

Abstract
The molecular mechanism of endoplasmic reticulum (ER) stress in vascular pathophysiology remains inadequately understood. We studied the role of ER stress in homocysteine-induced impairment of coronary dilator function, with uncovering the molecular basis of the effect of ER stress on smooth muscle large-conductance Ca2+-activated K+ (BKCa) channels. The vasodilatory function of BKCa channels was studied in a myograph using endothelium-denuded porcine small coronary arteries. Primary cultured porcine coronary artery smooth muscle cells were used for mRNA and protein measurements and current recording of BKCa channels. Homocysteine inhibited vasorelaxant response to the BKCachannel opener NS1619, lowered BKCa β1 subunit protein level and suppressed BKCa current. Inhibition of ER stress restored BKCa β1 protein level and NS1619-evoked vasorelaxation. Selective blockade of the PKR-like ER kinase (PERK) yielded similarly efficient restoration of BKCa β1, preserving BKCa current and BKCa-mediated vasorelaxation. The restoration of BKCa β1 by PERK inhibition was associated with reduced atrogin-1 expression and decreased nuclear localization of forkhead box O transcription factor 3a (FoxO3a). Silencing of atrogin-1 prevented homocysteine-induced BKCa β1 loss and silencing of FoxO3a prevented atrogin-1 upregulation induced by homocysteine, accompanied by preservation of BKCa β1 protein level and BKCa current. ER stress mediates homocysteine-induced BKCa channel inhibition in coronary arteries. Activation of FoxO3a by PERK branch underlies the ER stress-mediated BKCa inhibition through a mechanism involving ubiquitin ligase-enhanced degradation of the channel β1 subunit.
Keywords: cardiovascular risk factors, coronary circulation, endoplasmic reticulum stress, homocysteine, smooth muscle cell
INTRODUCTION
Calcium-activated potassium (KCa) channels contri-bute to the control of vascular tone. Being abundantly expressed in vascular smooth muscle cells, functional large-conductance KCa (BKCa) channel is a tetrameric assembly of pore-forming α-subunits in complex with auxiliary β-subunits by which the channel activity is tightly regulated [1]. Activation of BKCachannels opposes vasoconstriction via hyperpolarizing cell membrane thus limiting extracellular Ca2+ entry through voltage-dependent Ca2+ channels in smooth muscle cells [2]. Moreover, as a target site of endothelium-derived relaxing factors, i.e., nitric oxide (NO) [3, 4] and endothelium-derived hyperpolarizing factor [5, 6], BKCa channels are of importance in mediating endothelium-dependent relaxation in vasculatures. Disturbed function of BKCa channel has been found to be associated with various cardiovascular disorders such as hypertension [7] and diabetes [8].
Hyperhomocysteinemia is a prevalent and independent risk factor for atherosclerotic vascular disease and thromboembolism. In patients with coronary artery disease, plasma total homocysteine level is considered as a strong predictor of cardiovascular mortality [9]. Homocysteine causes vascular dysfunction through mechanisms of endothelial impairment including inhibition of endothelial NO synthase (eNOS) [10–12], induction of oxidative stress [13, 14], and activation of iNOS and arginase [15], etc. Previous studies reported the inhibitory effect of homocysteine on smooth muscle BKCa channels. The BKCa current was observed to be significantly suppressed in animal and human vascular smooth muscle cells subjected to homocysteine exposure [16, 17]. Nevertheless, the mechanisms by which homocysteine inhibits BKCa channels remain poorly studied.
Recent studies concerning the role of endoplasmic reticulum (ER) stress in vascular dysfunction highlighted endothelial mechanism. ER stress causes inhibition of eNOS-NO pathway and increases production of endothelium-derived contracting factors [18–20]. In homocysteine-induced endothelial dysfunction, ER stress was found to be involved in NADPH activation and reactive oxygen species (ROS) production as well as NF-κB activation [21]. We recently demonstrated that ER stress induced by homocysteine compromises endothelial function through suppression of intermediate- and small-conductance KCa (IKCa and SKCa) channels [22]. Whether ER stress inhibits smooth muscle BKCa channels and BKCa-mediated vasodilatory function so far remains uninvestigated.
Forkhead box o transcription factors (FoxOs) serve essential roles in maintaining vascular stability with significance in differentiation, proliferation, and survival of endothelial and smooth muscle cells [23, 24]. The finding that FoxOs may negatively regulate eNOS expression [25] supports a link between endothelial dysfunction and FoxOs dysregulation. Recent studies demonstrated that increase of FoxO3a transcriptional activity contributes to BKCa channel dysfunction in vessels of diabetic animals, which was believed to be mediated by muscle-specific E3 ubiquitin ligase, i.e., atrogin-1 [8, 26]. It was reported that FoxO activity is modulated by ROS [27]. Nevertheless, whether FoxOs respond to ER stress and how the response alters vascular dilator function are barely studied.
The present study aimed to investigate whether ER stress is involved in homocysteine-induced BKCa channel inhibition in coronary arteries. Further exploration of the molecular determinants of homocysteine-induced BKCa channel inhibition aimed to advance our understanding of the cellular mechanisms of ER stress signaling in vascular disorders associated with hyperhomocysteinemia.
RESULTS
ER stress mediates homocysteine-induced loss of dilatory function of BKCa channels in PCAs
Homocysteine induced ER stress in PCASMCs, evidenced by increased expression of ER stress molecules GRP78, ATF6 and enhanced phosphorylation of PERK, eIF2α, and IRE1. Both 4-PBA and TUDCA effectively inhibited ER stress in homocysteine-exposed PCASMCs (Figure 1). The BKCa channel activator NS1619 induced dose-dependent relaxation in endothelium-denuded PCAs. Exposure to homocysteine for 24 h did not alter the resting force of coronary arteries and there were no significant differences among groups with regard to U46619-induced pre-contraction (data not shown), whereas the NS1619-induced vasorelaxation was attenuated (Rmax: 61.9±2.3% vs. 80.6±3.6% in control, p<0.01), which was restored by ER stress inhibitors, 4-PBA (85.4±1.7%) and TUDCA (77.8±3.3%) (p<0.05 vs. homocysteine) (Figure 2A). In addition, exposure of PCAs to a chemical ER stress inducer tunicamycin also significantly attenuated the vasorelaxant response to NS1619 and such attenuation was prevented by either 4-PBA or TUDCA (Figure 2B). In arteries without homocysteine or tunicamycin exposure, the ER stress inhibitor itself did not alter BKCa channel-mediated vasorelaxation (Figure 2A & 2B).
Figure 1. Homocysteine exposure induces ER stress in PCASMCs.
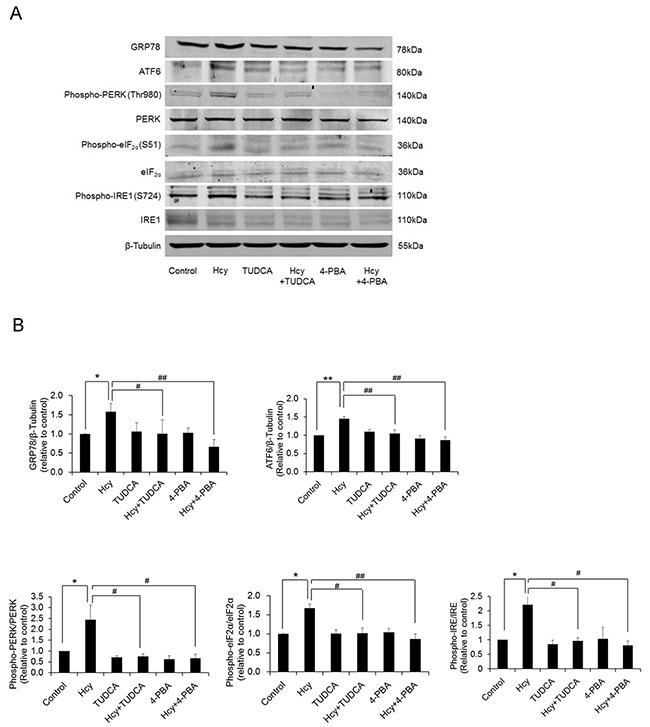
(A) representative blots of ER stress molecules; (B) expression levels of ER stress molecules from 4 independent experiments, each obtained from cell isolates of different heart. *p<0.05, **p<0.01; #p<0.05, ##p<0.01. Hcy: homocysteine, TUDCA and 4-PBA: ER stress inhibitors.
Figure 2. Vasorelaxant responses of PCAs to the BKCa channel activator NS1619 are attenuated after exposure to homocysteine (A, n=8) and tunicamycin (B, n=8).
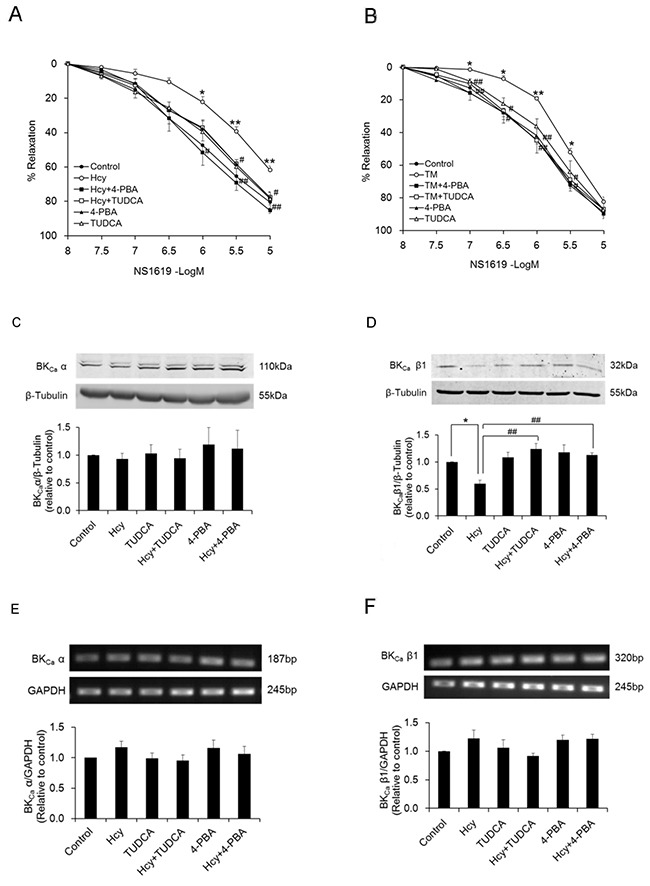
Treatment with ER stress inhibitors, either 4-PBA or TUDCA preserves the relaxant response in both homocysteine- (A) and tunicamycin- (B) exposed arteries. Homocysteine exposure lowers the protein level of BKCa β1 subunit in PCASMCs (n=5) whereas shows no significant effect on the protein content of α subunit (n=8) (C & D). Inhibition of ER stress during homocysteine exposure restores BKCa β1 protein level (D). Homocysteine does not alter mRNA expression levels of both α and β1 subunits of BKCa (E & F, n=7). N indicates the number of independent experiments, each obtained from vessels (a & b) or cell isolates (c-f) of different heart. *p<0.05, **p<0.01 vs. control; #p<0.05, ##p<0.01 vs. Hcy or TM. Hcy: homocysteine, TM: tunicamycin.
Homocysteine reduces protein level of BKCa β1 subunit in PCASMCs via an ER stress-dependent mechanism
Homocysteine exposure lowered the protein level of β1 subunit of BKCa in PCASMCs whereas showed no significant effect on the protein content of α subunit (Figure 2C & 2D). Inhibition of ER stress with either TUDCA or 4-PBA during homocysteine exposure preserved the protein level of BKCa β1 (Figure 2D). Exposure to homocysteine did not alter the mRNA expression of either α or β1 subunit of BKCa in PCASMCs (Figure 2E & 2F).
PERK activation in response to ER stress mediates homocysteine-induced BKCa channel dysfunction
The selective PERK inhibitor GSK2606414 effectively suppressed the activation of PERK branch of ER stress in homocysteine-exposed PCASMCs. Phosphorylation of PERK and its downstream molecule eIF2α was inhibited (Figure 3A). Inhibition of PERK activation with GSK2606414 preserved the relaxant response to NS1619 in PCAs subjected to homocysteine exposure. The maximal response was 77.0±2.8% in homocysteine-exposed arteries with GSK2606414 treatment, in comparison with 62.2±3.0% in those without GSK2606414 treatment (Figure 3C). Further patch clamp recording showed that inhibition of PERK protects smooth muscle BKCa channels from homocysteine-induced inhibition. Both basal K+ current (76.74±5.30 vs. 103.37±7.99 pA/pF in control, p<0.05) and the current in response to NS1619 (137.57±7.67 vs. 195.59±11.59 pA/pF in control, p<0.05) (Figure 4A & 4B) were significantly suppressed in PCASMCs subjected to homocysteine exposure, which largely resulted from BKCa channel inhibition. Differentiation of BKCa component with the specific BKCa channel blocker iberiotoxin showed that after homocysteine exposure basal BKCa channel current decreases from 70.07±8.69 pA/pF to 42.83±5.50 pA/pF (p<0.05) and the BKCa current in response to NS1619 declines from 156.62±10.34 pA/pF to 92.76±3.65 pA/pF (p<0.05) (Figure 4A & 4B). Compared with homocysteine-exposed PCASMCs, cells exposed to homocysteine and co-treated with GSK2606414 exhibited significant (p<0.05) larger current density of BKCa channels under both basal (55.46±4.32 pA/pF) and NS1619-stimulated (126.37±11.89 pA/pF) conditions (Figure 4A & 4B). The enhancement of BKCa channel current by GSK2606414 in homocysteine-exposed PCASMCs was observed to be associated with restoration of the protein level of BKCa β1 (Figure 3B). These data indicated that the PERK branch of ER stress mediates homocysteine-induced BKCa channel inhibition, which is attributed to the loss of β1 subunit of the channel.
Figure 3. Inhibition of activation of PERK branch of ER stress by GSK2606414, evidenced by decreased phosphorylation of PERK and eIF2α (A, n=4), prevents homocysteine-induced loss of BKCa β1 protein in PCASMCs (B, n=4), which is accompanied by an improved vasorelaxant response to NS1619 (C, n=8).
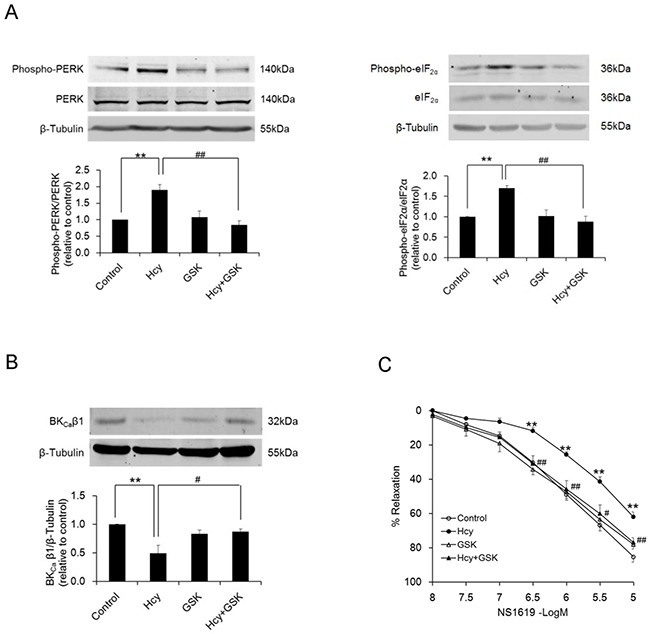
**p<0.01 vs. control; #p<0.05, ##p<0.01 vs. Hcy. N indicates the number of independent experiments, each obtained from cell isolates (a & b) or vessels (c) of different heart. Hcy: homocysteine, GSK: GSK2606414.
Figure 4. Homocysteine exposure suppresses BKCa channel currents in PCASMCs.
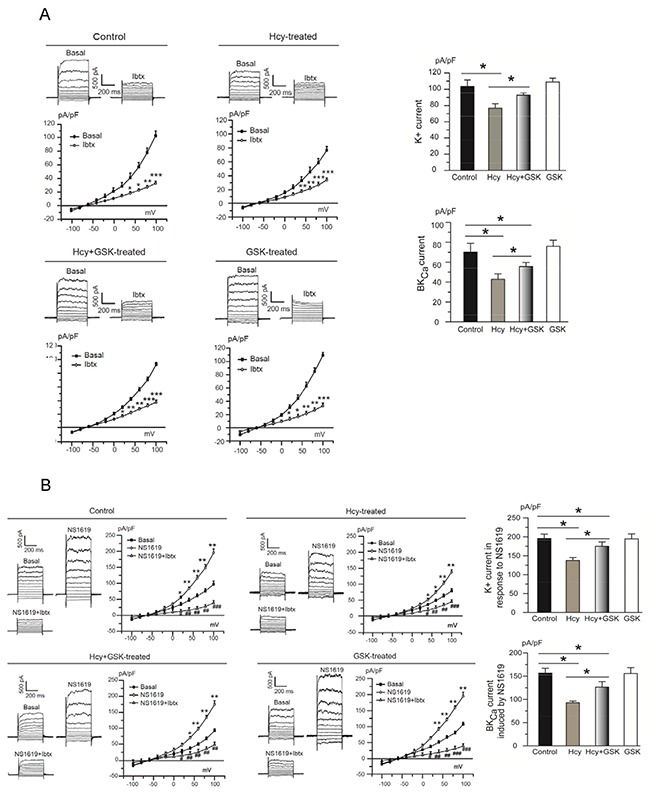
Treatment of the cells with the selective PERK inhibitor GSK2606414 protects BKCa channels against homocysteine-induced inhibition. Original traces and I-V curves of whole-cell K+ current under unstimulated condition (A - left panel) and in response to NS1619 (B - left panel) in cells from different treatment groups before and after application of the BKCa channel blocker iberiotoxin. *p<0.05, **p<0.01, ***p<0.001 vs. basal, #p<0.05, ##p<0.01, ###p<0.001 NS1619+Ibtx vs. NS1619. Summarized data of BKCa channel current from 5 independent experiments under conditions without (A - right panel) or with NS1619 stimulation (B - right panel), each obtained from cell isolates of different heart. *p<0.05. Hcy: homocysteine, GSK: GSK2606414, Ibtx: iberiotoxin.
Activation of PERK branch of ER stress promotes nuclear translocation of FoxO3a in homocysteine-exposed PCASMCs
Homocysteine promoted the translocation of FoxO3a from cytoplasm to nucleus in PCASMCs. While the whole cell protein level of FoxO3a remained unchanged (Figure 5A), the nuclear/cytoplasmic expression ratio of FoxO3a significantly increased in PCASMCs subjected to homocysteine exposure (p<0.01 vs. control). Inhibition of ER stress prevented the increase of nuclear/cytoplasmic FoxO3a ratio induced by homocysteine (Figure 5B). Further experiments showed that activation of the PERK branch of ER stress drives nuclear translocation of FoxO3a in homocysteine exposure. As compared with homocysteine-exposed PCASMCs, cells co-treated with homocysteine and the PERK inhibitor GSK2606414 showed significantly reduced nuclear/cytoplasmic expression ratio of FoxO3a, which does not differ from that of the cells without homocysteine exposure (Figure 5C).
Figure 5. Homocysteine exposure promotes nuclear translocation of FoxO3a and upregulates atrogin-1 expression in PCASMCs.
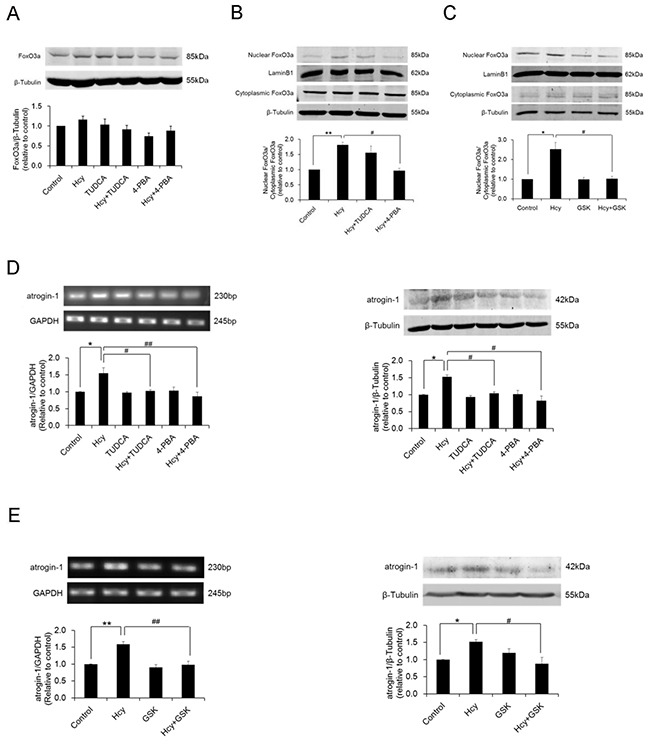
While the overall level of FoxO3a expression remains unchanged (A, n=7), nuclear to cytoplasmic expression ratio of FoxO3a significantly increases in homocysteine-exposed PCASMCs (B, n=4). Inhibition of ER stress and selective blockade of the PERK branch of ER stress both effectively inhibit homocysteine-induced FoxO3a nuclear translocation (B & C, n=4) and atrogin-1 upregulation (D & E, n=4). *p<0.05, #p<0.05; **p<0.01, ##p<0.01. Hcy: homocysteine, TUDCA and 4-PBA: ER stress inhibitors, GSK: GSK2606414, PERK selective blocker.
Activation of FoxO3a by PERK-ER stress signaling mediates homocysteine-induced loss of BKCa β1 via upregulation of atrogin-1
Homocysteine exposure increased mRNA and protein expressions of atrogin-1 in PCASMCs, which were prevented by co-incubation of the cells with ER stress inhibitors TUDCA and 4-PBA (Figure 5D). Prevention of atrogin-1 upregulation was also achieved by the PERK selective inhibitor GSK2606414 (Figure 5E). These results suggest that activation of PERK pathway of ER stress is responsible for homocysteine-induced atrogin-1 expression in PCASMCs.
Silencing of FoxO3a with siRNA normalized atrogin-1 expression in homocysteine-exposed PCASMCs (Figure 6A). Meanwhile, FoxO3a knockdown restored homocysteine-induced reduction of BKCa β1 protein (Figure 6B), with consequent enhancement of BKCa channel currents. Compared with homocysteine-exposed control cells, basal BKCa currents increased from 41.37±5.39 pA/pF to 58.77±2.92 pA/pF and BKCa currents activated by NS1619 enhanced from 86.77±6.49 pA/pF to 119.19±11.25 pA/pF in homocysteine-exposed cells that transfected with FoxO3a siRNA (Figure 6D & 6E). These results in conjunction with the finding of preserved BKCa β1 protein level in atrogin-1-silenced PCASMCs therefore unraveled atrogin-1 as a critical link between FoxO3a activation and BKCa β1 loss in homocysteine-induced BKCa channel dysfunction (Figure 6C).
Figure 6.
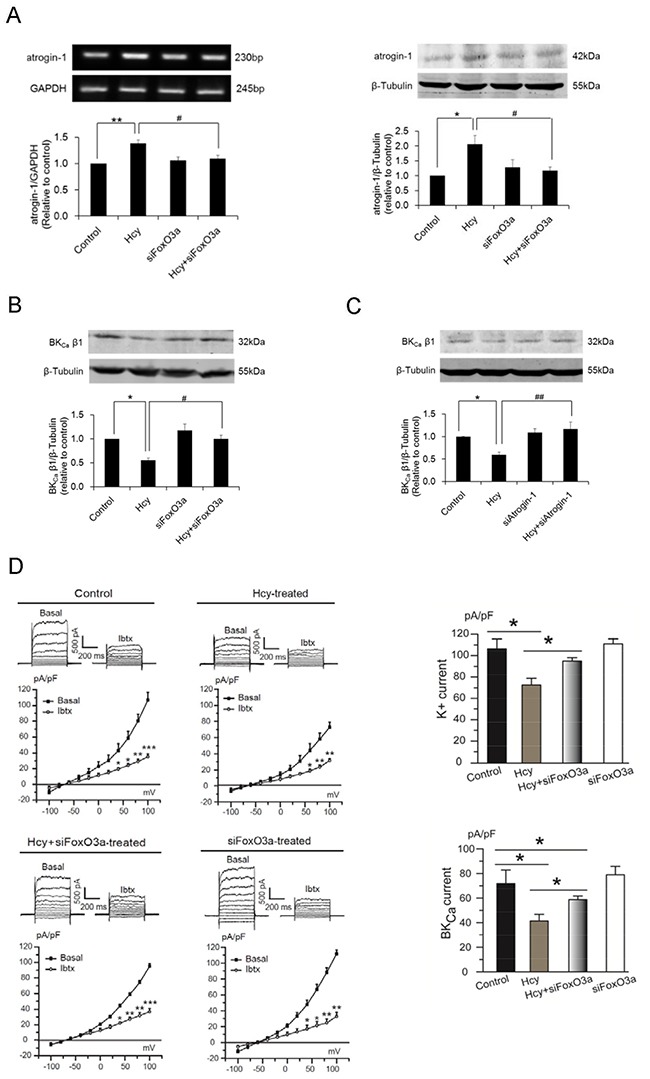
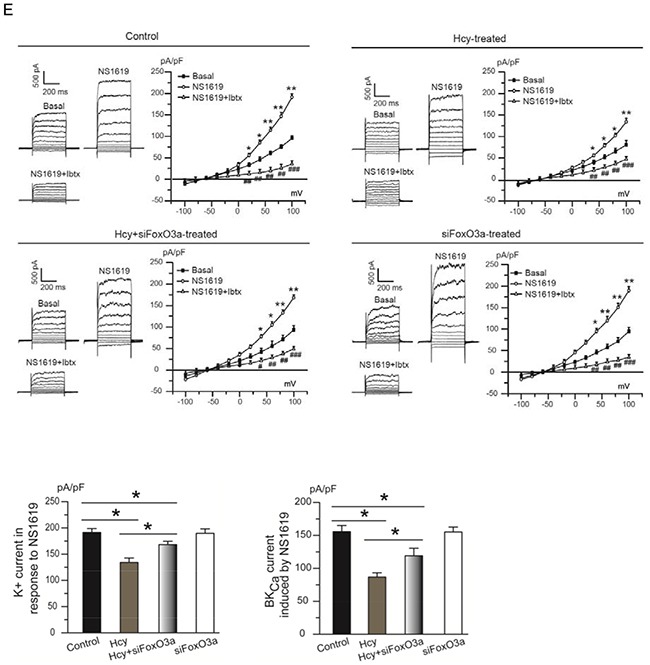
Silencing of the FoxO3a gene by siRNA prevents atrogin-1 upregulation (A) and BKCa β1 downregulation (B) in homocysteine-exposed PCASMCs. The role of atrogin-1 as a critical mediator of homocysteine-induced BKCa β1 loss is evidenced by the preserved protein level of BKCa β1 in homocysteine-exposed cells that are transfected with atrogin-1 siRNA (C). *p<0.05, **p<0.01; #p<0.05, ##p<0.01. Silencing of the FoxO3a gene prevents homocysteine-induced inhibition of BKCa channel currents (D & E). Original traces and I-V curves of whole-cell K+ current under unstimulated condition (D - left panel) and in response to NS1619 (E - left panel) in cells from different treatment groups before and after application of the BKCa channel blocker iberiotoxin. *p<0.05, **p<0.01, ***p<0.001 vs. basal, #p<0.05, ##p<0.01, ###p<0.001 NS1619+Ibtx vs. NS1619. Summarized data of BKCa channel currents from 5 independent experiments under conditions without (D - right panel) or with NS1619 stimulation. (E - lower panel), each obtained from cell isolates of different heart. *p<0.05. Hcy: homocysteine, Ibtx: iberiotoxin.
DISCUSSION
The present study for the first time demonstrated that in coronary arteries 1) ER stress mediates homocysteine-induced smooth muscle BKCa channel dysfunction; 2) loss of β1 subunit of BKCa triggered by the activation of PERK branch of ER stress is an underlying basis of the channel inhibition caused by homocysteine; 3) PERK-dependent activation of FoxO3a is responsible for the BKCa β1 loss and atrogin-1 acts as a critical downstream mediator of FoxO3a.
The ER of vascular cells becomes stressed when subjected to homocysteine exposure. As we previously observed in endothelial cells [22], homocysteine also induced ER stress in smooth muscle of coronary arteries. All three branches of the unfolded protein response (UPR), i.e., PERK, IRE1, and ATF6, were activated in homocysteine-exposed PCASMCs. Homocysteine caused significant loss of vasodilatory function of BKCa channels in endothelium-denuded coronary arteries and such loss was prevented by ER stress inhibitors, suggesting the role of ER stress in homocysteine-induced smooth muscle BKCa channel inhibition. Relaxation studies in arteries exposed to tunicamycin, a chemical ER stress inducer, provided parallel evidence that BKCa channels are susceptible to ER stress.
Further studies of channel expression demonstrated that decrease of β1 subunit resulting from ER stress is responsible for homocysteine-induced loss of vasodilatory function of BKCa channels. The β1 subunit has been well documented for its role in maintaining normal BKCa channel function in vasculature. Mice lacking BKCa β1 exhibits vascular smooth muscle hypercontractility and elevated blood pressure [28, 29]. In type 2 diabetes mellitus, downregulation of BKCa β1 was demonstrated to be a contributor to enhanced arterial tone [30]. The present study added one more piece of evidence supporting the significance of the β1 subunit of BKCa channels in vascular pathophysiology. More importantly, we for the first time demonstrated that ER stress is a key mediator of homocysteine-induced loss of BKCa β1, which provided new mechanistic insight into hyperhomocysteinemia-associated vascular dysfunction.
Protein homeostasis is maintained by a balanced regulation between synthesis and degradation. The reduced protein level whereas unchanged mRNA content of BKCa β1 following exposure to homocysteine indicates posttranscriptional regulation of the β1 subunit that might involve enhanced degradation of BKCa β1 protein. As a dominant mechanism in mammalian cells that controls the protein degradation, the ubiquitin-proteasome system is composed of an enzyme cascade involving E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) that facilitate the ubiquitination of the target protein for subsequent destruction by the proteasome [31]. Atrogin-1 (FbxO-32) is a muscle specific E3 ubiquitin ligase [32] that belongs to F-box only (FbxO) protein family [33]. The ability of atrogin-1 binding to the PDZ-binding motif in substrates [32] makes it potentially important in BKCa protein regulation since the PDZ-binding motif is present in most BKCa β1 isoforms in different species including human [26]. In fact, previous investigation in animal model of diabetes demonstrated that atrogin-1 facilitates BKCa β1 degradation in vascular smooth muscle cells [26]. In this study we observed that loss of BKCa β1 caused by homocysteine is also associated with an upregulation of atrogin-1, which suggests the role of E3 ubiquitin ligase in hyperhomocysteinemia-related vascular pathology.
FbxO expression is positively regulated by FoxOs [34]. In studies of diabetes, Zhang and colleges for the first time attributed atrogin-1-promoted BKCa β1 downregulation to FoxO activation in vascular smooth muscle cells [26]. We observed that in homocysteine-exposed PCASMCs, enhanced expression of atrogin-1 is associated with increased nuclear to cytoplasmic expression ratio of FoxO3a that implies increased FoxO3a transcriptional activity. siRNA knockdown of FoxO3a prevented homocysteine-induced atrogin-1 upregulation. In conjunction with the findings that silencing of FoxO3a or atrogin-1 normalized the protein level of BKCa β1 in homocysteine-exposed PCASMCs, these data provided additional proof of FoxO/atrogin-1 cascade in BKCa β1 regulation. The findings of restoration of BKCa channel currents by FoxO3a silencing further confirmed the significance of FoxO3a in homocysteine-induced BKCa channel inhibition.
Homocysteine-induced nuclear accumulation of FoxO3a and upregulation of atrogin-1 in PCASMCs were prevented by ER stress inhibitors and selective PERK inhibitor, suggesting that activation of the PERK branch of the UPR in response to ER stress stands upstream of FoxO3a activation. Our finding of PERK-dependent FoxO3a activation is in agreement with the report in human lung carcinoma cell line that depletion of PERK limits FoxO activity [35]. The essential role of PERK in FoxO/atrogin-1 activation therefore explains the capacity of the PERK inhibitor in restoring homocysteine-induced loss of β1 subunit of BKCa and the resulting enhancement of channel currents.
Limitations of the study
By uncovering the role of ER stress in BKCa channel inhibition, we identified a new mechanistic link between hyperhomocystinemia and vascular dysfunction that is characterized as PERK-FoxO3a-atrogin-1-BKCa β1 regulation. Although our results demonstrated that activation of PERK branch of the UPR increases nuclear localization of FoxO3a thereby enhancing its activity to promote atrogin-1-mediated BKCa β1 degradation, how PERK activation facilitates nuclear translocation of FoxO3a was not further elucidated. Akt-mediated phosphorylation of FoxO is known as an important regulatory mechanism controlling the subcellular localization of FoxO [36], whether Akt is a possible mediator of PERK-induced FoxO3a activation in our study subject warrants further investigation. Another issue worthy of attention is that although inhibition of PERK and silencing of FoxO3a both restored the protein level of BKCa β1 in homocysteine-exposed PCASMCs, these treatments were unable to fully recover the whole-cell BKCa channel currents. These results implied that mechanisms other than PERK-FoxO3a-mediated loss of β1 subunit may also take part in homocysteine-induced inhibition of BKCa channels. One potential possibility is inhibition of the activity of single BKCa channel. It will be interesting to further study whether PERK and/or the other two UPR branches, i.e. IRE1 and ATF6 have an impact on this by employing single-channel recording techniques. Moreover, in light of the evidence that ROS virtually eliminates activation of single BKCa channel by targeting a cysteine residue near the Ca2+ bowl of the BKCa α subunit [37], and the cross-talk between ER stress and oxidative stress [38], future studies aiming at signaling cascades linking ER stress and ROS may help provide profound knowledge on the role of ER stress in vascular BKCa channel dysfunction in hyperhomocysteinemia.
In conclusion, the present study demonstrated that homocysteine impairs coronary dilator function through a mechanism involving ER stress-mediated inhibition of smooth muscle BKCa channels. Atrogin-1-promoted degradation of BKCa β1 subunit plays a key role in the ER stress-mediated channel inhibition that could be attributed to PERK-dependent FoxO3a activation. This study provides a new understanding of the molecular mechanisms involved in homocysteine-induced vascular injury and offers new perspectives in the development of vascular protection strategies for patients with hyperhomocystinemia.
MATERIALS AND METHODS
Fresh hearts of young adult pigs (~35 kg) were collected from a local slaughterhouse. Once excised, the hearts were placed in cold (4°C), pre-oxygenated (95%O2/5%CO2) Krebs solution and immediately transferred to the laboratory for dissection of small coronary arteries as in our previous studies [15, 39, 40]. All experiments were in accordance with institutional guidelines. The study was approved by the Animal Experimentation Ethics Committee, and Safety Office of the Chinese University of Hong Kong (Ref. No. 14/010/GRF).
Isolation and primary culture of porcine coronary artery smooth muscle cells (PCASMCs)
PCASMCs were cultured after isolation by enzymatic digestion from porcine small coronary arteries [41]. In brief, small coronary arteries were cut open longitudinally and the endothelium was gently scraped off with a microsurgical blade. The tissue strip was then washed with cold Krebs and dissected into 1×1 mm2 strips, followed by 30-min digestion at 37°C in 2 ml of phosphate-buffered saline solution (PBS), containing 2 mg/ml of collagenase (type 2; Worthington Biochemicals, USA), 0.5 mg/ml of papain (Worthington Biochemicals, USA), 1.75 mg/ml of DL-Dithiothreitol (DTT, Sigma, USA), and 5 mg/ml albumin bovine (BSA, Amresco, USA). The enzymatic activity was stopped by Dulbecco's Modified Eagle Medium (DMEM, Thermo Fisher Scientific, USA) containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific, USA). The suspension was centrifuged at 1600 rpm for 5 min. Cells were then resuspended in 12 ml DMEM containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Thermo Fisher Scientific, USA). After 1-h incubation at 37°C, the medium was replaced once to remove unattached cells. Attached PCASMCs were cultured in a humidified incubator with 5%CO2 at 37°C. For maintaining electrophysiological properties of isolated PCASMCs, only primary cultured cells were used for experiments.
Patch-clamp recording of BKCa channel currents in PCASMCs
Whole-cell K+ current of PCASMCs were recorded by patch-clamp (EPC10, HEKA, Lambrecht, Germany) at room temperature (20-24°C) with further differentiation of the BKCa component [41]. Patch pipettes with resistance of 3-5 MΩ were filled with a solution containing (mmol/L): NaCl 10, KCl 110, MgCl2 4, CaCl2 7, ethylene-glycol tetraacetic acid (EGTA) 10, Mg-ATP 5, and HEPES 10. The bath solution contained (mmol/L): NaCl 130, KCl 5, MgCl2 1.2, CaCl2 1.5, Glucose 10, and HEPES 10. PCASMCs were held at -60 mV and voltage steps ranging from -100 to +100 mV were applied for 500 milliseconds in 20-mV step increments. K+ currents without and with stimulation of the BKCa channel activator NS1619 (3 μmol/L) were recorded with further application of the specific BKCa blocker iberiotoxin (100 nmol/L) to identify the BKCa current. Data were analyzed with PulseFit software (HEKA). The current was normalized by cell capacitance into current densities (pA/pF).
Isometric force study
Porcine small coronary arteries (PCAs) (300-500 μm in diameter) were dissected from the branches of left anterior descending artery (LAD) and cut into cylindrical rings with 2-mm in length. Prior to mounting, normalization, and equilibration in a four-channel Mulvany myograph (Model 610M, J.P.Trading, Aarhus, Denmark) as published elsewhere [15, 40], the rings were denuded of endothelium by gently rubbing the intraluminal surface with wires and successful removal of endothelium was confirmed by the lack of relaxant response to the endothelium-dependent vasodilators, as in our previous studies [42, 43]. Cumulative dose-response curve to the BKCa activator NS1619 (-8~-5 Log M) were then established following U46619 pre-contraction in the endothelium-denuded rings.
Reverse-transcriptase polymerase chain reaction (RT-PCR) analysis
Total RNA from PCASMCs was extracted using total RNA reagent (RNAiso Plus, TaKaRa Biotechnology, China) and mRNA was converted to cDNA using reverse transcriptase kit (PrimeScript™ RT Master Mix, TaKaRa Biotechnology, China) according to manufacturer's instructions. PCR amplification was performed with Taq DNA polymerase (GoTaq® G2 Flexi DNA Polymerase, Promega, USA) and the thermal cycling conditions were: 95°C for 30s, followed by 30 cycles of 95°C for 30s, 50°C for 1 min, 72°C for 1 min, and completed by 72°C for 5 min. PCR products were then separated by 1.5% agarose gel electrophoresis. Density analysis of the signals was evaluated using the GelQuant.NET 1.8.2 software (Biochem Lab Solutions, University of California, San Francisco). Primers used for PCR amplification were as follows: BKCa α, 5’-GCCAGCAACTTTCACTAC-3’ (forward) and 5’-CTGACAGGATAACGCACA-3’ (reverse); BKCa β1, 5’-TGTGCTGTCATCGCCTACT-3’ (forward) and 5’-ACCTGGTGCTCGTGGAAC-3’ (reverse); FoxO3a, 5’-ACATGGCCGGAACCATGAAT-3’ (forward) and 5’-GTCCAAACACTGTGCTGCTG-3’ (reverse); atrogin-1, 5’-TGGATGGCTGGGGATACAGA-3’ (forward) and 5’-TAAATTCCCCGCCAGTGTCC-3’ (reverse). GAPDH was amplified in parallel as an internal loading control with primers 5’-GGTCGGAGTGAACGGATTT-3’ (forward) and 5’-ATTTGATGTTGGCGGGAT-3’ (reverse).
Western blot analysis
Protein expression at whole cell level
Whole cell lysates of PCASMCs were used for protein expression analysis of 1) α and β1 subunits of BKCa channels; 2) ER stress molecules including 78-kDa glucose regulated protein (GRP78), protein kinase RNA-like ER kinase (PERK), phosphorylated PERK, inositol-requiring enzyme 1 (IRE1), phosphorylated IRE1, activating transcription factor 6 (ATF6), eukaryotic translation initiation factor 2α (eIF2α) and phosphorylated eIF2α; 3) FoxO3a; and 4) atrogin-1.
Whole cell protein was extracted with lysis buffer containing protease and phosphatase inhibitor cocktail (Roche Diagnostic). The protein extraction was centrifuged at 4°C for 20 min at 12000 rpm and mixed with loading buffer, heated up to 100°C for 10 min, then fractionated by a denaturing 8% sodium-dodecyl-sulfate polyacrylamide-gel electrophoresis (30 μg per lane) for 90 min at 120V and electro-transferred to polyvinylidene difluoride (PVDF) membrane (Thermo Scientific) for 90 min at 100V. The membrane was blocked with 5% non-fat milk/TBST for 1 h at room temperature and incubated with the primary antibody in 5% non-fat milk/TBST overnight at 4°C. Primary antibodies used include BKCa α (1:1000 dilution, Abcam), BKCa β1 (1:500, Abcam), GRP78 (1:1000, Abcam), PERK (1:1000, Cell Signaling), phosphorylated (Thr980) PERK (1:500, Bioss), IRE1 (1:1000, Abcam), phosphorylated (Ser724) IRE1 (1:1000, Abcam), eIF2α (1:1000, Abcam), phosphorylated (Ser51) eIF2α (1:500, Abcam), ATF6 (1:1000, Abnova), FoxO3a (1:500, Abcam), and atrogin-1 (1:500, Abcam). The membrane was then washed in TBST followed by incubation with secondary IRDye800®-infrared fluorescent dye-conjugated goat anti-rabbit or rabbit anti-mouse antibody (1:10000, Rockland) in TBST for 1 h at room temperature. Imaging was performed at a wavelength of 800 nm by using Odyssey gel imaging scanner (Li-Cor Biosciences). The intensity of the bands was analyzed by Quantity One imaging system (Version 4.6.6, Bio-Rad) and β-tubulin (1:2500, Abcam) was used as internal loading control.
Protein expression at subcellular level
The NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, USA) were used to separate nuclear and cytoplasmic fractions of PCASMCs. Briefly, primary cultured PCASMCs were harvested with trypsin-EDTA (Thermo Fisher Scientific, USA) and centrifuged at 500×g for 5 min to collect the cell pellet. After wash in PBS, cells were centrifuged to remove the wash buffer and ice-cold cytoplasmic extraction reagents I and II were then added to the cell pellet in proportion of 200:11 (μl). The supernatant was collected as cytoplasmic lysate after 5-min centrifugation at 16,000×g. Nuclear extraction reagent (100 μl) was then added to the precipitate for further centrifugation (16,000×g, 10 min) to collect the supernatant as the nuclear lysate. Western blotting was then performed using proteins from nuclear and cytoplasmic preparations for expression analysis of FoxO3a. Lamin B1 (1:2000, Abcam) and β-tubulin (1:2500, Abcam) were used as nuclear and cytoplasmic loading control respectively.
Gene silencing by siRNAs
Primary cultured PCASMCs were transfected with specific siRNA targeting porcine FoxO3a and atrogin-1, respectively. For each target, three pairs of siRNAs were designed along with a scramb-led control siRNA: siFoxO3a-1: 5’-CCGGCUGGA AGAACUCUAUTT-3’ (sense), 5’-AUAGAGUUCUU CCAGCCGGTT-3’(antisense); siFoxO3a-2: 5’-CCGGA ACCAUGAAUCUCAATT-3’ (sense), 5’-UUGAGAUUC AUGGUUCCGGTT-3’ (antisense); siFoxO3a-3: 5’-GG AGCUUGGAAUGUGACAUTT-3’ (sense), 5’-AUGUC ACAUUCCAAGCUCCTT-3’ (antisense); siAtrogin-1-1, 5’-CCUUCAAAGGCCUCACCUUTT-3’ (sense), 5’-AAG GUGAGGCCUUUGAAGGTT-3’ (antisense); siAtrogin-1 -2, 5’-GCAACUGAACAUCAUGCAATT-3’ (sense), 5’-UUGCAUGAUGUUCAGUUGCTT-3’ (antisense); siAtrogin-1-3, 5’-GCCACAUUCUUUCCUGGAATT-3’ (sense), 5’-UUCCAGGAAAGAAUGUGGCTT-3’ (antisense). To perform transfection, PCASMCs were seeded in 6-well plate and cultured in DMEM containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin until they reached 70-80% confluence. Cells were then incubated in Opti-MEM® I Reduced Serum Media (Thermo Fisher Scientific, USA) for 24 h. The mixture of siRNA (gene-specific or corresponding scrambled control) (20 μmol/L, Shanghai GenePharma, China) and Lipofectamine® 2000 Transfection Reagent (Thermo Fisher Scientific, USA) in proportion of 5:2 was added with further incubation for 6 h. Culture medium was then changed to normal DMEM and siRNA knockdown efficiency was determined at both mRNA level by RT-PCR and protein level by western blotting after 24 and 48 h, respectively. For each target, the siRNA displaying the highest knockdown efficiency was chosen for subsequent experiments.
Experimental protocols
Studies of the role of ER stress in homocysteine-induced BKCa channel dysfunction
Effect of homocysteine on BKCa channel-mediated vasorelaxation – role of ER stress
PCA rings taken from the same branch of the LAD were scraped off endothelium before allocated to different treatment groups and treated for 24 h as: control, homocysteine (100 μmol/L), tauroursodeoxycholate (TUDCA, ER stress inhibitor, 200 mmol/L), homocysteine+TUDCA, 4-phenylbutyric acid (4-PBA, ER stress inhibitor, 2 mmol/L), homocysteine+4-PBA. After pre-constriction with U46619, vasorelaxant responses to the BKCa activator NS1619 (-8~-5 LogM) were studied.
Effect of ER stress inducer on BKCa channel-mediated vasorelaxation
Endothelium-denuded PCAs were allocated and treated for 4 h as: control, tunicamycin (chemical ER stress inducer, 5 μg/ml), TUDCA, tunicamycin+TUDCA, 4-PBA, tunicamycin+4-PBA. NS1619-induced vasorelaxation was then studied in U46619 pre-constriction.
Studies of the effect of homocysteine on BKCa channel expression – role of ER stress
PCASMCs were divided into six groups and treated for 24 h as: control, homocysteine, TUDCA, homocysteine+TUDCA, 4-PBA, and homocysteine+4-PBA. Cells were then collected for determination of 1) protein expression and phosphorylation of ER stress molecules, i.e., GRP78, ATF6, p-PERK/PERK, p-eIF2α/eIF2α, and p-IRE1/IRE1; and 2) mRNA and protein expressions of α and β1 subunits of BKCa.
Studies of the role of PERK branch of ER stress in homocysteine-induced BKCa channel dysfunction
Effect of pharmacological inhibition of PERK on BKCa channel-mediated relaxation in homocysteine-exposed PCAs
Endothelium-denuded PCAs were allocated and treated for 24 h as: control, homocysteine, GSK2606414 (PERK selective inhibitor [44], Selleckchem, USA, 500 nmol/L), and homocysteine+GSK2606414. NS1619-induced relaxation was studied after U46619 preconstriction.
Effect of pharmacological inhibition of PERK on BKCa channel expression and BKCa current in homocysteine-exposed PCASMCs
PCASMCs were grouped and treated as above, followed by determination of protein levels of BKCa, p-PERK/PERK, and p-eIF2α/eIF2α and recording of BKCa current.
Studies of the effect of homocysteine on FoxO3a and atrogin-1 – role of PERK branch of ER stress
Role of ER stress in the regulation of FoxO3a and atrogin-1 in homocysteine-exposed PCASMCs
PCASMCs were allocated to the following treatment groups: control with or without 4-PBA or TUDCA, and homocysteine with or without 4-PBA or TUDCA. After 24 h, mRNA and protein expressions of atrogin-1, protein expression of FoxO3a, and FoxO3a protein level in both the nucleus and the cytoplasm were examined.
Role of PERK branch of ER stress in homocysteine-induced FoxO3a and atrogin-1 regulation
PCASMCs were treated for 24 has: control, homo-cysteine, GSK2606414, and homocysteine+GSK2606414, followed by determination of mRNA and protein expressions of atrogin-1 and nuclear/cytoplasmic expression of FoxO3a.
Studies of the role and relations of FoxO3a and atrogin-1 in homocysteine-induced BKCa channel dysfunction
Effect of FoxO3a knockdown on atrogin-1 expression in homocysteine-exposed PCASMCs
PCASMCs were transfected with scrambled control or specific FoxO3a siRNA before subjected to 24-h exposure to homocysteine. mRNA and protein expressions of atrogin-1 were then examined.
Effect FoxO3a knockdown on BKCa channel expression and activity in homocysteine-exposed PCASMCs
PCASMCs transfected with scrambled control or specific FoxO3a siRNA were treated as above, followed by detection of protein expression and current recording of BKCa channels.
Effect of atrogin-1 knockdown on BKCa channel expression in homocysteine-exposed PCASMCs
PCASMCs were transfected with scrambled control or specific siRNA targeting atrogin-1 before subjected to 24-h exposure to homocysteine. Cells were then examined for BKCa protein level.
Data analysis
Relaxation was expressed as the percentage decrease of pre-contraction induced by U46619. All data were presented as mean±s.e.m. Statistical analyses were performed by one-way ANOVA followed by Scheffe tests (SPSS, version 20). Differences were considered as significant at p<0.05.
Abbreviations
- 4-PBA
4-phenylbutyric acid
- ATF6
activating transcription factor 6
- BKCa channel
large-conductance Ca2+-activated K+ channel
- eIF2α
eukaryotic translation initiation factor 2α
- ER
endoplasmic reticulum
- FoxO
forkhead box O transcription factor
- GRP78
78-kDa glucose regulated protein
- IRE1
inositol-requiring enzyme 1
- PCAs
porcine small coronary arteries
- PCASMCs
porcine small coronary artery smooth muscle cells
- PERK
protein kinase RNA-like ER kinase
- ROS
reactive oxygen species
- TUDCA
tauroursodeoxycholate
- UPR
unfolded protein response
Footnotes
CONFLICTS OF INTEREST
None.
FUNDING
This work was supported by grants from Research Grant Council of Hong Kong (GRF CUHK14118414), Lui Che Woo Institute of Innovative Medicine (CARE theme 8303303), and CUHK Direct Grant 4054182 & 4054273.
REFERENCES
- 1.Cox DH, Aldrich RW. Role of the beta1 subunit in large-conductance Ca(2+)-activated K(+) channel gating energetics. Mechanisms of enhanced Ca(2+) sensitivity. J Gen Physiol. 2000;116:411–32. doi: 10.1085/jgp.116.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- 3.Mistry DK, Garland CJ. Nitric oxide (NO)-induced activation of large conductance Ca2-dependent K channels (BKCa) in smooth muscle cells isolated from the rat mesenteric artery. Br J Pharmacol. 1998;124:1131–40. doi: 10.1038/sj.bjp.0701940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–3. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 5.Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation. 2003;107:769–76. doi: 10.1161/01.cir.0000047278.28407.c2. [DOI] [PubMed] [Google Scholar]
- 6.Weston AH, Feletou M, Vanhoutte PM, Falck JR, Campbell WB, Edwards G. Bradykinin-induced, endothelium-dependent responses in porcine coronary arteries: involvement of potassium channel activation and epoxyeicosatrienoic acids. Br J Pharmacol. 2005;145:775–84. doi: 10.1038/sj.bjp.0706256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Li PY, Cheng J, Mao L, Wen J, Tan XQ, Liu ZF, Zeng XR. Function of BKCa channels is reduced in human vascular smooth muscle cells from Han Chinese patients with hypertension. Hypertension. 2013;61:519–25. doi: 10.1161/HYPERTENSIONAHA.111.00211. [DOI] [PubMed] [Google Scholar]
- 8.Lu T, Chai Q, Yu L, d’Uscio LV, Katusic ZS, He T, Lee HC. Reactive oxygen species signaling facilitates FOXO-3a/FBXO-dependent vascular BK channel beta1 subunit degradation in diabetic mice. Diabetes. 2012;61:1860–8. doi: 10.2337/db11-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nygård O, Nordrehaug JE, Refsum H, Ueland PM, Farstad M, Vollset SE. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med. 1997;337:230–6. doi: 10.1056/nejm199707243370403. [DOI] [PubMed] [Google Scholar]
- 10.Chen C, Conklin BS, Ren Z, Zhong DS. Homocysteine decreases endothelium-dependent vasorelaxation in porcine arteries. J Surg Res. 2002;102:22–30. doi: 10.1006/jsre.2001.6304. [DOI] [PubMed] [Google Scholar]
- 11.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–92. doi: 10.1097/01.ASN.0000132474.50966.DA. [DOI] [PubMed] [Google Scholar]
- 12.Hedayati N, Annambhotla S, Jiang J, Wang X, Chai H, Lin PH, Yao Q, Chen C. Growth hormone-releasing peptide ghrelin inhibits homocysteine-induced endothelial dysfunction in porcine coronary arteries and human endothelial cells. J Vasc Surg. 2009;49:199–207. doi: 10.1016/j.jvs.2008.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antoniades C, Antonopoulos AS, Tousoulis D, Marinou K, Stefanadis C. Homocysteine and coronary atherosclerosis: from folate fortification to the recent clinical trials. Eur Heart J. 2009;30:6–15. doi: 10.1093/eurheartj/ehn515. [DOI] [PubMed] [Google Scholar]
- 14.Topal G, Brunet A, Millanvoye E, Boucher JL, Rendu F, Devynck MA, David-Dufilho M. Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free Radic Biol Med. 2004;36:1532–41. doi: 10.1016/j.freeradbiomed.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Yang Q, Xue HM, Underwood MJ, Yu CM. Mechanistic studies of AVE3085 against homocysteine in endothelial protection. Cardiovasc Drugs Ther. 2013;27:511–20. doi: 10.1007/s10557-013-6478-5. [DOI] [PubMed] [Google Scholar]
- 16.Au AL, Seto SW, Chan SW, Chan MS, Kwan YW. Modulation by homocysteine of the iberiotoxin-sensitive, Ca 2-activated K channels of porcine coronary artery smooth muscle cells. Eur J Pharmacol. 2006;546:109–19. doi: 10.1016/j.ejphar.2006.06.073. [DOI] [PubMed] [Google Scholar]
- 17.Cai B, Gong D, Pan Z, Liu Y, Qian H, Zhang Y, Jiao J, Lu Y, Yang B. Large-conductance Ca 2 -activated K currents blocked and impaired by homocysteine in human and rat mesenteric artery smooth muscle cells. Life Sci. 2007;80:2060–6. doi: 10.1016/j.lfs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Spitler KM, Matsumoto T, Webb RC. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. 2013;305:H344–53. doi: 10.1152/ajpheart.00952.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassan M, Galan M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652–61. doi: 10.1161/ATVBAHA.112.249318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galan M, Kassan M, Choi SK, Partyka M, Trebak M, Henrion D, Matrougui K. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60:71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu S, Gao X, Yang S, Meng M, Yang X, Ge B. The role of endoplasmic reticulum stress in endothelial dysfunction induced by homocysteine thiolactone. Fundam Clin Pharmacol. 2015;29:252–9. doi: 10.1111/fcp.12101. [DOI] [PubMed] [Google Scholar]
- 22.Wang XC, Sun WT, Yu CM, Pun SH, Underwood MJ, He GW, Yang Q. ER stress mediates homocysteine-induced endothelial dysfunction: modulation of IKCa and SKCa channels. Atherosclerosis. 2015;242:191–8. doi: 10.1016/j.atherosclerosis.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 23.Paik JH. FOXOs in the maintenance of vascular homoeostasis. Biochem Soc Trans. 2006;34:731–4. doi: 10.1042/BST0340731. [DOI] [PubMed] [Google Scholar]
- 24.Oellerich MF, Potente M. FOXOs and sirtuins in vascular growth, maintenance, and aging. Circ Res. 2012;110:1238–51. doi: 10.1161/CIRCRESAHA.111.246488. [DOI] [PubMed] [Google Scholar]
- 25.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382–92. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang DM, He T, Katusic ZS, Lee HC, Lu T. Muscle-specific f-box only proteins facilitate bk channel beta(1) subunit downregulation in vascular smooth muscle cells of diabetes mellitus. Circ Res. 2010;107:1454–9. doi: 10.1161/CIRCRESAHA.110.228361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klotz LO, Sanchez-Ramos C, Prieto-Arroyo I, Urbanek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72. doi: 10.1016/j.redox.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–6. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 29.Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87:E53–60. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- 30.Nystoriak MA, Nieves-Cintron M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell’acqua ML, Scott JD, Santana LF, Navedo MF. AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res. 2014;114:607–15. doi: 10.1161/CIRCRESAHA.114.302168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463–78. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–5. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kipreos ET, Pagano M. The F-box protein family. Genome Biol. 2000;1:REVIEWS3002. doi: 10.1186/gb-2000-1-5-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, Cohen SM. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013;27:441–9. doi: 10.1101/gad.201731.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–87. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 37.Tang XD, Garcia ML, Heinemann SH, Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol. 2004;11:171–8. doi: 10.1038/nsmb725. [DOI] [PubMed] [Google Scholar]
- 38.Dandekar A, Mendez R, Zhang K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol Biol. 2015;1292:205–14. doi: 10.1007/978-1-4939-2522-3_15. [DOI] [PubMed] [Google Scholar]
- 39.Huang JH, He GW, Xue HM, Yao XQ, Liu XC, Underwood MJ, Yang Q. TRPC3 channel contributes to nitric oxide release: significance during normoxia and hypoxia-reoxygenation. Cardiovasc Res. 2011;91:472–82. doi: 10.1093/cvr/cvr102. [DOI] [PubMed] [Google Scholar]
- 40.Yang Q, Huang JH, Man YB, Yao XQ, He GW. Use of intermediate/small conductance calcium-activated potassium-channel activator for endothelial protection. J Thorac Cardiovasc Surg. 2011;141:501–10. 510.e1. doi: 10.1016/j.jtcvs.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 41.Han JG, Yang Q, Yao XQ, Kwan YW, Shen B, He GW. Role of large-conductance calcium-activated potassium channels of coronary arteries in heart preservation. J Heart Lung Transplant. 2009;28:1094–101. doi: 10.1016/j.healun.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 42.Chen ZW, Huang Y, Yang Q, Li X, Wei W, He GW. Urocortin-induced relaxation in the human internal mammary artery. Cardiovasc Res. 2005;65:913–20. doi: 10.1016/j.cardiores.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 43.Bai XY, Zhang P, Yang Q, Liu XC, Wang J, Tong YL, Xiong SJ, Liu LH, Wang L, He GW. Suxiao jiuxin pill induces potent relaxation and inhibition on contraction in human artery and the mechanism. Evid Based Complement Alternat Med. 2014;2014:956924. doi: 10.1155/2014/956924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl) phenyl] acetyl}-2, 3-dihydro-1 H-indol-5-yl)-7 H-pyrrolo [2, 3-d] pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) J Med Chem. 2012;55:7193–207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]