

Significance
The poor prognosis of epithelial ovarian cancer (EOC) has not improved for several decades because of drug resistance to current anticancer drugs. Furthermore, few molecularly targeted agents are effective for EOC, likely because EOC has high tumor heterogeneity. Discovering new drug targets and mechanisms involved in the progression of EOC is therefore sorely needed. Our multiple CRISPR and RNAi-based in vivo loss-of-function screens have identified multiple new EOC candidate drug targets, including the druggable oncogene KPNB1, whose inhibition caused multiphased cell cycle arrest and induced apoptosis. Ivermectin, a Food and Drug Administration-approved antiparasitic drug, exerts KPNB1-dependent antitumor effects and synergistically inhibits tumor growth in combination with paclitaxel, and therefore represents a new potential combinatorial therapy for EOC through drug repositioning.
Keywords: ovarian cancer, KPNB1, loss-of-function screen, CRISPR/Cas, RNAi
Abstract
Epithelial ovarian cancer (EOC) is a deadly cancer, and its prognosis has not been changed significantly during several decades. To seek new therapeutic targets for EOC, we performed an in vivo dropout screen in human tumor xenografts using a pooled shRNA library targeting thousands of druggable genes. Then, in follow-up studies, we performed a second screen using a genome-wide CRISPR/Cas9 library. These screens identified 10 high-confidence drug targets that included well-known oncogenes such as ERBB2 and RAF1, and novel oncogenes, notably KPNB1, which we investigated further. Genetic and pharmacological inhibition showed that KPNB1 exerts its antitumor effects through multiphase cell cycle arrest and apoptosis induction. Mechanistically, proteomic studies revealed that KPNB1 acts as a master regulator of cell cycle-related proteins, including p21, p27, and APC/C. Clinically, EOC patients with higher expression levels of KPNB1 showed earlier recurrence and worse prognosis than those with lower expression levels of KPNB1. Interestingly, ivermectin, a Food and Drug Administration-approved antiparasitic drug, showed KPNB1-dependent antitumor effects on EOC, serving as an alternative therapeutic toward EOC patients through drug repositioning. Last, we found that the combination of ivermectin and paclitaxel produces a stronger antitumor effect on EOC both in vitro and in vivo than either drug alone. Our studies have thus identified a combinatorial therapy for EOC, in addition to a plethora of potential drug targets.
Epithelial ovarian cancer (EOC) is the fifth leading cause of cancer-related deaths among women in the United States and the most lethal gynecological cancer (1). Because of its rapid growth and lack of effective early detection strategies, 70% of EOC patients are diagnosed at an advanced stage, accompanied by peritoneal carcinomatosis (PC) (2). Most EOC patients also eventually become refractory to platinum, the major drug for treating EOC (3). Importantly, there are few molecularly targeted agents available for treating EOC, likely because EOC has high intertumor and intratumor heterogeneity at the molecular and epigenetic levels (4). Therefore, the mortality rate of EOC has not been significantly changed for several decades (5). To improve the poor prognosis of EOC, the discovery of new drug targets is sorely needed.
To elucidate the molecular pathogenesis of EOC, the Cancer Genome Atlas project has performed whole-genome sequencing and DNA copy number analysis of 489 patients with high-grade serous ovarian cancer, the most common type of cancer found in EOC patients. Sequencing revealed that almost all tumors (96%) had mutations in TP53, which serves as a major driver of this cancer (6). Low-prevalence but statistically significant mutations in nine other genes including NF1, BRCA1, BRCA2, RB1, and CDK12 were also identified, but the majority of genes were mutated at low frequency, making it difficult to distinguish between driver and passenger mutations.
To overcome this problem, several alternative methods have recently been used for cancer gene discovery, including insertional mutagenesis and RNAi/shRNA/CRISPR/Cas9-based screens. In our laboratory, we have performed transposon-based mutagenesis screens for a variety of cancer types and successfully identified many candidate cancer genes in a high-throughput manner (7, 8). However, the lack of EOC mouse models that recapitulate human EOC has hampered our use of this technology for cancer gene discovery in EOC. Although several groups have used pooled RNAi library screens for identifying new drug targets for EOC, most of these screens were performed in vitro (9, 10), which may not represent the clinical setting. To overcome these limitations, we performed an in vivo loss-of-function screen in human tumor xenografts using a pooled shRNA library targeting thousands of druggable genes. Then, in follow-up studies, we performed a second screen using a genome-wide CRISPR/Cas9 library. These screens identified 10 high-confidence candidate drug targets for EOC. In addition, we functionally validated a potent EOC oncogene, KPNB1, and showed its clinical relevance to human EOC. We also showed that a well-established antiparasitic drug, ivermectin, has antitumor effects on EOC through its inhibition of KPNB1, and describe a combinational therapy for treating EOC.
Results
An in Vivo Loss-of-Function Screen Identifies Potent Drug Targets for EOC.
To identify new drug targets for EOC, we conducted an in vivo dropout screen using a pooled shRNA library targeting 7,490 druggable genes (Fig. 1A). We performed this screen using an established EOC PC model (11), since PC is the biggest contributing factor to the lethality of EOC. In this screen, the human serous ovarian cancer cell line, SKOV3, was lentivirally transduced with the shRNA library and then i.p. injected into female nude mice. Genomic DNA from the PC tumors was PCR-amplified and sequenced to identify shRNAs depleted in tumors compared with transduced cells (Fig. 1A and Dataset S1). By focusing on depleted shRNAs, we hoped to identify drug targets whose inhibition would efficiently suppress PC formation. Thousands of shRNAs were depleted in tumors through in vivo selective pressure (Fig. 1B). The top 10 genes, which had multiple shRNAs showing substantial depletion in PC tumors, are shown in Fig. 1C (Methods for detailed criteria). To confirm the validity of these results, we repeated the screen and confirmed that most of the shRNAs targeting the top 10 genes were consistently depleted in tumors (Fig. 1C), excluding the possibility that this depletion was due to random chance.
Fig. 1.

An in vivo loss-of-function screen identifies potent drug targets for EOC. (A) Diagram of the in vivo pooled library screen. A large-scale pooled shRNA library targeting 7,490 druggable genes with 42,450 shRNAs was transduced into SKOV3 cells at an MOI of 0.2. Five million transduced cells then were injected into 12 female nude mice i.p., and peritoneal tumors were harvested 7 wk after injection. Genomic DNA extracted from referential transduced cells, and the 12 biggest tumors, were amplified and sequenced. (B) The average numbers of detected shRNAs from the first and second screen are shown. (C) The top 10 genes, which had multiple shRNAs showing substantial depletion in PC tumors, are ranked according to the average FC of shRNAs read counts in tumors relative to cells.
We conducted another screen using a genome-scale CRISPR/Cas9 knockout library (GeCKOv2) (12). Interestingly, most of the single-guide RNAs (sgRNAs) targeting the top 10 genes identified in our initial shRNA library screen were also depleted in the CRISPR screen (Fig. S1 and Dataset S1), further confirming that these genes are essential for PC tumors to survive. Among the top 10 genes, ERBB2 and RAF1 are well-known oncogenes in many solid tumors (13, 14) and several HER2 inhibitors have already been used in the clinic for multiple cancer types (15), confirming that our screen is capable of identifying promising drug targets.
Fig. S1.

(A) Genome-wide pooled CRISPR/Cas library targeting 19,050 genes with 65,383 sgRNAs were transduced to SKOV3 cells at MOI 0.2. Seven million transduced cells were injected into 12 female nude mice i.p., and peritoneal tumors were harvested 6 wk after injection. Genomic DNA extracted from referential transduced cells, and the 12 biggest tumors were amplified and sequenced. Average numbers of detected sgRNAs during CRISPR screen are shown. (B) For 10 candidate genes (shown in Fig. 1C), numbers of sgRNA showing 10-fold decrease of read counts in tumors relative to those in cells and numbers of designed sgRNA targeting each gene, were shown.
To verify the potential of these genes as drug targets, we assessed the effect of their depletion on cell proliferation/survival, which is highly related to tumor growth (16). In agreement with the results of our in vivo screen, individual knockdown of all but one gene decreased cell proliferation/survival (Fig. S2). Taken together, these results suggest that our screen provides a good resource to identify potential drug targets for EOC.
Fig. S2.

For individual validation of 10 candidate genes, cell proliferation/survival was assessed by WST-1 3 d after transfection of target siRNA or NC siRNA into SKOV3 cells. Among three siRNAs targeting each gene, the numbers of siRNA showing ≥10% decrease compared with NC siRNA, and the average proliferation rate relative to NC siRNA, are shown (n = 4).
KPNB1 Is a Drug Target Candidate for EOC.
Since Food and Drug Administration (FDA)-approved drugs are already in clinical use for our top-ranked gene ERBB2, we decided to test the potential of our second-ranked gene, KPNB1 (karyopherin beta-1 or, alternatively, importin beta-1), as a new drug target for EOC. First, to validate our initial screening results, we individually transduced KPNB1 shRNAs or negative control (NC) shRNA into SKOV3 cells and then i.p. injected them into nude mice. KPNB1 knockdown by any of the three shRNAs (Fig. S3A) significantly reduced tumor size and numbers in their peritoneal cavity (Fig. 2A). We also assessed the effect of KPNB1 knockdown on cell proliferation/survival in three human EOC cell lines, SKOV3, CAOV3, and OVCAR3. The siRNA-mediated knockdown of KPNB1 (Fig. S3 B–D) significantly decreased cell proliferation/survival in all three lines (Fig. 2 B–D). We then examined the antitumor effect of pharmacological inhibition of KPNB1 using importazole, a small molecule that specifically inhibits the importin β family (17). Consistent with our siRNA results, blockade of KPNB1 by importazole also significantly reduced cell proliferation/survival in a dose-dependent manner (Fig. 2E). Collectively, these results identify KPNB1 as a promising therapeutic target for EOC.
Fig. S3.

(A) SKOV3 cells were transduced with NC shRNA or KPNB1 shRNA. After puromycin selection, expression levels of KPNB1 or β-actin were assessed by Western blots. (B) SKOV3, (C) CAOV3, and (D) OVCAR3 cells were transfected with NC siRNA or KPNB1 siRNA. Three days after transfection, expression levels of KPNB1 and β-actin were assessed by Western blots.
Fig. 2.

KPNB1 is a drug target candidate for EOC. (A) SKOV3 cells were lentivirally transduced with NC shRNAs or KPNB1 shRNAs, and the transduced cells were i.p. injected into female nude mice. Total tumor weights (Left) and tumor numbers (Right) in the peritoneum of each mouse were then measured 6 wk after injection (n = 5 each, *P < 0.05 vs. all). (B and C) SKOV3 (Left), CAOV3 (Middle), and OVCAR3 (Right) cells were transfected with NC siRNA or KPNB1 siRNA. (B) Cell proliferation/survival was then assessed by colorimetric measurement via WST-1 3 d after transfection (n = 4 each, *P < 0.05 vs. all). (C) Cell numbers were counted 3 d after transfection with NC siRNA or KPNB1 siRNA (n = 3 each, *P < 0.05 vs. all). (D) Five hundred SKOV3 cells were transduced with NC shRNA or three KPNB1 shRNAs and seeded into a six-well plate. Cells were stained with crystal violet 14 d after transfection. (E) SKOV3 (Left), CAOV3 (Middle), and OVCAR3 (Right) cells were treated with different doses of importazole (IMZ) for 3 d. Cell proliferation/survival was assessed by WST-1 (n = 4 each, *P < 0.05 vs. all).
KPNB1 Inhibition Induces Apoptosis.
To investigate the mechanism of the antitumor effect resulting from KPNB1 inhibition, we first focused on apoptotic cell death, which is one of the major determinants of cell proliferation/survival. KPNB1 inhibition via any of three KPNB1 siRNAs or importazole treatment induced apoptosis in human EOC cell lines (Fig. 3 A–F and Fig. S4), and was accompanied by an increase in the expression levels of the proapoptotic proteins BAX and cleaved caspase-3 (Fig. 3G). Furthermore, immunohistochemical staining showed an increase in the number of cleaved PARP-positive tumor cells in tissue sections (18) from xenografted tumors containing KPNB1 shRNAs (Fig. 3H), further confirming that KPNB1 inhibition induces apoptosis both in vitro and in vivo, and providing a mechanism for its antitumor effect in EOC.
Fig. 3.

KPNB1 inhibition induces apoptosis. (A) SKOV3 cells were transfected with NC siRNA or KPNB1 siRNA. Apoptosis was then assessed by caspase-3/7 activity in cell supernatants 3 d after transfection (n = 3 each, *P < 0.05 vs. all). (B and C) SKOV3 cells were transfected with NC siRNA or KPNB1 siRNA. Three days after transfection, cells were stained for annexin V and propidium iodide (PI), and the proportion of apoptotic cells was then measured by flow cytometry. (B) Representative dot plots of flow cytometry and (C) the percentage of apoptotic cells are shown. (D) SKOV3 cells were treated with different doses of IMZ for 3 d. Apoptosis was assessed by caspase-3/7 activity of cell supernatants (n = 3 each, *P < 0.05 vs. all). (E and F) SKOV3 cells were treated with vehicle or 16 μM IMZ for 3 d. Cells were then stained with annexin V and PI, and the population of apoptotic cells was measured by flow cytometry. (E) Representative dot plots of flow cytometry and (F) percentage of apoptotic cells are shown. (G) SKOV3 cells were transfected with NC siRNA or KPNB1 siRNA. Three days after transfection, expression levels of apoptosis-related proteins were assessed by Western blots. (H) SKOV3 cells were lentivirally transduced with NC shRNAs or KPNB1 shRNAs, and the transduced cells were i.p. injected into female nude mice. Six weeks after injection, i.p. tumors were collected, and apoptosis was assessed by the number of tumor cells with immunopositivity for cleaved PARP (n = 5 each, *P < 0.05 vs. NC shRNA).
Fig. S4.

SKOV3 (Left) and CAOV3 (Right) cells were treated with vehicle or IMZ at different dosages for 3 d. Apoptosis was assessed by caspase-3/7 activity of the supernatant of each cell (n = 3 each, *P < 0.05 vs. all).
KPNB1 Inhibition Induces Multiphase Cell Cycle Arrest.
We next examined the effect of KPNB1 inhibition on the cell cycle, another strong determinant of cell proliferation/survival. Knockdown of KPNB1 elevated the population of cells at the G2/M phase as measured by flow cytometric analysis (Fig. 4 A and B). To assess each stage of the cell cycle individually, we treated cells with nocodazole, an inhibitor of microtubule polymerization (19), to induce mitotic arrest. Twenty-four hours later, while the majority of the cells treated with NC siRNA were accumulated at the G2/M phase, a large fraction of cells treated with KPNB1 siRNAs still remained at the G1/S phase (Fig. 4 A and B), suggesting that KPNB1 inhibition delays the G1/S transition. In addition, 24 h after the withdrawal of nocodazole, the G2/M population in KPNB1 knockdown cells further increased, in sharp contrast to the rapid decrease of the G2/M population in control cells (Fig. 4 A and B), further suggesting that KPNB1 inhibition also delays the G2/M transition. We also confirmed that pharmacological inhibition of KPNB1 by importazole produced a similar effect (Fig. 4 C and D). We then checked the expression levels of a number of cell cycle regulators and found that KPNB1 inhibition increased p21 and p27 protein levels (Fig. 4E and Fig. S5). We also assessed the cell cycle in xenografted tumors derived from SKOV3 cells transduced with KPNB1 shRNAs. Immunohistochemical staining showed that the number of Ki67 positive cells was significantly lower in tumors transduced with KPNB1 shRNA (Fig. 4F), suggesting that KPNB1 inhibition also delayed cell cycle progression in vivo. Taken together, these results suggest that KPNB1 inhibition exerts its antitumor effect, in part, by inducing apoptosis and cell cycle arrest in human EOC.
Fig. 4.

KPNB1 inhibition induces multiphase cell cycle arrest. (A and B) SKOV3 cells were transfected with NC siRNA or KPNB1 siRNA. (A) Two days later, cell cycles were assessed by flow cytometry (Left); then, to induce G2/M arrest, cells were treated with 500 ng/mL of nocodazole (NOC) for 24 h, and cell cycles were again assessed by flow cytometry (Middle); cells were then further incubated for 24 h after withdrawal of NOC and analyzed by flow cytometry (Right). (B) Cell populations at each stage of cell cycle are shown. (C and D) SKOV3 cells were treated with vehicle or 16 μM IMZ. (C) Two days later, cell cycles were assessed by flow cytometry (Left); then, to induce G2/M arrest, cells were treated with 500 ng/mL of NOC for 24 h, and cell cycles were again assessed by flow cytometry (Middle); cells were further incubated for 24 h after withdrawal of NOC and analyzed by flow cytometry (Right). (D) Cell populations at each stage of the cell cycle are shown. (E) SKOV3 cells were transfected with NC siRNA or KPNB1 siRNA. Three days after transfection, the expression levels of cell cycle-related proteins were assessed by Western blots. (F) SKOV3 cell were lentivirally transduced with NC shRNAs or KPNB1 shRNAs, and the transduced cells were i.p. injected into female nude mice. Six weeks after injection, i.p. tumors were collected and stained with Ki-67 (×40, scale bar, 100 μm) (Left). The proportions of Ki-67 positive cells are shown (n = 5 each, *P < 0.05 vs. NC shRNA) (Right).
Fig. S5.
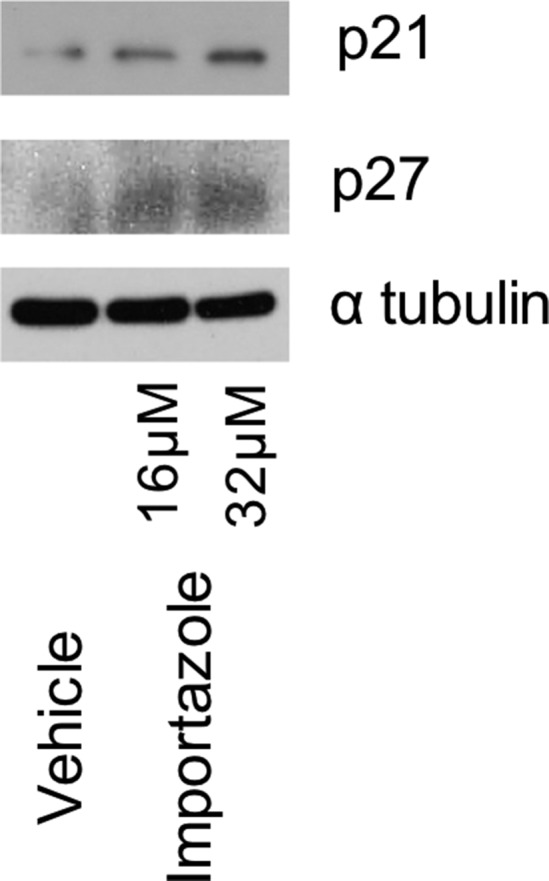
SKOV3 cells were treated with vehicle, or 16 μM or 32 μM IMZ for 3 d. Expression levels of cell cycle-related proteins were assessed by Western blots.
KPNB1 Functions as an Oncogene in EOC.
These results prompted us to examine whether KPNB1 acts as an oncogene in EOC. Stable overexpression of KPNB1 in SKOV3 and OVCAR3 (Fig. S6) significantly accelerated cell proliferation/survival (Fig. 5 A–C), confirming that KPNB1 functions as an oncogene in EOC. In contrast to the apoptosis induced by KPNB1 inhibition (Fig. 3), KPNB1 overexpression significantly decreased caspase-3/7 activity (Fig. 5D), in addition to the expression levels of cleaved caspase-3 and BAX proteins (Fig. 5E). KPNB1 overexpression also decreased p21 and p27 protein levels (Fig. 5E), as opposed to their increase by KPNB1 inhibition (Fig. 4E). Collectively, these results indicate that KPNB1 functions as an antiapoptotic and proproliferative oncogene in EOC.
Fig. S6.

(A) SKOV3 and (B) OVCAR3 cells were lentivirally transduced with control vector or KPNB1 expression vector. After blasiticidin selection, expression levels of KPNB1 or β-actin were assessed by Western blots.
Fig. 5.

KPNB1 functions as an oncogene in EOC. (A and B) SKOV3 (Left) and OVCAR3 (Right) cells were lentivirally transduced with a KPNB1 expression vector or control vector. (A) Cell proliferation was then assessed by WST-1 (n = 4 each, *P < 0.05 vs. all). (B) Cell numbers were counted 3 d after seeding equal numbers of cells in six-well plates and are shown as ratio relative to control cells (n = 3 each, *P < 0.05 vs. all). (C) Five hundred SKOV3 cells were seeded into a six-well plate after lentiviral transduction of a KPNB1 expression vector or control vector. Cells were stained with crystal violet 14 d after transfection. (D) SKOV3 (Left) and OVCAR3 (Right) cell lines were lentivirally transduced with a KPNB1 expression vector or control vector. Apoptosis was assessed by caspase-3/7 activity in cell supernatants (n = 3 each, *P < 0.05 vs. all). (E) SKOV3 cells were lentivirally transduced with a KPNB1 expression vector or control vector. Expression levels of apoptosis- and cell cycle-related proteins were assessed by Western blots.
To assess the clinical relevance of its oncogenic function, we analyzed the relationship between KPNB1 expression and EOC patient prognosis using a publicly available microarray dataset (GSE 9891) (20). Patients with higher expression levels of KPNB1 showed earlier recurrence and worse prognosis than those with lower expression levels of KPNB1 (Fig. S7 A and B), further confirming that KPNB1 acts as an oncogene in human EOC and represents a promising therapeutic target.
Fig. S7.

(A and B) The correlation between KPNB1 mRNA levels and patient prognosis in human EOC using a publically available microarray dataset (GSE 9891) (21). Patients with higher expression levels of KPNB1 showed (A) earlier recurrence and (B) worse prognosis than those with lower expression levels of KPNB1.
KPNB1 Positively Regulates Members of the Anaphase-Promoting Complex/Cyclosome.
KPNB1 functions as a nucleocytoplasmic transporter to import proteins containing nuclear localization signals into the nucleus (21). We thus decided to perform stable isotope labeling with amino acids in cell culture (SILAC) and mass spectrometric (MS) analysis to identify proteins shuttled by KPNB1 into the nucleus in a high-throughput manner. SKOV3 and OVCAR3 cells were transfected with NC siRNA or KPNB1 siRNA, labeled with light or heavy amino acids, respectively, and analyzed by quantitative MS (Fig. 6A). Comprehensive analysis revealed that protein levels of multiple anaphase-promoting complex/cyclosome (APC/C) family members decreased in the nucleus of cells treated with KPNB1 siRNA (Fig. 6B). APC/C is a positive regulator of mitosis and functions as an E3 ubiquitin ligase to mark cell cycle-related proteins for proteasomal degradation (22). Loss of APC/C results in cell cycle arrest at metaphase and severe aberrations of the mitotic spindle (23). To further investigate the relationship between KPNB1 and APC/C protein levels, we analyzed liquid chromatography (LC)-MS data for various other human cancer cell lines (24). We observed a positive correlation in protein levels between KPNB1 and multiple APC/C family members in several different cancer types (Fig. 6C), suggesting that regulation of APC/C proteins by KPNB1 may not be unique to EOC. In addition, we found that KPNB1 inhibition significantly decreased cell proliferation/viability in many other human cancer cell lines including breast, pancreas, and liver (Fig. 6D), suggesting that KPNB1 could also be a potential therapeutic target for these cancers in addition to EOC.
Fig. 6.

KPNB1 positively regulates multiple APC/C family members. (A) Diagram of LC-MS analysis after SILAC. (B) Expression levels of KPNB1 and APC/C-related proteins in the nucleus of SKOV3 and OVCAR3 cells following transfection with KPNB1 siRNA or NC siRNA as assessed by LC-MS. Relative expression ratios in cells treated with KPNB1 siRNAs compared with those treated with NC siRNA are shown. (C) Expression levels of KPNB1 and APC/C-related proteins in 136 human cancer cell lines as assessed by LC-MS and plotted to analyze the correlation between expression levels of KPNB1 and APC/C-related proteins. Pearson’s correlation coefficients for APC8, APC5, APC2, and APC4 and Spearman’s correlation coefficients for APC7 and APC1 are shown. (D) MDA-MB-231 (breast adenocarcinoma), MCF-7 (breast adenocarcinoma), MIA PaCa-2 (pancreas ductal adenocarcinoma), and Huh-7 cells (well-differentiated hepatocellular carcinoma) were transfected with NC siRNA or KPNB1 siRNA. Cell proliferation/survival was then assessed by WST-1 3 d after transfection (n = 4 each, *P < 0.05 vs. all).
Ivermectin Exerts a Strong Antitumor Effect On EOC in a KPNB1-Dependent Manner.
Next, we searched clinically available drugs that can potentially target KPNB1 and found a recent report that ivermectin, an FDA-approved antiparasitic drug, inhibits importin α/β-mediated nuclear import (25). In our initial studies, we found that ivermectin treatment suppressed cell proliferation/viability in a dose-dependent manner (Fig. 7A), indicating that it exerts an antitumor effect on EOC. Similar to KPNB1 inhibition or importazole treatment, ivermectin also induced apoptosis (Fig. 7 B and C). We also found that ivermectin increased the expression levels of BAX, and cleaved PARP, as well as p21 and p27 (Fig. 7D). Cell cycle analysis with nocodazole also showed a cell cycle arrest upon ivermectin treatment, similar to what we observed with KPNB1 inhibition (Fig. 7 E and F). These findings suggest that ivermectin exerts its antitumor effect through the same mechanisms as KPNB1 inhibition. To provide additional validation, we treated SKOV3 and OVCAR3 cells with ivermectin in the absence of KPNB1. In sharp contrast to the strong antitumor effect we observed for ivermectin in NC siRNA-treated cells, we observed a severely diminished effect for ivermectin in KPNB1 siRNA-treated cells (Fig. 7G and Fig. S8), indicating that KPNB1 inhibition is responsible for the antitumor effect of ivermectin.
Fig. 7.

Ivermectin (IV) exerts KPNB1-dependent antitumor effects on EOC. (A and B) SKOV3 (Left), CAOV3 (Middle), and OVCAR3 (Right) cells were treated with different doses of IV for 3 d. (A) Cell proliferation/survival was then assessed by WST-1 proliferation (n = 4 each, *P < 0.05 vs. all). (B) Apoptosis was assessed by caspase-3/7 activity in cell supernatants (n = 4 each, *P < 0.05 vs. all). (C and D) SKOV3 cells were treated with vehicle or 16 μM IV for 3 d. (C) Cells were then stained with annexin V and PI, and the population of apoptotic cells was assessed by flow cytometry. (D) Expression levels of cell cycle- and apoptosis-related proteins were assessed by Western blots. (E and F) SKOV3 cells were treated with 8 μM IV. (E) Two days later, cell cycles were assessed by flow cytometry (Left); then, to induce G2/M arrest, cells were treated with 500 ng/mL of NOC for 24 h, and the cell cycles were assayed by flow cytometry (Middle); cells were further incubated for 24 h after withdrawal of NOC and analyzed by flow cytometry (Right). (F) Cell populations at each stage of the cell cycle are shown. (G) SKOV3 cells were transfected with NC siRNA or KPNB1 siRNA. Two days later, they were treated with vehicle or 12 μM IV for 2 d. Cell proliferation/survival was then assessed by WST-1 proliferation (n = 4 each, *P < 0.05). (H–J) SKOV3 cells were treated with vehicle, 2 μM IV, 5 nM PTX, or 2 μM IV + 5 nM PTX for 2 d. (H) Cell proliferation/survival was then assessed by WST-1 proliferation (n = 4 each, *P < 0.05 vs. all) and (I) apoptosis was assessed by caspase-3/7 activity in cell supernatants (n = 3 each, *P < 0.05 vs. all). (J) Cells were stained with annexin V and propidium iodide, and the number of apoptotic cells was assessed by flow cytometry. (K) SKOV3 cells were s.c. injected into immunodeficient NSG mice. One week later, mice were randomly assigned into four groups (control, IV, PTX, and IV+PTX). Mice were then treated with i.p. injection of PBS, or 1 mg/kg of IV and/or 15 mg/kg of PTX 5 d a week for 40 d. Xenograft tumor volumes were then measured at different times after the initiation of treatment (n = 4 each, *P < 0.05 vs. all).
Fig. S8.

OVCAR3 cells were transfected with NC siRNA or KPNB1 siRNA. Two days later, they were treated with vehicle or 4 μf IV for 2 d. Cell proliferation/survival was assessed by WST-1 assay (n = 4 each, *P < 0.05).
Ivermectin Synergistically Suppresses EOC Tumor Growth in Combination with Paclitaxel.
Paclitaxel is a drug currently in use for EOC. It is used as a first-line chemotherapy in combination with platinum, or as a second-line therapy for platinum/paclitaxel-refractory cancers (26). We thus explored the potential of ivermectin combined with paclitaxel for the treatment of EOC. Interestingly, we found that ivermectin synergistically reduced cell proliferation/viability in combination with paclitaxel in human EOC cells (Fig. 7H), via the induction of apoptosis and cell cycle arrest at the G2/M phase (Fig. 7 I and J and Fig. S9). Lastly, we tested this combination therapy in vivo. Single treatment of ivermectin or paclitaxel reduced tumor growth in nude mice, but, notably, combination treatment of ivermectin and paclitaxel almost completely suppressed tumor growth (Fig. 7K). These results identify ivermectin and paclitaxel as a promising combinatorial therapy for human EOC.
Fig. S9.

SKOV3 cells were treated with vehicle or 8 μM IV or/and 5 nM PTX for 3 d. Cell cycle was assessed by flow cytometry. Representative image and cell population were shown.
Discussion
We identified, in this study, several candidate drug targets for EOC using a combination of shRNA and CRISPR/Cas9 library screening. Among these drug targets, we showed that inhibition of one, KPNB1, by ivermectin has promising antitumor effects and synergizes with paclitaxel in the treatment in EOC. While RNAi/shRNA/CRISPR/Cas9 library screening was originally developed for use in vitro, more recently, these technologies have been used in vivo to mimic a more clinically relevant physiological environment (27). Several in vivo loss of function screens using RNAi or CRISPR-Cas9 have been reported that identified shRNAs or sgRNAs, which were enriched in tumors, but the targets of these screens are mostly undruggable tumor suppressor genes (27, 28). In contrast, our screen focused on shRNAs or sgRNAs (CRISPR/Cas9) that are depleted in tumors and thus potentially represent druggable oncogenes. We also performed multiple screens using the same experimental conditions, to help eliminate background noise. Interestingly, we observed depletion of many more genes in tumors using CRISPR/Cas9 compared with shRNA. We first excluded the possibility that cancer cells containing the CRISPR library were engrafted unsuccessfully, because the tumor size and growth were similar in both screens. We also confirmed that essential genes, identified by previously reported CRISPR screens (29, 30), were significantly more depleted than nonessential genes in peritoneal tumors in our CRISPR screen [depletion rate; essential genes vs. nonessential genes defined by Blomen et al. (29) and Wang et al. (30), 72.4% vs. 60.4%, P < 8.67 × 10−18, and 70.7% vs. 60.4%, P < 5.24 × 10−23, respectively], suggesting that genes were not depleted randomly but by in vivo selective pressure. The shRNA generally induces partial gene knockdown, and the residual activity of the wild-type protein may thus prevent a knockdown phenotype from being observed. This is in contrast to CRISPR/Cas9, where complete gene knockouts are usually produced. This functional difference between RNAi and CRISPR/Cas may be, at least in part, responsible for the different screening output. By analyzing both types’ loss-of-function screens, we hoped to provide a more highly reliable list of drug targets for EOC.
Our top-ranked gene, ERBB2, is amplified and overexpressed in many cancers, including breast (31), ovary (31), colon (32), bladder (33), non-small-cell lung (34), and gastric cancer (35), and is a poor prognostic factor in certain cancer types (36, 37). Several HER2 inhibitors, either tyrosine kinase inhibitors or monoclonal antibodies, are already in clinical use and are invaluable drugs in the treatment of breast cancer. Regarding their clinical implications for EOC, several clinical trials using HER2 inhibitors have already been conducted with unsatisfying results (38, 39), However, the latest phase III clinical trials (pertuzumab in platinum-resistant low human epidermal growth factor receptor 3 messenger ribonucleic acid epithelial ovarian cancer; PENELOPE) for EOC have shown favorable results for pertuzumab combined with gemcitabine and paclitaxel for platinum-resistant EOC with low HER3 mRNA levels (40). In accordance with these results, SKOV3 has the highest expression levels of HER2, and the fifth-lowest expression levels of HER3, among 52 EOC cell lines registered in the cancer cell line encyclopedia (41), which might have resulted in the identification of ERBB2 as the most potent drug target in our screen.
KPNB1 was the second-highest-ranked gene identified in our screen. Increased KPNB1 protein levels have been reported in several cancers, including cervical cancer (42), hepatocellular carcinoma (43), and glioma (44), suggesting KPNB1’s oncogenic potential in these tumor types. In this study, we validated an oncogenic function of KPNB1 in EOC through apoptosis inhibition and cell cycle progression. MS analysis also revealed the positive regulation of several APC/C family members by KPNB1, suggesting that KPNB1 might shuttle these nuclear proteins related to cancer progression. To determine whether these proteins directly interact, we further performed MS of proteins immunoprecipitated (IP-MS) with a KPNB1 antibody. Although IP-MS could identify known partner molecules of KPNB1, such as RAN and RAN-GTP, none of the APC/C members were identified in this assay (Dataset S1). This may be because interaction between APC/C and KPNB1 is not static, or KPNB1 might indirectly affect APC/C protein levels in the nucleus. Nonetheless, positive regulation of APC/C by KPNB1 could still be important for cell cycle control, since APC/C is known to regulate the cell cycle from late G2/M to G1. Specifically, APC/Ccdc20 is required for anaphase onset and mitotic exit, while APC/Ccdh1 is essential for mitotic exit to G1 (45). Taken together, our findings suggest that KPNB1 might serve as a master regulator of cell cycle by regulating several cell cycle-related proteins, including p21, p27, and APC/C family members.
Ivermectin has a long history of safe treatments for several parasitic infections in humans and mammals (46). Recently, Wagstaff et al. (25) has also identified ivermectin as a specific inhibitor of importin α/β-mediated nuclear import via high-throughput screening. Indeed, in this study, we discovered a KPNB1-dependent antitumor effect of ivermectin on multiple EOC cell lines. We found that ivermectin induces apoptosis and multiphase cell cycle arrest in EOC cells, accompanied by alterations in several molecules, all of which are consistent with the alterations we observed upon KPNB1 inhibition. The regimen used for the mice in our study was not the same as the one used for parasite treatment in humans. However, Guzzo et al. (47) reported that higher and/or more-frequent doses of ivermectin than currently approved for humans are well tolerated in humans. In addition, none of the mice in this study treated with the effective dosage of ivermectin for in vivo anticancer therapy showed severe adverse event. Therefore, although we need to optimize the regimen in the clinical settings, considering its strong antitumor effect and accumulated safety profile as an antiparasitic drug, our results identify ivermectin as an attractive alternative therapeutic toward EOC patients through drug repositioning. In addition, we found that the combination of ivermectin and paclitaxel produces a stronger antitumor effect on EOC cell lines than either drug alone. Recently, molecular therapies targeting cell cycle, including CDK4/6 inhibitors, have been approved by the FDA (48). In addition, cell cycle targeted therapy has been studied in a number of clinical trials, in combination with chemotherapeutic agents, to pursue synergistic antitumor effects. Indeed, the latest phase III clinical trials have shown efficacy for combination therapy of CDK4/6 inhibitors and hormone therapy for breast cancer (49), via overcoming the resistance to prior hormonal therapy. Although the precise molecular mechanism of the synergistic antitumor effect of ivermectin and paclitaxel needs further investigation, both drugs have been in clinical use for several decades, which should facilitate clinical trials assessing the combinatorial effects of these two drugs in EOC patients.
Methods
Cell Culture.
SKOV3, OVCAR3, and CAOV3 human liver cancer cell lines, MIA PaCa-2 human pancreatic cancer cell line, MDA-MB231 and MCF-7 human breast cancer cell lines, and HEK-293 cell line were purchased from the American Type Culture Collection. The Huh-7 human liver cancer cell line was purchased from the Japanese Collection of Research Bioresources Cell Bank. These cell lines were cultured in DMEM or RPMI-1640 medium supplemented with 10% FBS and penicillin−streptomycin, except for OVCAR3, which was supplemented with 0.01 mg/mL of bovine insulin, 20% FBS, and penicillin−streptomycin. All cell lines were maintained in a humid incubator (5% CO2, 37 °C) that was confirmed to be free from pathogens and mycoplasma.
Chemicals and Reagents for Biological Assays.
Ivermectin (Selleckchem), importazole (Selleckchem), paclitaxel (Sigma Aldrich), and nocodazole (Sigma Aldrich) were dissolved in DMSO to prepare stock solutions, and were diluted in cell growth medium for assays.
In Vivo Pooled shRNA Library Screen.
Decode pooled shRNA druggable library (#RHS6082) was purchased from GE Dharmacon. Female athymic nude mice [Crl:Nu(NCr)-Foxn1nu] were purchased from Charles River Laboratories. SKOV3 cells were lentivirally transduced with the shRNA library containing 42,450 shRNAs targeting 7,490 genes at a multiplicity of infection (MOI) of 0.2, and transduced cells were selected by 1.5 mg/mL puromycin treatment for 7 d. Pellets of transduced cells were stored at −80 °C as a reference for sequencing. Five million transduced cells were i.p. injected into female nude mice. Mice were monitored for tumor formation using the general animal health guidelines, and tumors bigger than 10 mm in diameter were collected 7 wk after injection and snap-frozen. The Institutional Animal Care and Use Committee of Houston Methodist Research Institute approved all mouse procedures.
Genomic DNA Sequence.
Genomic DNA was extracted from cells and xenograft tumors transduced with the pooled shRNA or CRISPR/Cas library, using the DNeasy Blood and Tissue kit (Qiagen), according to the manufacturer’s instructions. For shRNA library, shRNA sequences of extracted gDNA were then amplified using Illumina-adapted Decode Forward and Reverse indexed PCR primers, according to the protocol supplied with the Decode Pooled Lentiviral shRNA screening libraries. To keep the fold representation at 100 for each of the shRNAs, 27.8 μg of gDNA per each sample was used for the PCR. Indexed PCR products were purified using a PCR purification kit (Qiagen). Purified PCR amplicons were multiplexed and sequenced using Decode sequencing primers on an Illumina Hiseq. For CIRSPR/Cas library, sgRNA sequences were first amplified with forward primer AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG and reverse primer TCTACTATTCTTTCCCCTGCACTGTTGTGGGCGATGTGCGCTCTG. Then, they were secondarily amplified with forward primer AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT(1-8 bp stagger)(8 bp barcode)TCTTGTGGAAAGGACGAAACACCG and reverse primer CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTCTACTATTCTTTCCCCTGCACTGT. To keep the fold representation at 100 for each of the sgRNAs, 45 μg of gDNA per sample was used for the PCR. Purified PCR amplicons were multiplexed and sequenced using Illumina sequencing primers on an Illumina Hiseq. We obtained reads per tumor. The read counts for each unique shRNA/gRNA for a given sample were normalized as previously described (12). Then, these normalized read counts were converted into the fold change (FC) relative to those of referential cells. For dropout hit identification, we first selected genes targeted by more than or equal to four shRNAs showing an FC of less than 0.1. Genes were then ranked based on average FC of all designed shRNAs targeting each gene. The top 10 genes, which showed average FC of less than 0.2, are listed in Fig. 1C.
Flow Cytometry.
SKOV3 cells were transfected with NC siRNA or KPNB1 siRNA, or treated with anticancer drugs or vehicle. Cell cycle analysis was then performed 2 d later. Cells were first labeled with propidium iodinide (Sigma Aldrich), and their cell cycle was analyzed using a BD LSRFortessa cell analyzer (BD Biosciences). In some experiments, the cells were first synchronized by the addition of nocodazole for 24 h, and the cell cycle was then analyzed. Synchronized cells were also analyzed 24 h after the withdrawal of nocodazole. For apoptosis analysis, 2 d later, the cells were trypsinized and washed with PBS. Cell pellets were then diluted with annexin V binding buffer 10× concentrate (BD Biosciences) and incubated with BD phospholipid-binding protein APC Annexin V (BD Biosciences) and propidium iodinide (Sigma Aldrich). The cells were then analyzed using a BD LSRFortessa cell analyzer. Data were analyzed using BD FACSDiva 8.0.1.
In Vivo Xenografts.
Female NOD scid gamma (NSG) mice (NOD.Cg-PrkdcscidIl2rgtm1wjl/SzJ) were purchased from The Jackson Laboratory. Five million SKOV3 cells were injected s.c. into both flanks of 16 NSG mice. One week later, mice were randomly assigned into four groups [control, ivermectin (IV), paclitaxel (PTX), and IV+PTX]. Mice were then treated with i.p. injection of PBS, or 1 mg/kg of IV and/or 15 mg/kg of PTX, 5 d a week for 40 d. Xenograft tumor volumes were then measured at different times after the initiation of treatment. Tumor volumes were calculated via the formula of 1/2 (length × width2).
Proteomic Analysis.
SKOV3 and OVCAR3 cells were labeled with heavy isotope, 13C6 l-Lysine-2HCl, or light isotope, l-Lysine-2HCl, via Pierce SILAC Protein quantitation kit-RPMI 1640 containing 10% dialyzed FBS (ThermoFisher Scientific). Cells were passaged and expanded for at least 10 doublings, before transfection of KPNB1 siRNA or NC siRNA. For whole-cell extracts, cells were lysed in 1 mL of PBS containing detergent, protease inhibitor (Roche Diagnostics), and phosphatase inhibitor, 72 h after siRNA transfection, followed by sonication and centrifugation. For nuclear cell extracts, cells were lysed in cell lysis buffer with protease inhibitor and homogenized, followed by centrifugation. Nuclei resuspension buffer was then added to the pellets, followed by centrifugation. For the IP-MS experiment, nuclear cell extracts of SKOV3 were obtained as described above and immunoprecipitated by IgG or KPNB1 antibody (Abcam). MS analysis were performed as previously described (24).
Bioinformatic Analysis.
To investigate an association between expression of KPNB1 gene and prognosis in EOC patients, we downloaded GSE9891 dataset from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) (20). We obtained Affymetrix Human Genome U133 Plus 2.0 array data from 285 ovarian tumor samples, and their patient survival data. We used the data from 194 advanced stage high-grade serious ovarian cancer patients without chemotherapeutic treatment before the sample collection. We performed Kaplan−Meier survival analysis in R 2.15.3 (R Development Core Team 2013) using package “survival.”
Statistical Analysis.
All data were analyzed with Graphpad Prism 6 (GraphPad Software) and provided as mean ± SEM unless otherwise indicated. Statistical analyses were performed using an unpaired Student’s t test, Mann−Whitney U test, or a one-way ANOVA. For multiple comparisons, P value was adjusted by Bonferroni correction of the differences or the differences in the mean values among the groups were examined by Dunnett’s multiple comparison test. P < 0.05 was considered statistically significant. For all other information on materials and methods, please see Supporting Information.
Lentiviral Transduction
All lentiviral particles containing GIPZ shRNA or nontargeting shRNA (NC) were purchased from Thermo Fisher Scientific. shRNA clones used in this study are as follows: KPNB1-shRNA1; V2LHS_210254, KPNB1-shRNA2; V3LHS_353237, and KPNB1-shRNA3; and V3LHS_353239. Cells were plated at a density of 1.0 × 105 cells per six-well plate 1 d before the infection. The next day, the medium was changed into serum-free medium containing 8 μg/mL of polybrene (EMD Millipore) and transduced with lentivirus at an MOI of 2 to 3. The following day, cells were supplemented with complete medium containing 1.0 μg/mL to 1.5 μg/mL of puromycin (Thermo Fisher Scientific) to establish stable cell lines containing lentiviral integrations.
In Vivo Pooled CRISPR/Cas Library Screen
The genome-wide GeCKO v2 library A used in our screen contained 65,386 sgRNAs targeting 19,050 genes and 1,864 miRNAs, including “cell-essential genes.” To amplify the GeCKO v2 library A, it was electroporated and transformed following the protocol from the Zhang laboratory (50). To produce lentivirus, 200 μL of Plus reagent with 20 μg of lentiCRISPR plasmid library in OptiMEM media (Thermo Fisher Scientific), and 10 μg of pVSVg (Addgene), 15 μg of psPAX2 (Addgene), and 100 μL of Lipofectamine 2000 (Thermo Fisher Scientific) in 4 mL of OptiMEM, were mixed and added to HEK293 cells (50). The supernatant was then filtered and concentrated using a Lenti-X Concentrator (Takara Bio USA). After centrifugation and removing the supernatant, the pellet was resuspended in 5 mL of complete DMEM and stored at −80 °C. SKOV3 cells were subsequently transduced with this GeCKO library at an MOI of 0.2, and transduced cells were selected by 1.5 mg/mL puromycin treatment for 7 d. Seven million transduced cells then were i.p. injected into female nude mice, and tumors were harvested in the same manner as the shRNA library screen.
Cell Proliferation Assays
SKOV3, OVCAR3, and CAOV3 cells were transfected with silencer select NC siRNA or KPNB1 siRNA (Thermo Fisher Scientific), or treated with anticancer drugs or vehicle. For counting actual cell numbers, 2.0 × 105 transfected cells were plated into six-well plates, and cell numbers were counted using an automated cell counter, 72 h after transfection. For colorimetric assays, cells were transfected with siRNA in 96-well plates. Premix WST-1 (Takara Bio) was added to each well 72 h after transfection and incubated at 37° for 1.5 h. The plate was then read on Tecan i-control infinite 200Pro, using a test wavelength of 450 nm and a reference wavelength of 650 nm.
Caspase-3/7 Activity Assay
Five thousand SKOV3 cells, and 10,000 OVCAR3 or CAOV3 cells, were seeded in a 96-well plate. Anticancer drugs or vehicle were added into each well 1 d after cell seeding, and the cells were incubated for 72 h. Caspase-Glo reagent (Promega) was then added into cultured cell supernatant, and luminescence levels were measured using Tecan i-control infinite 200Pro. For siRNA experiments, cells were transfected with KPNB1 siRNA or NC siRNA, and caspase-3/7 activity was evaluated 96 h after transfection.
Western Blotting
Cells were lysed by RIPA buffer with 100× halt phosphatase inhibitor mixture (Thermo Fisher Scientific). Protein concentration was measured via Bicinchoninic acid assay (Thermo Fisher Scientific). Proteins were separated using polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane via a wet or dry system. The membrane was incubated overnight with primary antibodies, KPNB1, P21, P27, BAK, BAX, cleaved PARP, cleaved caspase-3, and β-actin (Cell Signaling). After incubation with anti-rabbit or anti-mouse secondary antibody (Bio-Rad Laboratories), detection of immunolabeled proteins was performed using a chemiluminescent substrate (Thermo Fisher Scientific). The blots were quantified using Image J software. Density values for each protein were adjusted by the value for β-actin. Relative values, calculated by dividing the value for each lane by the NC siRNA or vehicle value, are shown under the blots (Figs. 3G, 4E, 5E, and 7D).
Immunohistochemical Staining
Sections were deparaffinized and rehydrated. Slides were then soaked in citric acid buffer (Vector Laboratories) and microwaved for 15 min. Slides were subsequently washed with PBS, treated with 3% H2O2, and then washed with 0.2% Triton X-100 in PBS. Sections were blocked with 10% goat serum for 1 h and hybridized with primary antibodies in a humidity chamber at 4 °C overnight. Biotin-conjugated secondary antibodies (Vector Laboratories) were added for 1 h, followed by hybridization with ABC reagent (Vector Laboratories). DAB (3,3′-diaminobenzidine) was used for chromogen until the desired color appeared, depending on the antibody. Sections were counterstained in hematoxylin, dehydrated, and mounted.
Supplementary Material
Acknowledgments
We thank E. Freiter and H. Lee for monitoring the mice and animal technical assistance, David L. Haviland for technical assistance and discussion about flow cytometry experiments, and Ayako Okamura for the experiments for revision. The Cancer Prevention Research Institute of Texas (CPRIT) supported this research [Grants R1112 (N.G.C.) and R1113 (N.A.J.)]. N.G.C. and N.A.J. are both CPRIT Scholars in Cancer Research.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1705441114/-/DCSupplemental.
References
- 1.Group US Cancer Statistics Working Group 2016 United States cancer statistics: 1999–2013 incidence and mortality web-based report. Available at www.cdc.gov/uscs. Accessed October 31, 2016.
- 2.Rauh-Hain JA, Krivak TC, Del Carmen MG, Olawaiye AB. Ovarian cancer screening and early detection in the general population. Rev Obstet Gynecol. 2011;4:15–21. [PMC free article] [PubMed] [Google Scholar]
- 3.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: New opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blagden SP. Harnessing pandemonium: The clinical implications of tumor heterogeneity in ovarian cancer. Front Oncol. 2015;5:149. doi: 10.3389/fonc.2015.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Howlader N, et al. 2016 SEER Cancer Statistics Review, 1975–2013 (Natl Cancer Inst, Bethesda). Available at https://seer.cancer.gov/csr/1975_2013/. Accessed October 31, 2016.
- 6.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kodama T, et al. Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc Natl Acad Sci USA. 2016;113:E3384–E3393. doi: 10.1073/pnas.1606876113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda H, et al. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat Genet. 2015;47:142–150. doi: 10.1038/ng.3175. [DOI] [PubMed] [Google Scholar]
- 9.Sethi G, et al. An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. PLoS One. 2012;7:e47086. doi: 10.1371/journal.pone.0047086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arora S, et al. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol Oncol. 2010;118:220–227. doi: 10.1016/j.ygyno.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther. 2004;10:1032–1042. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011;30:3477–3488. doi: 10.1038/onc.2011.160. [DOI] [PubMed] [Google Scholar]
- 14.Hynes NE, Lane HA. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 15.Schroeder RL, Stevens CL, Sridhar J. Small molecule tyrosine kinase inhibitors of ErbB2/HER2/Neu in the treatment of aggressive breast cancer. Molecules. 2014;19:15196–15212. doi: 10.3390/molecules190915196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tubiana M. Tumor cell proliferation kinetics and tumor growth rate. Acta Oncol. 1989;28:113–121. doi: 10.3109/02841868909111193. [DOI] [PubMed] [Google Scholar]
- 17.Soderholm JF, et al. Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chem Biol. 2011;6:700–708. doi: 10.1021/cb2000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bressenot A, et al. Assessment of apoptosis by immunohistochemistry to active caspase-3, active caspase-7, or cleaved PARP in monolayer cells and spheroid and subcutaneous xenografts of human carcinoma. J Histochem Cytochem. 2009;57:289–300. doi: 10.1369/jhc.2008.952044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zieve GW, Turnbull D, Mullins JM, McIntosh JR. Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor nocodazole: Nocodazole accumulated mitotic cells. Exp Cell Res. 1980;126:397–405. doi: 10.1016/0014-4827(80)90279-7. [DOI] [PubMed] [Google Scholar]
- 20.Tothill RW, et al. Australian Ovarian Cancer Study Group Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 21.Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol. 2007;8:195–208. doi: 10.1038/nrm2114. [DOI] [PubMed] [Google Scholar]
- 22.Sivakumar S, Gorbsky GJ. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat Rev Mol Cell Biol. 2015;16:82–94. doi: 10.1038/nrm3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song L, Rape M. Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Mol Cell. 2010;38:369–382. doi: 10.1016/j.molcel.2010.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schliekelman MJ, et al. Molecular portraits of epithelial, mesenchymal, and hybrid states in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015;75:1789–1800. doi: 10.1158/0008-5472.CAN-14-2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem J. 2012;443:851–856. doi: 10.1042/BJ20120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markman M, et al. Gynecologic Oncology Group Phase II trial of weekly paclitaxel (80 mg/m2) in platinum and paclitaxel-resistant ovarian and primary peritoneal cancers: A Gynecologic Oncology Group study. Gynecol Oncol. 2006;101:436–440. doi: 10.1016/j.ygyno.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 27.Zender L, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell. 2008;135:852–864. doi: 10.1016/j.cell.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen S, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blomen VA, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 30.Wang T, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slamon DJ, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazières J, et al. Lung cancer that harbors an HER2 mutation: Epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31:1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 35.Jaehne J, et al. Expression of Her2/neu oncogene product p185 in correlation to clinicopathological and prognostic factors of gastric carcinoma. J Cancer Res Clin Oncol. 1992;118:474–479. doi: 10.1007/BF01629433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gravalos C, Jimeno A. HER2 in gastric cancer: A new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19:1523–1529. doi: 10.1093/annonc/mdn169. [DOI] [PubMed] [Google Scholar]
- 37.Brabender J, et al. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clin Cancer Res. 2001;7:1850–1855. [PubMed] [Google Scholar]
- 38.Gordon AN, et al. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: Results from a phase II multicenter study. Int J Gynecol Cancer. 2005;15:785–792. doi: 10.1111/j.1525-1438.2005.00137.x. [DOI] [PubMed] [Google Scholar]
- 39.Vergote IB, et al. Randomized phase III study of erlotinib versus observation in patients with no evidence of disease progression after first-line platin-based chemotherapy for ovarian carcinoma: A European Organisation for Research and Treatment of Cancer-Gynaecological Cancer Group, and Gynecologic Cancer Intergroup study. J Clin Oncol. 2014;32:320–326. doi: 10.1200/JCO.2013.50.5669. [DOI] [PubMed] [Google Scholar]
- 40.Kurzeder C, et al. Double-blind, placebo-controlled, randomized phase III trial evaluating pertuzumab combined with chemotherapy for low tumor human epidermal growth factor receptor 3 mRNA-expressing platinum-resistant ovarian cancer (PENELOPE) J Clin Oncol. 2016;34:2516–2525. doi: 10.1200/JCO.2015.66.0787. [DOI] [PubMed] [Google Scholar]
- 41.Barretina J, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Watt PJ, et al. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer. 2009;124:1829–1840. doi: 10.1002/ijc.24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, et al. Suppression of the nuclear transporter-KPNβ1 expression inhibits tumor proliferation in hepatocellular carcinoma. Med Oncol. 2015;32:128. doi: 10.1007/s12032-015-0559-1. [DOI] [PubMed] [Google Scholar]
- 44.Lu T, et al. Karyopherinβ1 regulates proliferation of human glioma cells via Wnt/β-catenin pathway. Biochem Biophys Res Commun. 2016;478:1189–1197. doi: 10.1016/j.bbrc.2016.08.093. [DOI] [PubMed] [Google Scholar]
- 45.García-Higuera I, et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802–811. doi: 10.1038/ncb1742. [DOI] [PubMed] [Google Scholar]
- 46.Omura S, Crump A. The life and times of ivermectin - A success story. Nat Rev Microbiol. 2004;2:984–989. doi: 10.1038/nrmicro1048. [DOI] [PubMed] [Google Scholar]
- 47.Guzzo CA, et al. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J Clin Pharmacol. 2002;42:1122–1133. doi: 10.1177/009127002401382731. [DOI] [PubMed] [Google Scholar]
- 48.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 49.Turner NC, Huang Bartlett C, Cristofanilli M. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:1672–1673. doi: 10.1056/NEJMc1510345. [DOI] [PubMed] [Google Scholar]
- 50.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.