

Abstract
The mechanism of clinical action for the FDA approved hypomethylating drugs azacitidine and decitabine remains unresolved and in this context the potential immunomodulatory effect of these agents on leukemic cells is an area of active investigation. Induced expression of methylated Cancer Testis Antigen (CTA) genes has been demonstrated in leukemic cell lines following exposure to hypomethylating drugs in vitro. SGI-110 is a novel hypomethylating dinucleotide with prolonged in vivo exposure and clinical activity in patients with MDS and AML. We demonstrate that this agent, like decitabine, produces robust re-expression of the CTAs NY-ESO-1 and MAGE-A, both in vitro and in leukemia-bearing AML xenografts. Upregulation of these genes in vitro was sufficient to induce cytotoxicity by HLA-compatible CD8+ T-cells specific for NY-ESO-1, a well-recognized and immunogenic CTA. Additionally, exposure to SGI-110 enhances MHC class I and co-stimulatory molecule expression, potentially contributing to recognition of CTAs. SGI-110, like the parent compound decitabine, induces expression of CTAs and might modulate immune recognition of myeloid malignancy.
Keywords: Acute Myeloid Leukemia, SGI-110, Cancer testis antigens, Epigenetics, DNA methylation, DNA methyltransferase inhibitors, cancer germline genes, epigenetics, immune modulation
Introduction
The currently available hypomethylating agents (HMAs), Decitabine (DAC) and Azacitidine (AZA) have changed the face of myelodysplastic syndrome (MDS) therapy, inducing hematological responses and improving quality of life for patients (1). Unfortunately, although these drugs provide significant benefit, responses are of limited duration and once a patient has failed an HMA, survival is dismal (2–5). HMAs may exert their effect through hypomethylation and induction of silenced tumor suppressor genes; however, when patients develop resistance to these drugs their hypomethylating effects are not lost (6). In addition to DNA hypomethylation, both drugs have also been shown to induce DNA damage as a result of double strand DNA breaks resulting from the bulky adducts formed when DNMTs become trapped on azanucleoside-substituted DNA (7–10). Despite clear demonstration of both events (i.e. tumor suppressor gene activation and DNA damage), neither mechanism has been reliably predictive of clinical response (7, 11). An additional less-examined potential mechanism of action for these drugs is the induction or alteration of autologous immune responses against induced neo-antigens (12–14). Numerous reports of correlation between auto-immunity and response to therapy with hypomethylating drugs in patients with MDS have been made, suggesting that the immune system may be aberrantly activated (15–17). Furthermore, patients with evidence of MDS and co-existing autoimmune phenomena demonstrate a more indolent clinical course (17).
We and others have shown both in vitro and in patients treated with the drugs AZA and DAC that among the genes induced by hypomethylation following HMA exposure are the Cancer Testis, (or Cancer Germline) antigens (CTAs) (18–20). CTAs are genes whose normal expression is limited to the adult testis and fetal ovary, although they can also be aberrantly expressed in cancer (21). Among the most studied CTAs are NY-ESO-1 and the MAGE-A gene family. Both NY-ESO-1 and MAGE-A proteins are intensely immunogenic; in solid tumors, their endogenous expression is associated with autologous anti-tumor immune responses as well as delayed tumor progression (22–24). In normal tissues, and in cancer cells that do not express them, CTAs demonstrate dense promoter methylation, associated with gene silencing (25). Cancer vaccine strategies targeting these genes are in active clinical development in phase I–III clinical trials, however a majority of these are limited to cancers where endogenous gene expression is common, such as lung cancer, melanoma and ovarian cancer (26). By contrast with solid tumors, hematological malignancies rarely if ever endogenously express NY-ESO-1 or MAGE-A, but recent efforts have demonstrated that expression of these genes is induced in AML blasts at conventional doses and schedules of the FDA approved hypomethylating agents, albeit at low level (12, 18, 27).
Further emphasizing the potential role of immune recognition in response to HMA treatment, a recent clinical study which enrolled 21 patients with MDS or Acute Myeloid Leukemia (AML) to receive combination therapy with AZA and valproic acid (a histone deacetylase inhibitor, HDACi), reported that of 15 patients with samples suitable for analysis, 11 demonstrated induction of MAGE-A specific CD8+ cytotoxic T cells (CTLs) (12). Most notably, 8 of the 11 patients with circulating CTLs achieved a major clinical response to HMA therapy suggesting that induction of CTL responses may play a role in the clinical outcome of patients treated with such drugs.
Efforts to improve the pharmacokinetics and clinical efficacy of HMA therapy have led to the development of SGI-110. SGI-110 is a rationally designed HMA which complexes DAC with deoxyguanosine, thereby producing a relative resistance to degradation by cytidine deaminase and providing a longer in vivo half-life in comparison with the parent compound (28). Due to differences in molecular weight, the molar equivalent of a 1mg dose of DAC is approximately 2.5 mg of SGI-110 (29). Recently, SGI-110 has been tested in phase I and II clinical trials of relapsed/refractory MDS and AML patients and has been demonstrated to result in longer exposure to active DAC as well as significant clinical responses, even in patients who had previously failed a first generation HMA (30).
In this study we examine the ability of SGI-110, relative to the conventional HMAs DAC and AZA, to induce hypomethylation and gene expression of the recognized immunotherapy targets NY-ESO-1 and the MAGE-A family in a variety of leukemia cell lines both in vitro and as tumor xenografts. We examined several different doses and schedules of SGI-110 in order to optimize the biological effects of this novel HMA to induce hypomethylation and gene expression. We demonstrate that SGI-110 induces the expression of both NY-ESO-1 and MAGE-A family genes in vitro and in vivo. We further show that such re-expression is sufficient to induce cytotoxicity in HLA compatible, NY-ESO-1 specific CD8+ T cell lines derived from ovarian cancer patients whose cancers spontaneously express NY-ESO-1 and who have been vaccinated with an NY-ESO-1 vaccine (31, 32). Additionally, we show that SGI-110 induces the expression of MHC class I and ICAM1 on the surface of leukemia cells, potentially contributing to their enhanced recognition by cytotoxic T-cells. Similar results have been previously reported in solid tumor cell lines, but this is among the first demonstration of such activity leukemia cells treated with SGI-110 (33–35). Evaluation of several different doses and schedules, in association with the demonstration of enhanced presentation of MHC class I and ICAM1, an important co-stimulatory molecule, provides insight into the duration of induced gene expression and will help to inform future combination strategies with CTA directed vaccination in leukemia.
Materials and Methods
Cell lines and in vitro drug treatments
HL60 and KG1a were propagated in Iscove’s Modified Dulbecco’s Medium supplemented with 20% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin (P/S); U937 was propagated in RPMI 1640, 10% FBS, 1% P/S. SGI-110 was provided under a research agreement with Astex Pharmaceuticals, Inc. (Dublin, CA). AZA and DAC were obtained from Sigma. Cell lines were treated with SGI-110 at concentration 0.1, 1.0 and 5 μmol/L (dissolved in PBS) on day 1 and day 3, and harvested on day 5. As controls, cells were treated with vehicle alone (PBS), 0.5 μmol/L DAC, and 2.0 μmol/L AZA using the same treatment schedule as SGI-110. Cell viability was measured using trypan blue exclusion (Supplemental Fig 1).
Xenograft experiments
10 million U937 cells were implanted subcutaneously into 6 week old female SCID mice using an IACUC-approved protocol. After ~2–3 weeks, when macroscopic tumors (~50mm3) formed, mice were assigned to groups for treatments (5 mice/group): Groups 1–5 were dosed daily for 5 days with vehicle (65% propylene glycol, 25% glycerol, 10% ethanol), SGI-110 3, 6.1 or 10 mg/kg/day, or DAC 2.5 mg/kg/day. Groups 6–10 were treated weekly for three weeks with vehicle, SGI-110 6.1, 12.2 or 24.4 mg/kg/day, or DAC 5.0 mg/kg/day. Groups 11–15 were treated twice weekly for three weeks, receiving vehicle, SGI-110 6.1, 12.2 or 24.4 mg/kg/day, or DAC 2.5mg/kg/day. All groups 1 to 15 were exposed to vehicle, SGI-110 or DAC subcutaneously. Tumors were excised for molecular analysis day 7 after treatment for Groups 1–5, and day 20 for Groups 6–15. For group 11–15 the time of harvest was variable due to toxicity of SGI-110 at doses of 12.2 and 24.4 mg/kg/day. For these samples, tissues were collected earlier than the planned endpoint. Mice were monitored daily for toxicity related to drug treatment; changes in activity, coat condition and weight loss were monitored. At the time of sacrifice or death, mice underwent necropsy and visible organ damage was noted and reported (Table 1).
Table 1.
SGI-110 toxicity in vivo
| Groups | Treatment mg/kg/d | Toxicity |
|---|---|---|
| G1–5 | Vehicle | 5/5 mice were alive till endpoint |
| SGI-110-3 | 2/5 mice were found slightly hunched | |
| SGI-110-6.1 | 5/5 mice showed gas filled intestines | |
| SGI-110-10 | 4/5 mice were dead on the day of endpoint. 1 mouse was moribund | |
| DAC-2.5 | 2/5 mice were slightly hunched and showed gas filled intestines | |
| G6–10 | Vehicle | 5/5 mice were alive till endpoint |
| SGI-110-6.1 | 5/5 mice were alive till endpoint | |
| SGI-110-12.2 | 5/5 mice were alive till endpoint | |
| SGI-110-24.4 | 5/5 mice were alive till endpoint | |
| DAC-5 | 5/5 mice were alive till endpoint | |
| G11–15 | Vehicle | 5/5 mice were alive till endpoint |
| SGI-110-6.1 | 1/5 mouse was dead | |
| SGI-110-12.2 | 1/5 mouse was found dead and remaining 4 tissue samples were harvested before the completion of last two treatments as the mice were sick. | |
| SGI-110-24.4 | 1/5 mouse was found dead and rest 4 mouse tissue samples were harvested before the completion of last two treatment as they were moribund or sick. | |
| DAC-2.5 | 5/5 mice were alive till endpoint |
Quantitative Bisulfite-Pyrosequencing and Methylation-Specific PCR (MSP)
The Puregene kit (Qiagen) was used to isolate genomic DNA and sodium bisulfite conversion performed using the EZ DNA Methylation Kit (Zymo Research). Methylation of the NY-ESO-1 promoter and the LINE-1 repetitive elements were determined by sodium bisulfite pyrosequencing (25). Primer sequences are shown in Supplemental Table 1. MSP was performed to evaluate MAGE-A3/6 promoter methylation (36). Primers were designed using MethPrimer, and listed in Supplemental Table 2 (37). Unmethylated and methylated PCR products were amplified with initial denaturation at 95°C for 5 min, then 35 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 60 s, followed by a final 5 min extension at 72°C. PCR products were analyzed on 2% agarose gel by ethidium bromide staining. Using ImageJ software, densitometry was performed and %methylation calculated using the formula- (MP/(UP+MP)*100) where MP is density of methylated PCR product and UP is density of Unmethylated PCR product
Quantitative Reverse Transcriptase PCR (qRT-PCR)
Total RNA was purified using Trizol (Invitrogen) and cDNA made using iScript cDNA synthesis kit (Bio-Rad). Absolute quantification of RNA was performed using PCR Master Mix for SYBR Green assays (Eurogentech) on the 7300 Real-Time PCR System (Applied Biosciences). All samples were run in triplicate, and NY-ESO-1 and MAGE-A3/6 gene expression data were normalized to 18S rRNA. Sequences for primers used are listed in Supplemental Table 3.
Western blot analysis
Whole protein was extracted from cell lines and/or tissue samples (38) and quantitated. 30–50 μg of protein was loaded onto a NuPAGE® Novex® 4–12% Bis-Tris gel (Invitrogen) and transferred to a nitrocellulose membrane (Invitrogen). 5% blotting grade blocker (Bio-Rad) in phosphate-buffered saline was used to block nonspecific binding. Membranes were incubated overnight at 4°C with NY-ESO-1 (Invitrogen-cat no. 35-6200) or MAGE-A antibodies (Invitrogen-cat no. 35-6300) at 1:200, then incubated with secondary antibody at 1:3000 dilution for 1h. β-actin antibody (MP Biomedicals-cat no-691001) at 1:10,000 dilution was used as a loading control. Proteins were visualized using an enhanced chemiluminescence detection kit (GE Healthcare).
NY-ESO-1-specific CD8 T cell recognition
NY-ESO-1-specific HLA-B35-restricted CD8+ T cells were prepared (31, 39) and co-cultured with HLA-B35+ KG1a cells pretreated with AZA, DAC, or varying doses of SGI-110 as described above for 6 hours at 37C in the presence of anti-CD107a (clone H4A3) and CD107b (clone H4B4). Monensin and brefeldin A were added during the last 4 hours incubation to block cytokine secretion (31). Cells were fixed with 2% formaldehyde, followed by staining with IFN-γ (clone B27), TNF-α (clone MAb11) and granzyme B (clone GB11) in the presence of normal mouse IgG and permeabilization buffer (Invitrogen-Caltag). Negative and positive control stimulations with and without peptide (NY-ESO-194–102) or phorbol myristate acetate and ionomycin were set up in parallel.
Flow cytometry
Cells were harvested and stained with anti-human HLA-ABC (clone W6/32) or anti-human CD54, alternatively known as ICAM1 (clone HA58) antibodies (eBioscience). Live, single cells were identified by forward and side scatter. Changes in the expression of HLA-ABC and ICAM1 following HMA treatment were determined by calculating the log2 ratio of median fluorescence intensity (MFI) of HMA versus vehicle control treated cell populations (40).
Statistical analysis
Statistical analysis was performed by analysis of variance using GraphPad Prism. Differences between the two groups were analyzed using the t test. All data are presented as mean ± standard error of the mean.
Results
SGI-110 treatment induces DNA hypomethylation and CTA expression in leukemic cell lines in vitro
To evaluate the effect of SGI-110 treatment relative to the FDA approved HMAs on CTA methylation, HL60, U937, and KG1a leukemic cell lines were treated with 0.1, 1.0 and 5 μM SGI-110 and harvested on day 5. LINE1 was used as a surrogate marker for global DNA methylation (41). SGI-110 treatment resulted in significant reductions of LINE-1 and NY-ESO-1 promoter methylation in HL60, U937 and KG1a cells, as determined by quantitative bisulfite pyrosequencing (Fig 1A). As it was not feasible to perform pyrosequencing assays for the MAGE-A3/6 promoter (data not shown), we used MSP to analyze methylation of this locus. As expected, MAGE-A3/6 was also hypomethylated following SGI-110 treatment in all cell lines (Figure 1A and Supplemental Fig 2A). AZA and DAC treatment also resulted in hypomethylation of these regions (Fig 1A and Supplemental Fig 2A). To determine whether SGI-110 induced hypomethylation resulted in gene expression, we assessed mRNA and protein expression of NY-ESO-1 and MAGE-A. All cell lines demonstrated significant increases in NY-ESO-1 and MAGE-A3/6 mRNA following SGI-110 treatment, and this expression was at least as prominent as that observed following treatment with AZA or DAC (Fig 1B–C). Importantly, all cell lines also showed marked induction of NY-ESO-1 and MAGE-A proteins following SGI-110 exposure particularly at the 1uM dose level (Fig 1D). Low level baseline MAGE-A expression was apparent in KG1a cells (Fig 1D).
Figure 1. SGI-110 treatment induces hypomethylation and expression of NY-ESO-1 and MAGE-A3/6 in leukemic cell lines in vitro.
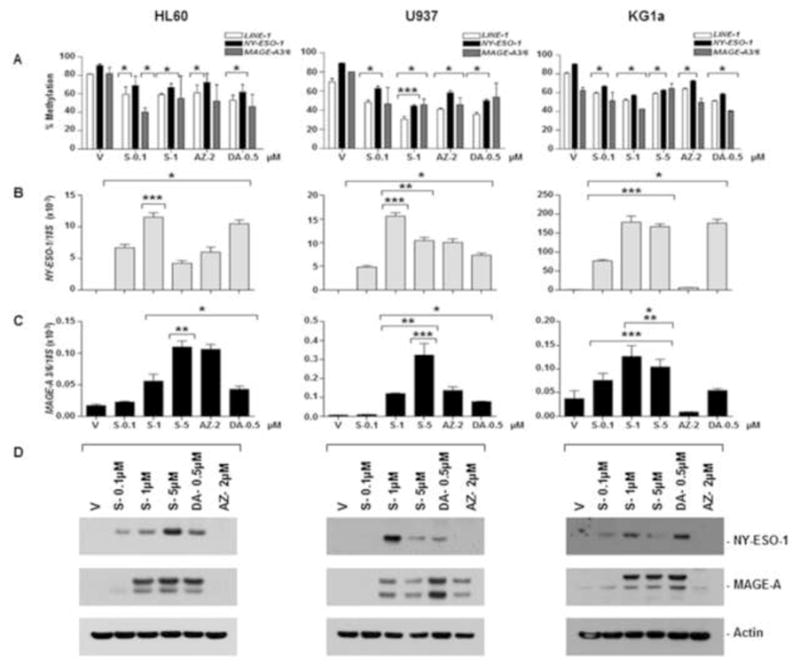
HL60, U937, or KG1a cells were treated with SGI-110, DAC and AZA as described. A) LINE-1 and NY-ESO-1 promoter methylation were determined by bisulfite pyrosequencing. MAGE-A3/6 methylation is expressed as a relative percentage of methylated product (MP/(UP+MP)*100) based upon densitometry from MSP (Representative of two experiments; primary data are shown in supplemental Figure 2) B) NY-ESO-1 and MAGE-A3/6 mRNA were quantified by qRT-PCR. C) NY-ESO-1 and MAGE-A protein expression was determined by western blot. All experiments were completed at least three times. * p<0.05 vs. vehicle, **p<0.05 vs. DAC, ***p<0.05 vs. AZA, S-SGI-110, V-Vehicle, DA-Decitabine, AZ-Azacytidine, MP-Density of methylated PCR product, UP- Density of unmethylated PCR product
SGI-110 treatment induces DNA hypomethylation and CTA expression in U937 xenografts
The following in vivo experiments were designed to re-capitulate the doses and schedules being tested in a currently enrolling phase I/II clinical trial of SGI-110 in patients with MDS and AML (NCT01261312). Given the longer in vivo half-life of SGI-110 relative to DAC we hypothesized that more distributed dosing schedules (weekly or twice weekly, rather than the 5-day continuous schedule currently the standard of care clinically), might be a feasible approach to modulate CTA genes.
Daily x 5d Treatment Schedule
Tumor-bearing immune-deficient mice were treated with a series of clinically relevant dosing schedules of SGI-110 or DAC (using the schedules currently in phase I/II testing for MDS or AML, see Materials and Methods). We did not include AZA treatment as AZA was less potent in vitro as compared to SGI-110 or DAC (Fig 1). We evaluated the effect of SGI-110 and DAC treatment on LINE-1, NY-ESO-1, and MAGE-A3/6 promoter methylation in excised U937 tumors as described above. Treatment groups 1 to 5 were exposed subcutaneously to SGI-110 at doses of 3, 6.1, or 10 mg/kg, or DAC at 2.5 mg/kg, daily for 5 days, with tumors harvested on day 7. Most mice (4/5) treated on the 5 day schedule with 10mg/kg/d SGI-110 died; all mice treated with 6.1mg/kg/day SGI-110 developed gastrointestinal toxicity (Table 1). In contrast, minimal toxicity was observed in mice treated with 3mg/kg/day SGI-110. SGI-110 treatment caused hypomethylation of LINE-1 and NY-ESO-1 at all doses (Fig 2A). MAGE-A3/6 hypomethylation was observed following SGI-110 (Fig 2A and Supplemental Fig 2B). Consistent with the observed hypomethylation, tumors excised on day 7 demonstrated a variable degree of NY-ESO-1 or MAGE-A3/6 mRNA expression (Fig 2B), along with robust CTA protein expression (Fig 2C).
Figure 2. SGI-110 treatment induces hypomethylation and expression of NY-ESO-1 and MAGE-A3/A6 in U937 xenografts treated using a daily x 5 schedule.
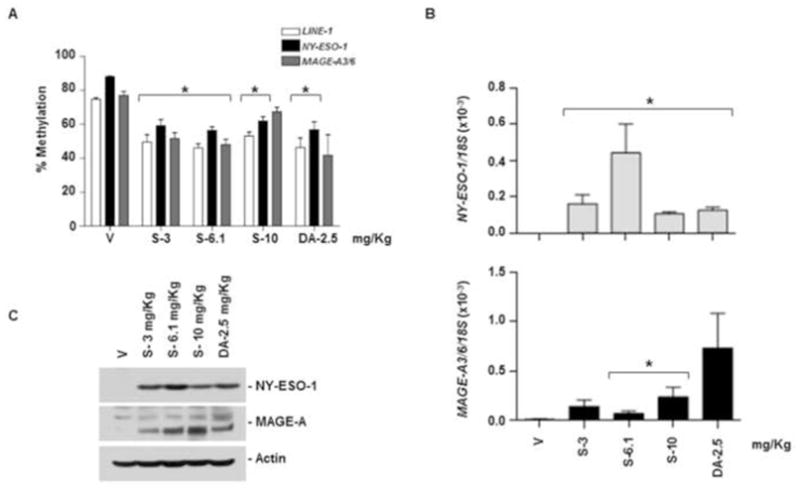
SGI-110 at varying doses (3, 6.1,10 mg/kg) or DAC (2.5 mg/kg) was administered for five days (days 1–5); on day 7 after treatment, tumors were harvested. A) LINE-1 and NY-ESO-1 promoter methylation bisulfite pyrosequencing; MAGE-A3/6 by MSP (Representative of two experiments) B) NY-ESO-1 and MAGE-A3/6 mRNA quantified by qRT-PCR. C) NY-ESO-1 and MAGE-A protein expression by western blotting. Data is representative of 3 animals/group. *p<0.05 vs. vehicle.
Weekly x 3 Treatments Schedule
In treatment groups 6 to 10, mice received SGI-110 at 6.1, 12.2, or 24.4 mg/kg, or DAC at 5 mg/kg, once a week for 3 weeks. SGI-110 treatment resulted in significant LINE-1 and NY-ESO-1 hypomethylation on day 20 post-treatment. SGI-110 at the highest dose, 24.4 mg/kg, led to the greatest hypomethylation, and was not associated with overt toxicity in treated mice (Fig 3A, Table 1). Using MSP we evaluated MAGE-A3/6 promoter methylation and observed hypomethylation on day 20 (Fig 3A and Supplemental Fig 2C). Both DAC and SGI-110 treatments resulted in a significant increase in NY-ESO-1 mRNA expression; however MAGE-A3/6 mRNA expression was not consistently increased (Fig 3B). Similarly, NY-ESO-1 protein expression was enhanced (Fig 3C), but no changes in MAGE-A protein expression were consistently observed (Fig 3C).
Figure 3. SGI-110 treatment induces hypomethylation and expression of NY-ESO-1 and MAGE-A3/A6 in U937 xenografts treated using a weekly x 3 schedule.
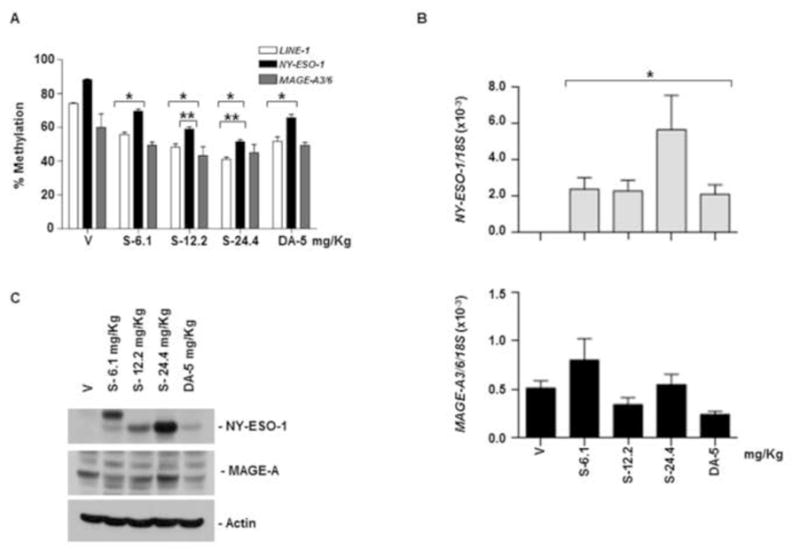
SGI-110 at varying doses (6.1, 12.2, 24.4 mg/kg) or DAC (5mg/kg) was administered once weekly for three weeks (days 1, 8, 15); on day 20 after treatment, tumors were harvested. A) LINE-1 and NY-ESO-1 promoter methylation by bisulfite pyrosequencing; MAGE-A3/6 by MSP (Representative of two experiments). B) NY-ESO-1 and MAGE-A3/6 mRNA by qRT-PCR. C) NY-ESO-1 and MAGE-A protein expression by western blotting. Data represent 3 animals/group. *p<0.05 vs. vehicle; **p<0.05 vs. DAC.
Twice Weekly x 3 Treatment Schedule
Mice in groups 11 to 15 received SGI-110 at doses of 6.1, 12.2, 24.4 mg/kg, or DAC at 2.5 mg/kg, twice per week for 3 weeks. Mice treated at the highest SGI-110 dose, 24.4mg/kg demonstrated significant mortality (Table 1). The 12.2 mg/kg dose was also excessively toxic with most mice demonstrating morbidity. All doses of SGI-110 elicited LINE-1 and NY-ESO-1 promoter hypomethylation (Fig 4A). SGI-110 at the 6.1 mg/kg dose resulted in greater hypomethylation than did DAC at 2.5mg/kg (near molar equivalent doses with respect to relative DAC content) (Fig 4A). Similarly MAGE-A3/6 hypomethylation was observed with SGI-110 treatment (Supplemental Fig 2D). As expected, NY-ESO-1 expression was enhanced with SGI-110 treatment (Fig 4B). In contrast, MAGE-A3/6 expression was not induced (Fig 4B). The 6.1mg/kg SGI-110 dose resulted in a significant increase in NY-ESO-1 as compared to DAC. NY-ESO-1 protein expression was induced by all doses of SGI-110, and to a limited extent with DAC (Fig 4C). No consistent induction of MAGE-A protein expression was observed with this treatment schedule (Fig 4C).
Figure 4. SGI-110 treatment induces hypomethylation and expression of NY-ESO-1 and MAGE-A3/A6 in U937 xenografts treated using a twice weekly x 3 week schedule.
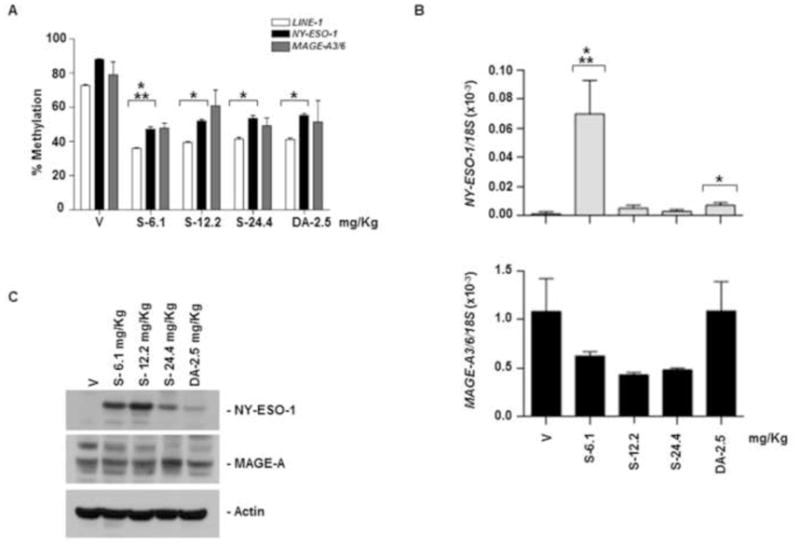
SGI-110 at varying doses (6.1,12.2, 24.4mg/kg) or DAC (2.5mg/kg) administered twice weekly for three weeks (days 1, 4, 8, 11, 15, 18); and tumors were harvested. A) LINE-1 and NY-ESO-1 promoter methylation by bisulfite pyrosequencing; MAGE-A3/6 by MSP (Representative of two experiments). B) NY-ESO-1 and MAGE-A3/6 mRNA by qRT-PCR. C) NY-ESO-1 and MAGE-A protein expression by western blotting. Data represent 3 animals/group. *p<0.05 vs. vehicle; **p<0.05 vs. DAC.
Taken together these data suggest that the daily x 5 schedule of SGI-110 results in the consistently robust hypomethylation and induction of CTA genes in leukemia xenografts.
SGI-110 treatment promotes NY-ESO-1-specific CD8+ T cell recognition of leukemic cells
To determine if HMA-mediated induction of NY-ESO-1 expression resulted in enhanced immune recognition, we co-cultured NY-ESO-1-specific CD8+ T cells with KG1a cells pretreated with AZA, DAC and varying doses of SGI-110. Expression of CD107a/b, INFγ, TNFα, IL-2 and Granzyme B expression were determined by flow cytometry. CD107a/b is well established as a surrogate marker for T cell mediated cytotoxicity (42). SGI-110 treated KG1a cells co-cultured with NY-ESO-1 specific CD8+ T cells demonstrated a marked increase in CD107a/b staining (Fig 5A). We also observed increases in the expression of IFN-γ, TNFα, and Granzyme B in T-cells co-cultured with SGI-110 treated KG1a cells compared to T-cells co-cultured with vehicle control treated KG1a cells (Figure 5B–E). Induction of these molecules is strongly associated with a cytotoxic T-cell response (43). No change in the expression of IL-2 was seen with any of the drugs tested (data not shown). These data suggest that SGI-110 enhances NY-ESO-1 antigen specific T-cell recognition of HLA compatible tumor cells.
Figure 5. SGI-110 treatment induces recognition by NY-ESO-1-specific, HLA compatible CD8+ T cells.
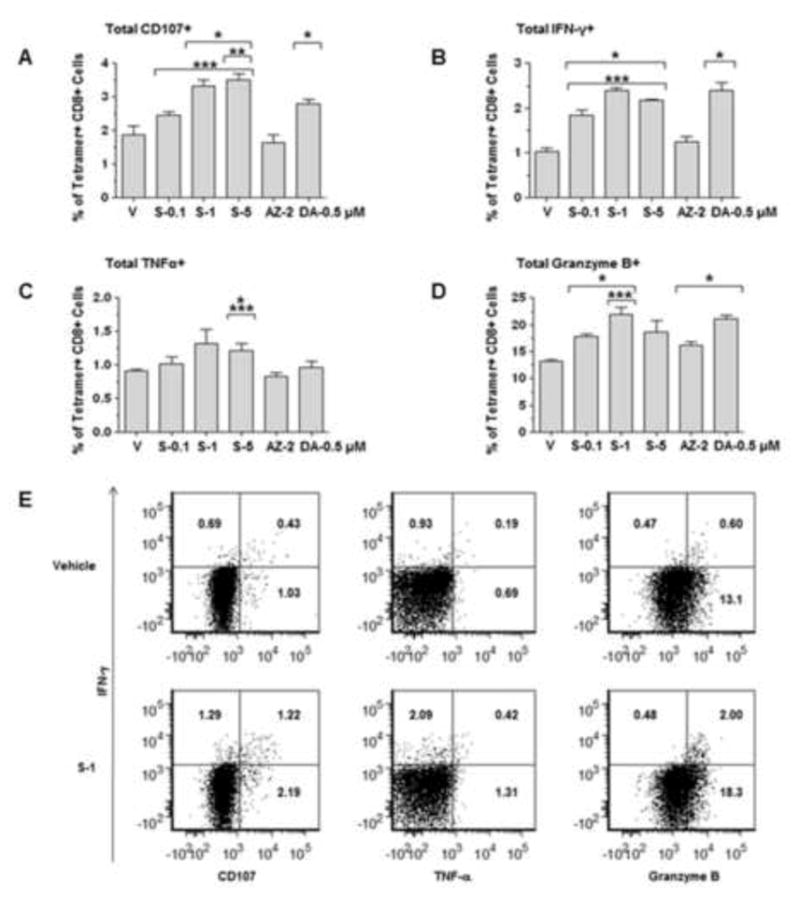
A–D) KG1a cells pretreated with SGI-110, DAC, or AZA at the concentrations shown were cultured with HLA-B35 restricted NY-ESO-1-specific CD8+ T cells. Expression of CD107a/b, IFN-γ, TNF-α, and granzyme B in HLA-B35/NY-ESO-194–102 tetramer+ CD8+ T cells was assessed by flow cytometry. All experiments were repeated three times. E) Representative flow plots for vehicle and SGI-110- 1 μM treated KG1a cells* p<0.05 vs. vehicle, **p<0.05 vs. DAC, ***p<0.05 vs. AZA.
SGI-110 treatment enhances cell surface expression of MHC class I and ICAM1 in leukemic cells
To determine whether SGI-110 treatment enhanced expression of additional machinery important for T-cell recognition of KG1a cells, we tested the relative expression of MHC class I and the co-stimulatory molecule ICAM1 following HMA exposure in vitro by flow cytometry. SGI-110 and DAC upregulated the expression of HLA-ABC, while the effect of AZA was much less substantial (Figures 6A, 6C). Treatment with all the tested HMAs resulted in a significant increase in the expression of ICAM1 (Figure 6B). Thus, in addition to upregulation of antigen expression, HMA treatment enhances expression of MHC class I and ICAM1, potentially contributing to the observed enhancement in immune recognition by HLA-compatible T-cells.
Figure 6. SGI-110 treatment enhances HLA-ABC and ICAM1 expression in KG1a cells treated in vitro.
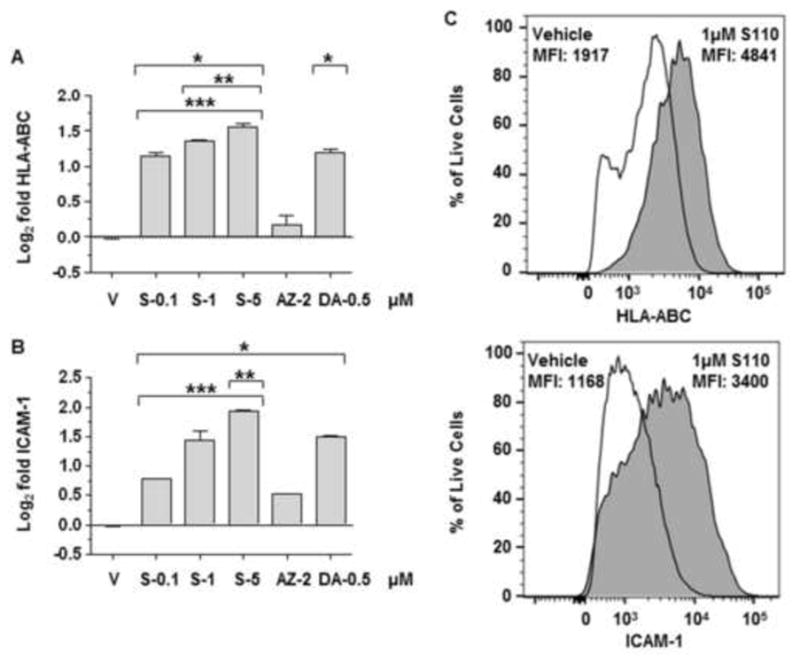
KG1a cells treated with SGI-110, DAC, or AZA, expression of A) HLA-ABC and B) ICAM1 determined by flow cytometry. Data represent the Log2 transformation of the MFI post treatment vs. average MFI vehicle control from three experiments. C) Representative flow plots for vehicle and SGI-110 5uM treated KG1a cells. Live, single KG1a cells were used to determine HLA-ABC expression or ICAM1. For simplicity, only KG1a cells treated with vehicle or 1 μM SGI-110 are shown. * p<0.05 vs. vehicle, **p<0.05 vs. DAC, ***p<0.05 vs. AZA, MFI- Median fluorescence intensity.
Discussion
Here we demonstrate the activity of SGI-110 to induce hypomethylation of CTA promoters and CTA gene expression in leukemia cell lines, and to enhance recognition of cancer cells by HLA compatible antigen specific T-cells. We further observe, as others have demonstrated, that the HMAs tested enhance the expression of MHC class I and the co-stimulatory molecule ICAM1 (44). These data show that SGI-110, like the parent compound DAC, can induce the expression of CTAs including MAGE-A and NY-ESO-1 at both the mRNA and protein levels. Re-expression of these genes is sufficient to trigger a cytotoxic T-cell response in HLA compatible, gene specific T-cell clones isolated from an NY-ESO-1 vaccinated ovarian cancer patient with an endogenously NY-ESO-1 expressing cancer (31). Expression of ICAM1 and HLA class I are enhanced by exposure to HMAs, likely through enhanced expression of transcription factors (45, 46). This study is the first to report such results using SGI-110 in leukemic cell lines and demonstrates that the drug is at least as potent as DAC at inducing these genes in a mouse model designed to re-capitulate treatment schedules of the currently enrolling clinical study in MDS and AML patients. These data support the hypothesis that SGI-110, like DAC, induces expression of CTA genes and that this induced expression is sufficient to activate HLA compatible cytotoxic T-cells.
DAC generated by the cleavage of subcutaneously administered SGI-110, benefits from a lower peak plasma concentration (limiting toxicity), in association with a longer plasma half-life when compared to DAC administered as an IV infusion. Both of these alterations provide a theoretical advantage over IV administration of DAC in enhancing the potential for more slowly dividing cells to incorporate the modified base, and thereby to improve hypomethylation. These differences may be particularly important for hypomethylation of methylated loci, such as those surrounding the CTA gene promoters. Our data demonstrate that at near-equivalent molar doses, SGI-110 is at least equivalent to Dac in inducing CTA and co-stimulatory molecule gene expression. It is also important to note that the level of cytidine deaminase in mice is significantly higher than that seen in primates and humans, and therefore the seeming equivalence in terms of gene expression observed between DAC and SGI-110 in mice may be more dramatic in humans (47).
It is notable that MAGE-A family members have significant homology in terms of protein sequence. In our mouse experiments we sometimes observed discordance between protein and mRNA expression. We suspect that this apparent discordance is reflective of the relative specificity of the MAGE-A3/6 primers when compared with the antibody (ie, the mRNA primers are specific for MAGE-A3/6 while the antibody detects many different members of the MAGE-A family (48). We further recognize that MAGE-A family members have variable baseline expression in leukemia cells. Some groups have demonstrated spontaneous expression of these genes (49, 50) while others have reported no expression (51). Chambost et al. reported that MAGE-A family members were not expressed in primary leukemia patient samples, but did note variable expression in leukemia cell lines (52). In our study, we demonstrate MAGE-A3/6 mRNA expression in all the tested leukemia cell lines. Furthermore, in our excised tumor xenografts we saw enhanced expression of MAGE-A3/6, particularly in tumors harvested at later time points (G1 to 5 at day 7 were less impressive than G6 to15 which were harvested on day 20). Our data support the previous reports of baseline expression of MAGE-A family members in the AML cell lines tested; expression appears to have been augmented by culture in the xenograft setting.
The role of the induced anti-tumor immunity in the observed responses to HMAs remains an area of active investigation, but emerging data support the suggestion that this may be an important and under-recognized aspect of their mechanism of action (32, 35, 53–55). Based upon this and other data supporting the hypothesis that NY-ESO-1 induced gene expression is sufficient to trigger T-cell cytotoxicity, a phase I study examining a combination of NY-ESO-1 protein vaccination in combination with conventional dose DAC is ongoing at our institution in patients with MDS and low blast count AML (NCT01834248).
From a clinical perspective, SGI-110 can be given subcutaneously and appears to be clinically active, thus a potential for combination therapy with SGI-110 and CTA vaccination would be relatively straightforward and provide patients with a completely subcutaneous regimen, free from the need for daily IV delivery. Furthermore, in patients for whom transplant is not an option, an immunotherapeutic strategy offers the potential for autologous anti-tumor immunity in the absence of systemic toxicity from graft versus host disease. Better understanding of the impact of these agents, both on the malignant clone, as well as on the immune cells themselves are necessary in order to improve our approach to patients with MDS associated malignancy. Such insights may allow us to offer our patients, many of whom are outside the age acceptable for transplant, hope for a curative strategy.
Supplementary Material
Highlights.
SGI-110 induces expression of NY-ESO-1 and MAGE-A in AML cells.
Induced expression of NY-ESO-1 in AML triggers cytotoxicity from T-cell clones.
Hypomethylating agents enhance HLA class I and ICAM1 expression in AML cells.
Induced Cancer Testis Genes may be a vaccine target in hematological malignancy.
Acknowledgments
The authors would like to thank Drs. Kunle Odunsi and Mohammad Azab for the intellectual support of this project and Dr. Michael Moser from the animal core facility at Roswell Park Cancer Institute. EAG and ARK designed the research study; PS, BJP, JM, SRJ, GCL and JK performed the research; EAG, ARK, PS, JM and MJN analyzed the data; PT contributed essential reagents and helped to design the mouse experiments; PS, BJP, EAG and ARK wrote the paper. This work was funded by a research grant from Astex Pharmaceuticals (to EAG and ARK) and by the NCI Cancer Center Support Grant CA016056. BJP was supported through an institutional training grant from the NCI (5T32CA009072-39). EAG and MN are supported by the Roswell Park Alliance Foundation and by institutional funds provided by Roswell Park Cancer Institute. EAG and ARK declare that funding for this work was provided through a research grant from Astex Pharmaceuticals, we therefore declare this as a conflict of interest, PT is an employee of Astex Pharmaceuticals and declares this as a conflict of interest, PS, BJP, JM, GCL, SRJ, JK, MJN have no conflicts to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Estey EH. Epigenetics in clinical practice: The examples of azacitidine and decitabine in myelodysplasia and acute myeloid leukemia. Leukemia. 2013 Sep;27(9):1803–12. doi: 10.1038/leu.2013.173. [DOI] [PubMed] [Google Scholar]
- 2.Jabbour E, Garcia-Manero G, Batty N, Shan J, O’Brien S, Cortes J, et al. Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer. 2010 Aug 15;116(16):3830–4. doi: 10.1002/cncr.25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prebet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol. 2011 Aug 20;29(24):3322–7. doi: 10.1200/JCO.2011.35.8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prebet T, Gore SD, Thepot S, Esterni B, Quesnel B, Beyne Rauzy O, et al. Outcome of acute myeloid leukaemia following myelodysplastic syndrome after azacitidine treatment failure. Br J Haematol. 2012 Jun;157(6):764–6. doi: 10.1111/j.1365-2141.2012.09076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prebet T, Thepot S, Gore SD, Dreyfus F, Fenaux P, Vey N. Outcome of patients with low-risk myelodysplasia after azacitidine treatment failure. Haematologica. 2013 Feb;98(2):e18–9. doi: 10.3324/haematol.2012.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itzykson R, Fenaux P. Optimizing hypomethylating agents in myelodysplastic syndromes. Curr Opin Hematol. 2012 Mar;19(2):65–70. doi: 10.1097/MOH.0b013e32834ff58a. [DOI] [PubMed] [Google Scholar]
- 7.Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009 Sep 24;114(13):2764–73. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fandy TE, Jiemjit A, Thakar M, Rhoden P, Suarez L, Gore SD. Decitabine induces delayed reactive oxygen species (ROS) accumulation in leukemia cells and induces the expression of ROS generating enzymes. Clin Cancer Res. 2014 Mar 1;20(5):1249–58. doi: 10.1158/1078-0432.CCR-13-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2′-deoxycytidine. Mol Pharmacol. 2001 Apr;59(4):751–7. [PubMed] [Google Scholar]
- 10.Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002 Aug 12;21(35):5496–503. doi: 10.1038/sj.onc.1205602. [DOI] [PubMed] [Google Scholar]
- 11.Zeidan AM, Linhares Y, Gore SD. Current therapy of myelodysplastic syndromes. Blood Rev. 2013 Sep;27(5):243–59. doi: 10.1016/j.blre.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010 Sep 16;116(11):1908–18. doi: 10.1182/blood-2009-11-249474. [DOI] [PubMed] [Google Scholar]
- 13.Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML) Blood. 2012 Apr 5;119(14):3361–9. doi: 10.1182/blood-2011-09-377044. [DOI] [PubMed] [Google Scholar]
- 14.Karpf AR, Lasek AW, Ririe TO, Hanks AN, Grossman D, Jones DA. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine. Mol Pharmacol. 2004 Jan;65(1):18–27. doi: 10.1124/mol.65.1.18. [DOI] [PubMed] [Google Scholar]
- 15.Braun T, Fenaux P. Myelodysplastic syndromes (MDS) and autoimmune disorders (AD): Cause or consequence? Best Pract Res Clin Haematol. 2013 Dec;26(4):327–36. doi: 10.1016/j.beha.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Al Ustwani O, Ford LA, Sait SJ, Block AM, Barcos M, Vigil CE, et al. Myelodysplastic syndromes and autoimmune diseases--case series and review of literature. Leuk Res. 2013 Aug;37(8):894–9. doi: 10.1016/j.leukres.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olnes MJ, Sloand EM. Targeting immune dysregulation in myelodysplastic syndromes. JAMA. 2011 Feb 23;305(8):814–9. doi: 10.1001/jama.2011.194. [DOI] [PubMed] [Google Scholar]
- 18.Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, Karbach J, Pfeifer D, Jager E, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk Res. 2010 Jul;34(7):899–905. doi: 10.1016/j.leukres.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Griffiths EA, James SR, Ford LA, Beck AF, Wetzler M, Odunsi K, et al. Conventional dose hypomethylating agents induce CG antigen genes in vivo. Blood. 2011;118(21):2441. [Google Scholar]
- 20.Srivastava P, Collamat G, James SR, Ford LA, Wetzler M, Karpf AR, et al. Decitabine treatment induces NY-ESO1 promoter hypomethylation, mRNA and protein expression in acute myeloid leukemia (AML) blasts harvested from peripheral blood. Leukemia Research. 2013;37(Supplement 1):S132. [Google Scholar]
- 21.Akers SN, Odunsi K, Karpf AR. Regulation of cancer germline antigen gene expression: Implications for cancer immunotherapy. Future Oncol. 2010 May;6(5):717–32. doi: 10.2217/fon.10.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stockert E, Jager E, Chen YT, Scanlan MJ, Gout I, Karbach J, et al. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med. 1998 Apr 20;187(8):1349–54. doi: 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grah JJ, Katalinic D, Juretic A, Santek F, Samarzija M. Clinical significance of immunohistochemical expression of cancer/testis tumor-associated antigens (MAGE-A1, MAGE-A3/4, NY-ESO-1) in patients with non-small cell lung cancer. Tumori. 2014 Jan-Feb;100(1):60–8. doi: 10.1700/1430.15817. [DOI] [PubMed] [Google Scholar]
- 24.Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003 Sep 15;63(18):6076–83. [PubMed] [Google Scholar]
- 25.Woloszynska-Read A, Mhawech-Fauceglia P, Yu J, Odunsi K, Karpf AR. Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clin Cancer Res. 2008 Jun 1;14(11):3283–90. doi: 10.1158/1078-0432.CCR-07-5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aranda F, Vacchelli E, Eggermont A, Galon J, Sautes-Fridman C, Tartour E, et al. Trial watch: Peptide vaccines in cancer therapy. Oncoimmunology. 2013 Dec 1;2(12):e26621. doi: 10.4161/onci.26621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atanackovic D, Luetkens T, Kloth B, Fuchs G, Cao Y, Hildebrandt Y, et al. Cancer-testis antigen expression and its epigenetic modulation in acute myeloid leukemia. Am J Hematol. 2011 Nov;86(11):918–22. doi: 10.1002/ajh.22141. [DOI] [PubMed] [Google Scholar]
- 28.Griffiths EA, Choy G, Redkar S, Taverna P, Azab M, Karpf AR. SGI-110. DNA methyltransferase inhibitor, oncolytic. Drugs Fut. 2013;38(8):535. [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007 Jul 1;67(13):6400–8. doi: 10.1158/0008-5472.CAN-07-0251. [DOI] [PubMed] [Google Scholar]
- 30.Kantarjian H, Jabbour E, Yee K, Kropf P, O’Connell C, Stock W, et al. First clinical results of a randomized phase 2 study of SGI-110, a novel subcutaneous (SQ) hypomethylating agent (HMA), in adult patients with acute myeloid leukemia (AML) Blood. 2013;122(21):497. [Google Scholar]
- 31.Odunsi K, Matsuzaki J, Karbach J, Neumann A, Mhawech-Fauceglia P, Miller A, et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc Natl Acad Sci U S A. 2012 Apr 10;109(15):5797–802. doi: 10.1073/pnas.1117208109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odunsi K, Matsuzaki J, James SR, Mhawech-Fauceglia P, Tsuji T, Miller A, et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol Res. 2014;2:37–49. doi: 10.1158/2326-6066.CIR-13-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coral S, Parisi G, Nicolay HJ, Colizzi F, Danielli R, Fratta E, et al. Immunomodulatory activity of SGI-110, a 5-aza-2′-deoxycytidine-containing demethylating dinucleotide. Cancer Immunol Immunother. 2013 Mar;62(3):605–14. doi: 10.1007/s00262-012-1365-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krishnadas DK, Bao L, Bai F, Chencheri SC, Lucas K. Decitabine facilitates immune recognition of sarcoma cells by upregulating CT antigens, MHC molecules, and ICAM-1. Tumour Biol. 2014 Jun;35(6):5753–62. doi: 10.1007/s13277-014-1764-9. [DOI] [PubMed] [Google Scholar]
- 35.Covre A, Parisi G, Nicolay HJ, Fonsatti E, Fratta E, Sigalotti L, et al. In vivo immunomodulatory activity of SGI-110, a second generation hypomethylating agent, in hematologic malignancies. Cancer Res. 2013;73(Suppl 8):680. [Google Scholar]
- 36.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996 Sep 3;93(18):9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li LC, Dahiya R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics. 2002 Nov;18(11):1427–31. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 38.Srivastava P, Yadav N, Lella R, Schneider A, Jones A, Marlowe T, et al. Neem oil limonoids induces p53-independent apoptosis and autophagy. Carcinogenesis. 2012 Nov;33(11):2199–207. doi: 10.1093/carcin/bgs269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benlalam H, Linard B, Guilloux Y, Moreau-Aubry A, Derre L, Diez E, et al. Identification of five new HLA-B*3501-restricted epitopes derived from common melanoma-associated antigens, spontaneously recognized by tumor-infiltrating lymphocytes. J Immunol. 2003 Dec 1;171(11):6283–9. doi: 10.4049/jimmunol.171.11.6283. [DOI] [PubMed] [Google Scholar]
- 40.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004 Jul 23;118(2):217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 41.Woloszynska-Read A, Zhang W, Yu J, Link PA, Mhawech-Fauceglia P, Collamat G, et al. Coordinated cancer germline antigen promoter and global DNA hypomethylation in ovarian cancer: Association with the BORIS/CTCF expression ratio and advanced stage. Clin Cancer Res. 2011 Apr 15;17(8):2170–80. doi: 10.1158/1078-0432.CCR-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003 Oct 1;281(1–2):65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 43.Sandberg JK, Fast NM, Nixon DF. Functional heterogeneity of cytokines and cytolytic effector molecules in human CD8+ T lymphocytes. J Immunol. 2001 Jul 1;167(1):181–7. doi: 10.4049/jimmunol.167.1.181. [DOI] [PubMed] [Google Scholar]
- 44.Coral S, Parisi G, Nicolay HJ, Colizzi F, Danielli R, Fratta E, et al. Immunomodulatory activity of SGI-110, a 5-aza-2′-deoxycytidine-containing demethylating dinucleotide. Cancer Immunol Immunother. 2013 Mar;62(3):605–14. doi: 10.1007/s00262-012-1365-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altmann DM, Hogg N, Trowsdale J, Wilkinson D. Cotransfection of ICAM-1 and HLA-DR reconstitutes human antigen-presenting cell function in mouse L cells. Nature. 1989 Apr 6;338(6215):512–4. doi: 10.1038/338512a0. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka Y, Fukudome K, Hayashi M, Takagi S, Yoshie O. Induction of ICAM-1 and LFA-3 by Tax1 of human T-cell leukemia virus type 1 and mechanism of down-regulation of ICAM-1 or LFA-1 in adult-T-cell-leukemia cell lines. Int J Cancer. 1995 Feb 8;60(4):554–61. doi: 10.1002/ijc.2910600421. [DOI] [PubMed] [Google Scholar]
- 47.Redkar S, Joshi R, Tang C, Sadikin S, Inloes R, Shi C, et al. Subcutaneous dosing of SGI-110, a second-generation hypomethylating agent, shows activity in mouse xenograft models. AACR Cancer Epigenetics. 2010 [Google Scholar]
- 48.De Plaen E, Arden K, Traversari C, Gaforio JJ, Szikora JP, De Smet C, et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics. 1994;40(5):360–9. doi: 10.1007/BF01246677. [DOI] [PubMed] [Google Scholar]
- 49.Martinez A, Olarte I, Mergold MA, Gutierrez M, Rozen E, Collazo J, et al. mRNA expression of MAGE-A3 gene in leukemia cells. Leuk Res. 2007 Jan;31(1):33–7. doi: 10.1016/j.leukres.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 50.Shichijo S, Tsunosue R, Masuoka K, Natori H, Tamai M, Miyajima J, et al. Expression of the MAGE gene family in human lymphocytic leukemia. Cancer Immunol Immunother. 1995 Aug;41(2):90–103. doi: 10.1007/s002620050205. [DOI] [PubMed] [Google Scholar]
- 51.Greiner J, Ringhoffer M, Simikopinko O, Szmaragowska A, Huebsch S, Maurer U, et al. Simultaneous expression of different immunogenic antigens in acute myeloid leukemia. Exp Hematol. 2000 Dec;28(12):1413–22. doi: 10.1016/s0301-472x(00)00550-6. [DOI] [PubMed] [Google Scholar]
- 52.Chambost H, van Baren N, Brasseur F, Olive D. MAGE-A genes are not expressed in human leukemias. Leukemia. 2001 Nov;15(11):1769–71. doi: 10.1038/sj.leu.2402278. [DOI] [PubMed] [Google Scholar]
- 53.Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013 Nov;4(11):2067–79. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest. 2014 Jan 2;124(1):56–63. doi: 10.1172/JCI69736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014 Feb 15;5(3):587–98. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.