

Summary
Neisseria gonorrhoeae initiates infection at the apical surface of columnar endocervical epithelial cells in the female reproductive tract. These cells provide a physical barrier against pathogens by forming continuous apical junctional complexes between neighbouring cells. This study examines the interaction of gonococci (GC) with polarized epithelial cells. We show that viable GC preferentially localize at the apical side of the cell–cell junction in polarized endometrial and colonic epithelial cells, HEC-1-B and T84. In GC-infected cells, continuous apical junctional complexes are disrupted, and the junction-associated protein β-catenin is redistributed from the apical junction to the cytoplasm and to GC adherent sites; however, overall cellular levels remain unchanged. This redistribution of junctional proteins is associated with a decrease in the ‘fence’ function of the apical junction but not its ‘gate’ function. Disruption of the apical junction by removing calcium increases GC transmigration across the epithelial monolayer. GC inoculation induces the phosphorylation of both epidermal growth factor receptor (EGFR) and β-catenin, while inhibition of EGFR kinase activity significantly reduces both GC-induced β-catenin redistribution and GC transmigration. Therefore, the gonococcus is capable of weakening the apical junction and polarity of epithelial cells by activating EGFR, which facilitates GC transmigration across the epithelium.
Introduction
Neisseria gonorrhoeae causes gonorrhea, a common sexually transmitted infection (STI). This Gram-negative, obligate human pathogen causes different disease sequelae in men and women. The highest reported cases of gonorrhea are among teenage girls and young women (CDC, 2011). Since most gonococcal (GC) infections in women are asymptomatic, the infections remain undiagnosed, thus predisposing women to pelvic inflammatory disease (PID) and disseminated gonococcal infection (DGI), which can lead to infertility and arthritis respectively (Holmes et al., 1971; Hook and Holmes, 1985; CDC, 2011). Clinical studies show an association of GC infection with an increased risk of acquiring HIV (Ghys et al., 1997; Sheung et al., 2008), highlighting the significance of GC infection in public health. No vaccine has been successfully developed, primarily due to high levels of antigenic variation of the GC surface components and our lack of understanding of the cellular mechanism underlying the interaction of this bacterium with the mucosal surface of the female genital tract.
In women, the primary target of GC is the epithelial cell monolayer that lines the reproductive tract (Harkness, 1948; Merz and So, 2000). The interaction of GC with epithelial cells has been extensively studied, primarily using non-polarized epithelial cell lines or organ culture. Initiation of colonization is mediated by pili, which binds to receptors on columnar endocervical epithelial cells. Subsequent contraction of pili brings the bacteria close to the epithelial cells (Stephens, 1989; Merz and So, 2000; Timmerman et al., 2005), allowing GC to establish a more intimate attachment to the host membrane via opacity proteins (Opa) and lipooligosaccharides (LOS) (Hook and Holmes, 1985; Merz and So, 2000). In epithelial cells, these interactions induce a variety of signalling cascades, including calcium flux, phosphoinositide 3-kinase (PI3K), phospholipase C (PLC) and the mitogen-activated protein kinase (MAPK) Erk, leading to actin reorganization, microvillus elongation and the subsequent engulfment of GC (Shaw and Falkow, 1988; Merz and So, 2000). We have shown that the interaction of GC with human endometrial epithelial cells, HEC-1-B, increases the phosphorylation of epidermal growth factor receptor (EGFR) by triggering the expression and surface cleavage of EGFR ligands. This GC-induced EGFR trans-activation is required for GC invasion into non-polarized HEC-1-B cells. In addition to EGFR phosphorylation, apical inoculation of GC leads to a redistribution of EGFR from the basolateral surface of polarized HEC-1-B cells to GC adherent sites at the apical surface (Swanson et al., 2011).
In addition to attachment and invasion, GC are capable of transmigrating across polarized epithelial cells cultured in vitro (Merz et al., 1996; Mosleh et al., 1997; Wang et al., 1998; 2008). Subepithelial bacteria have been found in organ culture models and clinical samples from patients (Lightfoot and Gotschlich, 1974; Draper et al., 1980; McGee et al., 1981). This suggests that GC transmigration is associated with the pathology of the disease; however the cellular mechanism underlying GC transmigration is largely unknown. Based on the invading capability of GC, it has been proposed that GC transmigrate via an intracellular pathway where apically internalized GC traverse to and exit from the basolateral membrane of epithelial cells (McGee et al., 1981; Merz et al., 1996).
The single layered endocervical columnar epithelial cells have been shown to be the preferred tissue target for GC infection. The monolayer of epithelial cells on the mucosal surface is held together via apical junction complexes formed continuously between neighbouring cells. The apical junction complexes seal the paracellular space between epithelial cells, creating a physical barrier against pathogen movement via the space between cells. The apical junction also limits the lateral movement between the apical and basolateral membrane, generating and maintaining the polarized distribution of proteins and lipids in the apical or basolateral membrane and their distinct physiological functions (Miyoshi and Takai, 2005; Niessen and Gottardi, 2008; Koch and Nusrat, 2009). The actin cytoskeleton provides scaffolding supports for microvilli and the apical junction (Anderson and Van Itallie, 1995; Yonemura et al., 1995; Mitic and Anderson, 1998; Hartsock and Nelson, 2008; Miyoshi and Takai, 2008). The apical junction contains the tight junction and adherens junction (Farquhar and Palade, 1963; Hartsock and Nelson, 2008). The tight junction consists of transmembrane proteins such as occludin and claudins (Miyoshi and Takai, 2005; Koch and Nusrat, 2009) and associated protein ZO-1 that links the tight junction to the actin cytoskeleton (Mitic and Anderson, 1998; Hartsock and Nelson, 2008). The adherens junction is formed through calcium-dependent trans-homophilic interaction of E-cadherin on neighbouring cells (O’Keefe et al., 1987; Harris and Tepass, 2010) and is required for the assembly of the tight junction. Removing extracellular calcium leads to the disassembly of the apical junction (Rothen-Rutishauser et al., 2002). The apical junction is a dynamic structure and its barrier function as well as its assembly and disassembly are regulated by external and internal cell signalling through junction-associated proteins, such as β-catenin (Volberg et al., 1992; Nelson, 2008; Niessen and Gottardi, 2008; Koch and Nusrat, 2009). Signalling mediated by surface receptors, such as EGFR (Kuwada et al., 1998; Yasmeen et al., 2006), induces the phosphorylation of β-catenin, which causes the dissociation of β-catenin from the junctional complex and the actin cytoskeleton (Hoschuetzky et al., 1994; Takahashi et al., 1997; Roura et al., 1999; Rao et al., 2002). Endocytosis and lysosomal degradation of the junctional proteins can also lead to disassembly of the apical junction (Kowalczyk and Reynolds, 2004; Ivanov et al., 2005; Shen et al., 2008).
In order to establish infection, GC must attach to epithelial cells. Invasive diseases may require GC invasion into and/or transmigration across polarized epithelium. While the interaction of GC with non-polarized epithelial cells has been extensively studied, how GC interaction with polarized epithelial cells impacts the epithelial barrier has not been fully investigated. Many mucosal pathogens, including Clostridium difficile, Escherichia coli, Vibrio cholerae, Helicobacter pylori, Clostridium perfringens, Listeria monocytogenes, Salmonella enterica and Bacteroides fragilis, have developed means to weaken the epithelial barrier by directly or indirectly regulating the apical junction. The interaction of these pathogens with polarized epithelial cells disrupts the integrity of the junctional complex and increases epithelial permeability, which facilitate the passage of pathogens through the paracellular space (Finlay and Cossart, 1997; Amieva et al., 2003; Gruenheid and Finlay, 2003; Miyoshi and Takai, 2005; Sousa et al., 2005). Furthermore, N. meningidis, the other pathogenic species of Neisseria, transmigrates across the endothelia barrier by disrupting VE-cadherin-based intercellular junction (Coureuil et al., 2009; Schubert-Unkmeir et al., 2010).
In this study, we examine the interaction of GC with polarized epithelial cells. We found that apical inoculation of GC induces the disassembly of the apical junction, which is concurrent with an increase in the free lateral movement of membrane molecules between the apical and basolateral surface without affecting the permeability of polarized epithelial cells. GC-induced junction disassembly depends on the kinase activity of EGFR. Disrupting the apical junction using the calcium chelator, EGTA, increases GC transmigration. Inhibition of GC-induced junction disassembly by an EGFR kinase inhibitor significantly reduces GC transmigration. These results provide the first evidence that GC can negatively regulate the apical junction of polarized epithelial cells for its transmigration, and suggest that EGFR activation is required for this process.
Results
Preferential localization of live gonococci at the cell–cell junction of polarized epithelial cells
To investigate the interaction of gonococci (GC) with polarized epithelial cells, we used confocal microscopy to analyse the distribution of GC on polarized HEC-1-B and T84 cells, cells that have relatively low and high polarity respectively. Cell polarization was confirmed by a measured increase in the transepithelial electric resistance (TER) and the visualization of the polarized distribution of apical junction proteins. After apical inoculation of GC (live or gentamicin killed at a moi of 10 or 100 respectively) for 2, 4 and 6 h, the epithelial cells were stained for the apical junction protein ZO-1 and GC, and the distribution of GC relative to ZO-1 analysed using three-dimensional (3D) confocal fluorescence microscopy. The percentage of GC clusters in contact with the ZO-1 staining was determined by visual inspection (Fig. 1A). The data show that as early as 2 h post inoculation, ~ 80% of live GC clusters, but only ~ 50% of killed GC clusters, localized at the cell–cell junction of HEC-1-B (Fig. 1B). The percentages of both live and killed GC at the cell–cell junction did not increase over time. Similar results were seen for polarized T84 cells (Fig. 1C). These results indicate that GC preferentially localize at the apical cell–cell junction despite the different levels of polarity and tissue origins of epithelial cells, and this localization is dependent on the viability of the bacteria.
Fig. 1.

Live GC preferentially localize at the cell–cell junction of polarized epithelial cells. Polarized HEC-1-B and T84 cells were apically inoculated with live or gentamicin killed GC (P+O+ MS11) at a moi of 10 and 100 respectively for 2, 4 and/or 6 h. Cells were fixed, permeabilized and stained for ZO-1 and GC, and analysed using confocal microscopy. The number of GC clusters in contact with (long arrows) or away from (arrowhead) ZO-1 staining that marks the cell–cell junction was quantified by visual inspection. Shown are representative images (A) of T84 cells 6 h post inoculation and the average percentages (± SD) of GC clusters at the cell–cell junction of HEC-1-B (B) and T84 cells (C) from three independent experiments (~ 50 GC clusters per experiment). Scale bar, 10 μm. *P ≤ 0.05; ** P ≤ 0.01; and ***P ≤ 0.001.
GC inoculation disrupts the continuous apical junctional complexes between polarized epithelial cells
The preferential cell–cell junctional location of GC suggests a possible impact of GC on the apical junction of polarized epithelial cells, similar to what is observed in other mucosal bacterial pathogens (Finlay and Cossart, 1997; Katz et al., 2000; Ohl and Miller, 2001; Amieva et al., 2003; Gruenheid and Finlay, 2003; Soriani et al., 2006; Attali et al., 2008). To examine the effects of GC on the distribution of apical junctional proteins, polarized HEC-1-B and T84 cells were incubated with GC in the apical compartment for 2, 4 and/or 6 h and stained for the apical junctional protein, ZO-1, occludin or E-cadherin. Fluorescence intensity (FI) profiles of ZO-1 and occludin were generated from images acquired by confocal microscopy. In the absence of GC, ZO-1 (Fig. 2Aa and Ad) and occludin staining (Fig. 2Ba and Bd) continuously surrounded epithelial cells at the z-section close to the apical surface, indicating the formation of apical junctions between neighbouring cells and polarization of the monolayer. The level of the staining surrounding HEC-1-B cells was less intense and consistent than that observed in T84 cells and showed discontinuous staining in 14% of cells (Fig. 2 and Fig. S1). This indicates that the strength of the junctional complex in HEC-1-B cells is lower than that of T84 cells. The addition of live GC to polarized HEC-1-B cells resulted in redistribution of ZO-1 as early as 2 h, with 32% of HEC-1-B cells showing discontinuous ZO-1 staining (Fig. 2D). This percentage increased over time. By 6 h, 78% HEC-1-B cells showed discontinuous ZO-1 staining (Fig. 2Ac, Af, C and D), while 53% of cells showed discontinuous occludin staining (Fig. 2Bc, Bf and E). However, gentamicin killed GC that can attach to but not form tight microcolonies or invade into non-polarized epithelial cells (Bish et al., 2008) had no significant effect on ZO-1 (Fig. 2Ab, Ae and C) or occludin staining (Fig. 2Bb, Be and E). While the level of disruption of the apical junction in T84 cells appeared to be lower than that observed in HEC-1-B cells, GC interaction still increased the percentage of cells with discontinuous staining of ZO-1 and occludin from 0–2% to 18% and 33% respectively (Fig. S1). It was also noticed that most of the regions with discontinuous ZO-1 and occludin staining were underneath GC microcolonies (Fig. 2A and B and Fig. S1A and B).
Fig. 2.

Viable but not killed GC disrupt the continuous apical junction location of ZO-1 and occludin in polarized HEC-1-B cells. Polarized HEC-1-B cells were incubated with media only (a and d), gentamicin killed GC (b and e) or live GC (c and f) in the apical compartment for 6 h (A–C and E) or for 2, 4 and 6 h (D). Cells were fixed and stained for ZO-1 (A, C and D) or occludin (B and E) and GC, and then analysed using confocal microscopy. Shown are representative images (composites of 1 μm slices) (a–c) and their fluorescence intensity profiles (d–f) at 6 h. The percentage of cells with disrupted ZO-1 (C) and occludin (E) peripheral staining at 6 h or with disrupted ZO-1 peripheral staining over time (D) was quantified by visual inspection, and the average percentages (± SD) from three independent experiments (~50 cells per experiment) are shown. Scale bar, 5 μm. ***P ≤ 0.001. *P ≤ 0.05.
Similar to ZO-1 and occludin, E-cadherin staining was concentrated at the apical junction of polarized T84 cells in the absence of GC. A small amount of the E-cadherin staining appeared as punctate in the cytoplasm, indicating its vesicular location (Fig. 3A). After incubation of T84 cells with GC for 6 h, E-cadherin staining at the cell–cell junction decreased, while punctate E-cadherin staining in the cytoplasm increased (Fig. 3A). To quantify the translocation of E-cadherin from the apical junction to the cytoplasmic vesicles, we determined a fluorescence intensity ratio (FIR) of E-cadherin staining at the cell–cell junction to that in the cytoplasm using confocal image sectioning through the apical junction. In the absence of GC, the majority of the E-cadherin staining was detected at the apical junction region of epithelial cells with the FIR at ~ 3.75. After incubation with GC for 6 h, there was a significant decrease in the E-cadherin in the apical junction with a concomitant increase in its staining in the cytoplasmic vesicles, resulting in a reduction in the FIR to ~ 2 (Fig. 3B). The FIR of epithelial cells incubated with gentamicin killed GC was ~ 2.75, suggesting that killed GC have less impact on the redistribution of E-cadherin than live GC (Fig. 3B).
Fig. 3.

GC cause the translocation of E-cadherin from the apical junction to the cytoplasmic vesicles in polarized T84 cells. Polarized T84 cells were incubated with media only, live GC (10:1) or gentamicin killed GC (100:1) in the apical compartment for 6 h. Cells were fixed and stained for E-cadherin and GC and analysed using confocal microscopy. The cell–cell junction and the cytoplasmic regions were manually selected from confocal images sliced through the apical junction, and the fluorescence intensity of E-cadherin staining in each region was measured using the NIH ImageJ software. The fluorescence intensity ratio (FIR) at the apical junction to that in the cytoplasm was calculated. Shown are representative images (A) and the average FIR (± SD) (B) from three independent experiments (~ 50 cells per experiment). Scale bar, 5 μm. ***P ≤ 0.001. *P ≤ 0.05.
Taken together, these results indicate that the inoculation of viable GC, but not killed GC, induces the disassociation of the tight junctional proteins ZO-1 and occludin and the adherens junctional protein E-cadherin from the apical junction of polarized epithelial cells, suggesting a capability for GC to induce the disassembly of the apical junction during infection.
GC inoculation decreases the fence but not gate function of the apical junction
GC-induced redistribution of the apical junctional proteins potentially affects the functionality of the junctional complex. The apical junction performs two significant roles in epithelial cells. The ‘fence’ function prevents proteins and lipids in the apical and basolateral membrane from laterally moving to the other side, thereby maintaining the functional polarity of two surfaces. The ‘gate’ function controls the paracellular permeability of epithelial cells, preventing molecules and organisms such as mucosal pathogens from crossing the polarized monolayer through the paracellular space between cells. To examine the fence function, polarized HEC-1-B cells were grown on the underside of transwells and apically incubated with live GC at a moi of 10 for 4 h. The apical membrane was exclusively labelled with CellMask dye that only becomes fluorescent when inserted into membrane lipids. The distribution of the apical CellMask staining over time was analysed by time-lapse confocal fluorescence microscopy with xz images that reveal both the apical and basolateral surfaces. If the ‘fence’ function of the apical junction was intact, the labelling should remain on the apical surface. An increase of the labelling in the basolateral membrane indicates a decrease in the fence function. To quantify the lateral movement of the membrane dye from the apical to the basolateral surface, we determined the FIR of the dye in the apical surface to that in the basolateral surface. In the absence of GC, the majority of the apically labelled CellMask dye remained at the apical region of epithelial cells, with a FIR at ~ 33. This FIR decreased slowly over time and reduced to ~ 22 by 30 min post CellMask staining (Fig. 4A and B). In the presence of GC there was a rapid decrease in the Cell-Mask dye in the apical membrane, with a concomitant increase in the CellMask dye in the basolateral region, leading to a reduction in the FIR from 33 to 9 within the first 5 min of staining (Fig. 4A and B). The FIR further decreased over time and by 30 min, the FIR in GC-infected epithelial cells was reduced below 3 (Fig. 4A and B). These results show that GC inoculation significantly increases the lateral mobility from the apical to basolateral membrane, suggesting that GC induce a significant reduction in the fence function of the apical junction.
Fig. 4.

GC inoculation increases the lateral mobility between the apical and basolateral membrane, but not the permeability of the apical junction in polarized epithelial cells
A and B. Polarized HEC-1-B were incubated with medium alone (−) or GC (+) for 4 h. Cells were then apically exposed to the CellMask membrane dye, and live time-lapse images were acquired using a confocal microscope. The apical to basolateral fluorescence intensity ratio (FIR) of the CellMask dye over time was determined (B).
C. Epithelial cells were incubated with or without GC for 6 h in the presence of Lucifer yellow or fluorescein dye. The FIR of Lucifer yellow or fluorescein in the apical to that in the basal compartments was determined using a luminometer.
Shown are the representitive images and the average values (± SD) from three independent experiments (~ 10 cells per experiment for B). Scale bar, 5 μm.
The effect of GC on the gate function of the apical junction was determined by measuring the permeability of epithelial monolayers to Lucifer yellow and fluorescein, which are dyes with small molecular masses of 457 and 332 kDa respectively. Unlike the CellMask dye, these dyes are soluble in aqueous solution and incapable of incorporating into the cell membrane or binding to any proteins at the cell surface. The appearance of apically added dyes in the basolateral chamber indicates the diffusion of the dye through the apical junction. Polarized HEC-1-B cells were incubated with the Lucifer yellow or fluorescein apically in the absence or presence of GC for 6 h. The FIR of the dye in the basal to that in the apical media was determined and used as a quantitative measure of the apical to basal permeability. We found that GC inoculation had no significant effect on the amount of either Lucifer yellow or fluorescein diffusion to the basolateral medium (Fig. 4C). Consistent with this finding, we did not detect significant decreases in TER, which measures the permeability of the apical junction to charged molecules, after 6 h GC incubation. Disrupting the apical junction with the calcium chelator EGTA significantly increased the amount of the dyes in the basal medium (data not shown), demonstrating that the gate function of polarized epithelial cells is intact in the absence of GC. These results indicate that GC inoculation reduces the fence function of the apical junction, allowing membrane-associated molecules to move more freely between the apical and basolateral surfaces without significantly altering the permeability of the epithelium.
The phosphorylation and redistribution of β-catenin in GC-infected epithelial cells
The apical junction undergoes rapid assembly and disassembly in response to internal and external signals (Koch and Nusrat, 2009). β-Catenin provides a link between the apical junction and cellular signalling (Aberle et al., 1996; Moon et al., 2001; Niessen and Got-tardi, 2008; Yang et al., 2009). To understand how GC regulate the apical junction, we examined the effects of GC inoculation on the phosphorylation and cellular distribution of β-catenin. Polarized HEC-1-B cells were incubated with or without GC in the apical chamber for 4 h. Phosphorylated β-catenin was detected and quanti-fied using immunoprecipitation and Western blot analysis. We found that the presence of GC significantly increased the phosphorylation level of β-catenin without altering its total protein level (Fig. 5A and B), suggesting that GC stimulates β-catenin phosphorylation. We then used immunofluorescence microscopy to analyse the cellular distribution of β-catenin. The disassociation of β-catenin from the apical junction was quantified using the FIR of β-catenin at the cell–cell junction to that in the cytoplasm. β-Catenin was concentrated at the periphery of cells in the absence of GC in both polarized HEC-1-B and T84 cells (Fig. 5C–H). When exposed to GC, there were increases in the cytoplasmic levels with parallel decreases in the junctional level of β-catenin, leading to reductions in the junction to cytoplasm FIR of the β-catenin staining in both polarized HEC-1-B and T84 cells (Fig. 5C–H). Concurrent with this redistribution, a portion of β-catenin staining appeared to colocalize with GC (Fig. 5C and F), suggesting recruitment of β-catenin to GC adherent sites. In contrast, incubation with gen-tamicin killed GC for the same length of time did not affected the cellular distribution of β-catenin (Fig. 8D and G). These results demonstrate that the presence of GC induces the phosphorylation and redistribution of β-catenin from the apical junction to the cytoplasm, and suggest that GC regulate the apical junction via modulating the activity of β-catenin.
Fig. 5.

GC inoculation induces the phosphorylation and redistribution of β-catenin.
A and B. Polarized HEC-1-B cells were incubated apically with or without GC for 4 h. Cells were lysed and subjected to immunoprecipitation using β-catenin-specific antibodies. Immunoprecipitates were analysed by SDS-PAGE and Western blot probing for phosphotyrosine. The blot was quantified by densitometry to determine fold increase over no GC control.
C–H. Polarized HEC-1-B (C–E) and T84 (F–H) cells were incubated with or without GC apically for 6 h. Cells were stained for β-catenin and GC and analysed using confocal microscopy. Fluorescence intensity profiles along a line crossing cells (D and G) were generated to determine the β-catenin FIR at the membrane compared to the cytoplasm (E and H). Shown are representative blots, images, fluorescent intensity of the representative images, and the average FIR (± SD) from three independent experiments. Scale bar, 5 μm. ***P ≤ 0.001; ** P ≤ 0.01.
Fig. 8.

GC inoculation induces the phosphorylation of EGFR, which is required for GC-induced redistribution of β-catenin.
A and B. Polarized HEC-1-B cells were incubated with or without GC for 4 h and then lysed. Cell lysates were subjected to immunoprecipitation using a phosphotyrosine-specific antibody. Immunoprecipitates and the cell lysates were analysed using SDS-PAGE and Western blot, probing for EGFR and β-tubulin as loading controls. Shown are representative blots of three independent experiments (A). Densitometry analysis was performed to determine fold increase over control (B).
C–H. Polarized HEC-1-B (C–E) and T84 (F–H) cells were untreated or pre-treated with the EGFR kinase inhibitor AG1478 (10 μM) for 2 h then apically incubated with live GC in the absence or presence of the inhibitor or gentamicin killed GC for 6 h. Cells were stained for β-catenin and GC and analysed using confocal microscopy. The β-catenin FIR of membrane to cytoplasm was determined (D and G) as described in Fig. 5. Colocalization of GC microcolonies with β-catenin was analysed in HEC-1-B (E) and T84 (H) using Pearson correlation coefficients and the Zen software. Shown are representative images (C and F) and the average FIR (D and G) and coefficient (± SD) (E and H) of three independent experiments (~ 15 cells per experiment). Scale bar, 10 μm, ***P ≤ 0.001; **P ≤ 0.01.
GC inoculation does not change the cellular level of junctional proteins
A common mechanism underlying the disassembly of the apical junction is the endocytosis and lysosomal degradation of junctional proteins. To determine if GC trigger such a mechanism, we compared the cellular levels of the junctional proteins, occludin and ZO-1, in polarized HEC-1-B cells with and without 6 h GC incubation. Using Western blot analysis, we did not detect significant differences in the total protein levels of ZO-1 and occludin in GC inoculated epithelial cells in comparison with those without GC (Fig. 6A and B). This result suggests that GC inoculation does not lead to a significant degradation of junction proteins and that degradation of junction proteins is unlikely to be the mechanism for GC-mediated regulation of the apical junction.
Fig. 6.

GC inoculation does not change the cellular levels of junctional proteins. Polarized HEC-1-B cells were lysed and analysed by SDS-PAGE and Western blot, probing for junctional proteins, ZO-1 and occludin, and β-tubulin as the loading control. Shown are representative blots (A) and the average densitometry values (± SD) from three independent experiments (B).
Disrupting the apical junction with EGTA increases GC transmigration across polarized epithelial cells
The apical junction is essential for the mucosal epithelium’s barrier function. To investigate if junctional regulation is important for GC infection, we determined if disruption of the apical junction by EGTA has any effect on GC invasion and transmigration. EGTA is a Ca2+ chelator that induces the disassembly of the apical junction by inhibiting Ca2+-dependent trans-homophilic interaction of E-cadherin on neighbouring epithelial cells (Rothen-Rutishauser et al., 2002). Polarized HEC-1-B cells were pre-treated with EGTA in both the apical and basal compartments, and the EGTA was removed before GC inoculation in the apical compartment. EGTA pre-treatment dramatically reduced TER and caused a complete loss of the polarized distribution of ZO-1, but did not affect the viability of GC (data not shown), confirming the efficacy of EGTA in junction disruption. After the 6 h incubation with GC, the cfu of GC recovered from the basal medium was increased 100-fold in EGTA-treated HEC-1-B cells, compared with cells without EGTA treatment (Fig. 7A). However, EGTA treatment did not significantly change the level of GC invasion into HEC-1-B cells (Fig. 7B). Therefore, disrupting the apical junction increases GC transmigration across polarized epithelial monolayers but not their invasion into the cells.
Fig. 7.
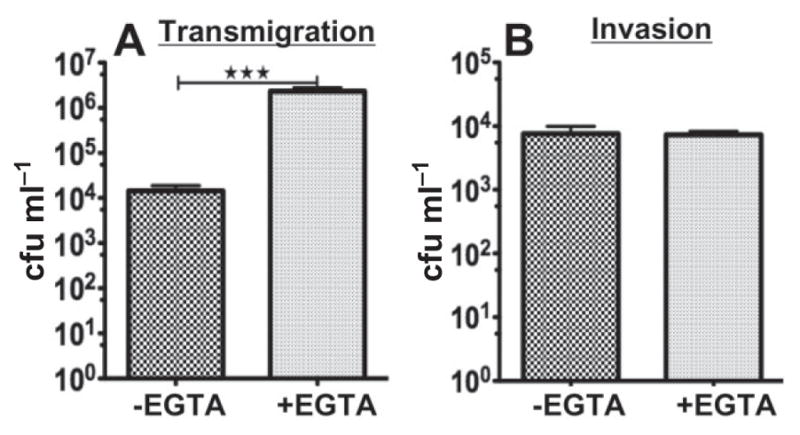
Disrupting the apical junction by EGTA increases GC transmigration. Polarized HEC-1-B cells were pre-treated with or without EGTA (5 mM) apically and basally for 10 min. Cells were then washed and inoculated with GC for 6 h. The basal medium was collected and plated to determine the transmigrated GC (A). Cells were washed, incubated with gentamicin and lysed, and the bacteria collected and plated to determine invaded GC (B). Shown are the average values (± SD) from more than three independent experiments. ***P ≤ 0.001.
GC-induced β-catenin redistribution depends on the kinase activity of EGFR
The increased phosphorylation level and redistribution of the junction signalling connecter, β-catenin, in GC-inoculated epithelial cells suggest that GC potentially regulate the apical junction via activating host signalling cascades. Based on previous findings that GC inoculation increases EGFR phosphorylation (Swanson et al., 2011) and that EGFR activation has been shown to lead to β-catenin phosphorylation (Hoschuetzky et al., 1994; Takahashi et al., 1997; Yasmeen et al., 2006), we hypothesized that GC-induced redistribution of β-catenin is related to EGFR activation. We examined whether GC inoculation impacts EGFR phosphorylation in polarized epithelial cells and if the EGFR kinase inhibitor AG1478 has any effect on GC-induced redistribution of β-catenin. The phosphorylation of EGFR was determined by immunoprecipitation and Western blot. Similar to our previous observations in non-polarized cells (Swanson et al., 2011), the level of phosphorylated EGFR in polarized HEC-1-B cells was increased after interaction with GC for 4 h, compared with media only controls (Fig. 8A and B). When EGFR activation was inhibited with the EGFR kinase inhibitor AG1478, we detected a significant reduction in GC-induced redistribution of β-catenin from the membrane to the cytoplasm in both polarized HEC-1-B and T84 cells, increasing the membrane to cytoplasm FIR of β-catenin from ~ 3 back to 5 (Fig. 8C, D, F and G). Additionally, treatment with the EGFR inhibitor reduced the colocalization between GC microcolonies and β-catenin for both polarized HEC-1-B and T84 cells (Fig. 8E and H). The inhibitory effect of the EGFR kinase inhibitor on GC-induced redistribution of β-catenin and β-catenin colocalization with GC microcolonies suggests that GC-induced EGFR activation and its downstream signalling lead to the disassembly of the apical junction in GC-infected epithelial cells.
Negative regulation of the apical junction by GC facilitates GC transmigration
To investigate whether GC-induced redistribution of junctional proteins and reduction in the fence function of the apical junction contribute to GC infection, we utilized EGF and the EGFR kinase inhibitor to manipulate the effect of GC on the apical junction. Treatment with EGF, which activates EGFR signalling cascades and promotes junction disassembly (Ray et al., 2007; Ji et al., 2009) resulted in an approximately fivefold increase in GC transmigration across polarized HEC-1-B cells, as compared with the untreated control cells (Fig. 9A). In contrast, the treatment of the EGFR kinase inhibitor, which inhibits the negative effects of GC on the apical junction, led to an approximately fivefold decrease in GC transmigration compared with untreated control cells and a ~10-fold decrease when compared with EGF-treated cells (Fig. 9A). However, both EGF and EGFR kinase inhibitor did not significantly change the level of GC invasion (Fig. 9B). These results suggest that GC-induced EGFR activation and the consequent disassembly of the apical junction facilitate the transmigration of GC across the epithelial monolayer.
Fig. 9.
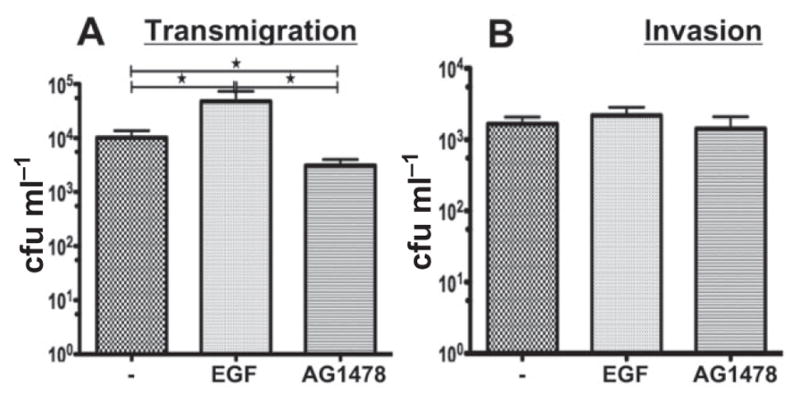
GC transmigration is EGFR dependent. Polarized HEC-1-B cells were untreated or pre-treated with the EGFR kinase inhibitor AG1478 (10 μM) or EGF (10 nM) for 2 and 1 h respectively, and then apically incubated with GC for 6 h in the presence or absence of AG1478 or EGF. The basal medium was collected and plated to determine transmigrated cfu (A). Cells were washed and incubated with gentamicin and lysed, and the bacteria collected and plated to determine invaded GC (B). Shown are the average values (± SD) of more than three independent experiments. *P ≤ 0.05.
Discussion
GC establish infection in the female genital tract primarily by interacting with the endocervical epithelial monolayer. This monolayer of columnar epithelial cells is highly polarized and held together by the apical junction. This study provides the first detailed examination of the interaction of GC with polarized epithelial cells and reveals the cellular mechanisms underlying this interaction. Our results demonstrate that GC interaction with polarized epithelial cells induces the disassembly of the apical junction, weakening its barrier function against the free lateral movement of membrane-associated molecules between the apical and basolateral surfaces, but not its barrier function against paracellular permeability. The negative effect of GC on the apical junction is dependent on GC-induced EGFR activation, which leads to the phosphorylation and disassociation of β-catenin from the apical junction. The weakening of the apical junction by GC facilitates their transmigration across polarized epithelial cells, contributing to GC pathogenicity.
To establish infection, mucosal pathogens have to overcome our body’s first line of defence, the epithelium. Therefore, it is not surprising that many mucosal pathogens, such as enteric bacterial pathogens enterohaemor-rhagic E. coli, Salmonella and H. pylori, are capable of disrupting the apical junction that secures the epithelial physical barrier, thereby increasing the permeability of epithelial monolayers lining the lumen of the intestine (Simonovic et al., 2000; Ohl and Miller, 2001; Amieva et al., 2003; Muza-Moons et al., 2003; Miyoshi and Takai, 2005; Sousa et al., 2005). Meningococci have been shown to disrupt both the occludin-based tight junction and the VE-cadherin-based intercellular junction of endothelial cells, providing a mechanism for meningococci to cross the brain blood barrier (Coureuil et al., 2009; 2012; Schubert-Unkmeir et al., 2010). GC have been shown to induce the redistribution of E-cadherin but not the apical junctional proteins ZO-1 and occludin in immortalized endometrial epithelial cells and isolated primary fallopian epithelial cells that are not polarized (Rodríguez-Tirado et al., 2012). This study shows for the first time that the apical incubation of GC with polarized epithelial cells induces the protein redistribution of both adherens and tight junctions. Such redistribution was not only observed in endometrial epithelial cells HEC-1-B that have a relatively low polarity, but also in colonic epithelial cells T84 that are highly polarized.
Our studies found that the viability of GC was required for both GC localization at the cell–cell junction and GC-induced disassembly of the apical junction. Gentamicin-killed GC used in this study have been shown to be capable of attaching to epithelial cells but are unable to form tight microcolonies (LeVan et al., 2012) and to reprogramme their gene expression in response to the interaction with host cells. Therefore, the formation of tight microcolonies, which depends on pili retraction and LOS–Opa interaction (Merz et al., 1996; Ilver et al., 1998; LeVan et al., 2012), at the cell–cell junction and/or gene expression induced by GC–epithelial cell interaction are potentially involved in the disassembly of the apical junction.
The dynamics of assembly and disassembly of the apical junction is tightly controlled by cell signalling (Ivanov et al., 2005; Miyoshi and Takai, 2005). While many mucosal pathogens are capable of inducing junction disassembly, different pathogens appear to use different mechanisms, probably due to their intrinsic properties and/or the type of epithelial cells that the bacteria interact with. For example, H. pylori injects CagA and other proteins via the type IV secretory apparatus into epithelial cells (Guttman and Finlay, 2009), where CagA targets to PAR1/MARK kinase complexes that play essential roles in epithelial cell polarity (Saadat et al., 2007). S. enterica injects SopB, SopE, SopE2 and SipA and enteropathogenic E. coli EspF and EspG into epithelial cells through their type III secretory system. Injected effectors cause junction disruption by interfering with host signalling and cytoskeleton (Bertelsen et al., 2004; Viswanathan et al., 2004; Matsuzawa et al., 2005; Boyle et al., 2006). In the case of meningococcus, the bacterium disrupts the intercellular junction by activating β2-adrenoceptor/β-arrestin pathway in endothelial cells (Coureuil et al., 2010). Here we show that GC induces EGFR activation in polarized epithelial cells, and that GC induced redistribution of β-catenin and GC transmigration across polarized epithelial cells depend on EGFR activation. These data suggest an involvement of EGFR in GC-induced junctional disassembly.
EGFR activation is known to activate β-catenin by inducing its phosphorylation and its release from the junctional complex. This leads to the disassembly of the apical junction and frees epithelial cells from cell–cell contact inhibitory mechanisms, which are required for cell proliferation and migration (Ji et al., 2009). While other mucosal bacterial pathogens, like H. pylori, also induce the transactivation of EGFR (Wallasch et al., 2002), the effector molecules secreted by these pathogens can directly interact with junctional proteins or regulators (Guttman and Finlay, 2009). Because GC lack the ability of injecting effector molecules into host cells, they may utilize EGFR transactivation as a primary mechanism to interfere with the integrity of the epithelial barrier.
The results from this study suggest that GC-induced junction disassembly facilitates GC transmigration across polarized epithelial cells. We found a significant increase in GC transmigration when the apical junction is disrupted by EGTA and a decrease in GC transmigration when GC-induced β-catenin dissociation from the apical junction is inhibited by the EGFR kinase inhibitor. However, the exact mechanism by which GC transmigrates across polarized genital epithelial cells remains unclear. There are two possible mechanisms for GC transmigration, an intracellular mechanism where GC invade epithelial cells from the apical surface, transcytose through the cells and exit from the basolateral membrane, and a paracellular mechanism where GC migrate through the apical junction. Our studies provide supporting evidence for the paracellular transmigration of GC. First, GC preferentially localize at the cell–cell junction. Second, the increase in GC transmigration is positively correlated with the junction disassembly. Last, EGF and EGFR inhibitor, which increase and decrease GC transmigration respectively, do not affect GC invasion. However, it is also possible that GC-induced junction disassembly increases GC transmigration through the intracellular pathway without increasing the number of gentamicin resistant, intracellular GC. GC-induced junctional disassembly is associated with actin reorganization and the redistribution of GC target molecules, such as CD46 (Källström et al., 1997) and EGFR (Swanson et al., 2011), from their restricted basolateral location to the apical surface (Maisner et al., 1997; Kuwada et al., 1998), which can facilitate GC invasion and intracellular transmigration. Since the EGFR kinase inhibitor does not completely block GC transmigration, it implies an EGFR independent mechanism for GC transmigration. Therefore, GC are likely to be able to transmigrate through both pathways, but may prefer one over the other under different environmental conditions and with different surface molecules expressed on their surface.
Different from the enteric bacterial pathogens and meningococcus, GC-induced redistribution of the junctional proteins does not lead to a significant increase in the permeability of epithelial monolayers (gate function). Instead, it only weakens the lateral mobility barrier between the apical and basolateral membrane (fence function). This allows proteins and lipids in the apical and basolateral membrane to laterally move more freely into each other, consequently reducing or losing the polarized functional domains in epithelial cells. How GC manage to affect the fence function of the apical junction more than its gate function is unclear. GC are known to form tight interactions with the plasma membrane of epithelial cells (Griffiss et al., 1999). Our finding that GC microcolonies preferentially localize at the cell–cell junction supports a hypothesis that the continuous interaction between the outer membrane of GC with the plasma membrane of epithelial cells at their junction may allow them to transmigrate through disassembled apical junction without opening the paracellular space for free diffusion. This is known to occur naturally during apical antigen sampling by mucosal dendritic cells, where the dendrites of the cells extend through the apical junction from the basolateral side to capture antigens at the apical surface, without increasing the permeability of the epithelium. This process is mediated by the direct interaction of junctional proteins expressed by dendritic cells with the apical junctional proteins of epithelial cells (Rescigno et al., 2001). Whether the cell–cell junctional location of GC is mediated by direct interaction of GC with junctional proteins and whether the tight interaction between the outer membrane of GC with the plasma membrane of epithelial cells prohibits paracellular diffusion remain to be determined.
Our previously published data show that GC induce EGFR activation by stimulating EGF ligand production and cleavage at the epithelial cell surface, but not by directly interacting with the receptor (Swanson et al., 2011). Since GC expressing either Opa or pili can activate EGFR but at a level lower than GC expressing both (Swanson et al., 2011), this suggests that EGFR activation does not depend on a specific surface molecule of GC. However, GC expressing different surface molecules, which interact with different host receptors, may induce EGFR transactivation to different levels, consequently causing different magnitudes of junction disruption. The stimulation of EGFR ligand production and surface cleave is known to depend on signalling (Kuwada et al., 1998). One possible source of this signal is Toll-like receptors (TLR), since neisserial porin has been shown to be a potent activator of TLR2 (Massari et al., 2003) and TLR2 has been shown to be expressed by HEC-1-B endometrial epithelial cells (Aboussahoud et al., 2010). While this hypothesis needs to be further examined, TLR-induced signalling via MyD88 has been reported to regulate the production of EGFR ligand in intestinal epithelial cells (Shaykhiev et al., 2008). Additionally, signalling induced by mechanical force generated by pilus retraction and the formation of GC microcolonies (Howie et al., 2005) that killed GC are incapable of may contribute to the induction of EGFR ligands.
In polarized epithelial cells, EGFR is recruited from the basolateral surface to GC adherent sites at the apical surface, which is independent of the kinase activity of EGFR (Swanson et al., 2011). The depolarization of EGFR surface distribution may be the result of GC induced decrease in the fence function of the apical junction. It can be also caused by the mistargeting of newly synthesized EGFR from the trans-Golgi network and/or internalized EGFR from early endosomes to the apical surface. The molecular mechanisms by which GC induce the redistribution of EGFR and the impact of EGFR redistribution on EGFR activation, the integrity of the apical junction, and GC infectivity remain to be investigated.
We previously showed that EGFR inhibition reduces GC invasion into non-polarized cells (Swanson et al., 2011). This is contrary with our finding that inhibition of EGFR activation reduces GC transmigration across but not invasion into polarized epithelial cells. This contradiction implies that EGFR in polarized and non-polarized epithelial cells plays different roles during GC infection. Polarized and non-polarized epithelial cells are known to have different cellular distributions of EGFR and its effector molecules (Kuwada et al., 1998) and different organization of the actin cytoskeleton (Yonemura et al., 1995). Such differences may cause GC to change their mechanisms to interact with and to infect polarized and non-polarized epithelial cells.
Since EGFR is essential for the survival and polarization of epithelial cells, we were unable to use other approaches, such as si/shRNA knock-down, to confirm the results from the EGFR kinase inhibitor. Therefore, we utilized the approach of activating EGFR by addition of EGF. Our results show that EGF-induced EGFR activation increases GC transmigration, opposite of the effect of the EGFR kinase inhibitor that blocks EGFR activation and decreases GC transmigration, supporting the hypothesis that GC-induced EGFR transactivation is involved in GC transmigration.
Our results demonstrate that GC utilize the EGFR signalling pathway to breach the epithelial barrier for their transmigration. Further studies are required to define the cellular mechanisms by which GC transactivate EGFR and GC transmigrate across polarized epithelial cells. The resulting mechanistic knowledge will expand our understanding of GC pathogenesis and provide new ideas for preventive measures against GC infection.
Experimental procedures
Epithelial cells
The human endometrial adenocarcinoma cell line, HEC-1-B (ATCC# HTB-113, Manassas, VA, USA), was maintained in Eagles MEM, alpha medium supplemented with 10% heat-inactivated fetal bovine serum (FBS). The human colorectal carcinoma cell line, T84 (ATCC# CCL-248), was maintained in Dulbecco’s modified Eagle’s medium:Ham F12 (1:1) supplemented with 7% heat-inactivated FBS. Cells were maintained at 37°C and 5% CO2. Cells were seeded at 6 × 104 (6.5-mm-diameter transwell) or at 1 × 105 (24-mm-diameter transwell) per transwell (3 μm pore size, polyester transwells inserts, Corning, Lowell, MA, USA) and cultured for ~ 10 days until transepithelial resistance (TER) reached ~ 400 Ω (HEC-1-B) and ~ 2000 Ω (T84). TER was measured using a Millicell ERS volt-ohm meter (Millipore, Bedford, MA, USA).
Neisseria strains
Neisseria gonorrhoeae strain MS11 that expressed both pili and Opa (Pil+ Opa+) was used. Bacteria were grown on GC media base plates with 1% Kellogg’s supplement (GCK) (White and Kellogg, 1965) for 15–18 h before inoculation. Pil+ Opa+ colonies were acquired based on their morphology using a dissecting light microscope. Bacteria were placed in suspension and the concentration determined using a spectrophotometer. Gentamicin killed bacteria were generated by incubating the bacteria with 100 μg ml−1 gentamicin sulfate for 4 h at 37°C and then overnight at 4°C. Bacteria were inoculated with epithelial cells at moi of 10:1 for viable GC and 100:1 for killed GC.
Immunofluorescence analysis
Cells were serum starved overnight, pre-treated with or without the EGFR inhibitor AG1478 (10 μM, Calbiochem, San Diego, CA, USA) for 2 h, and incubated with GC in the presence or absence of the inhibitor for 6 h. Cell were washed and fixed using a pH shift method (Berod et al., 1981), permeabilized, and stained with anti-ZO-1 (BD Bioscience, Bedford, MA, USA), anti-occludin (Invitrogen, Camarillo, CA, USA), anti-β-catenin (Millipore, Temecula, CA, USA) and anti-GC (Bish et al., 2008) antibodies. Cells were analysed by confocal fluorescence microscopy (Zeiss LSM 510 or 710, Carl Zeiss Microscopy LLC, Thornwood, NY, USA). Z-series of images were obtained in 0.5 μm slices from the top to the bottom of cells, and 3D composites obtained.
To analyse the location of GC relative to the cell–cell junction, three Z-series of images around the apical surface were merged together. From merged images, GC clusters that were in contact with ZO-1 staining were counted visually as localizing at the cell–cell junction, no matter if GC staining did or did not colocalize with ZO-1 staining or if there was discontinuous ZO-1 staining. To analyse the distribution of the tight junctional proteins, ZO-1 and occludin, we generated fluorescence intensity profiles using the NIH ImageJ software on images merged from three Z-slices around the apical surface. Cells showing discontinuous staining of junctional proteins in merged images were identified and counted by visual inspection. The redistribution of E-cadherin was quantified by its FIR at the apical junction to that in the cytoplasm. The cell–cell junction and the cytoplasmic regions were manually selected from confocal image sliced through the apical junction, and the fluorescence intensity of E-cadherin staining in each region was measured using the NIH ImageJ software. The redistribution of β-catenin from the apical junction to the cytoplasm was quantified by the FIR of β-catenin at the cell–cell junction to that in the cytoplasm using fluorescence intensity profiles generated using the ImageJ software. The colocalization of GC with β-catenin was quantified using Pearson correlation coefficient between two staining in single slice images at GC clusters (Manders et al. 1993). All the quantified data were generated from three or more independent experiments with 50 individual GC clusters or epithelial cells per condition and per experiment.
Immunoprecipitation and immunoblotting analyses
Polarized epithelial cells apically incubated with bacteria for varying lengths of time were lysed using RIPA buffer (Swanson et al., 2011). The cell lysates were incubated with protein A Sepha-rose beads (GE Healthcare, Piscataway, NJ, USA) and anti-β-catenin (Millipore) or anti-phosphotyrosine (mAb 4G10) (Millipore) antibodies. Immunoprecipitates were resolved by SDS/PAGE and analysed using Western blotting. For anti-β-catenin immunoprecipitation, the blots were probed for phosphotyrosine using 4G10 mAb to determine phosphorylated β-catenin. For phosphotyrosine immunoprecipitation, the blots were probed for EGFR (Santa Cruz, CA, USA). The total β-catenin and EGFR in the cell lysates were detected by Western blotting using specific antibodies. The blots were imaged using Fujifilm’s LAS-3000 (Valhalla, NY, USA) and quantified by Fujifilm’s MultiGuage software.
Functional analyses of the apical junction
To determine the effect of GC on the fence function of the apical junction, HEC-1-B cells were seeded at 1 × 105 on the underside of transwells and cultured for 10 days until TER reached the optimal level. GC were added to the apical surface and incubated for 4 h. Time-lapse xz images (a frame every 10 s) were acquired in the presence of the CellMask dye (5 μg ml−1, Invitrogen) in the apical chamber for 30 min using Leica TCS SP5 X confocal microscope (Leica Microsystems, Buffalo Grove, IL, USA). The fluorescence intensity profile of the dye along a line across the apical and basolateral membrane of a cell was generated and the fluorescence intensity of the dye in the apical and basolateral membrane was determined using the NIH ImageJ software. The FIR of the CellMask dye at the apical to basolateral membrane was calculated. To determine the effect of GC on the gate function of polarized epithelial cells against the diffusion of molecules through the paracellular spaces, cells were incubated with GC and Lucifer Yellow (500 μM, Sigma, Saint Louis, MO, USA) or fluorescein (50 μM, Acros Organics, Geel, Belgium) in the apical chamber for 6 h. The fluorescence intensity of Lucifer Yellow and fluorescein in the apical and basolateral media was determined using a fluorometer. The FIR of the two dyes at the apical to basolateral medium was determined.
GC invasion and transmigration assays
Polarized epithelia cells were incubated apically with GC for 6 h at 37°C. Where the EGFR inhibitor was used, cells were pre-treated with AG1478 (10 μM) for 2 h and incubated with GC in the presence of the inhibitor. Media from the basal compartment was collected and plated onto GCK to determine the number of transmigrated bacteria. Cell-associated bacteria that were resistant to gentamicin treatment were counted as invaded GC.
Statistical analysis
Statistical significance was assessed using the Student’s t-test by Prism software (GraphPad Software, San Diego, CA). P-values were determined in comparison with controls.
Supplementary Material
GC inoculation disrupts the continuous apical junction location of ZO-1 and occludin in polarized T84 cells. Polarized T84 cells were incubated with media only (a) or GC (b–c) in the apical compartment for 6 h. Cells were stained for ZO-1 or occludin and GC and analysed using confocal microscopy. Shown are representative images of ZO-1 (A) and occludin (B) and their fluorescence intensity profiles (d–e). Cells with disrupted ZO-1 (C) and occludin (D) peripheral staining were quantified by visual inspection, and the average percentages (± SD) from three independent experiments are shown. Scale bar, 10 μm. **P ≤ 0.01.
Acknowledgments
We acknowledge the UMCP-CBMG Imaging Core, where all confocal microscopy experiments were performed. This work was supported by a grant from the National Institutes of Health, AI068888, to D.C.S. and W.S.
Footnotes
Additional Supporting Information may be found in the online version of this article
References
- Aberle H, Schwartz H, Kemler R. Cadherin–catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61:514–523. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C514::AID-JCB4%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Aboussahoud W, Aflatoonian R, Bruce C, Elliott S, Ward J, Newton S, et al. Expression and function of Toll-like receptors in human endometrial epithelial cell lines. J Reprod Immunol. 2010;84:41–51. doi: 10.1016/j.jri.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical–junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol. 1995;269:G467–G475. doi: 10.1152/ajpgi.1995.269.4.G467. [DOI] [PubMed] [Google Scholar]
- Attali C, Durmort C, Vernet T, Di Guilmi AM. The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect Immun. 2008;76:5350–5356. doi: 10.1128/IAI.00184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berod A, Hartman BK, Pujol JF. Importance of fixation in immunohistochemistry: use of formaldehyde solutions at variable pH for the localization of tyrosine hydroxylase. J Histochem Cytochem. 1981;29:844–850. doi: 10.1177/29.7.6167611. [DOI] [PubMed] [Google Scholar]
- Bertelsen LS, Paesold G, Marcus SL, Finlay BB, Eckmann L, Barrett KE. Modulation of chloride secretory responses and barrier function of intestinal epithelial cells by the Salmonella effector protein SigD. Am J Physiol Cell Physiol. 2004;287:C939–C948. doi: 10.1152/ajpcell.00413.2003. [DOI] [PubMed] [Google Scholar]
- Bish SE, Song W, Stein DC. Quantification of bacterial internalization by host cells using a beta-lactamase reporter strain: Neisseria gonorrhoeae invasion into cervical epithelial cells requires bacterial viability. Microbes Infect. 2008;10:1182–1191. doi: 10.1016/j.micinf.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EC, Brown NF, Finlay BB. Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell Microbiol. 2006;8:1946–1957. doi: 10.1111/j.1462-5822.2006.00762.x. [DOI] [PubMed] [Google Scholar]
- CDC. Sexually Transmitted Disease Surveillance 2010. Atlanta: Centers of Disease Control and Prevention; 2011. [Google Scholar]
- Coureuil M, Mikaty G, Miller F, Lécuyer H, Bernard C, Bourdoulous S, et al. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science. 2009;325:83–87. doi: 10.1126/science.1173196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coureuil M, Lécuyer H, Scott MG, Boularan C, Enslen H, Soyer M, et al. Meningococcus hijacks a β2-adrenoceptor/β-arrestin pathway to cross brain microvasculature endothelium. Cell. 2010;143:1149–1160. doi: 10.1016/j.cell.2010.11.035. [DOI] [PubMed] [Google Scholar]
- Coureuil M, Join-Lambert O, Lécuyer H, Bourdoulous S, Marullo S, Nassif X. Mechanism of meningeal invasion by Neisseria meningitidis. Virulence. 2012;3:164–172. doi: 10.4161/viru.18639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper DL, James JF, Brooks GF, Sweet RL. Comparison of virulence markers of peritoneal and fallopian tube isolates with endocervical Neisseria gonorrhoeae isolates from women with acute salpingitis. Infect Immun. 1980;27:882–888. doi: 10.1128/iai.27.3.882-888.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BB, Cossart P. Exploitation of mammalian host cell functions by bacterial pathogens. Science. 1997;276:718–725. doi: 10.1126/science.276.5313.718. [DOI] [PubMed] [Google Scholar]
- Ghys PD, Fransen K, Diallo MO, Ettiègne-Traoré V, Coulibaly IM, Yeboué KM, et al. The associations between cervicovaginal HIV shedding, sexually transmitted diseases and immunosuppression in female sex workers in Abidjan, Côte d’Ivoire. AIDS. 1997;11:F85–F93. doi: 10.1097/00002030-199712000-00001. [DOI] [PubMed] [Google Scholar]
- Griffiss JM, Lammel CJ, Wang J, Dekker NP, Brooks GF. Neisseria gonorrhoeae coordinately uses Pili and Opa to activate HEC-1-B cell microvilli, which causes engulfment of the gonococci. Infect Immun. 1999;67:3469–3480. doi: 10.1128/iai.67.7.3469-3480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, Finlay BB. Microbial pathogenesis and cytoskeletal function. Nature. 2003;422:775–781. doi: 10.1038/nature01603. [DOI] [PubMed] [Google Scholar]
- Guttman JA, Finlay BB. Tight junctions as targets of infectious agents. Biochim Biophys Acta. 2009;1788:832–841. doi: 10.1016/j.bbamem.2008.10.028. [DOI] [PubMed] [Google Scholar]
- Harkness AH. The pathology of gonorrhoea. Br J Vener Dis. 1948;24:137–147. [PMC free article] [PubMed] [Google Scholar]
- Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11:502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KK, Counts GW, Beaty HN. Disseminated gonococcal infection. Ann Intern Med. 1971;74:979–993. doi: 10.7326/0003-4819-74-6-979. [DOI] [PubMed] [Google Scholar]
- Hook EW, Holmes KK. Gonococcal infections. Ann Intern Med. 1985;102:229–243. doi: 10.7326/0003-4819-102-2-229. [DOI] [PubMed] [Google Scholar]
- Hoschuetzky H, Aberle H, Kemler R. Beta-catenin mediates the interaction of the cadherin–catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie HL, Glogauer M, So M. The N. gonorrhoeae type IV pilus stimulates mechanosensitive pathways and cytoprotection through a pilT-dependent mechanism. PLoS Biol. 2005;3:e100. doi: 10.1371/journal.pbio.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilver D, Källström H, Normark S, Jonsson AB. Transcellular passage of Neisseria gonorrhoeae involves pilus phase variation. Infect Immun. 1998;66:469–473. doi: 10.1128/iai.66.2.469-473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AI, Nusrat A, Parkos CA. Endocytosis of the apical junctional complex: mechanisms and possible roles in regulation of epithelial barriers. Bioessays. 2005;27:356–365. doi: 10.1002/bies.20203. [DOI] [PubMed] [Google Scholar]
- Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell. 2009;36:547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Källström H, Liszewski MK, Atkinson JP, Jonsson AB. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol Microbiol. 1997;25:639–647. doi: 10.1046/j.1365-2958.1997.4841857.x. [DOI] [PubMed] [Google Scholar]
- Katz J, Sambandam V, Wu JH, Michalek SM, Balkovetz DF. Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect Immun. 2000;68:1441–1449. doi: 10.1128/iai.68.3.1441-1449.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Nusrat A. Dynamic regulation of epithelial cell fate and barrier function by intercellular junctions. Ann N Y Acad Sci. 2009;1165:220–227. doi: 10.1111/j.1749-6632.2009.04025.x. [DOI] [PubMed] [Google Scholar]
- Kowalczyk AP, Reynolds AB. Protecting your tail: regulation of cadherin degradation by p120-catenin. Curr Opin Cell Biol. 2004;16:522–527. doi: 10.1016/j.ceb.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Kuwada SK, Lund KA, Li XF, Cliften P, Amsler K, Opresko LK, Wiley HS. Differential signaling and regulation of apical vs. basolateral EGFR in polarized epithelial cells. Am J Physiol. 1998;275:C1419–C1428. doi: 10.1152/ajpcell.1998.275.6.C1419. [DOI] [PubMed] [Google Scholar]
- LeVan A, Zimmerman L, Mahle A, Swanson K, DeShong P, Park J, et al. Construction and characterization of a derivative of Neisseria gonorrhoeae strain MS11 devoid of opa genes. J Bacteriol. 2012;194:6468–6478. doi: 10.1128/JB.00969-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfoot RW, Gotschlich EC. Gonococcal disease. Am J Med. 1974;56:327–356. doi: 10.1016/0002-9343(74)90616-0. [DOI] [PubMed] [Google Scholar]
- McGee ZA, Johnson AP, Taylor-Robinson D. Pathogenic mechanisms of Neisseria gonorrhoeae: observations on damage to human fallopian tubes in organ culture by gonococci of colony type 1 or type 4. J Infect Dis. 1981;143:413–422. doi: 10.1093/infdis/143.3.413. [DOI] [PubMed] [Google Scholar]
- Maisner A, Zimmer G, Liszewski MK, Lublin DM, Atkinson JP, Herrler G. Membrane cofactor protein (CD46) is a basolateral protein that is not endocytosed. Importance of the tetrapeptide FTSL at the carboxyl terminus. J Biol Chem. 1997;272:20793–20799. doi: 10.1074/jbc.272.33.20793. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. Measurement of colocalization of object in dual-colour confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Massari P, Ram S, Macleod H, Wetzler LM. The role of porins in neisserial pathogenesis and immunity. Trends Microbiol. 2003;11:87–93. doi: 10.1016/s0966-842x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- Matsuzawa T, Kuwae A, Abe A. Enteropathogenic Escherichia coli type III effectors EspG and EspG2 alter epithelial paracellular permeability. Infect Immun. 2005;73:6283–6289. doi: 10.1128/IAI.73.10.6283-6289.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz AJ, So M. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu Rev Cell Dev Biol. 2000;16:423–457. doi: 10.1146/annurev.cellbio.16.1.423. [DOI] [PubMed] [Google Scholar]
- Merz AJ, Rifenbery DB, Arvidson CG, So M. Traversal of a polarized epithelium by pathogenic Neisseriae: facilitation by type IV pili and maintenance of epithelial barrier function. Mol Med. 1996;2:745–754. [PMC free article] [PubMed] [Google Scholar]
- Mitic LL, Anderson JM. Molecular architecture of tight junctions. Annu Rev Physiol. 1998;60:121–142. doi: 10.1146/annurev.physiol.60.1.121. [DOI] [PubMed] [Google Scholar]
- Miyoshi J, Takai Y. Molecular perspective on tight-junction assembly and epithelial polarity. Adv Drug Deliv Rev. 2005;57:815–855. doi: 10.1016/j.addr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Miyoshi J, Takai Y. Structural and functional associations of apical junctions with cytoskeleton. Biochim Biophys Acta. 2008;1778:670–691. doi: 10.1016/j.bbamem.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Moon HS, Choi EA, Park HY, Choi JY, Chung HW, Kim JI, Park WI. Expression and tyrosine phosphorylation of E-cadherin, beta- and gamma-catenin, and epidermal growth factor receptor in cervical cancer cells. Gynecol Oncol. 2001;81:355–359. doi: 10.1006/gyno.2001.6163. [DOI] [PubMed] [Google Scholar]
- Mosleh IM, Boxberger HJ, Sessler MJ, Meyer TF. Experimental infection of native human ureteral tissue with Neisseria gonorrhoeae: adhesion, invasion, intracellular fate, exocytosis, and passage through a stratified epithelium. Infect Immun. 1997;65:3391–3398. doi: 10.1128/iai.65.8.3391-3398.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muza-Moons MM, Koutsouris A, Hecht G. Disruption of cell polarity by enteropathogenic Escherichia coli enables basolateral membrane proteins to migrate apically and to potentiate physiological consequences. Infect Immun. 2003;71:7069–7078. doi: 10.1128/IAI.71.12.7069-7078.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ. Regulation of cell–cell adhesion by the cadherin–catenin complex. Biochem Soc Trans. 2008;36:149–155. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen CM, Gottardi CJ. Molecular components of the adherens junction. Biochim Biophys Acta. 2008;1778:562–571. doi: 10.1016/j.bbamem.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe EJ, Briggaman RA, Herman B. Calcium-induced assembly of adherens junctions in keratinocytes. J Cell Biol. 1987;105:807–817. doi: 10.1083/jcb.105.2.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohl ME, Miller SI. Salmonella: a model for bacterial pathogenesis. Annu Rev Med. 2001;52:259–274. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- Rao RK, Basuroy S, Rao VU, Karnaky KJ, Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002;368:471–481. doi: 10.1042/BJ20011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RM, Vaidya RJ, Johnson LR. MEK/ERK regulates adherens junctions and migration through Rac1. Cell Motil Cytoskeleton. 2007;64:143–156. doi: 10.1002/cm.20172. [DOI] [PubMed] [Google Scholar]
- Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Tirado C, Maisey K, Rodríguez FE, Reyes-Cerpa S, Reyes-López FE, Imarai M. Neisseria gonorrhoeae induced disruption of cell junction complexes in epithelial cells of the human genital tract. Microbes Infect. 2012;14:290–300. doi: 10.1016/j.micinf.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Rothen-Rutishauser B, Riesen FK, Braun A, Günthert M, Wunderli-Allenspach H. Dynamics of tight and adherens junctions under EGTA treatment. J Membr Biol. 2002;188:151–162. doi: 10.1007/s00232-001-0182-2. [DOI] [PubMed] [Google Scholar]
- Roura S, Miravet S, Piedra J, García de Herreros A, Duñach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. 2010;6:e1000874. doi: 10.1371/journal.ppat.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JH, Falkow S. Model for invasion of human tissue culture cells by Neisseria gonorrhoeae. Infect Immun. 1988;56:1625–1632. doi: 10.1128/iai.56.6.1625-1632.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaykhiev R, Behr J, Bals R. Microbial patterns signaling via Toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PLoS ONE. 2008;3:e1393. doi: 10.1371/journal.pone.0001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Hirsch DS, Sasiela CA, Wu WJ. Cdc42 regulates E-cadherin ubiquitination and degradation through an epidermal growth factor receptor to Src-mediated pathway. J Biol Chem. 2008;283:5127–5137. doi: 10.1074/jbc.M703300200. [DOI] [PubMed] [Google Scholar]
- Sheung A, Rebbapragada A, Shin LY, Dobson-Belaire W, Kimani J, Ngugi E, et al. Mucosal Neisseria gonorrhoeae coinfection during HIV acquisition is associated with enhanced systemic HIV-specific CD8 T-cell responses. AIDS. 2008;22:1729–1737. doi: 10.1097/QAD.0b013e32830baf5e. [DOI] [PubMed] [Google Scholar]
- Simonovic I, Rosenberg J, Koutsouris A, Hecht G. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol. 2000;2:305–315. doi: 10.1046/j.1462-5822.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- Soriani M, Santi I, Taddei A, Rappuoli R, Grandi G, Telford JL. Group B Streptococcus crosses human epithelial cells by a paracellular route. J Infect Dis. 2006;193:241–250. doi: 10.1086/498982. [DOI] [PubMed] [Google Scholar]
- Sousa S, Lecuit M, Cossart P. Microbial strategies to target, cross or disrupt epithelia. Curr Opin Cell Biol. 2005;17:489–498. doi: 10.1016/j.ceb.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Stephens DS. Gonococcal and meningococcal pathogenesis as defined by human cell, cell culture, and organ culture assays. Clin Microbiol Rev. 1989;2(Suppl):S104–S111. doi: 10.1128/cmr.2.suppl.s104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Griffiss JM, Edwards VL, Stein DC, Song W. Neisseria gonorrhoeae-induced transactivation of EGFR enhances gonococcal invasion. Cell Microbiol. 2011;13:1078–1090. doi: 10.1111/j.1462-5822.2011.01603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Suzuki K, Tsukatani Y. Induction of tyrosine phosphorylation and association of beta-catenin with EGF receptor upon tryptic digestion of quiescent cells at confluence. Oncogene. 1997;15:71–78. doi: 10.1038/sj.onc.1201160. [DOI] [PubMed] [Google Scholar]
- Timmerman MM, Shao JQ, Apicella MA. Ultrastructural analysis of the pathogenesis of Neisseria gonorrhoeae endometrial infection. Cell Microbiol. 2005;7:627–636. doi: 10.1111/j.1462-5822.2005.00491.x. [DOI] [PubMed] [Google Scholar]
- Viswanathan VK, Koutsouris A, Lukic S, Pilkinton M, Simonovic I, Simonovic M, Hecht G. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect Immun. 2004;72:3218–3227. doi: 10.1128/IAI.72.6.3218-3227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberg T, Zick Y, Dror R, Sabanay I, Gilon C, Levitzki A, Geiger B. The effect of tyrosine-specific protein phosphorylation on the assembly of adherens-type junctions. EMBO J. 1992;11:1733–1742. doi: 10.1002/j.1460-2075.1992.tb05225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallasch C, Crabtree JE, Bevec D, Robinson PA, Wagner H, Ullrich A. Helicobacter pylori-stimulated EGF receptor transactivation requires metallo-protease cleavage of HB-EGF. Biochem Biophys Res Commun. 2002;295:695–701. doi: 10.1016/s0006-291x(02)00740-4. [DOI] [PubMed] [Google Scholar]
- Wang J, Gray-Owen SD, Knorre A, Meyer TF, Dehio C. Opa binding to cellular CD66 receptors mediates the transcellular traversal of Neisseria gonorrhoeae across polarized T84 epithelial cell monolayers. Mol Microbiol. 1998;30:657–671. doi: 10.1046/j.1365-2958.1998.01102.x. [DOI] [PubMed] [Google Scholar]
- Wang JA, Meyer TF, Rudel T. Cytoskeleton and motor proteins are required for the transcytosis of Neisseria gonorrhoeae through polarized epithelial cells. Int J Med Microbiol. 2008;298:209–221. doi: 10.1016/j.ijmm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- White LA, Kellogg DS. Meisseria gonorrhoeae identification in direct smears by a fluorescent antibody-counterstain method. Appl Microbiol. 1965;13:171–174. doi: 10.1128/am.13.2.171-174.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Guo X, Debnath G, Mohandas N, An X. Protein 4.1R links E-cadherin/beta-catenin complex to the cytoskeleton through its direct interaction with beta-catenin and modulates adherens junction integrity. Biochim Biophys Acta. 2009;1788:1458–1465. doi: 10.1016/j.bbamem.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasmeen A, Bismar TA, Al Moustafa AE. ErbB receptors and epithelial-cadherin–catenin complex in human carcinomas. Future Oncol. 2006;2:765–781. doi: 10.2217/14796694.2.6.765. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Itoh M, Nagafuchi A, Tsukita S. Cell-to-cell adherens junction formation and actin filament organization: similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J Cell Sci. 1995;108(Part 1):127–142. doi: 10.1242/jcs.108.1.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GC inoculation disrupts the continuous apical junction location of ZO-1 and occludin in polarized T84 cells. Polarized T84 cells were incubated with media only (a) or GC (b–c) in the apical compartment for 6 h. Cells were stained for ZO-1 or occludin and GC and analysed using confocal microscopy. Shown are representative images of ZO-1 (A) and occludin (B) and their fluorescence intensity profiles (d–e). Cells with disrupted ZO-1 (C) and occludin (D) peripheral staining were quantified by visual inspection, and the average percentages (± SD) from three independent experiments are shown. Scale bar, 10 μm. **P ≤ 0.01.