability to pass through the plant cell wall. Plant cell wall degrading enzymes (PCWDEs) are a subset of carbohydrate-activated enzymes (CAZy) that are produced by plant pathogens to degrade plant cell walls. There was no information about PCWDEs in P. longicolla. Further, horizontal gene transfer or lateral gene transfer has been inferred to be the movement of genetic material between different organisms [23, 24]. If true it is a major force driving the evolution of both bacteria and eukaryotes [25, 26]. To date there was no report inferring the possibility of HGT in P. longicolla. Understanding the nature of the pathogen and mechanisms of PSD development in host plants will help us develop better disease management strategies. In recent years, genomic studies have made important contributions to research and disease management in plant pathology [27]. The next-generation sequencing technology has facilitated the genomics-based approached to both improve disease resistance in crops and enhance our understanding the mechanism of pathogenicity. The genomic approaches could provide an alternative way to identify host resistances.
To facilitate investigation of the genomic basis of pathogenicity in P. longicolla and to understand the mechanism of the disease development, the genome of isolate MSPL10–6 was sequenced and de novo assembled [28]. This research was conducted to analyze the genome sequences of the P. longicolla isolate MSPL10–6. The aims here were to understand the genome features of P. longicolla, identify genes encoding plant cell wall degrading enzymes, discover and classify the repeat elements in the genome, and investigate the potentially horizontally transferred genes in the P. longicolla genome.
Results
General genome features
The genome of the P. longicolla isolate MSPL 10–6 was assembled from both the paired-end and mate-pair libraries with the short oligonucleotide assembler package (SOAP), a denovo assembler. The oligonucleotides formed 108 scaffolds of 500 bases or larger. As reported in previous studies, the N50 length was 1,039,102 bp, and the largest scaffold contained 6,247,470 bp. The genome size was estimated to be approximately 62 Mb with an overall G + C content of 48.6% [28]. Statistics of genome sequencing and assembly are summarized in Table 1.
Table 1.
Statistics of genome sequencing and assembly of Phomopsis longicolla isolate MSPL 10–6
| Sequencing Statistics Library | Paired End (0.5 Kb inserts) | Mate Pair (3.9 Kb inserts) | Total |
|---|---|---|---|
| Raw data | |||
| Size | 6.9 Gb | 16.2 Gb | 23.1 Gb |
| Coverage | 108 X | 253 X | 361 X |
| Processed data | |||
| Size | 6.2 Gb | 8.2 Gb | 14.4 Gb |
| Coverage | 97 X | 128 X | 225 X |
| Assembly Statistics | Contigs | Scaffolds | |
| Total assembly size | 62 Mb | 66.7 Mb | |
| Total assembled sequences | 12,329 | 108 | |
| Longest sequence length | 215 Kb | 6.2 Mb | |
| Average sequence length | 5054 bp | 618 Kb | |
| N90 index | 2900 | 62 | |
| N90 length | 3.21 Kb | 299 Kb | |
| N50 index | 662 | 17 | |
| N50 length | 26.3 Kb | 1.04 Mb | |
Gene prediction and annotation
Gene prediction analysis yielded a total of 16,597 genes (Average Length was 1704 bp, Total Length was 28,287,360 bp, Total Coding Length was 24,840,981 bp), of which 4334 genes where found to consist of a single exon (Average Length = 1219 bp). The total number of exons in all predicted genes was 47,213 (Average Length was 3622 bp, Total Length = 4,435,952 bp).
Of 16,597 genes predicted, 9866 genes (59.45%) had significant matches to genes in the NCBI nr database, while 18.01% of them did not link to any gene ontology (GO) classification. Further, 9.64% of the genes did not significantly match any known genes. Enzyme codes were assigned to 15.45% of the genes. The gene prediction statistics are summarized in Table 2. Functional categorization and distribution of potential genes in the P. longicolla genome are shown in Fig. 1.
Table 2.
Statistics of genome annotation of Phomopsis longicolla isolate MSPL 10–6
| Genes | |||
| Single Exon Genes | All Genes | ||
| Count | 4334 | Count | 16,597 |
| Average Length | 1219.1 | Average Length | 1704.37 |
| Median Length | 1026 | Median Length | 1411 |
| Total Length | 5,283,582 | Total Length | 28,287,360 |
| Average Coding Length | 1216.07 | Average Coding Length | 1496.72 |
| Median Coding Length | 1023 | Median Coding Length | 1236 |
| Total Coding Length | 5,270,457 | Total Coding Length | 24,840,981 |
| Average Score | 0.82 | Average Score | 2.25 |
| Total Score | 3560.51 | Total Score | 37,365.46 |
| Ave Exons Per | 1 | Ave Exons Per | 2.84 |
| Med Exons Per | 1 | Med Exons Per | 2 |
| Total Exons | 4334 | Total Exons | 47,213 |
| Exons | |||
| Initial | Terminal | ||
| Count | 12,262 | Count | 12,262 |
| Average Length | 361.76 | Average Length | 636.18 |
| Median Length | 214 | Median Length | 434 |
| Total Length | 4,435,952 | Total Length | 7,800,818 |
| Average Score | 0.8 | Average Score | 0.79 |
| Total Score | 9765.44 | Total Score | 9696.51 |
| Internal | Single | ||
| Count | 18,355 | Count | 4334 |
| Average Length | 399.55 | Average Length | 1216.07 |
| Median Length | 233 | Median Length | 1023 |
| Total Length | 7,333,754 | Total Length | 5,270,457 |
| Average Score | 0.78 | Average Score | 0.82 |
| Total Score | 14,343 | Total Score | 3560.51 |
Fig. 1.
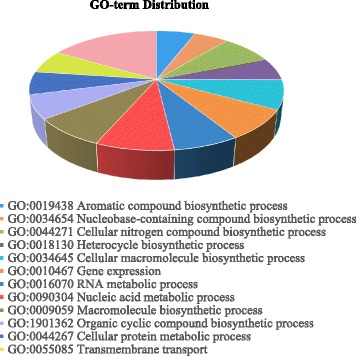
Functional categorization and distribution of potential genes in the genome of Phomopsis longicolla isolate MSPL 10–6
Plant cell-wall degrading enzymes
The enzyme classification code (EC number) class distributions (level 3) are summarized in Fig. 2. Of 2674 EC enzyme-like orthologs identified, 1184 (44.3%) were related to hydrolases or hydrolytic enzyme, while 63 (2.4%) sequences were related to isomerases. An abundance of genes encoding plant cell-wall degrading enzymes (PCWDEs) were found in the P. longicolla genome. Of 1221 putative genes that encode carbohydrate-activated enzymes (CAZys), (Additional file 1: Table S1), 199 genes encoded carbohydrate esterases (CE), 471 encoded glycoside hydrolases (GH), and 45 encoded polysaccharide lyases (PL) (Table 2). In the CAZy family, enzymes that have the same substrate and description could have different “domain” structures and coding sequences, such as CE1 – CE5, GH 10 and GH11 (Table 3). Three other classes of CAZys with indirect roles on degrading carbohydrates were auxiliary activities (AAs), carbohydrate-binding modules (CBMs), and glycosyl-transferases (GTs). The number of putative genes identified in the AA, CBM, and GT classes were 259, 113, and 134, respectively (Table 4). Comparisons of the numbers of CAZys in P. longicolla with other Ascomycete fungi are shown in Fig. 3.
Fig. 2.
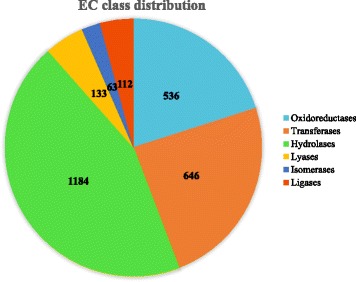
Enzyme code (EC) class distributions in the genome of Phomopsis longicolla isolate MSPL 10–6 based on the gene ontology classification
Table 3.
A list of the carbohydrate-activated enzymes (CAZy) identified in the genome of Phomopsis longicolla isolate MSPL 10–6
| CAZy familya | Substrate | Description | EC | Copy number |
|---|---|---|---|---|
| CE1 | Hemicellulose (xylan) | Acetyl xylan esterase | 3.1.1.72 | 33 |
| Feruloyl esterase | 3.1.1.73 | |||
| CE2 | Hemicellulose (xylan) | Acetyl xylan esterase | 3.1.1.72 | 2 |
| CE3 | Hemicellulose (xylan) | Acetyl xylan esterase | 3.1.1.72 | 10 |
| CE4 | Hemicellulose (xylan) | Acetyl xylan esterase | 3.1.1.72 | 8 |
| CE5 | Hemicellulose (xylan) | Acetyl xylan esterase | 3.1.1.72 | 18 |
| Cutin | Cutinase | 3.1.1.74 | ||
| CE7 | Hemicellulose (xylan) | Acetyl xylan esterase | 3.1.1.72 | 1 |
| CE8 | Pectin (homogalacturonan) | Pectin methylesterase | 3.1.1.11 | 6 |
| CE9 | Polysaccharides | N-acetylglucosamine 6-phosphate | 3.5.1.25 | 1 |
| Deacetylase | 3.5.1.80 | |||
| CE10 | Polysaccharides | Arylesterase | 3.1.1.- | 105 |
| CE12 | Pectin(homogalacturonan, rhamnogalacturonan I) | Pectin acetylesterase | 3.1.1.- | 5 |
| Hemicellulose | Acetyl pectin esterase | 3.1.1.72 | ||
| CE14 | Polysaccharides | N-acetylglucosaminylphosphatidy-linositol deacetylase | 3.5.1.89 | 1 |
| CE15 | Polysaccharides | 4-O-methyl-glucuronoyl methylesterase | 3.1.1.- | 2 |
| CE16 | Polysaccharides | Acetylesterase | 3.1.1.6 | 7 |
| GH1 | Cellulose | β-glucosidase | 3.2.1.21 | 6 |
| Hemicellulose (xylan, xyloglucan) | β-xylosidase | 3.2.1.37 | ||
| Pectin (rhamnogalacturonan I) | β-galactosidase | 3.2.1.23 | ||
| GH2 | Hemicellulose (xylan, xyloglucan, galactomannan) | β-mannosidase | 3.2.1.25 | 10 |
| Pectin (rhamnogalacturonan I) | β-glucuronidase | 3.2.1.31 | ||
| GH3 | Cellulose | β-glucosidase | 3.2.1.21 | 23 |
| Hemicellulose | β-xylosidase | 3.2.1.37 | ||
| (xylan, xyloglucan) | 3.2.1.74 | |||
| Pectin | exo-β-1,4-glucanase | |||
| GH5 | Cellulose | endo-β-1,4-glucanase | 3.2.1.4 | 27 |
| Hemicellulose (galactomannan) | endo-β-1,4-xylanase | 3.2.1.8 | ||
| Pectin (rhamnogalacturonan I) | exo-β-1,4-glucanase | 3.2.1.74 | ||
| GH6 | Cellulose | Cellobiohydrolase | 3.2.1.91 | 4 |
| endo-β-1,4-glucanase | 3.2.1.4 | |||
| GH7 | Cellulose | endo-β-1,4-glucanase | 3.2.1.4 | 9 |
| Cellobiohydrolase | 3.2.1.176 | |||
| GH9 | Cellulose | Cellusae | - | 1 |
| GH10 | Hemicellulose (xylan) | endo-β-1,4-xylanase | 3.2.1.8 | 7 |
| GH11 | Hemicellulose (xylan) | endo-β-1,4-xylanase | 3.2.1.8 | 4 |
| GH12 | Cellulose | endo-β-1,4-glucanase | 3.2.1.4 | 6 |
| Hemicellulose (xyloglucan) | Xyloglucanase | 3.2.1.151 | ||
| GH13 | Polysaccharides | α-amylase | 3.2.1.1 | 17 |
| GH15 | Polysaccharides | Glucoamylase | 3.2.1.3 | 1 |
| GH16 | Hemicellulose | Xyloglucanase | 3.2.1.151 | 21 |
| GH17 | Polysaccharides | endo-1,3-β-glucosidase | 3.2.1.39 | 7 |
| GH18 | Polysaccharides | Chitinase | 3.2.1.14 | 27 |
| endo-β-N-acetylglucosaminidase | 3.2.1.96 | |||
| GH20 | Polysaccharides | β-hexosaminidase | 3.2.1.52 | 3 |
| GH26 | Polysaccharides | beta-mannanase | 3.2.1.78 | 1 |
| GH27 | Hemicellulose (xylan, xyloglucan, galactomannan) | α-galactosidase | 3.2.1.22 | 2 |
| α-N-acetylgalactosaminidase | 3.2.1.49 | |||
| GH28 | Pectin (homogalacturonan, rhamnogalacturonan I) | Polygalacturonase | 3.2.1.15 | 21 |
| GH29 | Oligosaccharides | alpha-L-fucosidase | 3.2.1.51 | 4 |
| GH30 | Polysaccharides | Glucosylceramidase | 3.2.1.45 | 4 |
| GH31 | Hemicellulose (xyloglucan) | α-xylosidase | 3.2.1.177 | 7 |
| GH32 | Sucrose | Invertase | 3.2.1.26 | 6 |
| GH33 | Oligosaccharides | exo-α-sialidase | 3.2.1.18 | 1 |
| GH35 | Hemicellulose (xylan, xyloglucan, galactomannan) | β-galactosidase | 3.2.1.23 | 7 |
| Pectin (rhamnogalacturonan I) | exo-β-1,4-galactanase | 3.2.1.- | ||
| GH36 | Hemicellulose (xylan, xyloglucan, galactomannan) | α-galactosidase | 3.2.1.22 | 1 |
| α-N-acetylgalactosaminidase | 3.2.1.49 | |||
| GH37 | Trehalose | α,α-trehalase | 3.2.1.28 | 2 |
| GH38 | Oligosaccharides | α-mannosidase | 3.2.1.24 | 1 |
| GH39 | Oligosaccharides | alpha-L-iduronidase | 3.2.1.76 | 2 |
| GH42 | Oligosaccharides | beta-galactosidase | 3.2.1.23 | 1 |
| GH43 | Hemicellulose (xylan) | β-xylosidase | 3.2.1.37 | 40 |
| Pectin (rhamnogalacturonan I) | α-L-arabinofuranosidase | 3.2.1.55 | ||
| GH45 | Cellulose | endo-β-1,4-glucanase | 3.2.1.4 | 2 |
| GH47 | Oligosaccharides | α-mannosidase | 3.2.1.113 | 12 |
| GH51 | Cellulose | endo-β-1,4-glucanase | 3.2.1.4 | 4 |
| Hemicellulose (xylan,xyloglucan) | β-xylosidase | 3.2.1.37 | ||
| GH53 | Pectin (rhamnogalacturonan I) | endo-β-1,4-galactanase | 3.2.1.89 | 4 |
| GH54 | Pectin | alpha-L-arabinofuranosidase | 3.2.1.55 | 1 |
| GH55 | Polysaccharides | endo-1,3-β-glucosidase | 3.2.1.39 | 6 |
| GH62 | Polysaccharides | alpha-L-arabinofuranosidase | 3.2.1.55 | 1 |
| GH63 | Oligosaccharides | α-glucosidase | 3.2.1.106 | 4 |
| GH64 | Polysaccharides | endo-1,3-β-glucosidase | 3.2.1.39 | 4 |
| GH65 | Polysaccharides | alpha,alpha-trehalase | 3.2.1.28 | 2 |
| GH67 | Polysaccharides | alpha-glucuronidase | 3.2.1.139 | 1 |
| GH71 | Polysaccharides | α-1,3-glucanase | 3.2.1.59 | 9 |
| GH72 | Polysaccharides | β-1,3-glucanosyltransglycosylase | 2.4.1.- | 10 |
| GH74 | Cellulose | endo-β-1,4-glucanase | 3.2.1.4 | 11 |
| Hemicellulose (xyloglucan) | Xyloglucanase | 3.2.1.151 | ||
| GH76 | Oligosaccharides | α-1,6-mannanase | 3.2.1.101 | 14 |
| GH78 | Pectin | α-L-rhamnosidase | 3.2.1.40 | 14 |
| GH79 | Pectin (rhamnogalacturonan I) | β-glucuronidase | 3.2.1.31 | 9 |
| GH81 | Polysaccharides | endo-1,3-β-glucosidase | 3.2.1.39 | 2 |
| GH88 | Polysaccharides | β-glucuronyl hydrolase | 3.2.1.- | 1 |
| GH92 | Oligosaccharides | Mannosyl-oligosaccharide alpha-1,2-mannosidase | 3.2.1.113 | 8 |
| GH93 | Pectin (rhamnogalacturonan I) | exo-α-L-1,5-arabinanase | 3.2.1.- | 7 |
| GH94 | Cellulose | cellobiose phosphorylase | 2.4.1.20 | 1 |
| GH95 | Hemicellulose (xyloglucan) | α-1,2-L-fucosidase | 3.2.1.63 | 3 |
| GH105 | Pectin | Rhamnogalacturonyl hydrolase | 3.2.1.172 | 8 |
| GH106 | Polysaccharides | alpha-L-rhamnosidase | 3.2.1.40 | 4 |
| GH109 | Polysaccharides | α-N-acetylgalactosaminidase | 3.2.1.49 | 20 |
| GH114 | Polysaccharides | endo-α-1,4-polygalactosaminidase | 3.2.1.109 | 3 |
| GH115 | Hemicellulose (xylan) | Xylan α-1,2-glucuronidase | 3.2.1.131 | 4 |
| GH125 | Oligosaccharides | exo-α-1,6-mannosidase | 3.2.1.- | 4 |
| GH127 | Oligosaccharides | β-L-arabinofuranosidase | 3.2.1.185 | 3 |
| GH128 | Polysaccharides | endo-1,3-β-glucosidase | 3.2.1.39 | 6 |
| GH131 | Cellulose | exo-β-1,3/1,4/1,6-glucanase | 3.2.1.- | 8 |
| Hemicellulose | ||||
| GH132 | Polysaccharides | Activity on β-1,3glucan | – | 2 |
| GH133 | Polysaccharides | amylo-α-1,6-glucosidase | 3.2.1.33 | 1 |
| GH134 | Polysaccharides | endo-β-1,4-mannanase | 3.2.1.78 | 1 |
| GH135 | Polysaccharides | α-1,4-galactosaminogalactan hydrolase | 3.2.1.- | 4 |
| PL1 | Pectin (homogalacturonan) | Pectate lyase | 4.2.2.2 | 21 |
| PL3 | Pectin | Pectate lyase | 4.2.2.2 | 10 |
| PL4 | Pectin (rhamnogalacturonan I) | Rhamnogalacturonan lyase | 4.2.2.- | 8 |
| PL9 | Pectin | Pectate lyase | 4.2.2.2 | 2 |
| Exopolygalacturonate lyase | 4.2.2.9 | |||
| PL11 | Pectin | Rhamnogalacturonan endolyase | 4.2.2.23 | 2 |
| PL22 | Pectin | Oligogalacturonate lyase | 4.2.2.6 | 1 |
| PL24 | Pectin | Ulvan lyase | 4.2.2.- | 1 |
a CE Carbohydrate esterases, GH Glycoside hydrolases, PL polysaccharide lyases
Table 4.
Classes of auxiliary activity (AA), carbohydrate-binding module (CBM), and glycosyl-transferase (GT) enzymes in the genome of Phomopsis longicolla isolate MSPL 10–6
| CAZya family | Description | Copy Number |
|---|---|---|
| AA1 | Multicopper oxidases | 6 |
| AA2 | Lignin peroxidase | 16 |
| AA3 | Glucose-methanol-choline (GMC) oxidoreductases | 75 |
| AA4 | vanillyl-alcohol oxidase | 7 |
| AA5 | radical-copper oxidases | 3 |
| AA6 | 1,4-benzoquinone reductases | 1 |
| AA7 | Glucooligosaccharide oxidase | 96 |
| AA8 | Iron reductase | 7 |
| AA9 | Copper-dependent lytic polysaccharide monooxygenases | 35 |
| AA11 | monooxygenase | 10 |
| AA12 | The pyrroloquinoline quinone-dependent oxidoreductase activity was demonstrated for the CC1G_09525 protein of Coprinopsis cinerea | 2 |
| AA13 | Monooxygenase | 1 |
| CBM1 | Cellulose-binding | 21 |
| CBM6 | Amylase | 1 |
| CBM13 | Cellulose-binding | 2 |
| CBM18 | Chitin-binding | 12 |
| CBM20 | Starch-binding | 6 |
| CBM21 | Starch-binding | 1 |
| CBM23 | Mannan-binding | 1 |
| CBM24 | Alpha-1,3-glucan (mutan)-binding | 12 |
| CBM32 | Binding to LacNAc (beta-D-galactosyl-1,4-beta-D-N-acetylglucosamine) | 2 |
| CBM35 | Xylan-binding | 4 |
| CBM37 | Xylanase | 1 |
| CBM42 | Binding to arabinofuranose | 1 |
| CBM43 | Beta-1,3-glucan binding | 2 |
| CBM48 | Amylase | 1 |
| CBM50 | Peptidoglycan-binding (LysM domain) | 37 |
| CBM63 | Cellulose-binding | 1 |
| CBM66 | β-fructosidase | 2 |
| CBM67 | L-rhamnose-binding | 6 |
| GT1 | UDP-glucuronosyl-transferase | 12 |
| GT2 | Cellulose/chitin synthase | 16 |
| GT3 | Glycogen synthase | 1 |
| GT4 | Sucrose synthase | 7 |
| GT5 | Glycogen glucosyltransferase | 4 |
| GT8 | Lipopolysaccharide glucosyl-transferase | 6 |
| GT15 | α-1,2-mannosyl-transferase | 5 |
| GT17 | β-1,4-N-acetyl-glucosaminyl-transferase | 1 |
| GT20 | α,α-trehalose-phosphate synthase | 3 |
| GT21 | Ceramide β-glucosyl-transferase | 3 |
| GT22 | Man6GlcNAc2-PP-Dol α-1,2-mannosyl-transferase | 4 |
| GT24 | Glycoprotein α-glucosyl-transferase | 1 |
| GT25 | Lipopolysaccharide beta-1,4-galactosyltransferase | 6 |
| GT28 | Digalactosyl-diacyl-glycerol- synthase | 1 |
| GT31 | Fucose-specific β-1,3-N-acetylglucosaminyl-transferase | 4 |
| GT32 | α-1,6-mannosyl-transferase | 13 |
| GT33 | Chitobiosyl-diphosphodolichol β-mannosyl-transferase | 1 |
| GT34 | α-1,2-galactosyl-transferase | 2 |
| GT35 | Starch phosphorylase | 1 |
| GT39 | Protein α-mannosylt-ransferase | 3 |
| GT41 | Beta-N-acetylglucosaminyltransferase | 1 |
| GT48 | 1,3-β-glucan synthase | 1 |
| GT50 | α-1,4-mannosyl-transferase | 1 |
| GT55 | GDP-Man: mannosyl-3-phosphoglycerate synthase | 2 |
| GT57 | α-1,3-glucosyl-transferase | 3 |
| GT58 | Man5GlcNAc2-PP-Dol α-1,3-mannosyl-transferase | 1 |
| GT59 | Glc2Man9GlcNAc2-PP-Dol α-1,2-glucosyl-transferase | 1 |
| GT61 | Xylanase | 1 |
| GT62 | α-1,2-mannosyl-transferase | 3 |
| GT66 | dolichyl-diphospho-oligosaccharide-protein Glycotransferase | 1 |
| GT68 | O-alpha-fucosyltransferase | 1 |
| GT69 | α-1,3-mannosyl-transferase | 4 |
| GT71 | α-mannosyl-transferase | 8 |
| GT76 | α-1,6-mannosyl-transferase | 1 |
| GT77 | Xylanase | 1 |
| GT90 | Xylanase | 9 |
| GT92 | Glycanase | 1 |
a Carbohydrate-activated enzymes
Fig. 3.
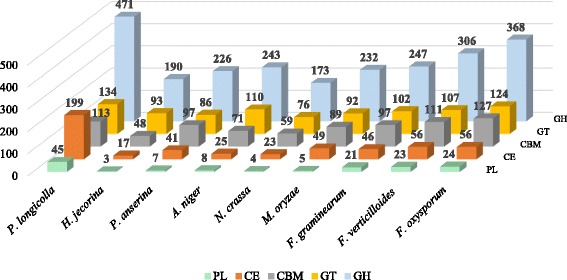
The number of different classes of the carbohydrate-activated enzymes (CAZy) in Phomopsis longicolla and other ascomycete fungi (Aspergillus niger, Hypocrea jecorina, Fusarium oxysporum, F. virguliforme, F. verticilloides, Magnaporthe oryzae, Neurospora crassa, and Podospora anserine)
Repetitive elements and transposase
Classification of the repetitive elements can be generally divided into two classes: Class I elements (Retrotransposons) and Class II elements (DNA Transposons). Of 12,322 repetitive elements identified in the genome of the MSPL 10–6 isolate, 7036 (57.1%) of the repetitive elements were Class I, while 5249 (42.6%) belonged to Class II. There were 370 (0.3%) unknown/unclassified repetitive elements (Table 5). The major transposons were DNA/TcMar-Fot1 (41.8%), LTR/ Copia (30.7%), and LTR/Gypsy (24.4%) like.
Table 5.
Classification of repetitive elements identified in the genome of Phomopsis longicolla isolate MS 10–6
| Total length (bp) | Repetitive content (%) | Genome content (%) | |
|---|---|---|---|
| Class I (Retrotransposon) | |||
| LTR retrotransposona | |||
| LTR/Copia | 2,408,959 | 30.71 | 3.89 |
| LTR/Gypsy | 1,914,239 | 24.40 | 3.09 |
| LTR/Other | 80,954 | 1.03 | 0.13 |
| Subtotal | 4,404,152 | 56.14 | 7.11 |
| Non-LTR retrotransposon | |||
| LINEb | 74,280 | 0.95 | 0.12 |
| Total Class I | 4,478,432 | 57.09 | 7.23 |
| Class II (DNA transoposon) | |||
| DNA/TcMar | 3,277,211 | 41.77 | 5.29 |
| DNA/Other | 65,709 | 0.84 | 0.11 |
| Total Class II | 3,342,920 | 42.61 | 5.4 |
| Other | |||
| Satellites, rRNA, Unknown repeats | 23,808 | 0.30 | 0.04 |
| Total | 7,845,160 | 12.67 | |
along terminal repeat retrotransposon
blong interspersed nuclear elements
Horizontal gene transfers
A total of 510 potential horizontal gene transfers (HGTs) were identified in the genome of the MSPL 10–6 isolate (Additional file 2: Table S2). They were originally from 53 species including 22 fungi, 26 eubacteria and 5 archaebacteria (Table 6). The majority of HGTs were from fungal origins (85.3%), while 13.3% and 1.4% of the HGTs were from eu- and archae- bacterial origins, respectively. Results of annotation of the HGTs based on gene ontology analysis are shown in Fig. 4. Over 70% of the HGTs were related to molecular functions.
Table 6.
The donor, taxonomy, and number of proteins encoded by the horizontal transferred genes in in the genome of Phomopsis longicolla isolate MSPL 10–6
| Donor | Taxonomy | Number of Proteins |
|---|---|---|
| Acidobacteriales | Bacteria | 1 |
| Agaricales | Fungus | 1 |
| Auriculariales | Fungus | 4 |
| Bacillales | Bacteria | 5 |
| Boletales | Fungus | 2 |
| Botryosphaeriales | Fungus | 24 |
| Burkholderiales | Bacteria | 8 |
| Capnodiales | Fungus | 28 |
| Chaetothyriales | Fungus | 89 |
| Corynebacteriales | Bacteria | 5 |
| Cytophagales | Bacteria | 2 |
| Deinococcales | Bacteria | 1 |
| Dictyosteliida | Fungus | 1 |
| Dothideales | Fungus | 12 |
| Enterobacterales | Bacteria | 2 |
| Eurotiales | Fungus | 73 |
| Haemosporida | Parasites | 1 |
| Helotiales | Fungus | 66 |
| Hymenochaetales | Fungus | 1 |
| Lactobacillales | Bacteria | 1 |
| Micrococcales | Bacteria | 4 |
| Micromonosporales | Bacteria | 1 |
| Myxococcales | Bacteria | 3 |
| Nostocales | Bacteria | 1 |
| Oceanospirillales | Bacteria | 1 |
| Onygenales | Fungus | 8 |
| Orbiliales | Fungus | 5 |
| Oscillatoriales | Bacteria | 1 |
| Peniculida | Oligohymenophorea | 1 |
| Planctomycetales | Bacteria | 1 |
| Pleosporales | Fungus | 78 |
| Polyporales | Fungus | 3 |
| Pseudomonadales | Bacteria | 1 |
| Pseudonocardiales | Bacteria | 4 |
| Rhizobiales | Bacteria | 6 |
| Rhodobacterales | Bacteria | 1 |
| Rhodospirillales | Bacteria | 2 |
| Russulales | Fungus | 2 |
| Schizosaccharomycetales | Fungus | 1 |
| Sphingobacteriales | Bacteria | 2 |
| Sphingomonadales | Bacteria | 5 |
| Sporidiobolales | Fungus | 1 |
| Streptomycetales | Bacteria | 7 |
| Streptosporangiales | Bacteria | 1 |
| Sulfolobales | Crenarchaeota | 1 |
| Thermoproteales | Crenarchaeota | 1 |
| Tremellales | Fungus | 4 |
| Trichomonadida | Parabasalia | 3 |
| Venturiales | Fungus | 9 |
| Verrucariales | Fungus | 8 |
| Verrucomicrobiales | Bacteria | 1 |
| Xanthomonadales | Bacteria | 1 |
| Xylonomycetales | Fungus | 15 |
| Total | 510 |
Fig. 4.
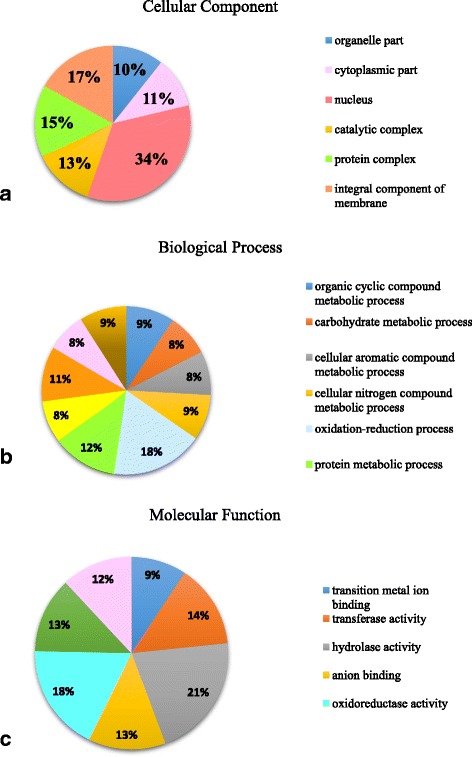
Distribution of annotated horizontal transferred genes in the genome of Phomopsis longicolla isolate MSPL 10–6 based on gene ontology analysis (a) cellular component, (b) biological process, and (c) molecular function
Discussion
In this study, the general genome feature of P. longicolla isolate MSPL10–6 from Mississippi, USA was described. The assembly of the genome was the result of combining the output from analyzing the sequences from both paired end and mate pair libraries. A de novo nuclear genome assembly was generated and characterized. The P. longicolla genome was estimated to be approximately 62 Mb, using both kmer and read coverage analyses. The size of the genome of P. longicolla MSPL 10–6 isolate appeared larger than other reported ascomycete soybean pathogens, such as Diaporthe aspalathi (55 Mb) [29], Fusarium virguliforme (50.5 Mb) [30] and Macrophomina phaseolina (49.3 Mb) [31]. The overall number of predicted gene in P. longicolla was 16,597, while it was 14, 962, 14,845, and 14,249 in D. aspalathi [29], F. virguliforme [30], and M. phaseolina [31], respectively. It is unknown whether the larger size of the genome and the bigger number of the predicted gene contributed in part to the fungal specialization and the pathogenicity of P. longicolla on soybean. Both P. longicolla and D. aspalathi are the members of Diaporthe-Phomopsis complex causing soybean diseases. They have similar culture morphology and very close relationships in taxonomy. However, P. longicolla is the primary cause of Phomopsis seed decay, while D. aspalathi primarily causes canker on soybean stem. It has been reported that soybean seeds, instead of stems and other soybean tissues, are more susceptible to P. longicolla than to other Diaphorthe species [14]. A comparative genome analysis is underway to address the questions about the pathogenicity mechanisms of those two species (Li et al., unpublished).
To investigate the genetic basis of pathogenicity, plant cell wall degrading enzymes (PCWDEs) in P. longicolla were identified and annotated. Results here indicated that P. longicolla contained abundance of genes encoding PCWDEs, which include all six classes of CAZys. For enzymes in the class of carbohydrate esterases (CE), the genome of P. longicolla contained 105 GH10 like genes that are absent in other pathogens, like F. virguliforme [32]. Plant cell walls contain abundant cellulose. Most of cellulose-degrading enzymes are classified into the glycoside hydrolase (GH) class. As indicated in Table 3, the substrates for the GH1, GH3, and GH5 degrading enzymes included not only cellulose, but also hemicellulose and pectin. It has been reported that most of biotrophic fungi do not have GH1 [33], while the genome of P. longicolla encodes six GH1 genes. Notably, there are 23 GH3 and 27 GH5 genes in the P. longicolla genome. Both GH3 and GH5 were common in pathogenic oomycetes, hemibiotrophic and necrotrophic fungi, which have more genes than biotrophic fungi and Pythium species [32]. The polysaccharide lyases (PL) are one of the important classes of PCWDEs. The PL class specializes in pectin degradation. It is well-known that pectin is the most divergent component of plant cell walls with different modifications on the side chains. The microbial pectinolytic enzymes have been studied and reviewed [34]. PL1 and PL3 were the most common pectin lyases found in plant pathogens. In this study, there were 21 and 10 copies of PL1 and PL3 in P. longicolla, respectively. Three other PL members, PL9, PL 20, and PL22, have been thought previously to be unique to N. haematococca and F. virguliforme, and were not found in other plant fungal pathogens or oomycetes [32], but both PL9 and PL22 domains were present in the P. longicolla genome. F. graminearum [35], F. fujikuroi [36], and F. virguliforme [32], P. longicolla had PL1, PL3, PL4, PL9, and PL22. They all had PL11 and PL24, except F. virguliforme [32]. However, P. longicolla did not have PL20 as F. virguliforme had.
Significantly, a novel fungal ulvan lyase (PL24; EC 4.2.2.-) was found in the P. longicolla genome. Ulvan lyases degrade ulvan, an anionic polysaccharide. This enzyme has only been reported recently, as the first members of a new polysaccharide lyase family in bacteria [37]. Ulvan is the most abundant component of the green algal cell wall. The role of ulvan lyases in the pathogenicity of P. longicolla is unknown. It will require further investigation. The PCWDE is one of the most important factors associated with pathogenicity of fungal pathogens. It could play a crucial role in infecting plants and influencing host resistance. The list of PCWDEs identified in P. longicolla laid the foundation for dissecting the mechanisms of the fungal pathogenicity through further functional analyses of genes encoding PCWDEs. Those analyses should aid in developing new strategies for breeding for resistance to Phomopsis seed decay in soybean.
To examine the genome architecture of P. longicolla, repetitive elements were analyzed. As noted, members of both class I and class II repetitive elements were found in the genomes of filamentous fungi [38]. Approximately 13% of the P. longicolla genome consists of repetitive elements. This is greater than the 10% and 1% in other ascomycetes Magnaporthe oryzae and M. poae genomes, respectively [39–41]. The majority (57.1%) of repetitive elements in the P. longicolla genome are categorized as Class I elements (retrotransposons). They are transcribed from DNA to RNA, and the RNA produced is then reverse transcribed into DNA. Moreover, retrotransposons mobilize via a “copy-and-paste”, which allow for many copies to be inserted throughout the genome. Thus, retrotransposons are the most common transposon in eukaryotes [42, 43] including P. longicolla. In addition, the long terminal repeat (LTR) is one of the main groups of retrotransposons [44]. The two main superfamilies of LTR retrotransposons found in fungi are Gypsy and Copia. In P. longicolla there were 30.7% of LTR/Copia and 24.4% of LTR/Gypsy among the repetitive elements. Since there are abundances of LTR/Copia present in the genomes of plants, fungi, animals, algae and several protists, it has been proposed that the ancestors of the LTR/Copia family probably co-existed with the ancestors of LTR/Gypsy before the separation between plants and other kingdoms [45].
The DNA transposons (class II elements) mobilize via a cut-and-paste mechanism that use a DNA intermediate, in which, the DNA itself is excised from the genome and integrated elsewhere. Mariner-like elements are one of prominent classes of the DNA transposons found in multiple species, including humans. This Class II transposable element is known for its uncanny ability to be transmitted horizontally between many species [46, 47]. It estimated that there were 14,000 copies of mariner in the human genome encompassing 2.6 million base pairs [48]. The first mariner-element transposons outside of animals were found in Trichomonas vaginalis, the most common pathogenic protozoan infection of humans [49]. Interestedly, the most major transposon in the P. longicolla genome was DNA/TcMar-Fot1 (41.8%). The function of this transposon in P. longicolla is unknown.
In the past, many documented HGT events inferred to fungi involved bacterial donors. For example, in a search for eubacterial-derived HGTs in 60 fully sequenced fungal species, Marcet-Houben and Gabaldon detected 713 transfer genes from bacteria [50]. Gene transfer between fungi has already been reported [51]. In a comparative genomic study of Fusarium species, four of F. oxysporum’s 15 chromosomes inferred to have been acquired through HGT from a fungal source [52]. Notably, chromosome 14, which is essential for pathogenicity of tomato, could be transferred between pathogenic and non-pathogenic strains of F. oxysporum resulting in conversion of non-pathogenic strains into pathogenic strains. Here, the majority of HGTs in the P. longicolla genome were of fungal origin (85.3%), while only 13.3% of the HGTs were from bacteria. Almost half of the HGT genes in the P. longicolla genome were related to molecular function. Further research will be necessary to address many open questions such as the impact of HGTs on the genome structure, gene function, and pathogenicity of P. longicolla.
The genome of the isolate MSPL10–6 was the first reported genome sequence in the fungal Diaphothe-Phomopsis complex causing soybean diseases. Our study represents the first genomic effort to discover the genome structure of P. longicolla. The genome data provide new insights into the gene repertoire and physiological potential of seed-borne pathogens. Additionally, the genomic resources presented here, including the genome sequences and annotations, detail lists of cell wall degrading enzymes, repetitive elements and horizontal transferred genes, enhance our knowledge about the biology and genetics of P. longicolla. These discoveries will facilitate further research on the genomic basis and pathogenicity mechanism of P. longicolla, and aid in development of improved strategies for efficient management of this pathogen.
Conclusions
Phomopsis seed decay of soybean is one of the most devastating diseases affecting soybean seed quality worldwide. However, genomic basis and mechanism of the pathogenicity of P. longicolla on soybean was lacking. The draft genome of the P. longicolla isolate MSPL10–6 represents the first reported genome sequence in the fungal Diaporthe-Phomopsis complex causing soybean diseases. The MSPL 10–6 genome contains a number of unique genomic features, including an abundance of genes encoding cell-wall degrading enzymes, numerous repetitive elements, as well as horizontal transferred genes from eubacteria, fungi and archaebacteria. Information obtained from this study enhances our knowledge about the biology and genetics of the seed-borne pathogen will facilitate further research on the genomic basis and pathogenicity mechanism of P. longicolla. The study will aid in development of improved strategies for efficient management of this pathogen.
Methods
Isolation, identification, and cultivation of P. longicolla isolate
A P. longicolla isolate MSPL10–6 was isolated from field-grown soybean seed in Mississippi, USA in 2010 using the seed plating method. Briefly, over 100 randomly chosen soybean seeds that were harvested from the field were surface-disinfected in 0.5% sodium hypochlorite for 3 min, rinsed in sterile distilled water 3 times (3 min each time), and then placed on potato dextrose agar (Difico Laboratories, Detroit, MI) that was acidified (pH 4.8) with 25% lactic acid after autoclaving (APDA). Five seeds were plated on each 100 mm-diameter Petri dish. After 4 days of incubation at 24 °C in the dark, putative/potential P. longicolla was isolated, streaked to the new APDA plates and incubated under 12-h light-and dark cycles. After 4–6 days, monoconidial cultures were obtained.
Identification of P. longicolla was first based on morphological characteristics according to Hobbs et al. [1] and then was confirmed by analysis of the ITS region of rDNA amplified by PCR with primers ITS1, 5′-TCCGTAGGTGAACCTGCGG-3’and ITS4, 5′-TCCTCCGCTTATTGATATGC-3′ [53] and the translation elongation factor 1-α gene primer set EF1-728F, 5′- CAT CGA GAA GTT CGA GAA GG -3′, and EF1-986R, 5′-TAC TTG AAG GAA CCC TTA CC -3′ [54, 55]. Pathogenicity tests were performed using a cut-seedling inoculation method as described by Li et al. [56]. Isolate MSPL 10–6 was one of the most aggressive isolates causing severe soybean stem lesion in the greenhouse tests (data not shown). This isolate has also been used to screen soybean germplasm and successfully identified 23 new sources of resistance to PSD [17, 57].
Genomic DNA extraction and sequencing
For DNA extraction, mycelial plugs (3-mm in diameter) from the margin of a 10-day old culture of MSPL 10–6 on APDA were cut and placed in potato dextrose broth (Difico Laboratories, Detroit, MI). After 4 days of incubation at 24 °C under 12-h light-and dark cycles, mycelia were collected on sterile cheesecloth, washed with sterile water, immediately frozen with liquid nitrogen, and lyophilized with a freeze-drier (IMC Instruments, Inc., Wisconsin, USA). Fungal mycelia were ground with a mortar and pestle and pulverized in liquid nitrogen. The genomic DNA was extracted using a Qiagen DNeasy Plant Mini Kit (Qiagen Inc., Valencia, CA) following the manufacturer’s instruction and qualified with Nanodrop (Thermo Scientific, Waltham, MA, USA).
Genomic DNA of the P. longicolla MSPL10–6 isolate was used to generate sequencing libraries as previous described [28]. Briefly, paired-end libraries were made with the TruSeq DNA PCR-Free Sample Preparation kit (Illumina San Diego, CA), while the no-gel mate-pair libraries were generated with the Nextera Mate-Pair Sample Preparation kit (Illumina San Diego, CA) according to the manufacturer’s protocols. All libraries were sequenced in separate lanes on an Illumina HiSeq 2500 sequencer using a TruSeq SBS sequencing kit (version 3, Illumina) at the Genomics Core Facility, Purdue University, West Lafayette, IN.
De novo genome assembly
Adapter sequences and poor quality bases (Phred score < 20) for each sequence read were trimmed using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). A total of 72,216,734 mate-pair reads with a total of 8.2 billion bp representing 128-fold coverage, and 63,763,666 paired-end reads with a total of 6.2 billion bp, representing 97-fold coverage, were generated. The P. longicolla genome was assembled from both libraries using the software SOAPdenovo assembler version 2.04 [58]. Raw sequence data was deposited into NCBI’s SRA database, under accession number: AYRD00000000 (1) (http://www.ncbi.nlm.nih.gov/nuccore/AYRD00000000).
Gene prediction and annotation
Gene prediction analysis was performed using a combination of homology searching and de novo prediction using Augustus web server [59, 60] with complete gene option enabled and default for the rest of the parameters. F. graminearum [31] was used as the reference species due to the relatively close phylogenetic relationship to P. longicolla MSPL10–6 among the genome sequences available in GenBank.
Predicted genes were functionally annotated using Blast2GO [61]. The gene models were BLAST-ed (BLASTx) [62] against the NCBI non-redundant protein database. Then domain finding searches were done using InterProScan [63]. Enzyme codes and GO ontologies were then assigned to the gene models as described by the Gene Ontology Consortium (http://geneontology.org).
Identification of carbohydrate-activated enzymes (CAZys) in the P. longicolla genome
To identify CAZys in the genome of P. longicolla isolate MSPL 10–6, Augustus program (http://bioinf.uni-greifswald.de/webaugustus/prediction/create) [59] trained with the parameters of the species Fusarium graminearum was used to predict putative proteins of the CAZy family. Using the web resources of dbCAN, CAZy domains in the genome of P. longicolla were identified with a cutoff E value of 10−3 [64]. Classification of CAZy was conducted as described by Chang et al., 2016 [32].
Identification and classification of repeat elements in the P. longicolla genome
Repeat elements were identified using two well-cited software packages, RepeatMasker (v. 4.0.5) and Censor (v. 4.2.29) [65, 66]. The analysis was partially carried out at the Bioinformatics Core at Purdue University. For RepeatMasker, the optimized default parameters were utilized with the ‘-lib’ option to find repeats associated with the RepBase file (v. 20.03) above. Additionally, the ‘-species Fungi’ option was also used in a separate analysis to find fungal repeats based on RepBase-derived libraries provided by RepeatMasker. Similarly, for Censor, with the ‘censor.ncbi’ module, the optimized default parameters were utilized and the ‘-lib’ option to find repeats.
The predicted protein sequences were compared against the NCBI non redundant database using BLASTP to find the top hits. The genes from contigs larger than 500 bp were used since the same contigs were used for repeat prediction. Similarly, the predicted protein sequences were compared against protein sequences of the closest relative (Diaporthe ampelina) by creating a BLAST-able database. The combined annotation file containing the annotation of each predicted genes and D. ampelina database was generated using in house scripts.
Identification of horizontal gene transfers
To identify putatively horizontally transferred genes in the P. longicolla isolate MSPL 10–6, the HGTector software (http://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-717) was used, which follows a hybrid approach between “BLAST-based” and phylogenetic. The software was setup with the following stringency parameters: threads = 12, selfTax = 1,230,121, closeTax = 147,550, searchTool = blastp, e-value cutoff = 1 × 10−5 for the BLAST hits, and default values for the rest of the parameters. The P. longicolla isolate MSPL 10–6 predicted proteins were blasted against a local NCBI nr database. NCBI Taxonomy database (downloaded on February 20, 2017) was used to classify BLAST matches.
Additional files
A list of putative genes encoding cell wall degrading enzymes. (TXT 73 kb)
A list of horizontal transferred genes identified in the genome of Phomopsis longicolla isolate MSPL 10–6. (XLSX 75 kb)
Acknowledgments
We thank Phillip SanMiguel at Purdue Genomics Core Facility for genome sequencing and David Lightfoot for valuable suggestions and discussion of this research. We also greatly appreciate Ning Jiang for the summary and classification of the repetitive elements. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Funding
This research was funded by the United States Department of Agriculture, Agricultural Research Service (USDA-ARS), Crop Genetics Research Unit at Stoneville, MS. Project number: 6402–21,220-012-00D.
Availability of data and materials
All sequence data are deposited into NCBI’s SRA database, under accession number: AYRD00000000 (1) (http://www.ncbi.nlm.nih.gov/nuccore/AYRD00000000). Putative genes encoding cell wall degrading enzymes and horizontal transferred genes identified in the genome of Phomopsis longicolla isolate MSPL 10–6 are listed in Additional file 1: Table S1 and Additional file 2, Table S2, respectively.
Abbreviations
- AA
Auxiliary activity
- CAZy
Carbohydrate-activated enzymes
- CBM
Carbohydrate-binding module
- CE
Carbohydrate esterases
- GH
Glycoside hydrolases
- GO
Gene ontology
- GT
Glycosyl-transferase
- HGT
Horizontal gene transfer
- LINE
Long interspersed nuclear elements
- LTR
Long terminal repeat
- PCWDE
Plant cell wall degrading enzyme
- PL
Polysaccharide lyases
- PSD
Phomopsis seed decay
Authors’ contributions
SL conceived and led the project, prepared fungal culture and DNA for sequencing; OD assembled the genome, predicted, annotated genes, identified HGT under the guidance of NA; BM identified and analyzed plant cell wall degrading enzymes; BFM acted as a scientific consultant. SL wrote the manuscript. All authors contributed to prepare tables and figures, edited and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4075-x) contains supplementary material, which is available to authorized users.
References
- 1.Hobbs TW, Schmitthenner A, Kuter GA. A new Phomopsis species from soybean. Mycologia. 1985;77:535–44. doi: 10.2307/3793352. [DOI] [Google Scholar]
- 2.Li S, Chen P, Hartman G. Phomopsis seed decay. Compendium of Soybean Diseases and Pests, Fifth Edition. vol. 5. Minnesota: APS Press; 2015. p47–48.
- 3.Santos JM, Vrandečić K, Ćosić J, Duvnjak T, Phillips AJL. Resolving the Diaporthe species occurring on soybean in Croatia. Persoonia-Molecular Phylogeny and Evolution of Fungi. 2011;27(1):9–19. doi: 10.3767/003158511X603719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinclair J. Phomopsis seed decay of soybeans: a prototype for studying seed disease. Plant Dis. 1993;77(4):329–34. doi: 10.1094/PD-77-0329. [DOI] [Google Scholar]
- 5.Li S. Phomopsis seed decay of soybean. In: Sudaric A, editor. Soybean – molecular aspects of breeding. Vienna: Intech Publisher; 2011. p. 277-92.
- 6.Kulik M, Sinclair J. Phomopsis seed decay. Compendium of soybean diseases. 1999;4:31–2.
- 7.Gleason ML, Ferriss RS. Influence of soil water potential on performance of soybean seeds infected by Phomopsis sp. Phytopathology. 1985;75(11):1236–41. doi: 10.1094/Phyto-75-1236. [DOI] [Google Scholar]
- 8.Hepperly P, Sinclair J. Quality losses in Phomopsis-infected soybean seeds. Phytopathology. 1978;68(12):1684–7. doi: 10.1094/Phyto-68-1684. [DOI] [Google Scholar]
- 9.Wrather J, Koenning S. Effects of diseases on soybean yields in the United States 1996 to 2007. Virus. 2009;2(2,144,000):9, 209,000 [PMC free article] [PubMed]
- 10.Koenning S: Southern United States soybean disease loss estimate for 2009. In: Proceedings of the Southern Soybean Disease Workers 37th Annual Meeting: 2010; 2010: 1.
- 11.Balducchi A, McGee D. Environmental factors influencing infection of soybean seeds by Phomopsis and Diaporthe species during seed maturation. Plant Dis. 1987;71(3):209–12.
- 12.Cross C, Wrather A, Fothergill K, Shannon G, Li S, Shumway C, Rupe J. Effect of lactofen, azoxystrobin, and genotypes on charcoal rot, Phomopsis seed decay, and pod and stem blight in soybean. Plant Dis. 2012;96(8):1154–8. doi: 10.1094/PDIS-09-11-0810-RE. [DOI] [PubMed] [Google Scholar]
- 13.Wrather J, Shannon J, Stevens W, Sleper D, Arelli A. Soybean cultivar and foliar fungicide effects on Phomopsis sp. seed infection. Plant Dis. 2004;88(7):721–3. doi: 10.1094/PDIS.2004.88.7.721. [DOI] [PubMed] [Google Scholar]
- 14.Xue A, Morrison M, Cober E, Anderson T, Rioux S, Ablett G, Rajcan I, Hall R, Zhang J. Frequency of isolation of species of Diaporthe and Phomopsis from soybean plants in Ontario and benefits of seed treatments. Canadian J Plant Patho. 2007;29(4):354–64.
- 15.Li S, Smith J. Evaluation of soybean breeding lines for resistance to Phomopsis seed decay in Stoneville Mississippi 2014. Plant Dis Manag Rep. 2016;10:FC045. [Google Scholar]
- 16.Brown E, Minor H, Calvert O. A soybean genotype resistant resistant to Phomopsis seed decay. Crop Sci. 1987;27(5):895–8. doi: 10.2135/cropsci1987.0011183X002700050012x. [DOI] [Google Scholar]
- 17.Li S, Rupe J, Chen P, Shannon G, Wrather A, Boykin D. Evaluation of diverse soybean germplasm for resistance to Phomopsis seed decay. Plant Dis. 2015;99(11):1517–25. doi: 10.1094/PDIS-04-14-0429-RE. [DOI] [PubMed] [Google Scholar]
- 18.Pathan M, Clark K, Wrather J, Sciumbato G, Shannon J, Nguyen H, Sleper D. Registration of soybean germplasm SS93–6012 and SS93–6181 resistant to Phomopsis seed decay. J Plant Reg. 2009;3(1):91–3. doi: 10.3198/jpr2008.01.0002crg. [DOI] [Google Scholar]
- 19.Smith S, Fenn P, Chen P, Jackson E. Inheritance of resistance to Phomopsis seed decay in PI 360841 soybean. J Heredity. 2008;99(6):588–92. doi: 10.1093/jhered/esn037. [DOI] [PubMed] [Google Scholar]
- 20.Jackson EW, Fenn P, Chen P. Inheritance of resistance to Phomopsis seed decay in soybean PI 80837 and MO/PSD-0259 (PI 562694) Crop Sci. 2005;45(6):2400–4. doi: 10.2135/cropsci2004.0525. [DOI] [Google Scholar]
- 21.Zimmerman MS, Minor HC. Inheritance of Phomopsis seed decay resistance in soybean PI 417479. Crop Sci. 1993;33(1):96–100. doi: 10.2135/cropsci1993.0011183X003300010017x. [DOI] [Google Scholar]
- 22.Jackson EW, Feng C, Fenn P, Chen P. Genetic mapping of resistance to Phomopsis seed decay in the soybean breeding line MO/PSD-0259 (PI562694) and plant introduction 80837. J heredity. 2009;100(6):777–83. doi: 10.1093/jhered/esp042. [DOI] [PubMed] [Google Scholar]
- 23.Doolittle WF. Lateral genomics. Trends Cell Bio. 1999;9(12):M5–M8. doi: 10.1016/S0962-8924(99)01664-5. [DOI] [PubMed] [Google Scholar]
- 24.Andersson JO. Gene transfer and diversification of microbial eukaryotes. Annul Rev Microbio. 2009;63:177–93. doi: 10.1146/annurev.micro.091208.073203. [DOI] [PubMed] [Google Scholar]
- 25.Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 2008;9(8):605–18. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- 26.Fitzpatrick DA. Horizontal gene transfer in fungi. FEMS Microbio Let. 2012;329(1):1–8. doi: 10.1111/j.1574-6968.2011.02465.x. [DOI] [PubMed] [Google Scholar]
- 27.Klosterman S, Rollins J, Sudarshana M, Vinatzer B. Disease Management in the Genomics era—Summaries of focus issue papers. Phytopathology. 2016;106(10):1068–70. doi: 10.1094/PHYTO-07-16-0276-FI. [DOI] [PubMed] [Google Scholar]
- 28.Li S, Darwish O, Alkharouf N, Matthews B, Ji P, Domier LL, Zhang N, Bluhm BH. Draft genome sequence of Phomopsis longicolla isolate MSPL 10-6. Genomics data. 2015;3:55–6. [DOI] [PMC free article] [PubMed]
- 29.Li S, Song Q, Martins AM, Cregan P. Draft genome sequence of Diaporthe aspalathi isolate MS-SSC91, a fungus causing stem canker in soybean. Genomics data. 2016;7:262–3. [DOI] [PMC free article] [PubMed]
- 30.Srivastava SK, Huang X, Brar HK, Fakhoury AM, Bluhm BH, Bhattacharyya MK. The genome sequence of the fungal pathogen Fusarium virguliforme that causes sudden death syndrome in soybean. PLoS One. 2014;9(1):e81832. doi: 10.1371/journal.pone.0081832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Islam MS, Haque MS, Islam MM, Emdad EM, Halim A, Hossen QMM, Hossain MZ, Ahmed B, Rahim S, Rahman MS. Tools to kill: genome of one of the most destructive plant pathogenic fungi Macrophomina phaseolina. BMC Genomics. 2012;13(1):493. doi: 10.1186/1471-2164-13-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang H-X, Yendrek CR, Caetano-Anolles G, Hartman GL. Genomic characterization of plant cell wall degrading enzymes and in silico analysis of xylanses and polygalacturonases of Fusarium virguliforme. BMC Microbiol. 2016;16(1):147. doi: 10.1186/s12866-016-0761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Z, Liu H, Wang C, Xu J-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013;14(1):274. doi: 10.1186/1471-2164-14-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jayani RS, Saxena S, Gupta R. Microbial pectinolytic enzymes: a review. Process Biochem. 2005;40(9):2931–44. doi: 10.1016/j.procbio.2005.03.026. [DOI] [Google Scholar]
- 35.Güldener U, Mannhaupt G, Münsterkötter M, Haase D, Oesterheld M, Stümpflen V, Mewes H-W, Adam G. FGDB: a comprehensive fungal genome resource on the plant pathogen Fusarium graminearum. Nucleic Acids Res. 2006;34(suppl 1):D456–8. [DOI] [PMC free article] [PubMed]
- 36.Jeong H, Lee S, Choi GJ, Lee T, Yun S-H. Draft genome sequence of Fusarium fujikuroi B14, the causal agent of the bakanae disease of rice. Genome announcements. 2013;1(1):e00035–13. [DOI] [PMC free article] [PubMed]
- 37.Kopel M, Helbert W, Belnik Y, Buravenkov V, Herman A, Banin E. New family of Ulvan Lyases identified in three isolates from the Alteromonadales order. J Biol Chem. 2016;291(11):5871–8. doi: 10.1074/jbc.M115.673947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–82. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 39.Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu J-R, Pan H. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature. 2005;434(7036):980–6. [DOI] [PubMed]
- 40.Xue M, Yang J, Li Z, Hu S, Yao N, Dean RA, Zhao W, Shen M, Zhang H, Li C. Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet. 2012;8(8):e1002869. doi: 10.1371/journal.pgen.1002869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okagaki LH, Nunes CC, Sailsbery J, Clay B, Brown D, John T, Oh Y, Young N, Fitzgerald M, Haas BJ. Genome sequences of three phytopathogenic species of the Magnaporthaceae Family of fungi. G3: Genes Genomes Genetics. 2015;5(12):2539–45. [DOI] [PMC free article] [PubMed]
- 42.Boeke J, Stoye J: Retrotransposons, endogenous retroviruses, and the evolution of retroelements. Retroviruses. In: Coffin JM, Hughes SH, Varmus HE, editors. Cold Spring Harbor Laboratory Press. 1997, 343–436. [PubMed]
- 43.Eickbush TH, Malik HS: Origins and evolution of retrotransposons. In: Mobile DNA ii. American Society of Microbiology; 2002: 1111–1144.
- 44.Havecker ER, Gao X, Voytas DF. The diversity of LTR retrotransposons. Genome Bio. 2004;5(6):225. doi: 10.1186/gb-2004-5-6-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Llorens C, Muñoz-Pomer A, Bernad L, Botella H, Moya A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol Direct. 2009;4(1):41. doi: 10.1186/1745-6150-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lohe AR, Moriyama EN, Lidholm D-A, Hartl DL. Horizontal transmission, vertical inactivation, and stochastic loss of mariner-like transposable elements. Mol Biol Evol. 1995;12(1):62–72. doi: 10.1093/oxfordjournals.molbev.a040191. [DOI] [PubMed] [Google Scholar]
- 47.Lampe DJ, Witherspoon DJ, Soto-Adames FN, Robertson HM. Recent horizontal transfer of mellifera subfamily mariner transposons into insect lineages representing four different orders shows that selection acts only during horizontal transfer. Mol Biol Evol. 2003;20(4):554–62. doi: 10.1093/molbev/msg069. [DOI] [PubMed] [Google Scholar]
- 48.Mandal PK, Kazazian HH: SnapShot: vertebrate transposons. Cell 2008, 135(1):192–192. e191. [DOI] [PubMed]
- 49.Carlton JM, Hirt RP, Silva JC, Delcher AL, Schatz M, Zhao Q, Wortman JR, Bidwell SL, Alsmark UCM, Besteiro S. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science. 2007;315(5809):207–12. [DOI] [PMC free article] [PubMed]
- 50.Marcet-Houben M, Gabaldón T. Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 2010;26(1):5–8. doi: 10.1016/j.tig.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Richards TA, Leonard G, Soanes DM, Talbot NJ. Gene transfer into the fungi. Fungal Bio Rev. 2011;25(2):98–110. doi: 10.1016/j.fbr.2011.04.003. [DOI] [Google Scholar]
- 52.Ma L-J, Van Der Does HC, Borkovich KA, Coleman JJ, Daboussi M-J, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464(7287):367–73. [DOI] [PMC free article] [PubMed]
- 53.White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications. 1990;18(1):315–22. [Google Scholar]
- 54.Carbone I, Kohn LM. A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia. 1999;91(3):553–6. doi: 10.2307/3761358. [DOI] [Google Scholar]
- 55.Li S, Hartman G. Molecular detection of Fusarium solani f. Sp. glycines in soybean roots and soil. Plant Pathol. 2003;52(1):74–83. doi: 10.1046/j.1365-3059.2003.00797.x. [DOI] [Google Scholar]
- 56.Li S, Hartman GL, Boykin DL. Aggressiveness of Phomopsis longicolla and other Phomopsis spp. on soybean. Plant Dis. 2010;94(8):1035–40. [DOI] [PubMed]
- 57.Li S, Smith JR, Nelson RL. Resistance to Phomopsis seed decay identified in maturity group V soybean plant introductions. Crop Sci. 2011;51(6):2681–8. doi: 10.2135/cropsci2011.03.0162. [DOI] [Google Scholar]
- 58.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoff KJ, Stanke M. WebAUGUSTUS—a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 2013;41(W1):W123–8. doi: 10.1093/nar/gkt418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24(5):637–44. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 61.Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–35. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Bio. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 63.Zdobnov EM, Apweiler R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17. [DOI] [PubMed]
- 64.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(W1):W445–51. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smit A, Hubley R, Green P: RepeatMasker Open-4.0. 2013–2015. Institute for Systems Biology http://repeatmasker.org 2015.
- 66.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and censor. BMC bioinformatics. 2006;7(1):474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of putative genes encoding cell wall degrading enzymes. (TXT 73 kb)
A list of horizontal transferred genes identified in the genome of Phomopsis longicolla isolate MSPL 10–6. (XLSX 75 kb)
Data Availability Statement
All sequence data are deposited into NCBI’s SRA database, under accession number: AYRD00000000 (1) (http://www.ncbi.nlm.nih.gov/nuccore/AYRD00000000). Putative genes encoding cell wall degrading enzymes and horizontal transferred genes identified in the genome of Phomopsis longicolla isolate MSPL 10–6 are listed in Additional file 1: Table S1 and Additional file 2, Table S2, respectively.