

Abstract
The histone acetyltransferase GCN5 has been suggested to be involved in promoting cancer cell growth. But its role in human colon cancer development remains unknown. Herein we discovered that GCN5 expression is significantly upregulated in human colon adenocarcinoma tissues. We further demonstrate that GCN5 is upregulated in human colon cancer at the mRNA level. Surprisingly, two transcription factors, the oncogenic c-Myc and the proapoptotic E2F1, are responsible for GCN5 mRNA transcription. Knockdown of c-Myc inhibited colon cancer cell proliferation largely through downregulating GCN5 transcription, which can be fully rescued by the ectopic GCN5 expression. In contrast, E2F1 expression induced human colon cancer cell death, and suppression of GCN5 expression in cells with E2F1 overexpression further facilitated cell apoptosis, suggesting that GCN5 expression is induced by E2F1 as a possible negative feedback in suppressing E2F1-mediated cell apoptosis. In addition, suppression of GCN5 with its specific inhibitor CPTH2 inhibited human colon cancer cell growth. Our studies reveal that GCN5 plays a positive role in human colon cancer development, and its suppression holds a great therapeutic potential in antitumor therapy.
Key words: E2F1, c-Myc, General control nonrepressed protein 5 (GCN5), Colon cancer, Gene transcription
INTRODUCTION
Acetylation of either histones or transcription factors is generally associated with transcriptional activation, which plays key roles in regulating cell growth and death. The acetylation is catalyzed by histone acetyltransferases (HATs) by adding acetyl groups to the lysine residues of their targets. Several HATs have been identified as transcription coactivators, including CBP, p300, and Tip60, all of which are involved in a variety of biological functions (1). The general control nonrepressed protein 5 (GCN5) was initially identified as member of the HAT superfamily that positively regulates the transcription of amino acid biosynthetic genes in yeast (2,3). Its mammalian ortholog was first cloned as the tetrahymena histone acetyltransferase A in regulating histone acetylation to gene activation (4). As a histone acetyltransferase, GCN5 has been shown to regulate gene transcription by catalyzing the acetylation of lysine residues on multiple histones including H2b, H3, and H4 (5–7). In addition to histones, GCN5 can directly interact with and acetylate transcription factors, such as FBP1 and N-Myc, in gene transcriptional regulation (8,9). GCN5 functions are required for mouse embryonic development as the genetic deletion of GCN5 leads to early embryonic lethality (10).
Recent studies have suggested that the elevated expression of HAT, including GCN5, can often be detected in human cancers and often predicts poor clinical outcome in cancer patients (11). On the other hand, suppression of the catalytic activity of HATs, either by genetic or pharmacological approaches, can inhibit cancer cell growth and induces their apoptosis, implying that HATs are potential therapeutic target for tumor chemotherapy (12). However, the roles of GCN5 in human colon cancer remain inclusive. In this study, we show that GCN5 expression is elevated in human colon cancer tissues. The elevated GCN5 expression is regulated at the mRNA levels, and the oncogenic transcription factor c-Myc is involved in GCN5 gene expression. In addition, suppression of GCN5 dramatically inhibited human colon cancer cell growth. Unexpectedly, the proapoptotic transcription factor E2F1 also promotes GCN5 expression, and suppression of E2F1-induced GCN5 transcription facilitated E2F1-induced cell death, implying a possible negative feedback in regulating cell apoptosis. Our study here identifies GCN5 as a possible oncogenic factor as well as a potential therapeutic target for human colon cancers.
MATERIALS AND METHODS
Cells, Reagents, and Antibodies
Human colon cancer HCT116 cells were cultured in DMEM containing 10% FBS. GCN5-specific inhibitor CPTH2 was purchased from Sigma-Aldrich Inc. c-Myc expression plasmid was used as reported. Antibodies (Abs, and their sources) used in this study included anti-GCN5 and anti-c-Myc (Santa Cruz Biotechnology) and anti-histone H3 acetyl-lysine 9 (Cell Signaling). shRNAs that specifically knock down c-Myc and control shRNA were purchased from Open Biosystems.
Immunohistochemical Analysis of GCN5 Expression in Human Colon Cancers
Micro tissue array (MTA) slides that carry paraffin-fixed human tumor tissues were purchased from Biomax (Rockville, MD, USA). A standard IHC procedure was used for the analysis as previously described (13). Briefly, after dewaxing with xylene followed by antigen retrieval, tissue sections were blocked by incubating them with 5% normal donkey serum. The slides were then incubated with primary antibodies against GCN5 (1:70 dilution) overnight at 4°C. The slides were then washed with PBST five times and incubated with biotinylated secondary antibodies (1:400; Vector Laboratories) followed by incubation with HRP-streptavidin. HRP activity was detected with the Dab Substrate Kit (Vector Laboratories). Tissues were scored in a double-blinded manner by the following criteria: 0, no specific staining; 1, less than 25% of cells with strong staining or with less than 50% cells with weak staining; 2, less than 50% cells with strong staining or more than 50% cells with weak staining; and 3, more than 50% cells with strong staining.
Analysis of GCN5 Subcellular Distribution in Human Colon Cancer Tissues
Human colon cancer tissues and adjacent normal controls were collected freshly following the guidelines of an approved Institutional Review Board for Health Sciences Research protocol. Subcellular cell fractionation was performed using Subcellular Protein Fractionation Kit for Tissues purchased from Life Technologies (Grand Island, NY, USA). Protein concentrations were measured using a Bio-Rad kit, and 10 μg of proteins from each fraction was used for the Western blotting analysis.
RNA Extraction and Real-Time PCR Analysis of Gene Expression
Total RNA was extracted using TRIzol reagent (Invitrogen, San Diego, CA, USA) as previously described (14). Quantitative real-time RT-PCR was performed using SYBR-Green qPCR master mix (Clontech, San Diego, CA, USA). The β-actin gene was used as a reference for sample normalization. Primers for mouse or human genes, including β-actin and Gcn5, were purchased from Real Time Primers (Elkins Park, PA, USA). A standard amplification protocol was used according to the manufacturer’s instructions.
Transfection and Western Blotting
HCT116 cells were transfected with 1 to 2 µg of shRNA plasmids specific to c-Myc using Lipofectamine 2000 reagent (Invitrogen). Western blotting was performed (15). The transfected cells were collected and lysed with RIPA buffer with protease inhibitor and incubated on ice for 15 min. Insoluble fractions were removed by centrifugation (15,000 × g for 15 min). Cell lysates were subjected to SDS-PAGE and transferred to nitrocellulose membrane. After blocking with 5% (w/v) skim milk in Tris-buffered saline containing 0.1% Tween 20, the membrane was incubated overnight at 4°C with the indicated primary Abs followed by horseradish peroxidase-conjugated secondary Ab. Membranes were then washed and visualized with enhanced chemiluminescence.
Cell Proliferation Assay and Analysis of Cell Apoptosis
In vitro cell proliferation was measured by using the colorimetric WST-1 assay (Cell proliferation reagent WST-1; Roche Diagnostics). Briefly, 4,000 cells were seeded in a 96-well plate with DMEM containing 10% FBS. Every 24 h, 10 µl of WST-1 reagent was added to each well followed by incubation for 2 h. The absorbance at 450 nm was measured using a microplate reader. To analyze apoptosis, single-cell suspension was stained with fluorescence-conjugated anti-annexin V on ice for 30 min, washed, fixed in 1% paraformaldehyde, and analyzed by flow cytometry as previously described (16).
Luciferase Assay
The human promoter region of GCN5 was cloned by PCR using the genomic DNA of HCT116 cells and subcloned into luciferase vector as recently reported (17). Luciferase experiments were performed as previously described (18). Briefly, HCT116 cells in 12-well plates were transfected with pRL-TK (Promega, Madison, WI, USA) and GCN5-luc plasmids, along with c-Myc expression plasmids, or with their specific shRNA. Transfected cells were lysed 2 days after transfection. The luciferase activities in the cell lysates were analyzed using a Dual Luciferase Reporter assay kit (Promega, Madison, WI, USA). Luciferase activity was measured using a luminometer (Turner BioSystems, Inc. Sunnyvale, CA, USA) and expressed in relative light units (RLUs).
RESULTS
GCN5 Protein Expression Is Elevated in Human Colon Cancers
To explore the involvement of GCN5 in colon cancer development, we utilized an immunohistological approach and analyzed the expression levels of GCN5 in human colon adenocarcinoma tissues and control normal colon tissues. As shown in Figure 1A, a low level of staining was observed in the tissue sections of normal human colons with GCN5-specific antibodies. In contrast, a dramatically increased GCN5 staining was detected in the adenocarcinoma tissue sections. We also observed that, in contrast to both cytoplasm and nuclear distribution of GCN5 proteins in normal human colons, GCN5 is largely localized to the nucleus in the colon cancer cells. A summarization of the immunohistological scores from 25 normal human colon tissues and 120 colon cancer tissues indicates a statistically significant increase in GCN5 expression in tumors (Fig. 1B). To further confirm our notion that GCN5 largely localizes in the nucleus of human colon cancer cells, subcellular fractionation was performed using freshly collected human colon cancer tissues. As shown in Figure 1C and D, in contrast to a higher cytoplasmic distribution of GCN5 in the control normal colon tissues, the levels of nuclei GCN5 in the colon cancer cells are significantly increased. These results indicate that GCN5 is upregulated and accumulated in the nucleus in human colon cancers.
Figure 1.

Elevated GCN5 expression in human colon cancer tissues. (A, B) The human colon carcinoma tissues were used for the immunohistological staining with GCN5-specific Abs. Normal adjacent tissues were used as controls. (A) Representative images are shown and (B) the histological scores from 120 adenocarcinoma and their adjacent control tissues are indicated. Student’s t-test was used for the statistic analysis. (C, D). Subcellular fractionation of freshly collected human colon cancer tissues and normal controls was performed, the expression levels of GCN5 and the cytoplasmic protein α-tubulin as a control.
GCN5 Is Upregulated at mRNA Level in Human Colon Cancers
In order to investigate at which steps GCN5 expression is upregulated in human colon cancers, we analyzed GCN5 mRNA levels in human colon cancer tissues. Consistent with our immunohistological data, the GCN5 mRNA could be detected at a low level in normal human colons analyzed by RT-PCR and real-time PCR (Fig. 2A and B). Notably, an average of at least fivefold increase in the GCN5 mRNA expression in human colon cancer tissues was detected (Fig. 2B), implying that GCN5 expression is upregulated at the mRNA level in human colon cancers. Consistent with our discoveries, analysis of the human colon cancer database (level 3) downloaded from published Cancer Genome Atlas (TCGA) Network (19), a statistical significant increase in GCN5 mRNA levels in colon cancers was confirmed (p = 5.4e-17) (Fig. 2C). As shown in Figure 2D, a similar result was further confirmed by analyzing another gene array dataset by Hong et al. (p = 2.1-10) in metastatic colon cancers (20). Collectively, our data indicate that GCN5 expression is upregulated in human colon cancers at the mRNA level.
Figure 2.

GCN5 is upregulated in human colon cancers at the mRNA level. (A, B) Total RNA was extracted from freshly prepared human colon cancer and their adjacent normal tissues. The levels of GCN5 mRNA were analyzed by (A) RT-PCR and (B) real-time PCR. (C, D) Dataset from TCGA colon cancer (C) and a microarray dataset (accession No. GSE9348) (D) were used. The levels of GCN5 in normal and colon cancer tissues were compared by two-tailed Student’s t-test.
The Transcription Factors c-Myc and E2F1 Are Involved in GCN5 Expression in Colon Cancer Cells
To further elucidate the underlying molecular mechanisms by which GCN5 gene expression is regulated in human colon cancers, we analyzed the conserved transcription factor binding sites in the promoter region of GCN5 gene. Notably, conserved binding sites of transcription factors c-Myc and E2F1 were identified. We then cloned the 3-kb region of the GCN5 promoter and generated GCN5 luciferase reporter plasmid (Fig. 3A). When transfected into human colon cancer HCT116 cells, a significantly increased luciferase activity was detected in the lysate of transfected cells, implying that this 3-kb region carries the transcription regulatory elements (Fig. 3B). We then asked whether c-Myc and E2F1 are involved in regulating GCN5 gene transcription. As expected, expression of either c-Myc or E2F1 dramatically promoted GCN5 reporter activity in HCT116 cells, and coexpression of both c-Myc and E2F1 further enhanced GCN5 reporter activity, indicating that c-Myc is a transcription factor for GCN5 gene transcription (Fig. 3C). Mutation of both the c-Myc and E2F1-binding sites in the GCN5 promoter region (Fig. 1A) resulted in a statistic significant reduction in GCN5 reporter activity. Notably, the mutation completely abolished the c-Myc/E2F1-induced GCN5 luciferase activity (Fig. 3C) Conversely, siRNA-mediated knockdown of either c-Myc or E2F1 led to a significant reduction in the expression levels of GCN5 in HCT116 cells, confirming that the transcription factors c-Myc and E2F1 mediates the mRNA transcription (Fig. 3D and E). Therefore, the transcription factors c-Myc and E2F1 mediate GCN5 gene expression in human colon cancers.
Figure 3.

The transcription factors c-Myc is involved in GCN5 mRNA transcription. (A) GCN5 promoter region was amplified by PCR using genomic DNA from PC12 cells as template. The amplified DNA fragment was subcloned into a luciferase reporter vector. Point mutations of the c-Myc and E2F1 binding sites are indicated. (B) GCN5 luciferase or control luciferase plasmid DNA was transfected into HCT116 human colon cancer cells. Forty-eight hours after transfection, the luciferase was determined, and the relative fold changes are shown. (C) GCN5 luciferase plasmid or its mutant was transfected with c-Myc or E2F1 expression plasmids or both. The luciferase activity was determined as in (B). (D, E) c-Myc-specific siRNAs were transfected into HCT116 cells. The protein expression levels of c-Myc (top) were determined by Western blotting using β-actin as a loading control (bottom) (D). The expression levels of GCN5 mRNA in the knockdown cells were analyzed by real-time PCR (E). Student’s t-test was used for the statistical analysis. **p < 0.01 and ***p < 0.001.
The Oncogenic Transcription Factor c-Myc Promotes Colon Cancer Cell Growth Through GCN5
The oncogenic transcription factor, c-Myc, facilitates tumor development and progression through enhancing cancer cell growth and suppressing apoptosis (21). We then asked whether c-Myc enhances human colon cell proliferation through GCN5. As expected, knockdown of c-Myc significantly inhibited HCT116 cell growth rate (Fig. 4A) and increased the percentage of annexin V-positive apoptotic cells (Fig. 4B). Importantly, expression of GCN5 in c-Myc knockdown cells largely rescued HCT116 cell growth and abolished apoptosis induced by c-Myc suppression (Fig. 4). Therefore, these data indicate that c-Myc promotes colon cancer cell growth in a GCN5-dependent manner.
Figure 4.
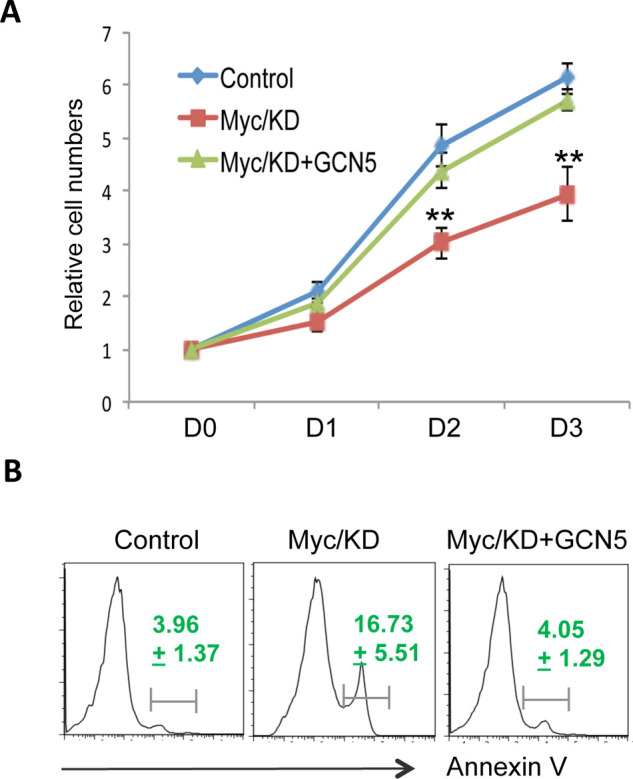
c-Myc promotes human colon cancer cell growth through GCN5. (A) HCT116 cells with c-Myc knockdown (KD) were transfected with or without GCN5. Their proliferation was measured by MTT assay. Error bars represent data from three independent experiments (mean ± SD). (B) Cells in (A) were collected at day 3. The apoptotic cells were analyzed by their expression of annexin V. Percentages of annexin V-positive cells are the average ± SD from three independent experiments. Student’s t-test was used for the statistical analysis. **p < 0.01.
E2F1-Mediated GCN5 Expression as a Possible Negative Feedback in Colon Cancer Cell Death
It is a surprise for us that E2F1, a proapoptotic transcription factor, promotes GCN5 expression in colon cancer cells (Fig. 4). As GCN5 expression is elevated in human colon cancers, we then asked whether the expression of its transcription factor E2F1, similar to c-Myc (22,23), is also increased. In an analysis of the human colon cancer database (level 3) downloaded from published Cancer Genome Atlas (TCGA) Network (19), a statistical significant increase in E2F1 mRNA levels in colon cancers was confirmed (p = 1.25e-9) (Fig. 5A). As shown in Figure 5B, a similar result was further confirmed by analyzing another gene array dataset by Hong et al. (p = 2.46e-5) in metastatic colon cancers (20). Together with our data in Figure 2C and D showing that the increased GCN5 in human colon cancer tissues analyzed from the same databases and E2F1 is a transcription factor of GCN5 (Figs. 3 and 4), these data suggest that E2F1 is a transcription factor of GCN5 in human colon cancers.
Figure 5.

GCN5 is a target of the transcription factor E2F1 to suppress E2F1-induced cell death. (A, B) HCT116 cells were transfected with E2F1 expression plasmids in the presence of GCN5 knockdown siRNA. The protein expression levels of E2F1 (top), GCN5 (middle), and tubulin control (bottom) were analyzed by Western blotting (A). The cell apoptosis was analyzed 5 days after transfection, and the average percentages of annexin V-positive cells from three independent experiments are indicated (B). (C, D) Dataset from TCGA colon cancer (C) and a microarray dataset (accession No. GSE9348) (D) were used. The levels of E2F1 in normal and colon cancer tissues were compared by two-tailed Student’s t-test.
To understand the functional consequences of GCN5 expression mediated by the proapoptotic transcription factor E2F1, we analyzed the role of E2F1-induced GCN5 expression on the survival of human colon cancer cells. As indicated in Figure 5C, transient expression of E2F1 in HCT116 cells resulted in a significant increase in GCN5 protein expression, further supporting our conclusion that E2F1 is a transcription factor of GCN5. Consistent with the previous studies (24), E2F1 expression increased HCT116 cell apoptosis (Fig. 5D). Notably, knockdown of GCN5 expression in HCT116 cells with E2F1 expression led to a dramatic increase in annexin V-positive apoptotic cells (Fig. 5D). These results indicate that GCN5 functions as a negative feedback to suppress E2F1-induced cell death.
GCN5 Suppression Inhibits Human Colon Cancer Cell Growth
The fact that GCN5 expression is elevated in human cancer cells implies that GCN5 suppression may hold therapeutic potentials in colon cancer treatment. To test this hypothesis, we then analyzed the therapeutic efficacy of pharmacological GCN5 suppression on human colon cancer growth. As shown in Figure 6A, treatment of HCT116 cells with a GCN5-specific inhibitor, CTPH2, dose-dependently attenuated H3K9 acetylation without affecting GCN5 protein expressions, indicating that CPTH2 suppresses the catalytic activity but not protein expression of GCN5. Importantly, pharmacological GCN5 suppression led to a significant reduction in the proliferation of human colon cancer cells, both HCT116 and HT9, in a dose-dependent manner (Fig. 6B). Analysis of the annexin V levels indicates that CPTH2 inhibited both HCT116 and HT9 cancer cell growth at 10 μg/ml, but a significant increase in annexin V-positive cells could only be detected when cells were treated with CPTH2 at 20 μg/ml (Fig. 6C). Collectively, our data indicate that GCN5 inhibition has a great therapeutic potential in human colon cancer therapy.
Figure 6.

GCN5 suppression inhibits human colon cancer cell growth. (A, B) HCT116 or HT9 colon cancer cells were treated with GCN5 inhibitor at each indicated concentration. The histone H3 acetylation at the lysine residue 9 (H3K9) was determined by Western blotting (B). The growth of treated cells in (A) was analyzed. Error bars represent data from three independent experiments (mean + SD). (C) The percentage of apoptotic cells in (B) was analyzed by annexin V staining and flow cytometry. Data are representative from three independent experiments (mean ± SD). Student’s t-test was used for the statistic analysis. *p < 0.05 and **p < 0.01. (D) A proposed model of GCN5 in colon cancer cell growth.
Based on our observations, we proposed a model for GCN5 in colon cancer cell growth (Fig. 6D): the oncogenic transcription factor c-Myc, which expression is often elevated in human colon cancer tissues, promotes GCN5 gene expression. The elevated GCN5 activity leads to colon cancer cell hyperproliferation and inhibits colon cancer cell apoptosis. Therefore, suppression of GCN5 inhibits colon cancer cell growth and induces their apoptosis. These discoveries imply that GCN5 is involved in promoting colon cancer development and progression, and suppression of GCN5 holds great therapeutic potentials in colon cancer treatment.
DISCUSSION
The current study demonstrates that the expression levels of GCN5 is elevated in primary human colon cancers and that GCN5 suppression, by both genetic and pharmacological approaches, inhibits human colon cancer cell proliferation. The oncogenic transcription factor c-Myc, whose expression is increased in human colon cancer and predicts the poor clinical outcomes of the patients, is the transcription factor responsible for GCN5 expression. These observations suggest that the elevated GCN5 function is involved in human colon cancer development, and GCN5 is a potential therapeutic target for the human colon cancer treatment.
It has been shown that GCN5 expression is elevated in multiple types of human cancers including lung cancers (11,25–29). This study is the first analysis of GCN5 expression in human primary colon cancers, and we observed a significant increase in both the protein and mRNA expression levels of GCN5, indicating that increased GCN5 expression is regulated at the transcription level. We further demonstrated that the transcription factor c-Myc, which have been shown as potential oncogene in human colon cancer development and progression (21), is involved in GCN5 gene transcription. This conclusion is supported by the following observations. First, expression of c-Myc promoted GCN5 reporter activity in human colon cells. Second, knockdown of c-Myc largely diminished GCN5 expression. More importantly, reconstitution of GCN5 rescues the growth of human colon cancer cell with c-Myc knockdown. GCN5 promotes human lung cancer cell growth through positively regulating the transcription of genes in cell cycle progression, gene transcription, and suppression of apoptosis (11,27–29). Therefore, c-Myc may achieve its oncogenic function through GCN5 gene transcription. Similarly, analysis of the potential influences of Myc on the global chromatin structure identified that N-Myc is a transcription factor for GCN5 gene expression (30). It will be interesting to further study whether the elevated expressions of c-Myc and GCN5 are positively correlated in human colon cancers.
It is a surprise that the proapoptotic transcription factor E2F1 also mediates the antiapoptotic gene GCN5 transcription. Analysis of two independent sets of database indicates that E2F1 expression levels are dramatically increased in human colon cancer tissues. While the regulatory mechanisms and functional consequences of E2F1 upregulation in human colon cancers are unclear, one would expect that the elevated E2F1 expression may promote colon cancer cell death. Our discord that the antiapoptotic lysine acetyltransferase GCN5 is a transcription target of E2F1 in colon cancer cells indicates that GCN5 functions as a negative feedback to antagonize the E2F1-induced cell death to promote colon cancer progression.
Histone acetyltransferase inhibitors have been extensively studied for their potentials in antitumor therapy. It has been shown that multiple inhibitors specific to HAT, such as p300 and CBP, suppress growth and induces apoptosis of multiple types of human cancers in vitro as well as tumors using the experimental mouse tumor models (31–34). Using a high-throughput screening approach, Chimenti et al. recently identified the chemical cyclopentylidene-[4-(4′-chlorophenyl)thiazol-2-yl]hydrazone (CPTH2) as a GCN5-specific inhibitor that suppresses the in vitro GCN5 catalytic activity and GCN5-mediated H3K9 acetylation in yeast and in human cancer cell lines (35). Our studies here show that treatment of human colon cancer HCT116 cells reduced H3K9 acetylation levels and inhibited the proliferation of HCT116 cells. Together with our observations that GCN5 expression is elevated in human colon cancers, our studies indicate that GCN5 is a potential therapeutic target for human colon cancer treatment.
ACKNOWLEDGMENTS
We thank Drs. Bert Vogelstein and Kenneth W. Kinzler (Johns Hopskins University) for the HCT116 human colon cancer cells. This research was supported by the Fundamental Research Funds for the Central Universities to J.Z. and D.F. as well as the NIH RO1 grants to D.F. This research is also partially supported by the NIH grant RO1 CA154377 to D.Z.
REFERENCES
- 1. Marmorstein R, Roth SY. Histone acetyltransferases: function, structure, and catalysis. Curr Opin Genet Dev 2001; 11:155–161. [DOI] [PubMed] [Google Scholar]
- 2. Greenberg ML, Myers PL, Skvirsky RC, Greer H. New positive and negative regulators for general control of amino acid biosynthesis in Saccharomyces cerevisiae. Mol Cell Biol 1986; 6:1820–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skvirsky RC, Greenberg ML, Myers PL, Greer H. A new negative control gene for amino acid biosynthesis in Saccharomyces cerevisiae. Curr Genet 1986; 10:495–501. [DOI] [PubMed] [Google Scholar]
- 4. Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, et al. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996; 84:843–851. [DOI] [PubMed] [Google Scholar]
- 5. Recht J, Osley MA. Mutations in both the structured domain and N-terminus of histone H2B bypass the requirement for Swi-Snf in yeast. EMBO J 1999; 18:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ikeda K, Steger DJ, Eberharter A, Workman JL. Activation domain-specific and general transcription stimulation by native histone acetyltransferase complexes. Mol Cell Biol 1999; 19:855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burgess SM, Ajimura M, Kleckner N. GCN5-dependent histone H3 acetylation and RPD3-dependent histone H4 deacetylation have distinct, opposing effects on IME2 transcription, during meiosis and during vegetative growth, in budding yeast. Proc Natl Acad Sci USA 1999; 96:6835–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Atanassov BS, Dent SY. USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Rep 2011; 12:924–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martinez-Cerdeno V, Lemen JM, Chan V, Wey A, Lin W, Dent SR, et al. N-Myc and GCN5 regulate significantly overlapping transcriptional programs in neural stem cells. PLoS One 2012; 7:e39456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phan HM, Xu AW, Coco C, Srajer G, Wyszomierski S, Evrard YA, et al. GCN5 and p300 share essential functions during early embryogenesis. Dev Dyn 2005; 233:1337–1347. [DOI] [PubMed] [Google Scholar]
- 11. Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem 2013; 288:14510–14521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dekker FJ, van den Bosch T, Martin NI. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today 2014; 19:654–660. [DOI] [PubMed] [Google Scholar]
- 13. Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell 2012; 46:484–494. [DOI] [PubMed] [Google Scholar]
- 14. Lee SM, Gao B, Fang D. FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood 2008; 111:3599–3606. [DOI] [PubMed] [Google Scholar]
- 15. Yang H, Qiu Q, Gao B, Kong S, Lin Z, Fang D. Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J Exp Med 2014; 211:2467–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao B, Calhoun K, Fang D. The proinflammatory cytokines IL-1beta and TNF-alpha induce the expression of Synoviolin, an E3 ubiquitin ligase, in mouse synovial fibroblasts via the Erk1/2-ETS1 pathway. Arthritis Res Ther 2006; 8:R172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li F, Gao B, Dong H, Shi J, Fang D. Icariin induces Synoviolin expression through NFE2L1 to protect neurons from ER stress-induced apoptosis. PloS One 2015; 10:e0119955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen A, Lee SM, Gao B, Shannon S, Zhu Z, Fang D. c-Abl-mediated tyrosine phosphorylation of the T-bet DNA-binding domain regulates CD4+ T-cell differentiation and allergic lung inflammation. Mol Cell Biol 2011; 31:3445–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hong Y, Downey T, Eu KW, Koh PK, Cheah PY. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin Exp Met 2010; 27:83–90. [DOI] [PubMed] [Google Scholar]
- 21. Garte SJ. The c-myc oncogene in tumor progression. Crit Rev Oncog 1993; 4:435–449. [PubMed] [Google Scholar]
- 22. Calabretta B, Kaczmarek L, Ming PM, Au F, Ming SC. Expression of c-myc and other cell cycle-dependent genes in human colon neoplasia. Cancer Res 1985; 45:6000–6004. [PubMed] [Google Scholar]
- 23. Finley GG, Schulz NT, Hill SA, Geiser JR, Pipas JM, Meisler AI. Expression of the myc gene family in different stages of human colorectal cancer. Oncogene 1989; 4:963–971. [PubMed] [Google Scholar]
- 24. Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci USA 1994; 91:3602–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Atanassov BS, Evrard YA, Multani AS, Zhang Z, Tora L, Devys D, et al. Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Mol Cell 2009; 35:352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X, Tesfai J, Evrard YA, Dent SY, Martinez E. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J Biol Chem 2003; 278:20405–12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin Q, Zhuang L, Lai B, Wang C, Li W, Dolan B, et al. Gcn5 and PCAF negatively regulate interferon-beta production through HAT-independent inhibition of TBK1. EMBO Rep 2014; 11:1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kurabe N, Katagiri K, Komiya Y, Ito R, Sugiyama A, Kawasaki Y, et al. Deregulated expression of a novel component of TFTC/STAGA histone acetyltransferase complexes, rat SGF29, in hepatocellular carcinoma: Possible implication for the oncogenic potential of c-Myc. Oncogene 2007; 26:5626–5634. [DOI] [PubMed] [Google Scholar]
- 29. Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol 2004; 24:10826–10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kloetzel PM, Ossendorp F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr Opin Immunol 2004; 16:76–81. [DOI] [PubMed] [Google Scholar]
- 31. Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014; 33:2169–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao XN, Lin J, Ning QY, Gao L, Yao YS, Zhou JH, et al. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLoS One 2013; 8:e55481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang H, Zhang C, Rorick A, Wu D, Chiu M, Thomas-Ahner J, et al. CCI-779 inhibits cell-cycle G2-M progression and invasion of castration-resistant prostate cancer via attenuation of UBE2C transcription and mRNA stability. Cancer Res 2011; 71:4866–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kindle KB, Collins HM, Heery DM. MOZ-TIF2-mediated destruction of CBP/p300 is blocked by calpain inhibitor 2. Leukemia 2010; 24:1359–1361. [DOI] [PubMed] [Google Scholar]
- 35. Chimenti F, Bizzarri B, Maccioni E, Secci D, Bolasco A, Chimenti P, et al. A novel histone acetyltransferase inhibitor modulating Gcn5 network: Cyclopentylidene-[4-(4’-chlorophenyl)thiazol-2-yl)hydrazone. J Med Chem 2009; 52:530–536. [DOI] [PubMed] [Google Scholar]