

Abstract
Introduction
Busulfan (Bu) is an alkylating agent with a limited therapeutic margin and exhibits inter-patient variability in pharmacokinetics (PK). Despite decades of use, mechanisms of Bu PK-based drug-drug interactions (DDIs), as well as the negative downstream effects of these DDIs, have not been fully characterized.
Areas covered
This article provides an overview of Bu PK, with a primary focus on how known and potentially unknown drug metabolism pathways influence Bu-associated DDIs. In addition, pharmacogenomics of Bu chemotherapy and Bu-related DDIs observed in the stem cell transplant clinic (SCT) are summarized. Finally the increasing importance of Bu therapeutic drug monitoring is highlighted.
Expert Opinion
Mechanistic studies of Bu metabolism have shown that in addition to GST isoenzymes, other oxidative enzymes (CYP, FMO) and ABC/MDR drug transporters likely contribute to the overall clearance of Bu. Despite many insights, results from clinical studies, especially in polypharmacy settings and between pediatric and adult patients, remain conflicting. Further basic science and clinical investigative efforts are required to fully understand the key factors determining Bu PK characteristics and its effects on complications after SCT. Improved TDM strategies are promising components to further investigate, for instance DDI mechanisms and patient outcomes, in the highly complex SCT treatment setting.
Keywords: busulfan, drug interaction, pharmacogenomics, pharmacokinetics, hematopoietic stem cell transplant, drug metabolism, GST, CYP, therapeutic drug monitoring
1. Introduction
Pre-transplant conditioning regimens with combination chemo- or chemo-radiotherapy followed by hematopoietic stem cell transplantation (HSCT) are widely used for curative treatment of several diseases, including hematological malignancies (e.g. leukemia, lymphoma), solid tumors, non-malignant disorders (e.g. thalassemia, sickle cell anemia) and other genetic diseases [1,2]. High-dose busulfan (Bu) has supplanted myeloablative total body irradiation (TBI) in many pre-transplant conditioning regimens used in the clinic today [3,4]. Although effective, high-dose Bu therapy, like all anticancer therapy based on DNA alkylation, has certain drawbacks. These include treatment-related toxicity, limited therapeutic interval, and inter-patient variability in pharmacokinetics (PK) [5]. Adverse drug toxicity associated with high exposure of Bu include mucositis, pulmonary toxicity, veno-occlusive disease (VOD), neurotoxicity (grand mal seizures), acute graft versus host disease (as a consequence of the cytokine storm induced from myeloablation and normal organ damage), and even death [6,7]. Low systemic exposure of Bu also leads to untoward effects such as disease relapse, failed engraftment and shortened survival [6]. HSCT patients with Bu exposure (AUC) residing outside its therapeutic range have shown inferior treatment outcomes [8]. Changing from the oral to IV formulation of Bu has, as expected [5,9], decreased the inter-dose and PK variability between patients; but on a global scale the inter-individual variation of IV Bu clearance still approaches 30% in some studies and treatment-related toxicities, including CNS toxicity and VOD, persist when Bu is utilized in high-dose therapy [8,10]. The risk increases when Bu is combined with (an-) other alkylating agents(s), or the dose administered is particularly high. Further improvements in Bu therapy are still desirable to overcome these obstacles and improve upon patient safety and treatment outcomes. One major advantage of Bu is the established analytical techniques to monitor its plasma concentrations, which lends itself to PK-based modified therapy with greater improvements in both the toxicity profile and tumor control [11,12].
Multiple drug therapy is common in today’s oncology medical practice, increasing the risk of single and/or multiplex drug-drug interactions (DDIs) – that often lead to adverse drug toxicities and unanticipated treatment outcomes [13]. HSCT patients are commonly subjected to poly-pharmacy, taking on average at least 8 or more drugs on admission, and they are exposed for lengthy periods of time to complex drug regimens consisting of multiple pharmacological classes, many of which interact with each other [13–15]. In HSCT patients, the signs/symptoms of a DDI may go undetected in the pre-HSCT phase and/or be dismissed as a symptom of the primary tumor, but may appear during later periods following the transplant [15]. Also, a greater risk of DDIs was correlated with worsened HSCT outcomes (e.g. increased morbidity), and especially among patients co-prescribed drugs with a narrow therapeutic index and therefore high risk of toxicity [13–15]. This is especially true in HSCT patients administered high-dose alkylating agent-based regimens (here, specifically busulfan), in which many medications administered with Bu influence multiple drug metabolism pathways, such as cytochrome P450 (CYP), glutathione S-transferase (GST), glutathione (GSH) conjugation and efflux/uptake transporters [16]. These factors combined place Bu patients in a “perfect storm” for unsafe DDIs.
Despite being in clinical use for several decades, Bu’s global metabolic profile remains unsettled. Therefore, it is difficult for clinicians to safely prescribe co-administered drugs, especially among heavily pre-treated patients, and there is always concern for one or more deleterious DDIs. Multiple in vitro and in vivo Bu DDI studies have been published; however, the molecular mechanisms of these DDIs have rarely been investigated. Several of these DDI studies report conflicting results for the same co-medication prescribed with Bu and incongruous mechanistic delineations of these important interactions in reference to Bu metabolism, creating further confusion regarding the clinical use of Bu. Package inserts for Bu provide little information regarding potential DDIs with co-administered drugs and mechanisms of importance for Bu metabolism [17,18]. Furthermore, the potential for serious DDIs with Bu are frustrating due to Bu’s noted use in high-dose therapy where there is a dose-response relationship favoring a beneficial effect for patients that achieve a high systemic exposure, yet the risk of overexposure/-dosing places the HSCT patient at risk for serious toxicity. To our knowledge, an exhaustive review of the PK properties of Bu combined with pharmacogenomics, drug metabolism characteristics and clinical DDIs has not been published. The purpose of this review is to discuss these important topics, which is a key step towards improving upon Bu therapeutics and clinical outcomes from a drug metabolism perspective, further supporting refinements to population PK modeling in conjunction with therapeutic drug monitoring (TDM) in Bu clinical scenarios.
The present paper reviews the literature concerning Bu PK, disposition, pharmacogenomics, in vitro/in vivo DDIs, and TDM. The searches were carried out using databases such as PUBMED, MEDLINE and SciFinder Scholar with no specific limit. Also included in our review was information obtained from citations of the articles that were retrieved during our searches. Only published information in peer-reviewed journals was included, whereas meeting abstracts and dissertations were excluded. Drug information was also obtained from the individual FDA-approved product inserts of Bu and other discussed drugs. Finally, only articles published in the English language were included in the review.
2. Busulfan pharmacology and pharmacokinetics
The chemical structure of Bu (Figure 1) contains 2 easily displaced methane sulfonate groups on opposite ends of a butane chain. Hydrolysis of these groups produces highly reactive, positively charged carbonium ions that alkylate and damage DNA molecules [18]. More precisely, Bu reacts with guanine molecules through a nucleophilic substitution reaction (SN2), forming DNA intra- or interstrand cross-links [19]. Bu’s in vitro toxicity is correlated with formation of these DNA lesions [20]. Bu also reacts with cysteine molecules on histone proteins, inducing DNA-protein binding [20].The alkylating activity of Bu produces alterations of cell replication, DNA damage repair, and gene transcription [19].
Figure 1. Metabolism of Bu (1,4-butanediol dimethanesulfonate).
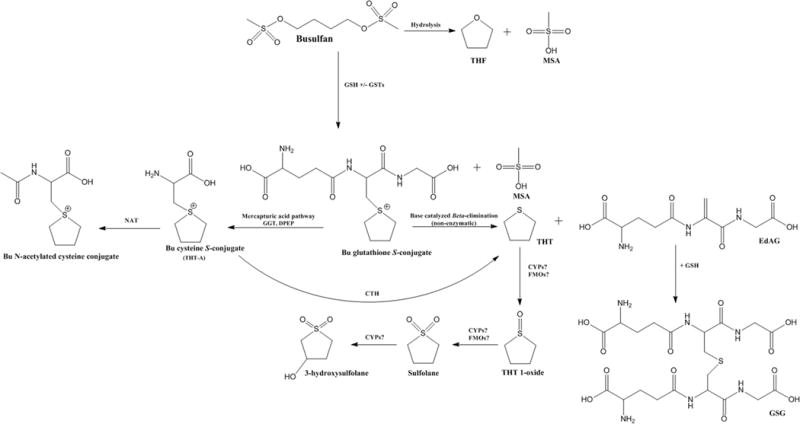
The formation of the Bu metabolites is depicted above. Bu is extensively metabolized in the liver with about 2% of unchanged drug excreted in the urine. Initial metabolism occurs primarily via conjugation with endogenous GSH (both spontaneously and by GST catalysis), and to a minor extent hydrolysis. Further metabolism of the Bu-GSH conjugate occurs via the mercapturic acid pathway or dissociation to THT and EdAG. THT is further metabolized by a sequence of oxidation steps, whereas EdAG binds with GSH to form GSG. The enzymes responsible for oxidative transformations are lesser understood.
Bu: Busulfan; CTH: Cystathionine gamma-lyase; DPEP: Dipeptidase; EdAG: γ-glutamyldehydroalanylglycine; GGT: Gamma-glutamyl transferase; GSG: EdAG glutathione conjugate; FMO: Flavin-containing monooxygenase; GSH: Glutathione; GST: Glutathione S-transferase; MSA: Methane sulfonic acid; THF: Tetrahydrofuran; THT: Tetrahydrothiophene; THT-A: Cysteine S-conjugate of Busulfan.
Furthermore, Bu reacts with the sulfhydryl groups of the endogenous tripeptide GSH, either spontaneously or via catalysis by GST enzymes, thus, disrupting the cellular redox equilibrium which results in oxidative stress [21]. These effects can be reversed by GST inhibitors and antioxidants [21]. Although many studies have shown that cellular toxicity of Bu is due to the parent drug, an emerging set of data suggests that a Bu (a) metabolite(s) formed from degradation of the Bu-GSH conjugate may play a hitherto unrecognized role [22–24].
Bu clearance following oral dosing can be best described by an open one-compartment model. Absorption of Bu following oral administration shows highly variable bioavailability and variable plasma peak concentrations in both adults and children [25–27]. The absorption process has been described as both zero-order or first-order kinetic mechanisms [28,29]. Contributing factors for variability in oral busulfan PK include circadian rhythmicity (chronopharmacological changes), disease states, age and dosage [28,29]. For instance, many patients ingesting high-dose Bu by the oral route (typically a bevy of tablets) experience substantial GI irritation, leading to nausea and vomiting, which ultimately leads to unpredictable absorption (bioavailability) of the drug. Erratic intestinal absorption coupled with GI complicating factors substantially increases the error margin in systemic dose delivery and has been attributed as a principle factor in oral PK variability, particularly in infants and children [30]. These unpredictable changes in Bu PK were a major impetus in developing an IV formulation of Bu [31], approved by the FDA in 1999, which when injected bypasses both the erratic intestinal absorption and hepatic first-pass metabolism and provides less challenges to TDM. The IV formulation IV Busulfex (Otsuka Pharmaceutical Co.) [18] marketed in the US and Busilvex™ in Europe, is the mainstay dosage form used clinically today. This IV Bu formulation virtually eliminated dose-to-dose variability in systemic exposure, as well as reduced the more than 10-fold variations in bioavailability of oral Bu to about the expected 2- to 2.5-fold variation that is due to inter-individual metabolic drug handling [10,32].
The percent coefficient of variation (CV) in IV Bu clearance, maximal plasma concentration (Cmax), AUC in 59 patients was 25%, 18%, and 20%, respectively [18]. Other studies have confirmed that IV Bu has a narrow therapeutic index, displaying wide inter-individual variability in its clearance and AUC [6,32,33]. Over the past 20 years, many studies have shown that Bu exposure is correlated with clinical outcomes [6,8,29]. For instance, the probability of developing Bu toxicity increases with rising Bu systemic exposure as measured by the AUC [6]. Moreover, the risk of death was significantly higher when per-dose AUC remained above or below a critical threshold level [6]. As a consequence, TDM of Bu in adults and children is practiced at many transplant facilities throughout the world, including our institute.
Approximately 32% of Bu irreversibly (covalently) binds to plasma proteins, primarily albumin [34]. The fraction of Bu irreversibly bound to erythrocytes is 47%, and the fraction of drug in plasma is inversely related to hematocrit in normal healthy subjects and cancer patients [35]. A small amount (<5%) is reversibly bound to plasma proteins, which is uncommon for alkylating drugs. Bu rapidly distributes into the cerebrospinal spinal fluid (CSF), achieving approximately equal concentrations to those in plasma [34]. This likely explains its major central nervous system (CNS) adverse drug effect, seizures [34].
Following intraperitoneal (i.p.) administration of 15 mg/kg of radiolabeled Bu to rats, approximately 70% of the total radioactivity was excreted in the urine over 72 hours, whereas negligible amounts of radioactivity were recovered in feces [36]. In humans, approximately 30% of total radioactivity was excreted in the urine over 48 hours, with negligible fecal excretion [18]. It is speculated that the incomplete recovery of radioactivity may be due to the formation of metabolites with extensive elimination half-lives, and/or due to irreversible alkylation of macromolecules [18]. The amount of intact Bu excreted unchanged in the urine is approximately 6% in rats and 2% in humans [36,37].
Bu may to some extent induce its own metabolism following repeated treatment in children and adults [27,37]. High-dose Bu was administered to five adult female patients (1 mg/kg p.o. every 6 hrs × 4 days) with acute myelogenous leukemia (AML) [37]. To evaluate temporal PK, blood samples were collected over the entire treatment course (10 samples each following the 1st and 2nd doses; 4–5 samples following the 3rd through 15th doses; and 16 samples following the last dose). In 3 of the 5 patients there was a continuous decrease in Bu steady-state concentrations (Css) over time, which were on average 32% lower compared to the first dose. The mean plasma elimination half-life (T1/2) was 3.4 hr after the first dose and 2.3 hr after the last dose, but the AUC decreased by only 7% between doses. In all patients, Css levels were uniformly lower than the Css levels predicted from the first dose AUC measurement [37].
In another study published from the same research group, high-dose Bu (1 mg/kg p.o. every 6 hrs × 4 days) was administered to 9 children (4 young children and 5 older children) and 18 adults [27]. Around 10 to 15 blood samples were collected following the 1st and 16th dose, whereas a trough plasma concentration (Ctr) was collected for the 2nd through 15th doses. About 35% of adult patients showed a continued decrease of 30%–60% in plasma Css between the 3rd and 16th doses. Only 2 of the young children and 1 of the older children showed a trend in continuous declines of Css [27].
Hassan et al. [27,37] have suggested that Bu may induce glutathione (GSH) synthesis and/or increase glutathione S-transferase (GST) activity, which are both responsible for part of parent Bu’s clearance pathways [38]. These mechanisms are indirectly supported by evidence of induction of GST activity by other reactive electrophilic compounds [39]. In mice administered Bu 16.5 mg/kg i.p. twice daily, an induction in Bu clearance was observed that was correlated with enhanced GSH synthesis rather than specific Bu GST activity [40]. Phenytoin is a common prophylactic anti-seizure medication used in Bu patients and is known to induce drug metabolizing enzymes [41]; however it is not known, to our best knowledge, if phenytoin induces GST enzymes in human clinical situations.
The chronopharmacokinetic (circadian rhythmicity) behavior of Bu has been reported in 2 studies [27,42]. In one study, which also studied potential Bu auto-induction, high-dose Bu (1 mg/kg p.o. every 6 hrs × 4 days) was administered to pediatric and adult patients [27]. Chronopharmacokinetics was evaluated by comparing the mean daytime concentrations with the mean nighttime concentrations at steady-state. In pediatric patients, especially those ≤ 5 years-old, up to a 3-fold variation between daytime and nighttime concentrations were observed, in which the nighttime concentrations were consistently higher than the daytime concentrations [27]. No circadian rhythmicity was observed in adult patients [27].
Chronopharmacokinetics of Bu was also studied in 21 pediatric patients with malignant solid tumors [42]. Children were administered Bu (37.5 mg/m2 p.o. every 6 hrs × 4 days) and Ctr levels were collected periodically over the 4-day treatment period. Mean Bu Ctr levels were higher at 06:00 compared to Ctr levels at 24:00, 12:00, 18:00 and 24:00, suggesting a significant chronopharmacokinetic variability [42]. In some children urine was collected during the 6 hr dosing periods for all treatment doses. Rates of urinary excretion of Bu also exhibited a significant circadian rhythm which was correlated to circadian changes in urinary output [42].
These diurnal variation in Bu PK are intriguing yet the cause(s) of this phenomenon remain unknown. A possible explanation is circadian variation of GST activity, which has been documented in mice, although not specifically studied in Bu-dosed animals [43]. Of note, the Bu chronopharmacokinetic studies were all performed in orally administered patients, which may have a bearing on the perceived chronopharmacological behavior since the intestinal absorption of oral Bu is highly variable over the 24-hour cycle. CYPs and other drug metabolizing enzymes also show a documented circadian rhythmicity [44]. The cytotoxic drugs doxorubicin and cisplatin have shown diurnal variations, and careful timing of their administration have led to improved treatment efficacy with lessened adverse effects [45].
Antineoplastic agents such as paclitaxel display significant variability in blood PK when the same drug and dose is administered by different infusion rates [46]. Bu clearance also appears to be dependent upon the infusion rate [47]. For instance, patients treated with an infusion rate that varies (2-fold to 4-fold) between the test dose of Bu and subsequent therapeutic dose, a significantly greater clearance was found following a lower test dose compared to the subsequent therapeutic dose [47]. However, when patients were administered Bu using the same infusion rate during test and therapeutic doses, the therapeutic dose clearance closely matched the test dose clearance. Similar infusion-rate dependent clearance rate(s) have been observed with paclitaxel [46] In addition, the authors conceived a threshold infusion rate of 45 mg/hr, above which the clearance of test and therapeutic doses is similar even if they were infused at different rates; however, there was insufficient data in their study to adequately confirm the threshold rate [47]. These findings underline the importance of a carefully standardized schedule for how to both administer the drug and collect blood samples if/when the physician plans to implement PK-guidance as part of an optimized treatment program.
3. Bu metabolism
Bu disposition is a multi-step, highly complex process that has been studied in rodents and humans. An overview of Bu’s currently known metabolic pathways can be found in Figure 1.
3.1 Bu metabolism in rats
One of the earlier known metabolites of Bu was methane sulfonic acid. This metabolite is chemically formed as a by-product following the reaction of Bu with GSH, and secondarily as a metabolite resulting from parent Bu hydrolysis (Figure 1) [48]. Methane sulfonic acid was detected in the blood, as well as many other tissues, of rats administered oral, i.p. and IV 35S Bu [48]. Methane sulfonic acid was almost completely eliminated from the blood within 10–15 minutes, and was found in large quantities in the urine during a 24 hour collection period [48].
Hassan M et al. [36] elucidated the urinary metabolites of Bu following i.p administration of [14C]Bu 15 mg/kg to rats. They found that Bu reacts with the endogenous tripeptide GSH, either spontaneously or via GST enzyme catalysis, forming a positively charged conjugate known as the sulfonium ion of GSH (Figure 1). The sulfonium ion undergoes further metabolism via the mercapturic acid pathway, or it undergoes a β-elimination reaction to tetrahydrothiophene (THT). Tetrahydrothiophene is converted to tetrahydrothiophene 1-oxide, which is further oxidized to sulfolane (tetrahydrothiophene-1,1-dioxide). Finally, sulfolane is oxidized to 3-hydroxysulfolane (tetrahydrothiophene-3-OH 1,1-dioxide). The major urinary metabolites detected in the urine of rats include 3-hydroxysulfolane (39%), tetrahydrothiophene 1-oxide (20%) and sulfolane (13%) [36]. Only 6% of unchanged Bu was excreted in the urine [36]. Another minor metabolite tetrahydrofuran (2%) was also excreted into rat urine [36].
3.2 Bu metabolism in humans
Bu metabolism in humans is similar to rats, and has been evaluated in multiple clinical studies. In adult acute myeloblastic leukemia (AML) patients, approximately 2% of unchanged Bu was found in the urine [37]. In this study, 3 major urinary metabolites (sulfolane, 3-hydroxysulfolane and tetrahydrothiophene 1-oxide) were detected [37], which is similar to rats [36]. Also similar to rats, tetrahydrothiophene was liberated following hydrolysis of urine samples with sodium hydroxide, further supporting the existence of a sulfonium ion conjugate of GSH [37]. Versace et al. [49] identified sulfolane in the plasma of children receiving intravenous Bu; however, the identification of other Bu metabolites was not reported. More recently, El-Serafi et al. [50] detected THT, tetrahydrothiophene 1-oxide, sulfolane and 3-hydroxysulfolane in the plasma and urine of a patient administered high-dose IV Bu.
3.3 Enzyme-mediated Bu metabolism
Overall 5 major metabolites, several minor and numerous unidentified metabolites of Bu have been found in rat urine and human plasma [50]. Bu metabolites are important since they possibly contribute to Bu’s toxic side effects (e.g. VOD) [21,24,51]. Formation of some Bu intermediate metabolites is enzyme driven, in which GSH conjugation via GST enzymes remains the most readily recognized first-step in its metabolism.
In an isolated perfused rat liver model, formation of the sulfonium ion of GSH was significantly reduced in the presence of the ethacrynic acid, a non-specific GST inhibitor [52] and recently found inhibitor of the MRP1-/ABCC1- transporter protein pump that participates in cellular outward transport of GSH conjugates of alkylating drugs [53]. Gibbs et al. [38] showed that human liver cytosolic, but not microsomal, GST enzymes metabolized Bu to the sulfonium ion of GSH, which accounted for 50% of its metabolism. They also demonstrated that ethacrynic acid inhibited the enzymatic formation of this metabolite in a concentration-dependent manner [38]. Furthermore, they showed that GST alpha (A1) was the predominant GSH catalyst for Bu-GSH conjugation in human liver, whereas GST mu (M1) and GST pi (P1) were minor contributors [54]. Bu conjugation in human intestine was also catalyzed by intestinal GST A1, and this activity was comparable to human liver GST A1 [55]. Age-dependent variation in intrinsic Bu clearance was associated with greater expression of intestinal GST A1, which was most pronounced in young children compared with older children [56]. The presence of highly-functional Bu conjugating GST enzymes in the intestine combined with age-related variances in enzyme expression, presumably, may be a contributing factor towards the up to, or more than, 10-fold variation in apparent oral Bu clearance observed in clinical trials.
The sulfonium ion of GSH (Figure 1) is further metabolized to THT by several routes: 1) The sulfonium ion of GSH (Bu-SG conjugate) undergoes metabolism in the mercapturic acid pathway by a series of enzymes, culminating with acetylation of the cysteine S-conjugate via N-acetyltransferase (NAT) enzymes to form the N-acetylcysteine S-conjugate [57]. Instead of acetylation, the cysteine S-conjugate can be metabolized by rat hepatic cytosolic cystathionine γ-lyases to form THT, pyruvate and ammonia [57]; or 2) The sulfonium ion of GSH (Bu-SG conjugate) undergoes a facile β-elimination reaction of the sulfonium group to form THT and γ-glutamyldehydroalanylglycine (EdAG) [24]. EdAG, which is later reviewed in this article, is an intriguing metabolite, with reactive electrophilic and free-radical scavenging properties [22,57], but has yet to be identified in vivo. The latter pathway (spontaneous elimination to THT and EdAG) is believed to be the most predominant mechanism of THT formation.
THT was not mutagenic to V79 Chinese hamster cell lines [36], but its potential contribution to adverse effects on organ systems in Bu-treated patients is not known. THT is a sulfur containing, heterocyclic compound that is commonly used as a natural gas odorant and industrial solvent [58]. The effects of THT on human beings have been sparsely recorded, with some evidence of neurological and pulmonary toxicity [58]. THT is also a volatile compound with a highly unpleasant odor, rendering quantification in metabolism studies challenging; thus, the functional roles of human oxidizing enzyme systems on THT metabolism have yet to be established. THT is a cyclic sulfur compound so, theoretically, it may be a substrate for human oxidases such as CYPs and/or flavin-containing monooxygenases (FMO). In vitro and in vivo data exist in animals supporting the enzyme-mediated clearance of THT. For instance, rat liver FMO, but not CYP, catalyzed the S-oxygenation of THT [59]. Krueger and Williams [60] reported that THT was a substrate for mammalian FMO. THT is also a substrate for purified mouse lung thioether methyltransferase, resulting in the formation of the THT methyl sulfonium ion [61]. This metabolite of THT, in the same study, was detected in the liver, lung, kidney and urine of THT-treated mice [61], but human data are lacking.
THT is metabolized by S-oxidation to THT 1-oxide (tetramethylene sulfoxide), which is a known electron acceptor that stimulates the growth of bacteria [62]. The next Bu metabolite, THT 1,1 oxide (sulfolane), is formed via S-oxidation of THT 1-oxide, which hypothetically is catalyzed by FMOs or CYPs. Based on its chemical structure, sulfolane has electrophilic properties capable of reacting with nucleophilic compounds [63]. Sulfolane is a polar solvent primarily used in the natural gas/petroleum industry as an odorant, and produces hypothermia and decreased oxygen consumption following acute inhalation exposure [64]. Oral and i.p. administration of sulfolane to rodents produces neurotoxicity, including generalized seizures [65,66]. Sulfolane and derivatives have affinity for the P2 ligands for HIV-1 protease inhibitors [67]. The overall contribution of THT 1-oxide or sulfolane to Bu treatment related toxicity is not known, but it is interesting to note that a major adverse effect of high-dose Bu therapy is generalized seizures, and sulfolane has shown seizure-inducing properties in animals, albeit at high doses [65]. In this context it is intriguing to note that the peak incidence of seizures after high-dose Bu typically occurs on the third to fifth day of treatment.
The last metabolite of Bu is believed to be 3-hydroxysulfolane, a more polar derivative of sulfolane. 3-hydroxysulfolane was found to be a contributing factor to hypothermic changes observed in rabbits administered sulfolane [68]. The mechanism of 3ʹ-oxidation of sulfolane, theoretically, could by mediated by CYPs or other oxidases, and conversion to a ketone group by alcohol dehydrogenases are plausible, however this remains to be determined.
Finally, Hassan and Andersson [16] speculated upon a novel model of Bu metabolism that involves drug transporters. They proposed that following conjugation with GSH, the newly formed Bu-GSH conjugate undergoes active cellular transport, which potentially involves one or more members of the ABC transporters that are normally involved in the transport of thiol-containing and glutathione conjugated molecules [16]. Furthermore, they suggested that the initial GSH conjugation step is subject to feedback inhibition by product accumulation (Bu-GSH conjugate), which ultimately would disrupt the PK, leading to intracellular accumulation of free, unconjugated Bu and possibly augmenting the cellular toxicity of Bu. Interestingly, GSH conjugates of several alkylating agents are substrates for MRP1 (ABCC1) [69], and the ethacrynic acid GSH adduct is both a substrate and inhibitor of MRP [70]. It is of importance to recognize that Hassan et al. [52] reported in an isolated perfused rat liver model that the hepatic excretion of the Bu-SG conjugate was impaired in the presence of ethacrynic acid. But, the relative distribution of alternative mechanisms, such as transporter inhibition, that contribute to the reduced hepatic excretion of the conjugate observed in this study cannot be distinctly elucidates since ethacrynic acid and its glutathionylated metabolite are potent inhibitors of GST enzymes [71]. Overall, further research is needed to confirm these interesting product-feedback mechanistic hypotheses. In this context, it is of interest that ethacrynic acid was recently reported as a most potent inhibitor of the ABCC1-transporter-/ MRP-mediated cellular efflux mechanism involved in cellular excretion of several different alkylating agent-SG conjugates [53].
3.4 Hydrolysis of busulfan
The chemical structure of Bu contains two sulfonic ester groups (Figure 1) which are susceptible to non-enzymatic, and possibly hydrolase-mediated breakdown. Minor metabolites of Bu found in rat urine and/or in aqueous solutions are the cyclic ether tetrahydrofuran (THF) [36], 1,4-butanediol [7] and methane sulfonic acid [48]. A mixture of Bu in water resulted in mono-sulfonyl hydrolysis to form methane sulfonic acid and the unstable intermediate 4-methanesulfonyloxybutanol [72]. This intermediate undergoes cyclization to THF via intramolecular alkylation [72]. Hydrolysis of busulfan in aqueous solution also resulted in THF and methane sulfonic acid [73]. THF has also been identified as a degradation impurity in Bu injectable products [74]. Also in aqueous solution, bi-sulfonyl hydrolysis of Bu produced 1,4-butanediol [75]. In human liver cytosol, 1,4-butanediol is further metabolized by aldehyde dehydrogenases to gamma-hydroxybutyric acid (GHB) [76], although these enzyme mechanisms have not been identified directly in the context of Bu metabolism.
The possible enzymatic formation of 1,4-butanediol from Bu is intriguing but has to our knowledge not been reported. It is interesting to note that 1,4-phenylene dimethanesulfonate, which is chemically similar to Bu, is hydrolyzed in human liver cytosol by carbonic anhydrases with sulfatase activity [77]. It is unknown if Bu is a substrate for carbonic anhydrase and/or sulfatase enzymes, and if so, what are the clinical consequences of this possible Bu metabolism step.
3.5 The busulfan metabolite γ-glutamyldehydroalanylglycine (EdAG)
The possibility of EdAG as a metabolite of Bu was first postulated by Roberts et al. [78]. In chemical stability studies, EdAG (along with THT) formed following the base catalyzed decomposition of the Bu-SG conjugate [79]. Further characterization of this compound, in regards to potential as an in vivo Bu metabolite, followed many years later [24]. EdAG was identified after co-incubation of Bu and GSH at pH 7.4 (37°C) in human liver cytosol [24]. EdAG, a dehydroalanine analog of GSH, is a Michael Acceptor compound with an electrophilic α,β-unsaturated carbonyl component [24]. This electrophilic moiety is capable of reacting with nucleophilic thiol compounds such as cysteine and GSH in vitro under aqueous conditions [24]. The work of Younis et al. [24] found that EdAG binds to GST A1, the major GST isoform responsible for Bu conjugation; per contra, Scian et al [23] demonstrated that EdAG does not bind to the GSH binding site on GST A1, but does irreversibly bind to a cysteine residue in the GSH binding pocket of glutaredoxin, resulting in a catalytically inactive protein [23].
Following exposure to C6 rat glioma cells, EdAG was modestly cytotoxic, exhibiting almost 2-fold less toxicity than Bu. LD50 values for EdAG and Bu were 880 μM and 460 μM, respectively [24]. In addition, Peer et al. [22] found that EdAG has free-radical scavenging properties in vitro, with an apparent second order rate of hydroxyl radical trapping of 8.4 × 109 M−1s−1 [22]. Interestingly, this apparent rate exceeds the rates for many other antioxidants, including resveratrol (9.45 × 108 M−1s−1) [80] and chlorophyllin (2.7 × 106 M−1s−1) [81], both of which have reported anti-cancer activity and other health benefits.
In vitro metabolism of EdAG, studied within simulated physiological aqueous conditions, includes a Michael addition reaction with GSH to produce 2-amino-5-[[3-[2-[[4-amino-5-hydroxy-5-oxopentanoyl]amino]-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-1-(carboxymethylamino)-1-oxopropan-2-yl]amino]-5-oxopentanoic acid (GSG) [24]. GSG is also putatively formed following the nucleophilic attack of GSH on the Bu-SG conjugate [78]. Additional metabolism of GSG likely proceeds through the enzymatic catalyzed mercapturic acid pathway, but this remains unconfirmed at this time.
3.6 Busulfan effects on CYP enzymes
Studies are few regarding Bu’s effects on the functionality and expression of hepatic CYP protein content. Bu did not inhibit CYP3A4 mediated oxidation of denitronifedipine in human liver microsomes (n=2 donors) at three tested concentrations (2.03 μM, 20.3 μM and 203 μM) [82]. A single dose of Bu 50 mg/kg, dissolved in corn oil and administered by oral gavage, to mice resulted in no changes in hepatic CYP protein content and not alterations in CYP functionality (as measured by NDMA-demethylase I and II and arylhydrocarbon hydroxylase activity) [83]. However the same dose administered once daily for 3 days resulted in an approximate 50% and 40% statistically significant increase in NDMA-demethylase-I and -II activity, respectively [83]. The 3 day dosing cycle had no effects on arylhydrocarbon hydroxylase activity, and exhibited no changes in hepatic CYP protein content [83]. Compared with Bu doses commonly prescribed in the HSCT clinic, the Bu dose (50 mg/kg) that increased enzyme activity in mice liver would approximate the upper-range of Bu regimens [84], so these findings are intriguing.
Demethylation of N-nitrosodimethylamine (NDMA) is an enzyme activity largely associated with CYP2A6 and CYP2E1 isoenzymes in human liver [85]. This suggests, although unconfirmed in humans, that Bu may accelerate the metabolism (and clearance) of drugs metabolized by these enzymes (e.g. acetaminophen, ethanol, theophylline, nicotine, metronidazole, ifosfamide, cyclophosphamide) – leading to unwanted changes in victim drug blood concentrations. This further highlights the need of TDM of these medications when co-administered with Bu-based regimens.
4. Pharmacogenomics of busulfan PK
An understanding of a drug’s link between its PK and pharmacogenomics provides indirect evidence towards its important, clinically relevant drug metabolism pathways. Numerous investigators have studied the effects of polymorphisms in drug disposition genes versus Bu PK (mostly clearance), and in some cases the effects on drug toxicity (e.g. VOD, graft versus host disease) and treatment outcomes (EFS and OS) [86–101]. Most of these studies have investigated PK correlations with GST isoenzyme genetic variants, while fewer studies have reported the influence of SNPs for CYP, other drug metabolizing enzymes, and drug transporter genes.
4.1 GST polymorphisms
Parent Bu is predominantly metabolized by GST isoenzymes in human liver cytosol [38,54], and GST generated metabolites are found in Bu-treated animals and humans [36,37,52]. Hence, it would be expected that polymorphisms in the GST isoenzymes would have a significant impact on Bu metabolism, thereby influencing its PK and potentially toxicities and treatment outcomes. Polymorphisms have been found in the GST isoforms responsible for Bu metabolism [102]. Numerous studies have investigated the potential link between GST polymorphisms and Bu clearance (and other PK parameters), a topic that has been extensively reviewed elsewhere [102]. It is the purpose of the following sections to briefly summarize studies that have contributed to the overall bulk of knowledge regarding pharmacogenomics of Bu therapy.
4.1.1 GSTA1 polymorphisms
GSTA1 is the predominant GST isoenzyme that catalyzes GSH conjugation of Bu [54]. Allelic variants of GSTA1 are known to cause altered GSTA1 activity and hepatic protein expression [102]. Correlative studies comparing GSTA1 polymorphisms vs. Bu PK have yielded inconsistent results. In most of the studies, as expected, patients with GST A1*A/*A genotype had higher Bu clearance (with consequent lower AUC), while patients with the GST*B/*B genotype had lower clearance (with consequent higher AUC). Changes in Bu clearance ranged, on average, from about ± 14% to 35%. The effects of heterozygous genotypes are conflicting, as Ansari et al. [88] observed a gene dose effect, while other studies found that the *A/*B genotype was associated with either higher [86,97] or lower [90,91,95] Bu clearance.
Although the majority of studies found an association between GSTA1 polymorphisms and Bu clearance, it is noteworthy that several studies found inferior (to no significant) clinical correlations. For example, Abbasi et al. [86] found that GSTA1 haplotypes were associated with oral, but not IV, Bu clearance. In children undergoing HSCT, GSTA1 polymorphisms did not influence population PK parameters of IV Bu [101]. Children with thalassemia expressing variants in GSTA1*B had a 10% lower IV Bu clearance compared with wild-type, although the clinical significance of this finding is probably limited at best [103]. Ansari et al. [87] reported that children undergoing HSCT exhibited no correlation between systemic IV Bu exposure and GSTA1 variants [87].
Similar to the GSTA1*A1 haplotype, pediatric patients undergoing HSCT expressing the GSTA1*A2 haplotype had significantly higher IV Bu clearance and lower Bu plasma levels, which was associated with HSCT treatment outcomes [88]. Moreover, children with the GSTA1*B and GSTA1*B1 haplotypes had a higher occurrence of VOD [88]. Bonifazi et al. [89] found that adult patients with the serine allele of the GSTA2 S112T (GSTA2*C) polymorphism exhibited lower IV Bu clearance and higher plasma levels compared with threonine carriers, and this polymorphism was also correlated with inferior survival and higher day 100 transplant-related mortality (TRM), as well as bilirubin levels. This latter finding is, however, is somewhat surprising since the GSTA2*C variant appears to confer normal enzyme activity in humans [104].
In pediatric patients with congenital hemoglobinopathies preconditioned with oral Bu prior to HSCT, the GSTA1 genotype was associated with a lower mean Cmax of Bu [90]. Furthermore, children with the GSTA1*B variant had a significantly higher IV Bu systemic exposure and Css compared with non-carriers [93], and adult patients with a GSTA1 variant showed a significantly lower Bu clearance [95]. Finally, ten Brink et al. [99,100] found that both adult and pediatric patients with the GSTA1*B variant had a significant reduction in IV Bu clearance and increase in Bu exposure; and adults with the GSTA5 variant had a significant reduction in IV Bu clearance [98].
Some studies have examined the effects of GSTA1 genotypes on Bu drug toxicities. Kim et al. [94] reported that the GSTA1*A/*A variant was an independent protective factor against graft vs host disease (GVHD), but other studies did not replicate these findings [88,90,100]. Ansari et al. [88] found that pediatric patients with the GSTA1*B/*B genotype incurred a higher risk of developing VOD, and this effect was more apparent in girls than in boys. However, other studies found no association between GSTA1 haplotypes and VOD in pediatric or adult patients [92,93,96,101].
4.2 GSTM1 polymorphisms
GSTM1 contributes to GSH conjugation of Bu at approximately 46% activity of the major facilitator GSTA1 in human liver cytosol [54]. Homozygous deletions of the GSTM1 genes (null genotypes) are associated with reduced clearance capacity [102]. The associated effects of Bu PK and polymorphisms in GSTM1 genotypes has been studied, with conflicting results between different investigators [86–88,90,92,95,96,98,100,101]. Although Abbasi et al. [86] found that oral Bu clearance was influenced by GSTA1 genotypes, there was no significant association between oral or IV Bu clearance and GSTM1 phenotypes. No significant association between GSTM1 polymorphisms and liver toxicity was found in patients receiving Bu based conditioning regimen prior to allogeneic HSCT [92]. Furthermore, the inter-individual variability of Bu PK in adults and children was not influenced by GSTM1 genotypes [98,100,101].
On the contrary, GSTM1-null children had a statistically significant lower Bu clearance and greater incidence of graft-versus-host disease [88]. The same group reported that children with GSTM1 polymorphisms had a higher Bu systemic exposure with significantly lower clearance [87]. Elhasid et al. [90] also found a lower oral Bu exposure in children with congenital hemoglobinopathies undergoing HSCT containing GSTM1-null genotype, compared to GSTM1-positive patients – which was associated with GVHD. Interestingly, beta-thalassemia patients with the GSTM1-null gene are predisposed to VOD, in which these patients had a higher Bu clearance, rather than lower Bu clearance, leading to a lower first-dose plasma concentrations [51]. These results may suggest a role of Bu metabolites, rather than parent drug, in treatment-related drug toxicity [51].
Several studies suggest that combined polymorphisms including GSTM1 genotypes are predictors of Bu clearance [95,96]. For instance, a combination of polymorphisms in ABCB1, a drug transporter that plays a role in multidrug resistance and drug interactions, and GSTM1 were associated with Bu clearance and drug exposure [96]. Further, we have published data demonstrating how corollary administration of HDAC-inhibitors influence cellular expression of both MDR1 and MRP1, both of which are important for metabolic handling of various cytotoxic agents [105].
4.3 GSTP1 polymorphisms
GSTP1 is less active than GSTA1 in catalyzing BU-GSH conjugation in human liver cytosol, showing approximately 18% activity of GSTA1 [54]. The GSTP1 gene has several variants that confer decreased enzyme activity [102]. The effects of polymorphisms in GSTP1 genotypes has been studied with inconclusive data [87,88,90,92,100,101]. GSTP1 polymorphisms influenced the PK of oral Bu in children with hemoglobinopathies undergoing HSCT [90], on the other hand no significant association was observed between GSTP1 polymorphisms and Bu PK [87,88,100,101]. Similar to GSTA1, GSTM1 and GSTT1, no significant associations between GSTP1 polymorphisms and liver toxicity were observed [92].
4.4 GSTT1 polymorphisms
Theoretically, GSTT1 may also facilitate Bu conjugation, but it has not been directly proven. Null genotypes in the GSTT1 gene are associated with defective enzyme activity [102]. However, pharmacogenomic studies have shown that genetic variations in GSTT1 are not associated with Bu clearance or liver toxicity [51,91,92,95].
4.5 Polymorphisms in CYPs and other drug disposition genes
The precise role of CYPs in the metabolism of Bu and its intermediates is not known at this time, but indirect evidence for their role has arisen from pharmacogenomic studies. For example, Uppugunduri et al. [106] demonstrated that children with allelic variants for the reduced-activity CYP2C9 gene exhibited a higher metabolic ratio of Bu:sulfolane compared to those with the high-activity allele. Furthermore, children with a higher metabolic ratio had a greater incidence of graft failure and therefore a lower event free survival [106]. However, there was no correlation between CYP2C9 genotypes and first dose BU clearance or treatment outcomes [106]. This suggests that CYP2C9 may not directly influence parent Bu disposition, but may play a role in downstream oxidation of intermediary metabolites that eventually culminate in the formation of sulfolane. The authors also found a correlation in children receiving cyclophosphamide (Cy) pre-conditioning between reduced event free survival and the reduced activity allele of CYP2B6 [106]. CYP2B6, as well as various other CYPs, mediate the bio-activation of Cy [107], but it is unknown towards its role in Bu metabolism. As pointed out by Hassan and Andersson [16], however, there may be indirect untoward effects created by CYP-interactions when Bu and Cy are combined in the commonly used 2- and 4-day variant Bu-Cy regimens.
The influence of polymorphisms in drug disposition genes on Bu clearance was explored in adult HSCT patients genotyped with a drug metabolizing enzyme and drug transporter chip array [98]. The author’s found seven markers in GSTA1, CYP2C19, CYP39A1, ABCB4, SLC22A4 and SLC7A8 that were associated with Bu clearance [98]. This same research group found that only genetic variants in GSTA1 and CYP39A1 were associated with Bu clearance in pediatric patients, and when combined they explained about 20% of variability in Bu clearance [99]. CYP39A1 is one of many CYP enzymes involved in bile acid synthesis from cholesterol [108]. Variants in CYP39A1 have shown a correlation with the incidence of certain adverse toxicities in patients treated with docetaxel [109]. At present further study is required to determine the functional role of CYP39A1 in Bu metabolism and/or treatment-related toxicities and outcomes.
Polymorphisms in the ABCB1 gene can influence Bu clearance [96]. Patients with variant alleles in ABCB1 had a significantly higher Bu clearance [96]. Also, patients with a combined GSTM1-null genotype and variant ABCB1 allele had a higher Bu clearance and lower AUC compared with carriers of the ABCB1 ancestral alleles [96]. Patients with a combined GSTM1-positive genotype and variant ABCB1 allele exhibited a reverse pattern, in which these individuals had a lower Bu clearance and higher AUC [96]. Ten Brink et al. [100] studied the correlation between SNPs in ABCB1 (although different SNPs from the Krivoy et al. study) and Bu clearance, but found no significant association [98]. They did find that ABCB4 variants were a marker of Bu clearance in their exploratory cohort [100], but not associated in the validation cohort group in the same study [98] and a future study [99]. Interestingly, patients (c.a. 65% received a Bu pre-conditioning regimen) carrying a SNP in the MDR1 gene receiving an allogeneic HLA-identical HSCT had worse overall survival and higher non-relapse mortality, but unfortunately Bu PK was not characterized in this study [110]. Unfortunately, the study results are further muddles by the fact that 65% (53/82) patients in that study received Bu-Cy variant regimens, and MDR1 is a major transmembrane transport protein involved with activated Cy [53]
5. Busulfan Drug-Drug Interactions
In accordance with its still unsettled drug metabolism pathways, Bu DDI studies have yielded contrasting results. A summary of the known DDI studies with Bu is included in Table 1, and the specific interacting characteristics of the possible perpetrating drug(s) is presented in Table 2. Specific information regarding these DDI studies is described in the text below.
Table 2.
Drug Interaction Characteristics of Co-administered Agents with Busulfan
| Co-administered Drug | Observed PK Interaction with Bu | Substrate | Inhibition Effect(s) | Induction/Activation Effect(s) | Other | References |
|---|---|---|---|---|---|---|
| Acetaminophen | N | CYP2E1; CYP3A4; CYP1A2; UGT1A1; UGT1A6; SULT1A1; SULT1A3/4; SULT1E1 | [135] | |||
| Acetaminophen metabolite – NAPQI | n.a. | GSTs | GS | Spontaneous GSH conjugation | [136,181] | |
| Ciprofloxacin | N | CYP1A2; BCRP; OAT; OCT | CYP1A2; CYP3A4 | [129,182,183] | ||
| Clonazepam | N | CYP3A4 | UGTd | UGTe | [144,145,184] | |
| Deferasiroxa | Y | UGT1A1; UGT1A3; CYP | CYP3A4; CYP2C8; CYP1A2; CYP2A6; CYP2D6; CYP2C19 | CYP3A4 | [169] | |
| Diazepam | N | CYP2B6; CYP2C19; CYP3A4 | CYP2C19; CYP2D6; CYP3A4 | [148,149] | ||
| Ethacrynic Acid | Y | GSTs | GSTs | Spontaneous GSH conjugation | [71] | |
| Flor-Essence (multiple constituents)b | Y | Likely | CYP1A2; CYP2C19; CYP2C9; CYP2D6; CYP3A4; CYP19; OAT1; OAT3; P-gp and others | GST | [171,185,186] | |
| Fluconazole | N | MDR1 (P-gp) | CYP3A4 CYP2C9 CYP2C19 UGT |
[114] | ||
| Fludarabine phosphatec | Y | 5′-nucleotidase; deoxycytidine kinase | [156] | |||
| Itraconazole | Y/N | CYP3A4; P-gP | BCRP; BSEP; CYP3A4; P-gp | [113,114,116] | ||
| Itraconazole metabolite hydroxy-itraconazole | n.a. | CYP3A4 | BCRP; BSEP; MATE1; CYP3A4; OATP1B1; OATP1B3; OAT3; OCT1; P-gp | [115,116] | ||
| Itraconazole metabolite keto-itraconazole | n.a. | CYP3A4 | BCRP; BSEP; CYP3A4; MATE1; OATP1B1; OATP1B3; OCT1; P-gp | [115,116] | ||
| Itraconazole metabolite N-desalkyl-itraconazole | n.a. | unknown | CYP3A4; P-gp | [115,116] | ||
| Ketobemidone | Y | CYP2C9; CYP3A4 | n.a. | n.a. | [133] | |
| Lorazepam | N | UGT1A7; UGT1A10; UGT2B4; UGT2B7; UGT2B15; | UGT | n.a. | [146,147] | |
| Metronidazole | Y | ADH; CYP2A6; CYP3A4; glucuronidation; sulfation | CYP2C9; CYP3A4 | n.a. | [121,125,187] | |
| N-acetyl-L-cysteine | N | Acylase I | n.a. | n.a. | Increases GSH synthesis | [137,188] |
| Phenytoin | Y | CYP2C9; CYP2C19; MRP2 | CYP2C9 | CYP2B6; CYP3A4; CYP2C9; C YP2C19 | [41,139,189] |
Abbreviations: ABC: ATP-binding cassette transporter; ADH: alcohol dehydrogenase; BSEP: bile salt export pump; CES: Carboxylesterase; CYP: Cytochrome P450; P-gp: P-glycoprotein; BCRP: Breast cancer resistance protein; FMO: Flavin-containing monooxygenase; GS: Glutathione synthetase; GSH: Glutathione; GST: Glutathione S-transferase; MATE: Multidrug and toxin extrusion protein; MRP: Multidrug resistance-associated protein; MDR: Multi-drug resistant protein; N: No; N.A.: Not applicable; NAPQI: N-acetyl-p-benzoquinone imine; OAT: Organic anion transporter; OATP: Organic anion transporting polypeptide; OCT: Organic cation transporter; SULT: Sulfotransferase; UGT: Uridine 5′diphospho-glucuronosyltransferase; Y: Yes
CYP isoform has not been identified; pathway only accounts for 8% of total intact drug metabolism [169].
Flor-essence is a CAAM product with many active compounds, which have been shown to individually modify drug metabolizing enzymes and transporters within in vitro and in vivo studies (see text for further explanation).
Fludarabine phosphate is a pro-drug that is rapidly de-phosphorylated by 5′-nucleotidase to 2F-ara-A (Flu). 2F-ara-A is phosphorylated intracellularly by deoxycytidine kinase to (2F-ara-ATP) [156].
Clonazepam inhibited glucuronidation of morphine in rat liver microsomes [145].
Clonazepam increased the glucuronidation of SN-38 (irinotecan metabolite) in human liver microsomes [144].
5.1 Pharmacokinetic Interaction Studies with Antifungals
Fungal infections incur a significant risk for morbidity and mortality in patients with hematological malignancies and recipients of HSCT [111]. Prophylaxis with triazole antifungals is a common treatment modality in these patients, despite imposing a significant risk of drug-drug interactions [111]. The work of Buggia et al. [112] demonstrated an interaction between oral Bu and itraconazole, but not fluconazole. Itraconazole or fluconazole were administered orally as a single daily dose of 6 mg/kg starting the day before initiation of the 4-day oral Bu conditioning regimen (1 mg/kg every 6 hours for 4 days). Compared with control patients (n=26), itraconazole treated patients (n=13) showed an average decrease in Bu clearance by 20% (p<0.5). Furthermore, itraconazole significantly increased Bu AUC and Css by 22% and 25%, respectively. There was no differences in Bu PK between fluconazole-treated patients (n=13) and control patients.
Itraconazole has a high potential for drug interactions, as it inhibits CYP3A4, P-glycoprotein (P-gp), breast cancer resistance protein (BCRP) and bile salt export pump (BSEP) (Table 2) [113,114]. Itraconazole is also a substrate for P-gp and highly metabolized by CYP3A4, producing hydroxyl-itraconazole, keto-itraconazole and N-desalkyl-itraconazole [115]. These itraconazole metabolites are equally or more potent CYP3A4 inhibitors compared to parent itraconazole, and also significantly inhibit numerous drug transporters (Table 2) [114–116]. Fluconazole, on the other hand, is cleared primarily by renal excretion and is a much less potent in vitro inhibitor of CYPs than itraconazole [114]. Fluconazole is a substrate but not inhibitor of P-gp, and inhibits CYP3A4, CYP2C9, 2C19 and uridine diphosphate glucuronyltransferases (UGTs). Buggia et al. [112] speculated that the mechanism behind the DDI between itraconazole and oral Bu is probably mediated by inhibition of CYP and/or inhibition of 5-lipoxygense (5-LO) enzymes. Interestingly itraconazole, but not fluconazole, is an inhibitor of 5-LO [117], an enzyme that catalyzes the formation of leukotrienes from arachidonic acid, and is involved in inflammatory processes [118]; but its role in DDIs is poorly known. Since oral Bu was administered in the Buggia et al. [112] study there is also a possibility of an interaction mediated by P-gp inhibition in the gastrointestinal tract by itraconazole, although this alternative mechanism was not addressed in the article. It is not fully known if Bu or any of its metabolites are substrates for P-gp or other transporter proteins located in the intestinal tract, liver, or other bodily tissues. More recent in vitro data suggest that while activated Cy and ifosfamide are excellent substrates for MDR1, GSH-conjugated Bu appears to be preferentially transported by MRP1.
Other studies have shown no significant interaction between Bu and antifungals [119,120]. For instance, in children, antifungal prophylaxis with itraconazole did not influence IV Bu (1.0 mg/kg or 0.8 mg/kg daily × 4 days) PK [120]. Also, children (n=13) co-administered IV Bu (0.8 mg/kg every 6 hours for 16 total doses) and fluconazole displayed no significant changes in total bodily clearance of Bu [119]. In the Madden et al. study [32], itraconazole or voriconazole (n=19 collectively) administered prior to IV Bu (130 mg/m2 infusion over 3 hours daily × 4 days) did not significantly alter the PK of Bu. The criteria for use of antifungals in this study included patients treated with voriconazole or itraconazole > 1 week, but discontinued drug at least 1 week prior to Bu based conditioning regimen [32]. Therefore, it is plausible that only sub-inhibitory concentrations (< IC50) of these drugs (and metabolites) were residing in the system at the time of Bu administration. Their findings did not confirm a drug interaction, but if an adverse drug interaction does indeed exist, the author’s results indicate that a “wash-out period” of at least one week is likely sufficient to avoid such interaction between these azole(s) and Bu.
5.2 Pharmacokinetic Interaction Studies with Antibiotics
Metronidazole is an imidazole antibiotic used to treat Helicobacter pylori, protozoal and anaerobic infections, and is commonly considered front-line therapy for Clostridium difficile related intestinal infections in HSCT patients [121]. Three separate clinical studies have documented a drug interaction between metronidazole and Bu (Table 1) [121–123]. In a case report, metronidazole 750 mg/day was administered for presumed C. difficile infection 1 day prior to IV Bu conditioning regimen (0.5 mg/kg followed by PK-guided IV dosing) to a 7 year-old child [121]. The first therapeutic dose PK of IV Bu demonstrated a pronounced reduction (approximately 46%) in clearance of Bu (as compared with the test dose clearance measured 24 hrs prior) [121]. This reduction in Bu clearance resulted in an increase of the daily predicted AUC and total course AUC of Bu by 86% [121]. Once the drug interaction was recognized, metronidazole was discontinued. The patient did not develop VOD but suffered recurrent AML about 3-months post-transplant [121]. The use of TDM in this patient case allowed the investigators to omit Bu after two of the planned three days regimen of IV Bu, and the total course AUC/systemic exposure exceeded the planned course AUC by only 24%, thereby protecting the patient from developing VOD.
In a case report, a child receiving once daily Bu (120 mg/m2 IV followed by PK-guided IV dosing) was co-administered a single IV dose of metronidazole 100 mg five hours prior to the 3rd dose of busulfan [122]. The patient further received 3 doses of metronidazole every 6 hours (total duration of metronidazole therapy was 23 hours) [122]. The estimated Bu clearance following the 3rd dose was reduced by 43% (compared to the calculated clearance after the 2nd dose). Due to the suspected DDI, metronidazole was discontinued prior to the 4th Bu dose. The Bu clearance on the 4th dose was further reduced by 20% compared to the 3rd Bu dose (or 55% compared to the 2nd Bu dose) [122]
Nilsson et al. [123] specifically studied the effects of metronidazole prophylaxis on Bu PK in adult HSCT patients. In five patients, the administration of concomitant oral Bu (1 mg/kg every 6 hrs × 4 days) plus metronidazole (400 mg × 3 p.o.) was compared with Bu alone. Patients receiving Bu and metronidazole had a statistically significant elevation in mean Bu Css trough concentrations (948 ng/ml vs. 507 ng/ml). Another group of patients (n=9) received metronidazole following 2 days of oral Bu, which resulted in a significant increase in mean Bu Css trough concentrations (807 ng/ml vs. 452 ng/ml) [123]. Furthermore, elevations in Bu concentrations were correlated with a greater incidence of hepatic toxicity in this study, imposing a greater risk of VOD when metronidazole and Bu were combined [123].
In a covariate analysis, the PK of oral Bu (1 mg/kg every 6 hrs × 4 days) was not associated with the co-administration of metronidazole [124] However, in this study the major focus was to study the influence of fludarabine on Bu PK, so the details of this sub-study were not clear [124].
Metronidazole is metabolized in the liver to 2-hydroxymetronidazole by CYP2A6, and to a lesser extent by CYP3A4, CYP3A5 and CYP3A7 (Table 2) [125]. Metronidazole is an inhibitor of CYP2C9, and possibly inhibits CYP3A4 [121] but not P-gp in humans [126]. Metronidazole’s modulation of GSH levels is not completely studied, but hypothetically metronidazole may decrease Bu clearance via a GSH mechanism − since the reduced nitroso-derivative of metronidazole presumably binds with GSH in rat and human tissues [127]. Taken together, the high propensity for metronidazole induced DDIs when co-administered with Bu highlights an increased importance of TDM when the 2 agents are co-administered.
Fluoroquinolone antibiotics (e.g. levofloxacin, ciprofloxacin) are commonly included within prophylactic regimens prior to HSCT. Styler et al. [128] found that prophylaxis with ciprofloxacin (CYP1A2 inhibitor [129]) was a significant risk factor for VOD in Bu patients, but Bu PK was not studied. Although not a major focus of their study, Bensinger et al. [130] evaluated the effect of the combination of pentoxifylline and ciprofloxacin on the PK of oral Bu (3.75–5.25 mg/kg/day) in breast cancer patients (n=11) undergoing autologous transplant [130]. A baseline Bu clearance for statistical comparisons was calculated following an oral test dose (0.25 mg/kg) in the absence of pentoxifylline and ciprofloxacin. During the therapeutic conditioning regimen the presence of pentoxifylline and ciprofloxacin did not significantly alter oral Bu clearance [130].
5.3 Pharmacokinetic Interaction Studies with Analgesics
HSCT patients prior to engraftment are commonly prescribed prescription and over-the-counter (OTC) analgesics for treatment of acute and chronic pain episodes. In a case report, a 33 year-old patient with rectal fissure was treated with the opioid agonist ketobemidone (through a self-manipulated pump) prior to initiation of an oral Bu conditioning regimen (1 mg/kg four times daily × 4 days) [131]. Bu AUC and trough levels were increased after the first, second and third Bu doses (Table 1) [131]. Further monitoring of ketobemidone plasma concentrations revealed a dose-dependent effect - higher ketobemidone levels were associated with higher Bu levels [131]. In this patient, substitution of ketobemidone with morphine resulted in a decrease in Bu steady-state trough concentrations [131]. Ketobemidone, which is mainly prescribed in Scandinavian countries, in humans undergoes several drug elimination pathways, including oxidation, methylation and glucuronidation pathways [132]. In human liver microsomes, ketobemidone is a substrate for CYP2C9 and CYP3A4, but not P-glycoprotein (Table 2) [133]. SNPs in CYP2D6 or CYP2C19 enzymes did not influence ketobemidone PK [134]. Otherwise, very little is known regarding clinical DDIs between ketobemidone and other therapeutic agents.
Acetaminophen, a popular OTC analgesic, is converted by CYP enzymes (mainly CYP2E1) to a toxic metabolite that can deplete GSH stores and is reactive with proteins in the liver [135]. Acetaminophen overdose, and at therapeutic doses, leads to measurable GSH depletion in animals and humans [135] Hypothetically, there is a strong likelihood of a DDI between acetaminophen and Bu that would involve perturbations in GSH levels and possibly GST activity. Many stem cell transplant centers avoid the co-use of acetaminophen during Bu conditioning regimen for preventive reasons. But the magnitude of this potential interaction is weak in clinical studies. In three published studies, patients (albeit small sample sizes) co-administered acetaminophen and Bu showed no abnormalities in Bu PK, however the extent of liver injury in these patients was not reported [32,119,120].
Acetaminophen undergoes extensive clearance in the body, including glucuronidation, sulfation and oxidation [135]. About 60% of drug is glucuronidated by UGT1A1 and UGT1A6, and about 30% of intact drug is sulfated by SULT1A1, SULT1A3/4 and SULT1E1 [135]. To a lesser extent (approximately 10%), acetaminophen is metabolized to a reactive, hepatotoxic metabolite N-acetyl-p-benzoquinone imine (NAPQI) by CYP enzymes (mainly CYP2E1) [135]. NAPQI is an electrophilic intermediate that reacts readily, both spontaneously and enzymatically, with nucleophilic sulfhydryl groups (including –SH groups on the GSH molecule) [136].
5.4 Pharmacokinetic Interaction Study with N-acetyl-L-cysteine (NAC)
N-acetyl-L-cysteine (NAC) is a precursor of GSH that repletes GSH stores, and is a key component in the mercapturate pathway that results in detoxification of highly reactive electrophilic compounds [137]. NAC is commonly prescribed to reduce hepatic toxicity following an acetaminophen overdose, and may have anecdotally reduced the incidence of Bu related VOD in a few high-risk patients [138]. In a single PK study, the effects of the administration of NAC (50 mg/kg twice daily) before and after oral Bu (initial dose 4 mg/kg/day twice daily followed by PK-guided dose adjustments) were studied in 10 adult patients [138]. The addition of NAC to the regimen was not reported to alter the AUC or myeloablative effects of Bu [138].
5.5 Pharmacokinetic Interaction Studies with Anticonvulsants
Bu rapidly crosses the blood-brain barrier and widely distributes into the CNS, achieving a concentration ratio between cerebrospinal fluid and plasma equal to or greater than 1 [34]. Therefore, high-dose Bu therapy is associated with neurological disturbances such as seizures, necessitating routine prophylaxis with anticonvulsants such as phenytoin. Phenytoin is metabolized by CYP2C9 and CYP2C19, inhibits CYP2C9 and induces CYP2B6, CYP3A4, CYP2C9, and CYP2C19 isoenzymes [41] (Table 2). Phenytoin is a substrate for multidrug resistance-associated proteins (MRPs) in the blood-brain barrier [139]. To our best knowledge phenytoin is not known to induce (or inhibit) GST activity in humans.
Since anticonvulsants (e.g. phenytoin, phenobarbital) are known inducers of hepatic drug metabolizing enzymes, potential drug interactions between Bu and anticonvulsants have been studied in humans [32,119,140–143]. The PK of oral Bu (1 mg/kg qid × 4 days) was studied in adult patients treated with phenytoin (n=9) or diazepam (n=8) as prophylactic anticonvulsant therapy [143]. Phenytoin treated patients (loading dose of 5 mg/kg/day × 4 days prior to high-dose Bu, followed by 2.5 mg/kg/day × 4 days during Bu therapy) demonstrated between the first and last dose a statistically significant elevation in Bu clearance (by 19%), lower AUC (by 20%), and reduced T1/2 (by 30%) [143].
Madden et al. [32] studied the influence of phenytoin on Bu PK in an unconventional, but clinically significant, manner. Firstly, Bu clearance was compared between a subset of patients (n=22) receiving phenytoin prophylaxis within 1 hr prior to the first therapeutic dose Bu infusion versus patients (n=38) who started phenytoin the evening prior to Bu infusion (≥ 12 hr). Secondly, the estimates of Bu clearance and AUC were compared between the first dose and final Bu dose. The use of phenytoin did not alter any PK parameters of Bu in this study [32].
Benzodiazepines are an alternative therapeutic option to replace phenytoin for seizure prophylaxis in HSCT patients receiving high-dose Bu. The influence of several benzodiazepines (clonazepam, lorazepam and diazepam) on Bu PK has been reported [140,141,143]. Compared to phenytoin treated patients, clonazepam (0.025–0.03 mg/kg/day as a continuous 12 hour IV infusion administered at least 12 hours prior to and 24 hours after last dose IV Bu) treated patients exhibited a 10% increase in IV Bu clearance [140]. In human liver microsomes, clonazepam was shown to increase the UGT-mediated conversion of SN-38 (irinotecan metabolite) to SN-38-glucuronide [144]. On the contrary, clonazepam inhibited the glucuronidation of morphine in rat liver microsomes [145], so overall its effects on the UGT enzyme system has still to be determined. Although lorazepam is an inhibitor of UGT activity [146] and substrate for several UGT1A and UGT1B isoenzymes [147], prophylactic use of IV or oral lorazepam (0.02 to 0.05 mg/kg i.v. or p.o., up to a maximum of 2 mg, every 6 h) with IV or oral high-dose Bu in pediatric patients undergoing HSCT did not result in any aberrant changes in Bu PK [141]. Finally, Bu PK in patients treated with diazepam (5 mg/day orally × 4 days) was unchanged [143]. In the context of drug interactions, diazepam is a substrate and inhibitor of multiple CYP isoenzymes [148,149].
5.6 Pharmacokinetic Interaction Studies with Fludarabine
Combinations of nucleoside analogs, such as fludarabine (Flu), and Bu are rapidly becoming part of standard conditioning therapy for acute myeloid leukemia/myelodysplastic syndrome (AML/MDS) patients undergoing HSCT [150,151]. DDI studies between Flu and Bu have been reported, with opposing findings [124,150,152–155] (Table 1).
Table 1.
Summary of Pharmacokinetic Drug-Drug Interaction Studies with Busulfan.
| Co-Administered Drug | Drug Class | PK Interaction | References |
|---|---|---|---|
| OBSERVED INTERACTIONS WITH BU | |||
| Deferasirox | Iron Chelating Agent | Increase in Bu AUC | [168] |
| Ethacrynic Acid | Diuretic | Inhibition of the rate of Bu-GSH conjugation | [38,52] |
| Flor-Essence | CAAM product | Decrease in Bu CL | [170] |
| Fludarabine | Chemotherapeutic (nucleoside analog) | Decrease in Bu CL Increase in Bu AUC Increase in Bu Css Increase in Bu Cmax |
[124,154,155] |
| Itraconazole | Anti-fungal | Decrease in Bu CL Increase in Bu AUC Increase in Bu Css |
[112] |
| Ketobemidone | Opioid Analgesic | Increase in Bu Ctrough | [131] |
| Metronidazole | Antibiotic | Decrease in Bu CL Increase in Bu AUC Increase in Bu Ctrough |
[121–123] |
| Phenytoin | Anti-convulsant | Increase in Bu CL Decrease in Bu AUC Decrease in Bu T1/2 |
[47,143] |
| NO PK INTERACTIONS WITH BU | |||
| Acetaminophen | Analgesic | None | [119,120] |
| Ciprofloxacin | Antibiotic | None | [130] |
| Clonazepam | Anti-convulsant | None | [140] |
| Diazepam | Anti-convulsant | None | [143] |
| Fluconazole | Anti-fungal | None | [112,119] |
| Fludarabine phosphate | Chemotherapeutic (nucleoside analog) | None | [150,152,153] |
| Itraconazole | Anti-fungal | None | [32,120] |
| Lorazepam | Anti-convulsant | None | [141,180] |
| Metronidazole | Antibiotic | None | [124] |
| N-acetyl-L-cysteine | GSH modifier | None | [138] |
| Oral contraceptives | Hormonal therapy | None | [32] |
Abbreviations: Bu: Busulfan; CAAM: Complementary and alternative medicine product; CL: Clearance; AUC: Area under the concentration-time curve; Css: Steady-state drug concentration; Ctrough: Trough drug concentration; GSH: Glutathione; T1/2: Elimination half-life
In patients who received IV Bu with or without Flu (or compared with IV Bu-Cy), no significant differences in Bu clearance, volume of distribution (Vd), Cmax/dose and T1/2 were observed between groups [150,152,153]. In contrast, some studies point toward a possible PK interaction between Bu and Flu [124,154,155]. De Castro et al. [124] compared the PK of oral Bu between patients (n=15) receiving a Bu-Cy (1 mg Bu/kg orally every 6 hrs × 4 days and 60 mg Cy/kg/day IV × 2 days after the end of Bu treatment) conditioning regimen, and patients (n=11) receiving Bu-Flu conditioning regimen (1 mg orally Bu/kg/day every 6 hrs × 4 days and 30 mg IV Flu/m2 × 5 days). On the fourth day of treatment, patients administered Bu-Flu had a higher Cmax, higher Css, higher AUC and lower clearance compared with patients receiving the Bu-Cy regimen [124]. Yet, the T1/2 when based on the Bu elimination phase only (approximately 2–6 hrs post-dose) appeared almost superimposable between treatment groups, suggesting that most, if not all, of the change in clearance was due to significant differences in intestinal absorption of Bu between the two cohorts [124].
Perkins et al. [154] studied the PK of IV Bu in adult patients concomitantly treated with IV Bu (initial dose of 170 mg/m2 to 220 mg/m2 followed by PK-guided dose adjustments) and IV Flu (40 mg/m2 × 4 days). The PK of Bu was evaluated following the first and fourth doses. The mean Bu clearance and T1/2 were significantly lower and higher, respectively, following the fourth dose compared to the first dose PK, -13% and 12%, respectively [154]. The author’s speculated that the observed alterations in clearance and T1/2 between doses one and four were possibly due to DDIs (e.g. concomitant medications that have unknown effects on Bu metabolism), changes in fluid status, and/or sample processing errors [154]. Similar to the Perkins study, Yeh et al. [155] found in patients (n=15) receiving IV Bu concomitantly with Flu a progressive decrease in Bu clearance, by on average 12%, from the first to the third daily doses, and a concomitant increase in systemic exposure between the first and third doses. These author’s suggested that clinicians using Bu TDM should consider targeting the lower Css (or AUC) range when treating patients receiving Flu concomitantly with Bu [155].
The mechanism describing a potential DDI between Bu and Flu is perplexing since Bu and Flu are metabolized by distinctly different enzyme systems. Flu is a prodrug that is rapidly dephosphorylated to F-ara-A by 5ʹ-nucleotidase followed by active transport into cells by nucleoside transporters [156]. Intracellularly, it is phosphorylated by deoxycytidine kinase to F-ara-ATP, an active metabolite [156]. Bu or its intermediate metabolites are not known to be metabolized or transported by any of these Flu-associated enzymes and transporters. It is possible that co-monitoring of both Bu and Flu, in both adults and children, may mitigate the risk of interactions and propagate better outcomes, although this remains to be studied.
5.7 Pharmacokinetic interaction studies with cyclophosphamide
Cyclophosphamide (Cy) is a prodrug that undergoes a labyrinth of drug metabolic activation and inactivation pathways. The effects of sequential administration of Bu then Cy, and the reverse sequence regimen of Cy then Bu, on the PK of Cy has been studied in patients receiving HSCT [157–159]. Patients administered Cy ≤ 24 hours after the last IV Bu dose (control group patients received Cy ≥ 24 hours after Bu) had a significantly lower clearance of Cy, longer T1/2 of Cy and lower AUC of its metabolite 4-hydroxycyclophosphamide [157]. These patients had a significantly greater incidence of drug toxicities – VOD and mucositis [157]. The work of Rezvani et al. [159] demonstrated that patients receiving IV Cy followed by IV Bu, compared with patients receiving the Bu then Cy regimen, had a greater AUC of Cy, lower AUC of 4-hydroxycyclophosphamide and a trend towards lower exposure of the carboxyethyl-phosphoramide mustard metabolite, as well as a lower incidence of VOD and lower day +100 mortality [159].
McCune et al. [158] showed that patients treated with oral Bu followed by IV Cy (compared to a control group that received Cy and total body irradiation, but no Bu) had a lower AUC of Cy, higher AUC of 4-hydroxycyclophosphamide and higher AUC of the carboxyethylphosphoramide mustard metabolite [158]. In this study, there was no statistically significant association between the AUC of Cy (and metabolites) and VOD, non-relapse mortality, relapse or survival [159]. Finally, patients receiving high-dose conditioning Bu and Cy regimens prior to HSCT have a decreased risk of hepatotoxicity when Bu is administered after Cy [160,161], although there is a possibility in these studies that hepatic CYP-induction by supportive care medications (e.g. phenytoin) or other interacting concomitant medications might have aggravated Cy toxicity, thereby contributing to the observed liver toxicities.
Cy is a prodrug that is metabolized in the liver to 4-hydroxycyclophosphamide (which is in equilibrium with its ring-open tautomer aldophosphamide) by mainly CYP2B6, with smaller contributions from CYP2A6, 3A4, 3A5, 2C9, 2C18 and 2C19 [107]. 4-hydroxy-cyclophosphamide, not cytotoxic in itself, readily diffuses into cells then spontaneously decomposes to the bioactive metabolite phosphoramide mustard. Cy is also directly metabolized by CYP3A4 to 2-dechloroethylcyclophosphamide (inactive) and the toxicant chloroacetaldehyde [107]. Detoxification of Cy is effected, in part, by aldehyde dehydrogenase type I class (ALDH1) catalyzed oxidation of aldophosphamide to the nontoxic metabolite, carboxyphosphamide [162]. Other metabolism pathways of the Cy metabolites include detoxification by GSH conjugation and further chemical decomposition [107]. The decreased patient safety observed when Bu is administered prior to Cy, rather than after, may hypothetically be explained by Bu’s (and/or metabolites) propensity to deplete hepatic GSH stores, causing oxidative stress and possibly reduced GSH conjugation (and reduced detoxification) of reactive Cy metabolites [21].
5.8 Pharmacokinetic interaction studies with melphalan
The combination of Bu and melphalan (Mel) prior to stem cell transplantation has become the standard high-dose chemotherapy regimen in patients with high-risk neuroblastoma [163]. However, high rates of extrahematologic toxicities – VOD, interstitial pneumonitis and pulmonary hypertension – are observed in patients receiving this regimen [163,164]. Since both drugs are metabolized through the GSH/GST system [165], there is a strong potential for pharmacological interactions. Dourthe et al. [163] compared toxicities in patients with Ewing sarcoma or neuroblastoma receiving either the Bu-Mel or Mel-Bu regimen. Unlike in the Bu-Cy scenario, the order of administration of Bu and Mel failed to show any benefit in reducing toxicities [163], suggesting no clinically significant interaction, although PK levels were not reported by the authors. Bouligand et al. [166] evaluated liver toxicities in children/adolescents treated with either a high-dose regimen of Bu-Mel or Bu plus thiotepa (an alkylating agent that also undergoes GSH/GST metabolism). Patients treated with Bu-Mel regimen experienced greater VOD but in contrast to the Bu-Thiotepa cohort, toxicity was not correlated with steady-state Bu exposure or trough levels [166]. Bouligand et al. [164] recently found in pre-clinical studies (mice) that Bu increased the systemic plasma exposure of Mel, which was further exacerbated in iron-overloaded mice. A possible mechanism of interaction was hypothesized that involves oxidative stress and Nrf2 (nuclear factor erythroid 2–related factor 2) [164]. This intriguing body of work [163,164,166] suggests that co-TDM of Bu and Mel may prove beneficial in lessening the risk of extreme treatment-related toxicity associated with this regimen.
5.9 Pharmacokinetic Interaction Studies with Other Drugs
Deferasirox is beneficial to treat iron overload prior to HSCT [167]. In a case report, the co-administration of deferasirox likely reduced the clearance and increased the AUC and plasma levels of Bu [168]. Deferasirox is glucuronidated by UGT1A1 and UGT1A3 (Table 2), but is not known to undergo GSH conjugation [169]. CYP-mediated oxidative metabolism of deferasirox is a minor elimination pathway that accounts for only 8% of its total elimination [169]. But interestingly in vitro and in vivo study data suggest a potential for interactions with co-administered CYP substrates. The manufacturer’s package insert states that deferasirox inhibits human CYP3A4, CYP2C8, CYP1A2, CYP2A6, CYP2D6, and CYP2C19 in vitro [169]. In clinical studies, deferasirox was demonstrated as a substrate, inhibitor and inducer of various CYP enzymes [169].
Co-administration of the complementary and alternative medicine (CAAM) product, Flor-Essence (manufactured by Flora Inc., Lynden WA, USA) resulted in a 40% reduction in IV Bu clearance and 67% higher AUC in a single patient with multiple myeloma undergoing autologous HSCT [170]. Flor-Essence contains a blend of eight herbal compounds [170], so it is not surprising that the co-administration of Flor-Essence with Bu resulted in a PK drug interaction, since many of these compounds have wide and varying effects on drug metabolizing enzymes and transporters [171].
A multivariate analysis revealed that the co-use of Bu and oral contraceptives (estrogens/progestins) was significantly associated with VOD [128]. Based on this report, Madden et al. [32] studied the potential PK interaction between Bu and oral contraceptive use. Bu PK parameters in women (n=12) administered oral contraceptives (estrogens/progestins) did not differ from women (n=16) not taking oral contraceptive therapy [32]. This does not, however, exclude an adverse interaction since the estrogen/progestin, like other non-cytotoxic agents, may interact with either cellular signaling, resulting in an imbalance of pro- and anti-apoptotic signals, and/or change in DNA damage recognition/repair status. These cellular changes may only be manifested in a situation of intense stress, such as during high-dose chemotherapy.
6. Therapeutic drug monitoring
TDM is the clinical practice of measuring drug(s) at designated time intervals to support PK-based dose adjustments for individual patients, thereby maintaining consistent concentrations in the patient’s bloodstream and reducing regimen-related toxicities and improving treatment efficacy. Bu satisfies several pharmacologic preconditions for TDM, in particular (a) Variability in PK across patients; (b) Narrow therapeutic window since plasma concentrations and exposure have been linked to both efficacy and toxicity; (c) Susceptible to multiplicative DDIs due to the heavily pre-treated tendencies of HSCT patients; (d) Administered in high-dose regimens, often combined with other drugs with a narrow therapeutic window; and (e) Readily available drug assays (e.g. HPLC, LC/MS, GC/MS) to measure circulating plasma concentrations.
Bu TDM has been utilized in clinical HSCT practice for over 15 years with notable international success (e.g. improved outcomes, reduced incidence of VOD, development of safer target exposures) across various hematologic malignant sub-types, differing myeloablative conditioning regimens, different HSCT procedures, and diverse patient populations [11,172,173]. One of the earlier clinical studies to document the improved utility of TDM was published in 1993, reporting that adult and pediatric patients receiving fixed Bu doses had a greater incidence of VOD compared to patients receiving PK-based dose adjustments [173]. Since then multitudes of other studies have documented improved patient safety, and in many cases superior outcomes, using TDM data. For instance, the work of Andersson et al. [11] showed that Flu with PK-guided IV busulfan therapy is superior to fixed-dose delivery, resulting in safer disease control and improved overall survival (OS) and progression free survival (PFS) in patients with active AML and MDS. When compared to patients receiving untargeted oral busulfan doses, patients receiving TDM guided dose adjustments had higher event-free survival (EFS) and OS rates [172].
Pediatric patients, when compared with adult patients, show surpassing inter-individual variability in PK and are at a greater risk of toxicities and inferior outcomes [26,174]. Thus, TDM in this special population is particularly important to optimize dosing. Not surprisingly, a number of rigorously tested Bu population PK models, constructed using extensive TDM, have been developed in pediatric patients [175–177]. For instance, instead of previous empirical age-based dosing, Nguyen et al. [176] employed population PK modeling to devise a new strategy for optimizing Bu doses by weight categories, significantly reducing the inter-patient coefficient of variation in IV Bu clearance. The work of Paci et al. [177] developed a population PK model that showed body weight, rather than BSA or age, was the predominant covariate effecting Bu PK in children < 9 kg (< 20 lbs). Using PK data acquired from 5 international pediatric transplant centers, Bartelink et al. [175] validated their previously published population PK model [178]; results showed that the model predicts busulfan concentrations in pediatric and young adult patients ranging between 3 and 86 kg without bias and with good precision [175]. Bartelink et al. [12] published a newer model that shows improved clinical outcomes by targeting a range of Bu AUC from 78 to 101 mg × h/L, a range that is somewhat higher than the oft currently applied historical target of 58–86 mg × h/L [179]. With risks of multiple drug therapy elicited DDIs and greater variability of PK in pediatric patients, TDM improved models, such as these, will likely see greater implementation into mainstream pediatric SCT practice.
7. Conclusion
Bu, an intriguing small molecule drug that has been used in the oncology clinic for over 4–6 decades, continues to be one of the most frequently prescribed drugs in high-dose conditioning regimens prior to HSCT. In spite of decades of use and risks of debilitating and sometimes life threatening toxicities, many questions remain regarding its disposition. Bu PK is variable between patients, especially in pediatric populations, but despite current TDM strategies, significant problems attributed to the use of Bu in pretransplant conditioning therapy still remain. To date, the liver is recognized as the major anatomic site for Bu metabolism, generating over 10 known metabolites and numerous unidentified metabolites. Besides GST enzymatic catalysis, there is indirect evidence for the role of other enzymes in the metabolism of Bu and its metabolites, most likely isoforms from the CYP and FMO gene families and possibly cellular efflux drug transporters. Although unequivocal data exists, most pharmacogenetic studies point toward a clinical influence of GST SNPs on Bu clearance and treatment efficacy. Some pharmacogenetic studies have shown an association between CYP polymorphisms and Bu clearance, and one study showed an association between Bu clearance and combined SNPs in GST/ABCB1. The effects of concomitant medications on Bu PK has been studied in a variety of clinical settings, but have generated inconsistent results (Table 1), which can be attributed to many known and unknown variable properties of Bu including use in a heavily pre-treated patient population, many of which present prior to HSCT with significant co-morbidities. Established medications that interfere with Bu PK include metronidazole, phenytoin, deferasirox, ketobemidone, itraconazole and Flu; whereas other azole antifungals, over-the counter medications (e.g. APAP), as well as natural products, may influence Bu PK and subsequent toxicity, but require further clinical study. Clinical DDI studies between Bu and metronidazole show that metronidazole has the greatest effect on reducing Bu clearance, suggesting refined methods of TDM in this clinical situation, as well as with other interacting perpetrator drugs, may be necessary. In summary, further understanding of the pharmacological mechanisms underlying Bu metabolism and PK variability, as well as pharmacogenomics, is warranted to further improve Bu chemotherapy in HSCT patients. Newer TDM strategies will greatly assist clinicians in avoiding or mitigating potential DDIs with Bu in patients (both adults and children) predisposed to organ injury and/or those prescribed polychemotherapy, leading to further optimization of Bu dosing, ultimately aimed at improving HSCT outcomes.
7. Expert Opinion
Administration of oral Bu results in highly variable PK both between patients and from dose-to-dose in the same patient – which leads to a greater risk of VOD and other potentially fatal adverse effects. The introduction of an IV formulation several years ago has mitigated much of the Bu PK variability and has largely supplanted prescribing of the oral formulation in the HSCT clinic today. This reduced the inter-patient variability in systemic drug exposure from more than 10-fold after oral Bu to currently 2- to 2.5-fold in patients administered the IV Bu formulation. TDM services at select hospitals and cancer institutes have been implemented, but on a global scale the facilities and expenses required for applying these TDM services in a clinical setting present a sizeable barrier to universal acceptance. In the authors’ expert opinion, this should not be an excuse for using TDM given the significant impact TDM has on improving patient survival. Centralization of TDM, which may decrease expenditures associated with this service, may be an alternative in these special scenarios. A further understanding of the variables that alter Bu clearance has many benefits, and will certainly continue to aid in improving treatment outcomes, mitigating treatment-related toxicities, and stimulate the development of more accurate population PK tools that produce precise Bu first dose calculations and estimates of individual patient clearance. Population PK of busulfan therapy in adults has been understudied compared with pediatric patients. Application of promising new pediatric models, such as those described herein, into clinical practice has the potential to improve survival and reduce toxicities in this special population already predisposed to greater treatment outcome variabilities. Refining these models with other covariates (e.g. co-administered drugs) may further improve clearance predictions and survival.
In general, the PK variability of anti-cancer drugs can be attributed to a multitude of factors, including but not limited to: nutritional state, food, ethnicity, age, body weight, sex, concomitant disease states, genetics, drug metabolism and DDIs. The focus of this review article was to describe the current literature regarding Bu pharmacological properties, drug metabolic pathways, findings from clinical DDI studies, therapeutic drug monitoring and a brief overview of Bu pharmacogenomics. Since the late 1990’s, it has been known that Bu-GSH conjugation is mediated by GST enzymes, mainly GSTA1, in human liver cytosol, and metabolites formed as a result of this reaction have been consistently found in animals and humans – although it should be mentioned that unknown metabolites of Bu may still exist. In the context of classical DDI mechanisms, one would predict that when Bu is co-administered with a drug(s) that is (are) a GSH and/or GST modulator, Bu metabolism and Bu-mediated cytotoxicity would be affected. However, within the few clinical studies that have been conducted, albeit with limited patient sample sizes, neither APAP (GSH depletor) nor N-acetyl-L-cysteine (GSH repletor) altered Bu clearance. Since APAP is a cost-effective, widely available OTC painkiller in HSCT patients, and its use is oftentimes hard to monitor in outpatient clinical settings, a future PK study should be conducted to directly study the effects of single-dose and chronic administration of APAP on Bu PK. Pharmacodynamic outcomes are of additional interest since both drugs are known hepatotoxins. At this time without further information, it appears prudent to warn clinicians about the dangers, such as the risk for accidental synergistic hepatic stress, when co-prescribing Bu with APAP. To minimize risk, APAP use should be discouraged shortly before, during and for at least 48 hrs after Bu administration. Safer alternatives include the synthetic opioid tramadol, although it should be noted that the opioid ketobemidone alters Bu clearance, so it can be generalized that structurally similar opioids may interfere with Bu clearance. Bu therapy, especially when administered in high doses in the SCT setting, incurs an increased risk of generalized seizures, likely due to drug penetration into the blood-brain barrier, achieving CSF concentrations equal to or greater than in plasma. Historically, phenytoin has been used for seizure prophylaxis prior to Bu conditioning regimens and was, and still is, beneficial in this setting. But since phenytoin induces various CYP enzymes (and drug transporters) there are concerns for a DDI between phenytoin and Bu, and without careful TDM dose adjustments patients may receive an unoptimized dose. Furthermore, hepatic induction is also a serious concern when Bu-Cy regimens are prescribed since phenytoin since phenytoin induces CYP2B6 and CYP3A4 enzymes, both of which are key catalysts in cyclophosphamide activation (toxification). An alternative anticonvulsant levetiracetam, with less risks of DDIs and in some cases adverse effects, is increasingly being prescribed prior to Bu conditioning regimens. Our recent data comparing phenytoin with levetiracetam for seizure prophylaxis in a reduced intensity Bu containing regimen showed that patients in the levetiracetam arm had minor changes in IV Bu clearance between test and first therapeutic doses, whereas patients in the phenytoin arm displayed significantly higher induction of clearance.
The association of Bu clearance with genetic variants of GST enzymes has been widely studied among many investigators, and the data leans toward an association between GST polymorphisms and Bu clearance, and in some cases treatment outcomes and liver toxicity. Differences among the myriad studies may be explained by varying routes of Bu administration, different patient populations (e.g. adults vs. pediatrics), disease state, concomitant medications, study designs, Bu’s noted variability in PK, single or multiple drug(s) interactions, and the way clearance is calculated based on body size. Some studies with positive associations have used clearance normalized to body weight, whereas other studies with a negative association have used clearance normalized to body surface area. But among the supportive studies, there are conflicting data on which GST isoenzyme (e.g. GSTM1 vs. GSTA1) is the best predictor of Bu clearance. Moreover, the majority of pharmacogenetic studies have focused on pediatric patients, with a paucity of data from adults. The missing data and lack of correlation(s) between genotype(s) at the DNA level highlights another problem, namely the post-transcriptional modification of both qualitative and quantitative phenotypic expression of such gene messages. This suggests that rather than DNA-gene messaging, one may have to consider measuring the final protein message(s) to find correlates with clinical outcome variabilities such as toxicities and survival. Unfortunately, even such investigations will not account for the potential impact of concomitant medications. All in all, in spite of its lengthy study, the clinically relevant role of GST SNPs on Bu clearance remains to be determined. Definitive studies could lead to real-time personalized therapy of Bu, such as pre-screening patients for specific SNPs in GST isoenzymes, then tailoring the Bu dose individually. This a potential new and exciting approach to Bu chemotherapy, but without further convincing data from large patient populations, the practical use of GST genotyping is not foreseen until the distant future.
Many HSCT patients are prescribed a plethora of concomitant medications for adjuvant cancer treatment, preparative therapy, and acute and chronic medical conditions. Besides GSTs, clinically relevant enzymes mediating the metabolism of Bu and/or its intermediate metabolites are still not fully described. There is suggestive evidence, and in some cases pharmacogenetic data, supporting the role of CYPs, FMOs, and efflux transporters in the metabolism of the major Bu intermediate metabolites (e.g. Bu-SG, THT, THT 1-oxide, sulfolane). There is also the hypothetical possibility of other phase II transformations (e.g. glucuronidation, sulfation) of these metabolites.
Bu is glutathionylated to form the sulfonium of GSH (Bu-SG conjugate), which is a relatively unstable intermediate that primarily undergoes non-enzymatic decomposition to THT and EdAG. A possible reversible reaction (THT + GSH) to the Bu-SG conjugate is not chemically favorable. For THT and further downstream intermediate metabolites, the most postulated disposition fates include oxidation via CYPs and/or FMOs and phase II metabolism pathways. The possible role of CYPs is clinically concerning since these enzymes are known to metabolize a significant number of prescription medicines, and interferences of their activity have led to detrimental, sometimes life-threatening, DDIs. Possible CYP generated metabolites of THT are hydroxyl-THT and hydroxyl-THT-1-oxide. Also some clinically pertinent antifungals (i.e. voriconazole) used in the HSCT setting inhibit FMO enzyme activity, and this has been studied within a Bu research group; however pharmacogenomic studies, albeit few in number, have not revealed a significant link between allelic variants in FMO enzymes and Bu PK.
The clinical relevance of CYP-mediated (or other enzyme/transporter) inhibition of downstream Bu metabolite formation is uncertain. Enzyme inhibition in this scenario would lead to an increased exposure of the substrate metabolite or decreased exposure of the formed metabolite. There has been a suggestion for a feedback mechanism in Bu metabolism, in which the amount of circulating metabolite in tissue influences the remaining conjugation of parent Bu. This would be an unusual DDI mechanism, but certainly an interesting and worthwhile pursuit for future Bu research. Furthermore, if accumulation of metabolites occurs due to enzyme/transporter inhibition, there are potential toxicological consequences. Several studies, although smaller in number, have pointed toward the role of Bu metabolites, in addition to the parent compound, in Bu treatment-related toxicity.
The role of the Bu metabolite EdAG in Bu-mediated cellular toxicity is an emerging area of research since EdAG, a reactive electrophilic (Michael acceptor) species, has been shown in vitro to irreversibly bind to cysteine residues on a glutathione modulating protein (glutaredoxins), resulting in a catalytically inactive protein. This suggests that EdAG may dysregulate the glutathiome in HSCT patients receiving high-dose Bu therapy, a potential new mechanism for Bu-mediated cellular toxicity. Additionally, EdAG has free-radical scavenging properties, which may contribute to or mitigate Bu redox related cytotoxicity. Clinically this is impactful since the second order rate constant of hydroxyl trapping of EdAG exceeds several other antioxidants, including resveratrol and chlorophyllin, both of which have shown anticancer activity and beneficial health properties. In addition, EdAG can bind with GSH forming the EdAG-SG conjugate [(2-amino-5-[[3-[2-[[4-amino-5-hydroxy-5-oxopentanoyl]amino]-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-1-(carboxymethylamino)-1-oxopropan-2-yl]amino]-5-oxopentanoic acid); GSG]. Thus, following high-dose Bu therapy in HSCT patients, EdAG formed from the degradation of the Bu-SG conjugate may contribute to GSH depletion, a widely known mechanism of liver and other organ injury, and may also lead to a disruption of metabolism of other GSH metabolized drugs often co-prescribed with Bu in conditioning regimens, including, but not limited to, both non-cytotoxic and cytotoxic agents such as Cy, ifosfamide and Mel. The metabolism of GSG likely proceeds through the mercapturic acid pathway, but this remains hypothetical. Finally, EdAG or GSG have yet to be identified in human fluids or tissues following Bu administration, and their possible enzymatic formation is speculative. If identified in clinical situations, EdAG has the potential to be a future marker for Bu toxicity.
An emerging set of data shows the critical role of iron status in Bu metabolism and toxicities, among such correlations, pre-transplant ferritin levels and an increased risk of VOD have been found in patients undergoing SCT. The oxidative stress induced by iron overload perturbs the glutathiome, which tightly regulates oxidation in the body, and may alter the PK of Bu and other GSH-metabolized agents in humans. This is concerning since many agents co-administered with Bu in pre-conditioning regimens also interact significantly with the GSH system (e.g. Mel, thiotepa, Cy) and also confer a risk for severe hepatic toxicities. Taken together, close tracking of iron status may prove beneficial in optimizing safer Bu doses, especially in patients pre-disposed to liver injury or those presenting with multiple co-morbidities.
Other major Bu metabolites (THT 1-oxide, sulfolane and 3-hydroxysulfolane) that have been identified in humans were not cytotoxic to Chinese hamster V79 cell lines in a single study. But other authors have indirectly reported cytotoxic effects of these individual metabolites in animal toxicological studies, such as sulfolane inducing seizures. It would be interesting to study the specific myelotoxic effects of Bu metabolites compared with parent Bu within in vitro and in vivo experimental models. Also future TDM approaches utilizing metabolite PK in conjunction with parent Bu PK are intriguing. Validation of a newer pragmatic TDM method, such as including a metabolomics approach, will require convincing clinical data to support the additional role of endogenous metabolite monitoring in tailoring Bu doses. Many Bu TDM laboratories utilize LC-MS/MS to measure parent Bu concentrations in plasma samples. Parallel measurement of Bu and its metabolites in the same assay run would likely require an ultra-sensitive GC-MS/MS assay – due to the low molecular weight and volatility of certain metabolites. Therefore, there is a need for further development of a rapid, cost-efficient and highly reproducible GC-MS/MS assay to support such an approach.
The pharmacogenomic and DDI data present in the literature provides some insight, although conflicting, regarding the pharmacological mechanism(s) underpinning non-GST perpetrated Bu DDIs. Besides GST enzymes, the other plausible mechanisms include CYP and/or FMO enzymes. A CYP mechanism is widely speculated upon but still remains a quandary. A few human clinical studies have shown a significant influence of itraconazole (increased Bu AUC and decreased clearance) and phenytoin (decreased Bu AUC and increased clearance) on parent Bu PK. Important to mention is that in the study which showed itraconazole increased Bu exposure, Bu was administered orally. Since itraconazole is also a strong inhibitor of P-gp, transporter related mechanisms in the GI tract and/or the liver cannot be ruled out. Bu’s second major metabolite THT is probably a substrate for human FMOs. Voriconazole is a substrate for human FMO3 and is prescribed in pre-transplant conditioning regimens for prophylaxis of fungal infections. There is a potential for a severe interaction, which is currently being studied, although available data from a small number of patients suggest no influence of voriconazole on parent Bu PK.
Some pharmacogenomic data points toward a role of CYP2C9 in sulfolane formation, but it should pointed out that fluconazole is a strong inhibitor of CYP2C9 and several clinical studies have failed to demonstrate an interaction between Bu and fluconazole. Pharmacogenetic data also shows that genetic variants in CYP39A1 are associated with Bu clearance. CYP39A1 is involved in the synthesis and excretion of bile acids and is not typically associated with clinically significant DDIs. CYP39A1’s direct or indirect role in Bu disposition is perplexing and clearly justifies further study.
A peculiar interaction between metronidazole and both orally and IV administered Bu has been firmly documented in three clinical studies, yet the mechanism is unclear. Metronidazole possibly inhibits CYP3A4 and CYP2C9, although the clinical impact of this inhibition has been questioned, and metronidazole has recently been shown to be a substrate for CYP2A6, forming the metabolite 2-hydroxymetronidazole. It is not known if metronidazole directly affects GST enzyme activity, but an electrophilic, highly unstable metronidazole metabolite reacts with GSH, which suggests that the co-administration of metronidazole with Bu may interfere with conjugation of GSH with Bu and consequent Bu PK. This mechanism is the best explanation for this DDI at this time. It is preferred that an alternative antibacterial agent with activity against Clostridium difficile and low potential for DDIs should be strongly considered during Bu preconditioning, such as oral vancomycin or fidoxomicin. Evolving data shows the superiority of vancomycin over metronidazole in treating Clostridium difficile infection, data which will perhaps influence emerging treatment guidelines regarding front line therapy. If an alternative antibacterial drug is not feasible, then an increased frequency of Bu TDM (e.g. for 3–4 days) should be strongly considered to avoid a severe reduction in Bu clearance and increased Bu exposure due to metronidazole co-use.
Of additional concern, future DDIs with Bu are highly probable, since a burgeoning number of approved drugs in the leukemia field (e.g. tyrosine kinase inhibitors) continue to reach FDA approval, but DDIs are not fully understood with these newer agents, some of which are cleared by (or inhibit) the CYP system and are prescribed immediately prior to Bu pre-transplant regimens. Also, administration of targeted covalent inhibitors in close conjunction with Bu is concerning since some of these compounds contain an acrylamide group that is highly reactive and can form covalent bounds with thiol-containing endogenous and exogenous compounds.
There are major obstacles to fully study Bu metabolism in human patients. One-on-one DDI studies between Bu and possible perpetrator drug(s), obviously, raise strong ethical concerns due to risks of life-threating toxicities and poor treatment outcomes. Discovery of unknown DDIs is daunting in the stem cell transplant clinic, since many DDIs may go undetected in this fast-paced environment, or are interpreted as manifestations of the cancer’s clinical development. Parent Bu may be a substrate for CYPs, such as undergoing hydroxylation or perhaps an obscure dehydrogenation reaction. Traditional models of drug metabolism, in the context of studying CYP and FMO mechanisms, are performed in human liver subcellular fractions, but would be difficult models to study Bu drug metabolism since first-pass metabolism of Bu through GSH/GSTs needs to occur first before subsequent phase I biotransformation. Alternative options include in vitro hepatocyte models and in vivo animal knock-out models. Also the effects of metronidazole and its reactive metabolites on human GST enzyme activity is a rather straightforward in vitro study, but in vivo models will require specialized knock-out animal models.
Meanwhile, clinicians cannot afford to wait for definitive PK DDI studies with Bu, which are slow to appear in the literature due, in part, to the historically insufficient funding trends of such studies and the limited number of investigators engaged in this area of research. At this time it is best, in the authors expert opinion, to inform clinicians about the risks of prescribing GST-, GSH-, CYP-, FMO-, efflux transporter – modulating agents in conjunction with Bu, which can potentially lead to over exposure (toxicity concerns) or under exposure (loss of efficacy) of Bu. There are dangers of incorrectly estimating Bu PK and dose adjustments when therapy of these potential enzyme/GSH modulating drugs is discontinued in close proximity to test or therapeutic Bu doses used for PK-guided dose adjustments, unless a sufficient washout period is allowed and/or multiple TDM-directed dose adjustments, or refined monitoring strategies, are an option for the patient. Clinicians should also be keen towards understanding that greater risks of unwanted outcomes is enhanced in patients exhibiting damage to organs following pre-treatment with multiple transfusions, since iron overload leads to oxidative stress and possible disruption of GSH-mediated drug clearance mechanisms. Also clinically significant, patients prescribed polychemotherapy, especially with other alkylating agents such as Mel or Cy, may require more advanced forms of TDM, such as co-monitoring of both drugs instead of solely Bu, the current method of practice. This co-monitoring strategy, perhaps coupled with definitive pharmacogenomic data, could be applied to other agents (e.g. Clofarabine, Flu, and Gemcitabine) in high-dose Bu containing conditioning regimens.
Article highlights.
Busulfan (Bu) is a preferred alkylating agent in myeloablative conditioning regimens prior to hematopoietic stem cell transplantation (HSCT), but carries a substantial risk for toxicities (high exposure) and poor efficacy (low exposure) when plasma concentrations are not properly monitored.
A bulk of knowledge shows that Bu has a fascinating, but highly complex, drug elimination profile that consists of both enzymatic and non-enzymatic processes; still, more basic scientific research is required to firmly establish the roles of non-GST enzyme(s) in Bu clearance. Such in vitro findings have potential benefits to support refinements of population PK models used in conjunction with therapeutic drug monitoring (TDM).
Despite decades of study, clinical PK drug-drug interaction (DDI) and pharmacogenomic findings show incongruous results, likely caused in part to polypharmacy (and polychemotherapy) tendencies of HSCT patients and their predisposition to organ toxicities resulting from inferior tumor control and/or multiple procedures prior to HSCT.
Newer published models of therapeutic drug monitoring (TDM) show a significantly greater benefit in improving survival in pediatric patients. The possible implementation of co-drug TDM strategies, in both pediatric and adult patients, may further improve outcomes and mitigate regimen-related toxicity in the complex HSCT treatment setting.
Acknowledgments
Funding
This paper was funded by Department Pharmacy Research (M.D. Anderson Cancer Center), National Cancer Institute P01-CA49639 and 2P30-CA16672, Stephen and Lavinia Boyd Fund for Leukemia Research and Various Donors’ Fund
Footnotes
Declaration of Interest
Y Nieto has received research support fortm Otsuka Pharmaceuticals Inc. BS Andersson is a former consultant to Otsuke Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Thomas ED. A history of allogeneic hematopoeitic cell transplanation. In: Appelbaum FR, Forman SJ, Negrin RS, Blume KG, editors. Thomas’ Hematopoietic Cell Transplantation. 4th. Wiley-Blackwell; Oxford, UK: 2009. pp. 3–7. [Google Scholar]
- 2.Thomas ED, Buckner CD, Banaji M, et al. One hundred patients with acute leukemia treated by chemotherapy, total body irradiation, and allogeneic marrow transplantation. Blood. 1977;49:511–533. [PubMed] [Google Scholar]
- 3.Bredeson C, LeRademacher J, Kato K, et al. Prospective cohort study comparing intravenous busulfan to total body irradiation in hematopoietic cell transplantation. Blood. 2013;122:3871–3878. doi: 10.1182/blood-2013-08-519009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Champlin RE. Busulfan or TBI: answer to an age-old question. Blood. 2013;122:3856–3857. doi: 10.1182/blood-2013-10-530006. [DOI] [PubMed] [Google Scholar]
- 5.Ciurea SO, Andersson BS. Busulfan in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2009;15:523–536. doi: 10.1016/j.bbmt.2008.12.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersson BS, Thall PF, Madden T, et al. Busulfan systemic exposure relative to regimen-related toxicity and acute graft-versus-host disease: defining a therapeutic window for i.v. BuCy2 in chronic myelogenous leukemia. Biol Blood Marrow Transplant. 2002;8:477–485. doi: 10.1053/bbmt.2002.v8.pm12374452. [DOI] [PubMed] [Google Scholar]
- 7.Vassal G, Deroussent A, Hartmann O, et al. Dose-dependent neurotoxicity of high-dose busulfan in children: a clinical and pharmacological study. Cancer Res. 1990;50:6203–6207. [PubMed] [Google Scholar]
- 8.McCune JS, Holmberg LA. Busulfan in hematopoietic stem cell transplant setting. Expert Opin Drug Metab Toxicol. 2009;5:957–969. doi: 10.1517/17425250903107764. [DOI] [PubMed] [Google Scholar]
- 9.Buxton ILO. Pharmacokinetics and Pharmacodynamics: the dynamics of drug absorption, distribution, metabolism, and elimination. In: Bruton LL, editor. Goodman & Gilmans’s The Pharmacological Basis of Therapeutics. 11th. McGraw-Hill Education; New York, NY: 2011. pp. 1–39. [Google Scholar]
- 10.Geddes M, Kangarloo SB, Naveed F, et al. High busulfan exposure is associated with worse outcomes in a daily i.v. busulfan and fludarabine allogeneic transplant regimen. Biol Blood Marrow Transplant. 2008;14:220–228. doi: 10.1016/j.bbmt.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 11*.Andersson BS, Thall PF, Valdez BC, et al. Fludarabine with pharmacokinetically-guided IV busulfan is superior to fixed-dose delivery in pretransplant conditioning of AML/MDS patients. Bone Marrow Transplant. 2017;52:580–587. doi: 10.1038/bmt.2016.322. An excellent article detailing the benefit of PK-guided Bu therapy vs. fixed dose Bu therapy in HSCT patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12**.Bartelink IH, Lalmohamed A, van Reij EM, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol. 2016;3:526–536. doi: 10.1016/S2352-3026(16)30114-4. A superb article that illustrates improved outcomes in children after employing a new therapeutic drug monitoring strategy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trevisan DD, Silva JB, Oliveira HC, et al. Prevalence and clinical significance of potential drug-drug interaction in hematopoietic stem cell transplantation. Cancer Chemother Pharmacol. 2015;75:393–400. doi: 10.1007/s00280-014-2657-8. [DOI] [PubMed] [Google Scholar]
- 14.Glotzbecker B, Duncan C, Alyea E, 3rd, et al. Important drug interactions in hematopoietic stem cell transplantation: what every physician should know. Biol Blood Marrow Transplant. 2012;18:989–1006. doi: 10.1016/j.bbmt.2011.11.029. [DOI] [PubMed] [Google Scholar]
- 15.Guastaldi RB, Reis AM, Figueras A, et al. Prevalence of potential drug-drug interactions in bone marrow transplant patients. Int J Clin Pharm. 2011;33:1002–1009. doi: 10.1007/s11096-011-9574-2. [DOI] [PubMed] [Google Scholar]
- 16.Hassan M, Andersson BS. Role of pharmacogenetics in busulfan/cyclophosphamide conditioning therapy prior to hematopoietic stem cell transplantation. Pharmacogenomics. 2013;14:75–87. doi: 10.2217/pgs.12.185. *A good article proposing a novel drug interaction mechanism of Bu. [DOI] [PubMed] [Google Scholar]
- 17.Myeleran (Busulfan) Prescribing Information. GlaxoSmith Kline; Greenville, NC, USA: 2002. [Google Scholar]
- 18.IV Busulfex (busulfan injection) Otsuka America Pharmaceutical Inc; Rockville, MD, USA: 2011. [Google Scholar]
- 19.Galaup A, Paci A. Pharmacology of dimethanesulfonate alkylating agents: busulfan and treosulfan. Expert Opin Drug Metab Toxicol. 2013;9:333–347. doi: 10.1517/17425255.2013.737319. [DOI] [PubMed] [Google Scholar]
- 20.Hartley JA, Fox BW. Cross-linking between histones and DNA following treatment with a series of dimethane sulphonate esters. Cancer Chemother Pharmacol. 1986;17:56–62. doi: 10.1007/BF00299867. [DOI] [PubMed] [Google Scholar]
- 21.DeLeve LD, Wang X. Role of oxidative stress and glutathione in busulfan toxicity in cultured murine hepatocytes. Pharmacology. 2000;60:143–154. doi: 10.1159/000028359. [DOI] [PubMed] [Google Scholar]
- 22.Peer CJ, Younis IR, Leonard SS, et al. Glutathione conjugation of busulfan produces a hydroxyl radical-trapping dehydroalanine metabolite. Xenobiotica. 2012;42:1170–1177. doi: 10.3109/00498254.2012.696740. [DOI] [PubMed] [Google Scholar]
- 23.Scian M, Atkins WM. The busulfan metabolite EdAG irreversibly glutathionylates glutaredoxins. Archives of biochemistry and biophysics. 2015;583:96–104. doi: 10.1016/j.abb.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Younis IR, Elliott M, Peer CJ, et al. Dehydroalanine analog of glutathione: an electrophilic busulfan metabolite that binds to human glutathione S-transferase A1-1. J Pharmacol Exp Ther. 2008;327:770–776. doi: 10.1124/jpet.108.142208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grochow LB, Jones RJ, Brundrett RB, et al. Pharmacokinetics of busulfan: correlation with veno-occlusive disease in patients undergoing bone marrow transplantation. Cancer Chemother Pharmacol. 1989;25:55–61. doi: 10.1007/BF00694339. [DOI] [PubMed] [Google Scholar]
- 26.Hassan M, Ljungman P, Bolme P, et al. Busulfan bioavailability. Blood. 1994;84:2144–2150. [PubMed] [Google Scholar]
- 27.Hassan M, Oberg G, Bekassy AN, et al. Pharmacokinetics of high-dose busulphan in relation to age and chronopharmacology. Cancer Chemother Pharmacol. 1991;28:130–134. doi: 10.1007/BF00689702. [DOI] [PubMed] [Google Scholar]
- 28.Ehrsson H, Hassan M, Ehrnebo M, et al. Busulfan kinetics. Clin Pharmacol Ther. 1983;34:86–89. doi: 10.1038/clpt.1983.134. [DOI] [PubMed] [Google Scholar]
- 29.Tran HT, Madden T, Petropoulos D, et al. Individualizing high-dose oral busulfan: prospective dose adjustment in a pediatric population undergoing allogeneic stem cell transplantation for advanced hematologic malignancies. Bone Marrow Transplant. 2000;26:463–470. doi: 10.1038/sj.bmt.1702561. [DOI] [PubMed] [Google Scholar]
- 30.Krivoy N, Hoffer E, Lurie Y, et al. Busulfan use in hematopoietic stem cell transplantation: pharmacology, dose adjustment, safety and efficacy in adults and children. Current drug safety. 2008;3:60–66. doi: 10.2174/157488608783333899. [DOI] [PubMed] [Google Scholar]
- 31.Bhagwatwar HP, Phadungpojna S, Chow DS, et al. Formulation and stability of busulfan for intravenous administration in high-dose chemotherapy. Cancer Chemother Pharmacol. 1996;37:401–408. doi: 10.1007/s002800050404. [DOI] [PubMed] [Google Scholar]
- 32.Madden T, de Lima M, Thapar N, et al. Pharmacokinetics of once-daily IV busulfan as part of pretransplantation preparative regimens: a comparison with an every 6-hour dosing schedule. Biol Blood Marrow Transplant. 2007;13:56–64. doi: 10.1016/j.bbmt.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 33.Andersson BS, Kashyap A, Gian V, et al. Conditioning therapy with intravenous busulfan and cyclophosphamide (IV BuCy2) for hematologic malignancies prior to allogeneic stem cell transplantation: a phase II study. Biol Blood Marrow Transplant. 2002;8:145–154. doi: 10.1053/bbmt.2002.v8.pm11939604. [DOI] [PubMed] [Google Scholar]
- 34.Hassan M, Ehrsson H, Smedmyr B, et al. Cerebrospinal fluid and plasma concentrations of busulfan during high-dose therapy. Bone Marrow Transplant. 1989;4:113–114. [PubMed] [Google Scholar]
- 35.Ehrsson H, Hassan M. Binding of busulfan to plasma proteins and blood cells. J Pharm Pharmacol. 1984;36:694–696. doi: 10.1111/j.2042-7158.1984.tb04847.x. [DOI] [PubMed] [Google Scholar]
- 36.Hassan M, Ehrsson H. Urinary metabolites of busulfan in the rat. Drug Metab Dispos. 1987;15:399–402. [PubMed] [Google Scholar]
- 37.Hassan M, Oberg G, Ehrsson H, et al. Pharmacokinetic and metabolic studies of high-dose busulphan in adults. Eur J Clin Pharmacol. 1989;36:525–530. doi: 10.1007/BF00558081. [DOI] [PubMed] [Google Scholar]
- 38.Gibbs JP, Czerwinski M, Slattery JT. Busulfan-glutathione conjugation catalyzed by human liver cytosolic glutathione S-transferases. Cancer Res. 1996;56:3678–3681. [PubMed] [Google Scholar]
- 39.Benson AM, Barretto PB. Effects of disulfiram, diethyldithiocarbamate, bisethylxanthogen, and benzyl isothiocyanate on glutathione transferase activities in mouse organs. Cancer Res. 1985;45:4219–4223. [PubMed] [Google Scholar]
- 40.Bouligand J, Deroussent A, Simonnard N, et al. Induction of glutathione synthesis explains pharmacodynamics of high-dose busulfan in mice and highlights putative mechanisms of drug interaction. Drug Metab Dispos. 2007;35:306–314. doi: 10.1124/dmd.106.012880. [DOI] [PubMed] [Google Scholar]
- 41.Nation RL, Evans AM, Milne RW. Pharmacokinetic drug interactions with phenytoin (Part I) Clin Pharmacokinet. 1990;18:37–60. doi: 10.2165/00003088-199018010-00003. [DOI] [PubMed] [Google Scholar]
- 42.Vassal G, Challine D, Koscielny S, et al. Chronopharmacology of high-dose busulfan in children. Cancer Res. 1993;53:1534–1537. [PubMed] [Google Scholar]
- 43.Inoue N, Imai K, Aimoto T. Circadian variation of hepatic glutathione S-transferase activities in the mouse. Xenobiotica. 1999;29:43–51. doi: 10.1080/004982599238803. [DOI] [PubMed] [Google Scholar]
- 44.Ferrell JM, Chiang JYL. Circadian rhythms in liver metabolism and disease. Acta Pharmaceutica Sinica B. 2015;5:113–122. doi: 10.1016/j.apsb.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hrushesky WJ. Circadian timing of cancer chemotherapy. Science. 1985;228:73–75. doi: 10.1126/science.3883493. [DOI] [PubMed] [Google Scholar]
- 46.Mross K, Haring B, Hollander N, et al. Comparison of 1-hour and 3-hours paclitaxel infusion pharmacokinetics: results from a randomized trial. Onkologie. 2002;25:503–508. doi: 10.1159/000068620. [DOI] [PubMed] [Google Scholar]
- 47.Kangarloo SB, Naveed F, Ng ES, et al. Development and validation of a test dose strategy for once-daily i.v. busulfan: importance of fixed infusion rate dosing. Biol Blood Marrow Transplant. 2012;18:295–301. doi: 10.1016/j.bbmt.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 48.Trams EG, Nadkarni MV, Dequattro V, et al. Dimethanesulphonoxybutane (Myleran): Preliminary studies on distribution and metabolic fate in the rat. Biochem Pharmacol. 1959;2:7–16. [Google Scholar]
- 49.Versace F, Uppugunduri CR, Krajinovic M, et al. A novel method for quantification of sulfolane (a metabolite of busulfan) in plasma by gas chromatography-tandem mass spectrometry. Analytical and bioanalytical chemistry. 2012;404:1831–1838. doi: 10.1007/s00216-012-6330-y. [DOI] [PubMed] [Google Scholar]
- 50.El-Serafi I, Terelius Y, Twelkmeyer B, et al. Gas chromatographic-mass spectrometry method for the detection of busulphan and its metabolites in plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2013:913–914. 98–105. doi: 10.1016/j.jchromb.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Srivastava A, Poonkuzhali B, Shaji RV, et al. Glutathione S-transferase M1 polymorphism: a risk factor for hepatic venoocclusive disease in bone marrow transplantation. Blood. 2004;104:1574–1577. doi: 10.1182/blood-2003-11-3778. [DOI] [PubMed] [Google Scholar]
- 52.Hassan M, Ehrsson H. Metabolism of 14C-busulfan in isolated perfused rat liver. Eur J Drug Me Pharmacokinet. 1987;12:71–76. doi: 10.1007/BF03189864. [DOI] [PubMed] [Google Scholar]
- 53.Valdez BC, Hassan M, Andersson BS. Development of an assay for cellular efflux of pharmaceutically active agents and its relevance to understanding drug interactions. Exp Hematol. 2017 doi: 10.1016/j.exphem.2017.04.011. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Czerwinski M, Gibbs JP, Slattery JT. Busulfan conjugation by glutathione S-transferases alpha, mu, and pi. Drug Metab Dispos. 1996;24:1015–1019. [PubMed] [Google Scholar]
- 55.Gibbs JP, Yang JS, Slattery JT. Comparison of human liver and small intestinal glutathione S-transferase-catalyzed busulfan conjugation in vitro. Drug Metab Dispos. 1998;26:52–55. [PubMed] [Google Scholar]
- 56.Gibbs JP, Liacouras CA, Baldassano RN, et al. Up-regulation of glutathione S-transferase activity in enterocytes of young children. Drug Metab Dispos. 1999;27:1466–1469. [PubMed] [Google Scholar]
- 57.Cooper AJ, Younis IR, Niatsetskaya ZV, et al. Metabolism of the cysteine S-conjugate of busulfan involves a beta-lyase reaction. Drug Metab Dispos. 2008;36:1546–1552. doi: 10.1124/dmd.108.020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baur X, Bittner C. Occupational obstructive airway diseases caused by the natural gas odorant tetrahydrothiophene–two case reports. American journal of industrial medicine. 2009;52:982–986. doi: 10.1002/ajim.20761. [DOI] [PubMed] [Google Scholar]
- 59.Damani LA, Houdi AA. Cytochrome P-450 and FAD-monooxygenase mediated S- and N-oxygenations. Drug Metabol Drug Interact. 1988;6:235–244. doi: 10.1515/dmdi.1988.6.3-4.235. [DOI] [PubMed] [Google Scholar]
- 60.Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106:357–387. doi: 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mozier NM, Hoffman JL. Biosynthesis and urinary excretion of methyl sulfonium derivatives of the sulfur mustard analog, 2-chloroethyl ethyl sulfide, and other thioethers. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1990;4:3329–3333. doi: 10.1096/fasebj.4.15.2253846. [DOI] [PubMed] [Google Scholar]
- 62.Meganathan R, Schrementi J. Tetrahydrothiophene 1-oxide as an electron acceptor for Escherichia coli. Journal of bacteriology. 1987;169:2862–2865. doi: 10.1128/jb.169.6.2862-2865.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roberts JJ, Warwick GP. The mode of action of alkylating agents. III. The formation of 3-hydroxytetrahydrothiophene-1:1-dioxide from 1:4-dimethanesulphonyloxybutane (myleran), S-beta-L-alanyltetrahydrothiophenium mesylate, tetrahydrothiopene and tetrahydrothiophene-1:1-dioxide in the rat, rabbit and mouse. Biochem Pharmacol. 1961;6:217–227. [Google Scholar]
- 64.Ruppert PH, Dyer RS. Acute behavioral toxicity of sulfolane: influence of hypothermia. Toxicology letters. 1985;28:111–116. doi: 10.1016/0378-4274(85)90018-9. [DOI] [PubMed] [Google Scholar]
- 65.Andersen ME, Jones RA, Kurlansik L, et al. Sulfolane-induced convulsions in rodents. Research communications in chemical pathology and pharmacology. 1976;15:571–580. [PubMed] [Google Scholar]
- 66.Burdette LJ, Dyer RS. Sulfolane effects on audiogenic, pentylenetetrazol and afterdischarge seizure activity. Neurobehavioral toxicology and teratology. 1986;8:621–626. [PubMed] [Google Scholar]
- 67.Ghosh AK, Lee HY, Thompson WJ, et al. The development of cyclic sulfolanes as novel and high-affinity P2 ligands for HIV-1 protease inhibitors. Journal of medicinal chemistry. 1994;37:1177–1188. doi: 10.1021/jm00034a016. [DOI] [PubMed] [Google Scholar]
- 68.Mohler FS, Gordon CJ. Thermoregulatory responses of the rabbit to central neural injections of sulfolane. Neurotoxicology. 1989;10:53–62. [PubMed] [Google Scholar]
- 69.Cole SP, Deeley RG. Transport of glutathione and glutathione conjugates by MRP1. Trends in pharmacological sciences. 2006;27:438–446. doi: 10.1016/j.tips.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 70.Zaman GJ, Cnubben NH, van Bladeren PJ, et al. Transport of the glutathione conjugate of ethacrynic acid by the human multidrug resistance protein MRP. FEBS Lett. 1996;391:126–130. doi: 10.1016/0014-5793(96)00718-1. [DOI] [PubMed] [Google Scholar]
- 71.Ploemen JH, van Ommen B, van Bladeren PJ. Inhibition of rat and human glutathione S-transferase isoenzymes by ethacrynic acid and its glutathione conjugate. Biochem Pharmacol. 1990;40:1631–1635. doi: 10.1016/0006-2952(90)90465-w. [DOI] [PubMed] [Google Scholar]
- 72.Feit PW, Rastrup-Andersen N. 4-Methanesulfonyloxybutanol: hydrolysis of busulfan. J Pharm Sci. 1973;62:1007–1008. doi: 10.1002/jps.2600620634. [DOI] [PubMed] [Google Scholar]
- 73.Hassan M, Ehrsson H. Degradation of busulfan in aqueous solution. J Pharm Biomed Anal. 1986;4:95–101. doi: 10.1016/0731-7085(86)80027-9. [DOI] [PubMed] [Google Scholar]
- 74.Reddy HR, Chandrasekhar N, Karigar CS. Gas Chromatographic Method for the Quantitative Determination of a Hydrolytic Degradation Impurity in Busulfan Injectable Products. J Chromatogr Sci. 2016;54:1475–1480. doi: 10.1093/chromsci/bmw117. [DOI] [PubMed] [Google Scholar]
- 75.Hudson RF, Timmis GM, Marshall RD. A physico-chemical investigation into the biological action of myleran and related sulphonic acid esters. Biochem Pharmacol. 1958;1:48–59. [Google Scholar]
- 76.Lenz D, Jubner M, Bender K, et al. Inhibition of 1,4-butanediol metabolism in human liver in vitro. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:647–654. doi: 10.1007/s00210-011-0627-9. [DOI] [PubMed] [Google Scholar]
- 77.Kazancioglu EA, Guney M, Senturk M, et al. Simple methanesulfonates are hydrolyzed by the sulfatase carbonic anhydrase activity. Journal of enzyme inhibition and medicinal chemistry. 2012;27:880–885. doi: 10.3109/14756366.2011.637202. [DOI] [PubMed] [Google Scholar]
- 78.Roberts JJ, Warwick GP. The mode of action of alkylating agents. II. Studies of the metabolism of myleran. The reaction of myleran with some naturally occurring thiols in vitro. Biochem Pharmacol. 1961;6:205–216. doi: 10.1016/0006-2952(61)90132-0. [DOI] [PubMed] [Google Scholar]
- 79.Marchand DH, Remmel RP, Abdel-Monem MM. Biliary excretion of a glutathione conjugate of busulfan and 1,4-diiodobutane in the rat. Drug Metab Dispos. 1988;16:85–92. [PubMed] [Google Scholar]
- 80.Leonard SS, Xia C, Jiang BH, et al. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochemical and biophysical research communications. 2003;309:1017–1026. doi: 10.1016/j.bbrc.2003.08.105. [DOI] [PubMed] [Google Scholar]
- 81.Kumar SS, Devasagayam TP, Bhushan B, et al. Scavenging of reactive oxygen species by chlorophyllin: an ESR study. Free Radic Res. 2001;35:563–574. doi: 10.1080/10715760100301571. [DOI] [PubMed] [Google Scholar]
- 82.Baumhakel M, Kasel D, Rao-Schymanski RA, et al. Screening for inhibitory effects of antineoplastic agents on CYP3A4 in human liver microsomes. International journal of clinical pharmacology and therapeutics. 2001;39:517–528. doi: 10.5414/cpp39517. [DOI] [PubMed] [Google Scholar]
- 83.Mostafa M, Chalvardjian K, Lami H, et al. Alteration in the capacities of carcinogen metabolizing system of mouse-livers during pretreatment with various antineoplastic agents. Oncology reports. 1994;1:651–656. doi: 10.3892/or.1.3.651. [DOI] [PubMed] [Google Scholar]
- 84.Freireich EJ, Gehan EA, Rall DP, et al. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer chemotherapy reports Part 1. 1966;50:219–244. [PubMed] [Google Scholar]
- 85.Yamazaki H, Inui Y, Yun CH, et al. Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis. 1992;13:1789–1794. doi: 10.1093/carcin/13.10.1789. [DOI] [PubMed] [Google Scholar]
- 86.Abbasi N, Vadnais B, Knutson JA, et al. Pharmacogenetics of intravenous and oral busulfan in hematopoietic cell transplant recipients. J Clin Pharmacol. 2011;51:1429–1438. doi: 10.1177/0091270010382915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ansari M, Lauzon-Joset JF, Vachon MF, et al. Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant. 2010;45:261–267. doi: 10.1038/bmt.2009.143. [DOI] [PubMed] [Google Scholar]
- 88.Ansari M, Rezgui MA, Theoret Y, et al. Glutathione S-transferase gene variations influence BU pharmacokinetics and outcome of hematopoietic SCT in pediatric patients. Bone Marrow Transplant. 2013;48:939–946. doi: 10.1038/bmt.2012.265. [DOI] [PubMed] [Google Scholar]
- 89.Bonifazi F, Storci G, Bandini G, et al. Glutathione transferase-A2 S112T polymorphism predicts survival, transplant-related mortality, busulfan and bilirubin blood levels after allogeneic stem cell transplantation. Haematologica. 2014;99:172–179. doi: 10.3324/haematol.2013.089888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Elhasid R, Krivoy N, Rowe JM, et al. Influence of glutathione S-transferase A1, P1, M1, T1 polymorphisms on oral busulfan pharmacokinetics in children with congenital hemoglobinopathies undergoing hematopoietic stem cell transplantation. Pediatric blood & cancer. 2010;55:1172–1179. doi: 10.1002/pbc.22739. [DOI] [PubMed] [Google Scholar]
- 91.Gaziev J, Nguyen L, Puozzo C, et al. Novel pharmacokinetic behavior of intravenous busulfan in children with thalassemia undergoing hematopoietic stem cell transplantation: a prospective evaluation of pharmacokinetic and pharmacodynamic profile with therapeutic drug monitoring. Blood. 2010;115:4597–4604. doi: 10.1182/blood-2010-01-265405. [DOI] [PubMed] [Google Scholar]
- 92.Goekkurt E, Stoehlmacher J, Stueber C, et al. Pharmacogenetic analysis of liver toxicity after busulfan/cyclophosphamide-based allogeneic hematopoietic stem cell transplantation. Anticancer Res. 2007;27:4377–4380. [PubMed] [Google Scholar]
- 93.Johnson L, Orchard PJ, Baker KS, et al. Glutathione S-transferase A1 genetic variants reduce busulfan clearance in children undergoing hematopoietic cell transplantation. J Clin Pharmacol. 2008;48:1052–1062. doi: 10.1177/0091270008321940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim I, Keam B, Lee KH, et al. Glutathione S-transferase A1 polymorphisms and acute graft-vs-host disease in HLA-matched sibling allogeneic hematopoietic stem cell transplantation. Clin Transplant. 2007;21:207–213. doi: 10.1111/j.1399-0012.2006.00624.x. [DOI] [PubMed] [Google Scholar]
- 95.Kim SD, Lee JH, Hur EH, et al. Influence of GST gene polymorphisms on the clearance of intravenous busulfan in adult patients undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2011;17:1222–1230. doi: 10.1016/j.bbmt.2010.12.708. [DOI] [PubMed] [Google Scholar]
- 96.Krivoy N, Zuckerman T, Elkin H, et al. Pharmacokinetic and pharmacogenetic analysis of oral busulfan in stem cell transplantation: prediction of poor drug metabolism to prevent drug toxicity. Current drug safety. 2012;7:211–217. doi: 10.2174/157488612803251324. [DOI] [PubMed] [Google Scholar]
- 97.Kusama M, Kubota T, Matsukura Y, et al. Influence of glutathione S-transferase A1 polymorphism on the pharmacokinetics of busulfan. Clinica chimica acta; international journal of clinical chemistry. 2006;368:93–98. doi: 10.1016/j.cca.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 98.Ten Brink MH, Swen JJ, Bohringer S, et al. Exploratory analysis of 1936 SNPs in ADME genes for association with busulfan clearance in adult hematopoietic stem cell recipients. Pharmacogenet Genomics. 2013;23:675–683. doi: 10.1097/FPC.0000000000000007. [DOI] [PubMed] [Google Scholar]
- 99.ten Brink MH, van Bavel T, Swen JJ, et al. Effect of genetic variants GSTA1 and CYP39A1 and age on busulfan clearance in pediatric patients undergoing hematopoietic stem cell transplantation. Pharmacogenomics. 2013;14:1683–1690. doi: 10.2217/pgs.13.159. [DOI] [PubMed] [Google Scholar]
- 100.ten Brink MH, Wessels JA, den Hartigh J, et al. Effect of genetic polymorphisms in genes encoding GST isoenzymes on BU pharmacokinetics in adult patients undergoing hematopoietic SCT. Bone Marrow Transplant. 2012;47:190–195. doi: 10.1038/bmt.2011.55. [DOI] [PubMed] [Google Scholar]
- 101.Zwaveling J, Press RR, Bredius RG, et al. Glutathione S-transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Ther Drug Monit. 2008;30:504–510. doi: 10.1097/FTD.0b013e3181817428. [DOI] [PubMed] [Google Scholar]
- 102**.Huezo-Diaz P, Uppugunduri CR, Tyagi AK, et al. Pharmacogenetic aspects of drug metabolizing enzymes in busulfan based conditioning prior to allogenic hematopoietic stem cell transplantation in children. Current drug metabolism. 2014;15:251–264. doi: 10.2174/1389200215666140202214012. This is a superb review article that discusses the pharmacogenomics of Bu therapy in children. [DOI] [PubMed] [Google Scholar]
- 103.Geller RB, Saral R, Piantadosi S, et al. Allogeneic bone marrow transplantation after high-dose busulfan and cyclophosphamide in patients with acute nonlymphocytic leukemia. Blood. 1989;73:2209–2218. [PubMed] [Google Scholar]
- 104.Tetlow N, Board PG. Functional polymorphism of human glutathione transferase A2. Pharmacogenetics. 2004;14:111–116. doi: 10.1097/00008571-200402000-00005. [DOI] [PubMed] [Google Scholar]
- 105.Valdez BC, Li Y, Murray D, et al. Differential effects of histone deacetylase inhibitors on cellular drug transporters and their implications for using epigenetic modifiers in combination chemotherapy. Oncotarget. 2016;7:63829–63838. doi: 10.18632/oncotarget.11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Uppugunduri CR, Rezgui MA, Diaz PH, et al. The association of cytochrome P450 genetic polymorphisms with sulfolane formation and the efficacy of a busulfan-based conditioning regimen in pediatric patients undergoing hematopoietic stem cell transplantation. The pharmacogenomics journal. 2014;14:263–271. doi: 10.1038/tpj.2013.38. [DOI] [PubMed] [Google Scholar]
- 107.de Jonge ME, Huitema AD, Rodenhuis S, et al. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet. 2005;44:1135–1164. doi: 10.2165/00003088-200544110-00003. [DOI] [PubMed] [Google Scholar]
- 108.Lorbek G, Lewinska M, Rozman D. Cytochrome P450s in the synthesis of cholesterol and bile acids–from mouse models to human diseases. The FEBS journal. 2012;279:1516–1533. doi: 10.1111/j.1742-4658.2011.08432.x. [DOI] [PubMed] [Google Scholar]
- 109.Uchiyama T, Kanno H, Ishitani K, et al. An SNP in CYP39A1 is associated with severe neutropenia induced by docetaxel. Cancer Chemother Pharmacol. 2012;69:1617–1624. doi: 10.1007/s00280-012-1872-4. [DOI] [PubMed] [Google Scholar]
- 110.Kim DH, Park JY, Sohn SK, et al. The association between multidrug resistance-1 gene polymorphisms and outcomes of allogeneic HLA-identical stem cell transplantation. Haematologica. 2006;91:848–851. [PubMed] [Google Scholar]
- 111.Akan H, Antia VP, Kouba M, et al. Preventing invasive fungal disease in patients with haematological malignancies and the recipients of haematopoietic stem cell transplantation: practical aspects. The Journal of antimicrobial chemotherapy. 2013;68(Suppl 3):iii5–iii16. doi: 10.1093/jac/dkt389. [DOI] [PubMed] [Google Scholar]
- 112.Buggia I, Zecca M, Alessandrino EP, et al. Itraconazole can increase systemic exposure to busulfan in patients given bone marrow transplantation. GITMO (Gruppo Italiano Trapianto di Midollo Osseo) Anticancer Res. 1996;16:2083–2088. [PubMed] [Google Scholar]
- 113.Lempers VJ, van den Heuvel JJ, Russel FG, et al. Inhibitory Potential of Antifungal Drugs on ATP-Binding Cassette Transporters P-Glycoprotein, MRP1 to MRP5, BCRP, and BSEP. Antimicrob Agents Chemother. 2016;60:3372–3379. doi: 10.1128/AAC.02931-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nivoix Y, Leveque D, Herbrecht R, et al. The enzymatic basis of drug-drug interactions with systemic triazole antifungals. Clin Pharmacokinet. 2008;47:779–792. doi: 10.2165/0003088-200847120-00003. [DOI] [PubMed] [Google Scholar]
- 115.Isoherranen N, Kunze KL, Allen KE, et al. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos. 2004;32:1121–1131. doi: 10.1124/dmd.104.000315. [DOI] [PubMed] [Google Scholar]
- 116.Vermeer LM, Isringhausen CD, Ogilvie BW, et al. Evaluation of Ketoconazole and Its Alternative Clinical CYP3A4/5 Inhibitors as Inhibitors of Drug Transporters: The In Vitro Effects of Ketoconazole, Ritonavir, Clarithromycin, and Itraconazole on 13 Clinically-Relevant Drug Transporters. Drug Metab Dispos. 2016;44:453–459. doi: 10.1124/dmd.115.067744. [DOI] [PubMed] [Google Scholar]
- 117.Jaschonek K, Steinhilber D, Einsele H, et al. 5-Lipoxygenase inhibition by antifungal azole derivatives: new tools for immunosuppression? Eicosanoids. 1989;2:189–190. [PubMed] [Google Scholar]
- 118.Samuelsson B, Dahlen SE, Lindgren JA, et al. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 119.Nguyen L, Leger F, Lennon S, et al. Intravenous busulfan in adults prior to haematopoietic stem cell transplantation: a population pharmacokinetic study. Cancer Chemother Pharmacol. 2006;57:191–198. doi: 10.1007/s00280-005-0029-0. [DOI] [PubMed] [Google Scholar]
- 120.Zwaveling J, Bredius RG, Cremers SC, et al. Intravenous busulfan in children prior to stem cell transplantation: study of pharmacokinetics in association with early clinical outcome and toxicity. Bone Marrow Transplant. 2005;35:17–23. doi: 10.1038/sj.bmt.1704707. [DOI] [PubMed] [Google Scholar]
- 121.Gulbis AM, Culotta KS, Jones RB, et al. Busulfan and metronidazole: an often forgotten but significant drug interaction. Ann Pharmacother. 2010;45:e39. doi: 10.1345/aph.1Q087. [DOI] [PubMed] [Google Scholar]
- 122.Chung H, Yu KS, Hong KT, et al. A Significant Influence of Metronidazole on Busulfan Pharmacokinetics: A Case Report of Therapeutic Drug Monitoring. Ther Drug Monit. 2017;39:208–210. doi: 10.1097/FTD.0000000000000395. [DOI] [PubMed] [Google Scholar]
- 123.Nilsson C, Aschan J, Hentschke P, et al. The effect of metronidazole on busulfan pharmacokinetics in patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant. 2003;31:429–435. doi: 10.1038/sj.bmt.1703896. [DOI] [PubMed] [Google Scholar]
- 124.de Castro FA, Lanchote VL, Voltarelli JC, et al. Influence of fludarabine on the pharmacokinetics of oral busulfan during pretransplant conditioning for hematopoietic stem cell transplantation. J Clin Pharmacol. 2013;53:1205–1211. doi: 10.1002/jcph.130. [DOI] [PubMed] [Google Scholar]
- 125.Pearce RE, Cohen-Wolkowiez M, Sampson MR, et al. The Role of Human Cytochrome P450 Enzymes in the Formation of 2-Hydroxymetronidazole: CYP2A6 is the High Affinity (Low Km) Catalyst. Drug Metab Dispos. 2013;41:1686–1694. doi: 10.1124/dmd.113.052548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim KA, Park JY. Effect of metronidazole on the pharmacokinetics of fexofenadine, a P-glycoprotein substrate, in healthy male volunteers. Eur J Clin Pharmacol. 2010;66:721–725. doi: 10.1007/s00228-010-0797-2. [DOI] [PubMed] [Google Scholar]
- 127.Larsson P, Cybulski W, Tjalve H. Binding of 3H-metronidazole in olfactory, respiratory and alimentary epithelia in rats. Pharmacol Toxicol. 1997;81:65–73. doi: 10.1111/j.1600-0773.1997.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 128.Styler MJ, Crilley P, Biggs J, et al. Hepatic dysfunction following busulfan and cyclophosphamide myeloablation: a retrospective, multicenter analysis. Bone Marrow Transplant. 1996;18:171–176. [PubMed] [Google Scholar]
- 129.Granfors MT, Backman JT, Neuvonen M, et al. Ciprofloxacin greatly increases concentrations and hypotensive effect of tizanidine by inhibiting its cytochrome P450 1A2-mediated presystemic metabolism. Clin Pharmacol Ther. 2004;76:598–606. doi: 10.1016/j.clpt.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 130.Bensinger WI, Buckner CD, Lilleby K, et al. Dose escalation of busulfan with pentoxifylline and ciprofloxacin in patients with breast cancer undergoing autologous transplants. Oncology. 2004;67:368–375. doi: 10.1159/000082920. [DOI] [PubMed] [Google Scholar]
- 131.Hassan M, Svensson JO, Nilsson C, et al. Ketobemidone may alter busulfan pharmacokinetics during high-dose therapy. Ther Drug Monit. 2000;22:383–385. doi: 10.1097/00007691-200008000-00003. [DOI] [PubMed] [Google Scholar]
- 132.Bondesson U, Hartvig P, Danielsson B. Quantitative determination of the urinary excretion of ketobemidone and four of its metabolites after intravenous and oral administration in man. Drug Metab Dispos. 1981;9:376–380. [PubMed] [Google Scholar]
- 133.Yasar U, Annas A, Svensson JO, et al. Ketobemidone is a substrate for cytochrome P4502C9 and 3A4, but not for P-glycoprotein. Xenobiotica. 2005;35:785–796. doi: 10.1080/00498250500183181. [DOI] [PubMed] [Google Scholar]
- 134.Al-Shurbaji A, Sawe J. The pharmacokinetics of ketobemidone are not affected by CYP2D6 or CYP2C19 phenotype. Eur J Clin Pharmacol. 2002;57:877–881. doi: 10.1007/s00228-001-0413-6. [DOI] [PubMed] [Google Scholar]
- 135.McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharmaceutical research. 2013;30:2174–2187. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Coles B, Wilson I, Wardman P, et al. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: a stopped-flow kinetic study. Archives of biochemistry and biophysics. 1988;264:253–260. doi: 10.1016/0003-9861(88)90592-9. [DOI] [PubMed] [Google Scholar]
- 137.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 138.Sjoo F, Aschan J, Barkholt L, et al. N-acetyl-L-cysteine does not affect the pharmacokinetics or myelosuppressive effect of busulfan during conditioning prior to allogeneic stem cell transplantation. Bone Marrow Transplant. 2003;32:349–354. doi: 10.1038/sj.bmt.1704143. [DOI] [PubMed] [Google Scholar]
- 139.Potschka H, Fedrowitz M, Loscher W. Multidrug resistance protein MRP2 contributes to blood-brain barrier function and restricts antiepileptic drug activity. J Pharmacol Exp Ther. 2003;306:124–131. doi: 10.1124/jpet.103.049858. [DOI] [PubMed] [Google Scholar]
- 140.Carreras E, Cahn JY, Puozzo C, et al. Influence on Busilvex pharmacokinetics of clonazepam compared to previous phenytoin historical data. Anticancer Res. 2010;30:2977–2984. [PubMed] [Google Scholar]
- 141.Chan KW, Mullen CA, Worth LL, et al. Lorazepam for seizure prophylaxis during high-dose busulfan administration. Bone Marrow Transplant. 2002;29:963–965. doi: 10.1038/sj.bmt.1703593. [DOI] [PubMed] [Google Scholar]
- 142.Fitzsimmons WE, Ghalie R, Kaizer H. Anticonvulsants and busulfan. Ann Intern Med. 1990;112:552–553. doi: 10.7326/0003-4819-112-7-552_2. [DOI] [PubMed] [Google Scholar]
- 143.Hassan M, Oberg G, Bjorkholm M, et al. Influence of prophylactic anticonvulsant therapy on high-dose busulphan kinetics. Cancer Chemother Pharmacol. 1993;33:181–186. doi: 10.1007/BF00686213. [DOI] [PubMed] [Google Scholar]
- 144.Charasson V, Haaz MC, Robert J. Determination of drug interactions occurring with the metabolic pathways of irinotecan. Drug Metab Dispos. 2002;30:731–733. doi: 10.1124/dmd.30.6.731. [DOI] [PubMed] [Google Scholar]
- 145.Pacifici GM, Gustafsson LL, Sawe J, et al. Metabolic interaction between morphine and various benzodiazepines. Acta Pharmacol Toxicol (Copenh) 1986;58:249–252. doi: 10.1111/j.1600-0773.1986.tb00103.x. [DOI] [PubMed] [Google Scholar]
- 146.Meacham RH, Jr, Sisenwine SF, Liu AL, et al. Inhibition of ciramadol glucuronidation by benzodiazepines. Drug Metab Dispos. 1986;14:430–436. [PubMed] [Google Scholar]
- 147.Uchaipichat V, Suthisisang C, Miners JO. The glucuronidation of R- and S-lorazepam: human liver microsomal kinetics, UDP-glucuronosyltransferase enzyme selectivity, and inhibition by drugs. Drug Metab Dispos. 2013;41:1273–1284. doi: 10.1124/dmd.113.051656. [DOI] [PubMed] [Google Scholar]
- 148.Jung F, Richardson TH, Raucy JL, et al. Diazepam metabolism by cDNA-expressed human 2C P450s: identification of P4502C18 and P4502C19 as low K(M) diazepam N-demethylases. Drug Metab Dispos. 1997;25:133–139. [PubMed] [Google Scholar]
- 149.Kenworthy KE, Clarke SE, Andrews J, et al. Multisite kinetic models for CYP3A4: simultaneous activation and inhibition of diazepam and testosterone metabolism. Drug Metab Dispos. 2001;29:1644–1651. [PubMed] [Google Scholar]
- 150.Andersson BS, de Lima M, Thall PF, et al. Once daily i.v. busulfan and fludarabine (i.v. Bu-Flu) compares favorably with i.v. busulfan and cyclophosphamide (i.v.BuCy2) as pretransplant conditioning therapy in AML/MDS. Biol Blood Marrow Transplant. 2008;14:672–684. doi: 10.1016/j.bbmt.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.de Lima M, Couriel D, Thall PF, et al. Once-daily intravenous busulfan and fludarabine: clinical and pharmacokinetic results of a myeloablative, reduced-toxicity conditioning regimen for allogeneic stem cell transplantation in AML and MDS. Blood. 2004;104:857–864. doi: 10.1182/blood-2004-02-0414. [DOI] [PubMed] [Google Scholar]
- 152.Almog S, Kurnik D, Shimoni A, et al. Linearity and stability of intravenous busulfan pharmacokinetics and the role of glutathione in busulfan elimination. Biol Blood Marrow Transplant. 2011;17:117–123. doi: 10.1016/j.bbmt.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 153.Bonin M, Pursche S, Bergeman T, et al. F-ara-A pharmacokinetics during reduced-intensity conditioning therapy with fludarabine and busulfan. Bone Marrow Transplant. 2007;39:201–206. doi: 10.1038/sj.bmt.1705565. [DOI] [PubMed] [Google Scholar]
- 154.Perkins JB, Kim J, Anasetti C, et al. Maximally tolerated busulfan systemic exposure in combination with fludarabine as conditioning before allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012;18:1099–1107. doi: 10.1016/j.bbmt.2011.12.584. [DOI] [PubMed] [Google Scholar]
- 155.Yeh RF, Pawlikowski MA, Blough DK, et al. Accurate targeting of daily intravenous busulfan with 8-hour blood sampling in hospitalized adult hematopoietic cell transplant recipients. Biol Blood Marrow Transplant. 2012;18:265–272. doi: 10.1016/j.bbmt.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet. 2002;41:93–103. doi: 10.2165/00003088-200241020-00002. [DOI] [PubMed] [Google Scholar]
- 157.Hassan M, Ljungman P, Ringden O, et al. The effect of busulphan on the pharmacokinetics of cyclophosphamide and its 4-hydroxy metabolite: time interval influence on therapeutic efficacy and therapy-related toxicity. Bone Marrow Transplant. 2000;25:915–924. doi: 10.1038/sj.bmt.1702377. [DOI] [PubMed] [Google Scholar]
- 158.McCune JS, Batchelder A, Deeg HJ, et al. Cyclophosphamide following targeted oral busulfan as conditioning for hematopoietic cell transplantation: pharmacokinetics, liver toxicity, and mortality. Biol Blood Marrow Transplant. 2007;13:853–862. doi: 10.1016/j.bbmt.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 159.Rezvani AR, McCune JS, Storer BE, et al. Cyclophosphamide followed by intravenous targeted busulfan for allogeneic hematopoietic cell transplantation: pharmacokinetics and clinical outcomes. Biol Blood Marrow Transplant. 2013;19:1033–1039. doi: 10.1016/j.bbmt.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cantoni N, Gerull S, Heim D, et al. Order of application and liver toxicity in patients given BU and CY containing conditioning regimens for allogeneic hematopoietic SCT. Bone Marrow Transplant. 2011;46:344–349. doi: 10.1038/bmt.2010.137. [DOI] [PubMed] [Google Scholar]
- 161.Kerbauy FR, Tirapelli B, Akabane H, et al. The effect of administration order of BU and CY on toxicity in hematopoietic SCT in humans. Bone Marrow Transplant. 2009;43:883–885. doi: 10.1038/bmt.2008.404. [DOI] [PubMed] [Google Scholar]
- 162.Dockham PA, Sreerama L, Sladek NE. Relative contribution of human erythrocyte aldehyde dehydrogenase to the systemic detoxification of the oxazaphosphorines. Drug Metab Dispos. 1997;25:1436–1441. [PubMed] [Google Scholar]
- 163.Dourthe ME, Ternes N, Gajda D, et al. Busulfan-Melphalan followed by autologous stem cell transplantation in patients with high-risk neuroblastoma or Ewing sarcoma: an exposed-unexposed study evaluating the clinical impact of the order of drug administration. Bone Marrow Transplant. 2016;51:1265–1267. doi: 10.1038/bmt.2016.109. [DOI] [PubMed] [Google Scholar]
- 164*.Bouligand J, Richard C, Valteau-Couanet D, et al. Iron Overload Exacerbates Busulfan-Melphalan Toxicity Through a Pharmacodynamic Interaction in Mice. Pharmaceutical research. 2016;33:1913–1922. doi: 10.1007/s11095-016-1927-z. An excellent article demonstrating that iron overload signficantly effects the PK and PD in a Bu-Mel regimen. [DOI] [PubMed] [Google Scholar]
- 165.Dirven HA, van Ommen B, van Bladeren PJ. Glutathione conjugation of alkylating cytostatic drugs with a nitrogen mustard group and the role of glutathione S-transferases. Chem Res Toxicol. 1996;9:351–360. doi: 10.1021/tx950143c. [DOI] [PubMed] [Google Scholar]
- 166.Bouligand J, Boland I, Valteau-Couanet D, et al. In children and adolescents, the pharmacodynamics of high-dose busulfan is dependent on the second alkylating agent used in the combined regimen (melphalan or thiotepa) Bone Marrow Transplant. 2003;32:979–986. doi: 10.1038/sj.bmt.1704275. [DOI] [PubMed] [Google Scholar]
- 167.Sheth S. Iron chelation: an update. Current opinion in hematology. 2014;21:179–185. doi: 10.1097/MOH.0000000000000031. [DOI] [PubMed] [Google Scholar]
- 168.Sweiss K, Patel P, Rondelli D. Deferasirox increases BU blood concentrations. Bone Marrow Transplant. 2012;47:315–316. doi: 10.1038/bmt.2011.75. [DOI] [PubMed] [Google Scholar]
- 169.EXJADE (deferasirox) tablets, for oral suspension. Novartis Pharmaceuticals; East Hanover, NJ, USA: 2013. [Google Scholar]
- 170.Carter J, Yeh RF, Braunschweig I, et al. Unreported use of an herbal supplement resulting in decreased clearance of intravenous busulfan in a patient undergoing auto-SCT. Bone Marrow Transplant. 2014;49:313–314. doi: 10.1038/bmt.2013.169. [DOI] [PubMed] [Google Scholar]
- 171.Yang AK, He SM, Liu L, et al. Herbal interactions with anticancer drugs: mechanistic and clinical considerations. Curr Med Chem. 2010;17:1635–1678. doi: 10.2174/092986710791111279. [DOI] [PubMed] [Google Scholar]
- 172.Bartelink IH, Bredius RG, Ververs TT, et al. Once-daily intravenous busulfan with therapeutic drug monitoring compared to conventional oral busulfan improves survival and engraftment in children undergoing allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2008;14:88–98. doi: 10.1016/j.bbmt.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 173.Grochow LB. Busulfan disposition: the role of therapeutic monitoring in bone marrow transplantation induction regimens. Semin Oncol. 1993;20:18–25. quiz 26. [PubMed] [Google Scholar]
- 174.Bostrom B, Enockson K, Johnson A, et al. Plasma pharmacokinetics of high-dose oral busulfan in children and adults undergoing bone marrow transplantation. Pediatric transplantation. 2003;7(Suppl 3):12–18. doi: 10.1034/j.1399-3046.7.s3.2.x. [DOI] [PubMed] [Google Scholar]
- 175.Bartelink IH, van Kesteren C, Boelens JJ, et al. Predictive performance of a busulfan pharmacokinetic model in children and young adults. Ther Drug Monit. 2012;34:574–583. doi: 10.1097/FTD.0b013e31826051bb. [DOI] [PubMed] [Google Scholar]
- 176.Nguyen L, Fuller D, Lennon S, et al. I.V. busulfan in pediatrics: a novel dosing to improve safety/efficacy for hematopoietic progenitor cell transplantation recipients. Bone Marrow Transplant. 2004;33:979–987. doi: 10.1038/sj.bmt.1704446. [DOI] [PubMed] [Google Scholar]
- 177.Paci A, Vassal G, Moshous D, et al. Pharmacokinetic behavior and appraisal of intravenous busulfan dosing in infants and older children: the results of a population pharmacokinetic study from a large pediatric cohort undergoing hematopoietic stem-cell transplantation. Ther Drug Monit. 2012;34:198–208. doi: 10.1097/FTD.0b013e31824c2f60. [DOI] [PubMed] [Google Scholar]
- 178.Bartelink IH, Boelens JJ, Bredius RG, et al. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet. 2012;51:331–345. doi: 10.2165/11598180-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 179.Slattery JT, Risler LJ. Therapeutic monitoring of busulfan in hematopoietic stem cell transplantation. Ther Drug Monit. 1998;20:543–549. doi: 10.1097/00007691-199810000-00017. [DOI] [PubMed] [Google Scholar]
- 180.Hamidieh AA, Hamedani R, Hadjibabaie M, et al. Oral lorazepam prevents seizure during high-dose busulfan in children undergoing hematopoietic stem cell transplantation: a prospective study. Pediatric hematology and oncology. 2010;27:529–533. doi: 10.3109/08880018.2010.496895. [DOI] [PubMed] [Google Scholar]
- 181.Walker V, Mills GA, Anderson ME, et al. The acetaminophen metabolite N-acetyl-p-benzoquinone imine (NAPQI) inhibits glutathione synthetase in vitro; a clue to the mechanism of 5-oxoprolinuric acidosis? Xenobiotica. 2016:1–12. doi: 10.3109/00498254.2016.1166533. [DOI] [PubMed] [Google Scholar]
- 182.Dautrey S, Felice K, Petiet A, et al. Active intestinal elimination of ciprofloxacin in rats: modulation by different substrates. British journal of pharmacology. 1999;127:1728–1734. doi: 10.1038/sj.bjp.0702703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Haslam IS, Wright JA, O’Reilly DA, et al. Intestinal ciprofloxacin efflux: the role of breast cancer resistance protein (ABCG2) Drug Metab Dispos. 2011;39:2321–2328. doi: 10.1124/dmd.111.038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Seree EJ, Pisano PJ, Placidi M, et al. Identification of the human and animal hepatic cytochromes P450 involved in clonazepam metabolism. Fundamental & clinical pharmacology. 1993;7:69–75. doi: 10.1111/j.1472-8206.1993.tb00219.x. [DOI] [PubMed] [Google Scholar]
- 185.Guo Z, Smith TJ, Wang E, et al. Effects of phenethyl isothiocyanate, a carcinogenesis inhibitor, on xenobiotic-metabolizing enzymes and nitrosamine metabolism in rats. Carcinogenesis. 1992;13:2205–2210. doi: 10.1093/carcin/13.12.2205. [DOI] [PubMed] [Google Scholar]
- 186.Lin SC, Chung TC, Lin CC, et al. Hepatoprotective effects of Arctium lappa on carbon tetrachloride- and acetaminophen-induced liver damage. The American journal of Chinese medicine. 2000;28:163–173. doi: 10.1142/S0192415X00000210. [DOI] [PubMed] [Google Scholar]
- 187.Edwards JA, Price J. Metronidazole and human alcohol dehydrogenase. Nature. 1967;214:190–191. doi: 10.1038/214190b0. [DOI] [PubMed] [Google Scholar]
- 188.Yamauchi A, Ueda N, Hanafusa S, et al. Tissue distribution of and species differences in deacetylation of N-acetyl-L-cysteine and immunohistochemical localization of acylase I in the primate kidney. J Pharm Pharmacol. 2002;54:205–212. doi: 10.1211/0022357021778394. [DOI] [PubMed] [Google Scholar]
- 189.Giancarlo GM, Venkatakrishnan K, Granda BW, et al. Relative contributions of CYP2C9 and 2C19 to phenytoin 4-hydroxylation in vitro: inhibition by sulfaphenazole, omeprazole, and ticlopidine. Eur J Clin Pharmacol. 2001;57:31–36. doi: 10.1007/s002280100268. [DOI] [PubMed] [Google Scholar]