

Abstract
The binding of human IgG1 to human Fc gamma receptors (hFcγR) is highly sensitive to the presence of a single N-linked glycosylation site at asparagine 297 of the Fc, with deglycosylation resulting in a complete loss of hFcγR binding. Previously, we demonstrated that aglycosylated human IgG1 Fc variants can engage the human FcγRII class of the low-affinity hFcγRs, demonstrating that N-linked glycosylation of the Fc is not a strict requirement for hFcγR engagement. In the present study, we demonstrate that aglycosylated IgG variants can be engineered to productively engage with FcγRIIIA, as well as the human Fc gamma RII subset. We also assess the biophysical properties and serum half-life of the aglycosylated IgG variants to measure stability. Aglycosylated constructs DTT-IYG and DAT-IYG optimally drove tumor cell phagocytosis. A mathematical model of phagocytosis suggests that hFcγRI and hFcγRIIIA dimers were the main drivers of phagocytosis. In vivo tumor control of B16F10 lung metastases further confirmed the variant DTT-IYG to be the best at restoring WT-like properties in prevention of lung metastases. While deuterium incorporation was similar across most of the protein, several peptides within the CH2 domain of DTT-IYG showed differential deuterium uptake in the peptide region of the FG loop as compared to the aglycosylated N297Q. Thus in this study, we have found an aglycosylated variant that may effectively substitute for wild-type Fc. These aglycosylated variants have the potential to allow therapeutic antibodies to be produced in virtually any expression system and still maintain effector function.
Keywords: directed evolution, yeast display, antibody engineering, Fc-gamma receptor, protein stability, mathematical modeling
Graphical abstract
Introduction
Over the past several decades, antibody-based therapy has emerged as a promising mode of treatment of human disease, and in particular in the treatment of human cancer [1,2]. While multiple mechanisms contribute to the efficacy of therapeutic antibodies [1,3], activation of immune effector functions has been shown to play a critical role in the efficacy of several therapeutic antibodies, in particular through an antibody’s engagement of the Fcγ receptors (FcγRs) of immune cells [4]. IgGs act as the adaptor between a target cell or pathogen and the immune response by simultaneously binding antigen through their variable regions and activating an immune response through interaction of their conserved Fc regions with FcγRs on immune cells.
In humans, the FcγR (hFcγR) family comprises of activating receptors and inhibitory receptors. Activating receptors include the high affinity FcγRI, and the low affinity FcγRIIA, FcγRIIIA, and GPI-linked FcγRIIIB, which require avid multivalent interactions for activation. The inhibitory receptor FcγRIIB binds IgG with micromolar affinity [5]. Studies have shown that the efficacy of therapeutic antibodies is strongly correlated to the allelic forms of FcγRIIIA that have varying binding affinity to IgG. Populations homozygous for a valine at position 176 of FcγRIIIA (FcγRIIIA176V), as opposed to a phenylalanine (FcγRIIIA176F), have dramatically improved objective response rates [6–9], likely due to a several-fold stronger binding of wild-type hIgG1 for the FcγRIIIA176V allele. Furthermore, DiLillo and Ravetch demonstrated that hFcγRIIIA is necessary and sufficient for mAb-mediated killing of tumor cells in vivo in FcγR-humanized mice, while FcγRIIA engagement can drive a vaccinal effect [10].
Previous studies have shown that the binding of IgG to FcγR is highly sensitive to the presence of a single N-linked glycosylation site at asparagine 297 (N297) of the Fc, with deglycosylation resulting in a complete loss of FcγR binding [11–16]. In the past, it has been demonstrated that aglycosylated protein mutants of human IgG1 Fc variants are capable of engaging a subset of the low-affinity FcγRs and high affinity FcγRI with approximately wild-type binding affinity. These aglycosylated IgG activated immune effector cells in vivo, demonstrating that N-linked glycosylation of the Fc is not a strict requirement for FcγR engagement [17–19]. Thus, aglycosylated variants that maintain engagement to FcγRs have the potential to open up therapeutic antibody production to virtually any expression system. One such organism could be the common prokaryotic expression host E. coli that completely lacks N-linked glycosylation, removing the post-translational variation in N-glycan synthesis that occurs across organisms. Such variation in the nature of the N-linked glycan imparts substantial changes in the affinity to FcγR and subsequent biological response [20,21].
Our initial screening methodology was focused on engineering the Fc C’/E loop, which contains the N-linked glycosylation site (Asn297-Ser298-Thr299) and makes direct contacts with FcγR. A library screen of all possible C’/E loop variants yielded a variant S298G/T299A (SGTA) that binds FcγRIIA and FcγRIIB with approximately wild-type affinity, but not FcγRIIIA. A second approach, based on screening each single point mutation within the C’/E loop, then combining candidate mutations, identified variants that weakly bind FcγRIIIA176V – T299A, N297D, N297H, and the double mutants N297D/S298T, N297D/S298A, and N297H/S298A – demonstrating that aglycosylated Fcs can engage this FcγR as well [17]. However, given the importance of FcγRIIIA to therapeutic outcome, it is likely that these variants would have limited therapeutic utility.
Building upon our previous work, we report here aglycosylated hIgG1 variants that can engage all of the low-affinity hFcγRs with approximately wild-type or improved binding affinity, thus identifying variants that might effectively substitute for the wild-type, glycosylated hIgG1. In doing so, we have focused on engineering additional loops of the Fc domain that make contact with FcγR, screening libraries that encode all possible amino acid diversity within segments of these loops to enrich variants with improved FcγRIIIA176F binding. Such variants, when placed in the previously identified aglycosylated backgrounds that allow for weak FcγRIIIA176V binding, yield fully FcγR competent aglycosylated Fcs with a range of affinities. In addition, we find that our approach of searching sequence space at contact loops in a focused manner, which allows us to theoretically screen all possible amino acid diversity at these sites in short segments, uncovered variants with mutations that act cooperatively, and thus not easily predicted by combining the properties of single point mutations identifiable through other protein engineering strategies.
Results
Engineering approach and screening methodology
In this new round of screening, saturation libraries of the lower hinge, B/C loop, and F/G loop were screened in a glycosylated background using full-length hIgG1 variants in a yeast secretion and surface capture system. Here, we constructed saturation libraries around three contact sites within the Fc – the lower hinge, the B/C loop, and F/G loop (Fig. 1a) – and screened them by displaying these full-length hIgG1 variants on the surface of yeast, as mentioned previously [17]. Contact interface libraries were constructed by fully-randomizing four amino acid stretches using degenerate NNK codons (N=ATCG, K=GT), which encode all 20 possible amino acids in 32 codons. This design approach allows for over-sampling the codon diversity (324 ~ 1×106), and thus amino acid diversity, in these yeast-based libraries, which often have a transformation efficiency on the order of 1×107. The following libraries were constructed and pooled by contact region: lower hinge (234–237, 236–239), B/C loop (265–268, 267–270), and F/G loop (326–329, 327–330, 329–332, 331–334). As a target we chose the FcγRIIIA176F allele, which, given its weaker binding for wild-type Fc, likely represents a more stringent, as well as therapeutically relevant, barrier for improved FcγRIIIA binding. Libraries were pooled by loop and individually screened by two rounds of enrichment on FcγRIIIA176F coated magnetic beads followed by three rounds of fluorescence activated cell sorting (FACS) at increasing stringency. From the yeast screens, only variants in the BC loop and FG loop were enriched (Supplementary Fig. S1a–d). Further binding studies from isolated HEK secretions suggested that two main F/G loop variants, K326I/A327Y/L328G (IYG) and K326I/A327E/L328E (IEA) contributed greatly to the increase of binding to hFcγRIIIA176F (Supplementary Fig. S2a).
Figure 1.

Characterization of aglycosylated antibodies. (a) Cartoon representation of the crystal structure of the hIgG1 Fc complex with hFcγRIII (PDB ID 1E4K). FcγRIII is shown in green, and both chains of the Fc in pale blue. Fc contact surfaces are colored: lower hinge (orange), B/C loop (blue), C’/E loop (red), and F/G loop (purple). Fc glycosylation is shown in yellow. Sequences identifying Fc mutagenized regions of the lower hinge, B/C loop, and C’/E loop. (b) SDS-PAGE gel of reduced antibodies. Aglycosylated heavy chains demonstrate higher mobility on the gel because of the lack of the large glycosylation group (c) SEC-HPLC profiles of aglycosylated constructs. (d) Aglycosylated constructs measured with DLS at 1 mg/ml in 1×PBS buffer. Hydrodynamic radii fall within the normal antibody range of 5–6nm. % Polydispersity demonstrates whether the protein preparations are monodisperse (below 20% is considered monodisperse).
Since these screens were initially on a glycosylated background, these F/G loop variants were transferred onto an aglycosylated background of T299A [17] and characterized for binding. The IYG aglycosylated mutant bound hFcγRIIIA176F to a slightly greater degree than wild-type hIgG1, whereas the IEA aglycosylated mutant bound hFcγRIIIA176F to a slightly lesser degree (Supplementary Fig. S2b), and both variants showed detectable binding to hFcγRIIIA176V, hFcγRIIA167R, and hFcγRIIB (Supplementary Fig. S2c–f). Given the large increase in binding to FcγRIIA and FcγRIIB imparted by using the T299A mutation to place the F/G loop variants in an aglycosylated background, we next sought to reduce the binding of the aglycosylated variant Fcs to these two receptors by placing the K326I/A327Y/L328G (IYG) F/G loop variant in alternative aglycosylated C’/E loop backgrounds. Aglycosylated double mutants, N297D/S298T (DTT), N297D/S298A (DAT), and N297H/S298A (HAT) that were previously isolated had weak binding to hFcγRIIIA176V, but no detectable binding to hFcγRIIA167R. Preliminary binding profiles of these aglycosylated variants suggest that DTT-IYG, DAT-IYG, and HAT-IYG have relatively similar binding to both alleles of hFcγRIIIA (Supplementary Fig. S3a–b) and have reduced binding to hFcγRIIA and the inhibitory hFcγRIIB (Supplementary Fig. S3c–d) compared to T299A-IYG.
Characterization of aglycosylated variants
The original hIgG1 variants were isolated as full antibodies in the yeast secretion and surface capture assay with a 4m5.3 variable region. In order to work with anti-tumor antibody variants, the 4m5.3 variable regions were switched out with the TA99 variable region, an antibody that targets the melanoma tumor antigen TYRP-1. The heavy chain bands of the aglycosylated variants exhibit higher mobility on a reducing SDS-PAGE gel, as compared to the glycosylated wild-type heavy chain (Fig. 1b). This extra mobility is consistent with the removal of the bulky N-linked Fc glycosylation. To investigate if the mutated residues on the aglycosylated variants impacted biophysical properties of the antibodies, purified protein was analyzed for elution times on size-exclusion chromatography. All aglycosylated TA99 variants in addition to the wild-type hIgG1 antibody ran at the expected elution time in single peaks on size-exclusion chromatography with less than 0.5% of high molecular weight species (Fig. 1c). Samples were analyzed with dynamic light scattering (DLS) to characterize protein size distribution. Variants had a hydrodynamic radius from the range of 5.1–5.6 nm (Fig. 1d), which was within the range of reported hydrodyanmic radii 5–6 nm of normal monoclonal antibodies [22]. When protein preparations were stored at 4°C for an extended period of time (approximately 2 weeks), the majority of the variants were monodisperse with a % polydispersity of < 20%, except for DAT-IYG and HAT-IYG (Fig. 1d). This suggested that some of the aglycosylated variants have increased heterogeneity and therefore may have decreased stability over an extended period of time unlike wild-type glycosylated antibodies. All other characteristics of the aglycosylated hIgG1 variants suggested that they behave similarly to wild-type antibodies.
Binding affinities of these TA99 aglycosylated variants to human FcγRI, FcγRIIA167H, FcγRIIA167R, FcγRIIB, FcγRIIIA176F, and FcγRIIIA176V, as well as glycosylated TA99 IgG1 and a human IgG1 as a control, were determined on Biacore (Table 1, Supplementary Fig. S4). The wild-type antibody has a KD of 3 nM to FcγRI and low levels of binding in the single-digit µM range for all of the remaining low-affinity FcγRs in agreement with literature [23]. As expected, the N297Q aglycosylated variant showed no detectable binding to the low affinity FcγRs and decreased binding by two orders of magnitude to the high-affinity FcγRI. The S298G/T299A (SGTA) variant only had detectable binding to the FcγRIIA and FcγRIIB receptors and not the FcγRIIIA receptor, in agreement with previous data [17]. The SGTA variant had a similar binding affinity as the aglycosylated N297Q variant to FcγRI. The N297D/S298T/K326I/A327Y/L328G (DTT-IYG) variant had no detectable binding to FcγRIIA167H or FcγRIIB, but had approximately a KD of 1–3 µM to both allelic variants of FcγRIIIA and FcγRIIA167R. Interestingly, DTT-IYG had restored binding to FcγRI with a measured KD of 5 nM, despite being aglycosylated. The N297D/S298A/K326I/A327Y/L328G (DAT-IYG) variant displayed single-digit µM binding affinity to both allelic variants of FcγRIIIA. This variant also had binding to the inhibitory FcγRIIB and slight binding to FcγRIIA167R, though no detectable binding to the FcγRIIA167H variant. DAT-IYG had reduced binding to FcγRI of approximately one order of magnitude at 29 nM, but is still one order of magnitude better than the aglycosylated N297Q mutant. The N297H/S298A/K326I/A327Y/L328G (HAT-IYG) variant had no detectable binding to both allelic variants of FcγRIIA or FcγRIIB, but had 1–2 µM binding affinity to both allelic variants of FcγRIIIA. Thus, the HAT-IYG variant could test the hypothesis of whether sole engagement of FcγRIIIA is sufficient for activity. The last aglycosylated variant T299A/K326I/A327Y/L328G (299A-IYG) had restored binding to all of the low affinity FcγRs with either wild-type or improved binding affinity. The binding affinity of 299A-IYG to the high-affinity FcγRI is on the same order of magnitude of N297Q, suggesting that binding to FcγRI was not restored. With this panel of aglycosylated variants we now have restored binding to the low affinity FcγR to wild-type or better affinities, along with different combinations of binding characteristics.
Table 1.
Binding affinities
| Sample | FcγRI Kinetic KD (nM) |
FcγRIIA 167H Steady State KD (nM) |
FcγRIIA 167R Steady State KD (nM) |
FcγRIIB Steady State KD (nM) |
FcγRIIIA 176F Steady State KD (nM) |
FcγRIIIA 176V Steady State KD (nM) |
|---|---|---|---|---|---|---|
| WT | 3 | 1148 | 8015 | 3516 | 4538 | 1703 |
| N297Q | 295 | n.d. | n.d. | n.d. | n.d. | n.d. |
| SGTA | 222 | 996 | >10,000 | 6447 | n.d. | n.d. |
| DTT-IYG | 5 | n.d. | 2730 | n.d. | 1807 | 1040 |
| DAT-IYG | 29 | n.d. | >10,000 | 4169 | 2227 | 1051 |
| HAT-IYG | 216 | n.d. | n.d. | n.d. | 1680 | 1793 |
| 299A-IYG | 243 | 605 | 2494 | 2101 | 1244 | 452 |
| hIgG1 | 3 | 1095 | 5160 | 6499 | 710 | 348 |
n.d. not detectable
Mathematical modeling of complete FcγR system
A mathematical model of phagocytosis was devised to understand the importance of each receptor in driving the phagocytosis of tumor cells. A previous model developed by the Georgiou group [19] to model the FcγRIIA167H, FcγRIIA167R and FcγRIIB contribution to phagocytosis was significantly modified and expanded to include all of the remaining FcγRs: FcγRI, FcγRIIIA176F, and FcγRIIIA176V, to represent all iterations of possible FcγR dimers. In clinical studies, therapeutic efficacy of antibodies correlated with their binding affinities to the allelic variant of FcγRIIIA [6,9]. By expanding the model to include all of the FcγR we can obtain a more complete representation of the driving force behind the phagocytosis results.
Our model represents the contact area of a tumor cell and a macrophage where phagocytosis is initiated (Fig. 2a). In this system, antibody (Ab) can bind to the antigen (Ag) with an equilibrium constant of KAb to become bound antibody (B). Free FcγR (Rf) can diffuse into the contact volume to become Rcv, and only FcγR in the contact volume (Rcv) can bind to bound antibody (B) with an equilibrium constant of KR to form complexes (C). Only complexes (C) can dimerize with an equilibrium constant of Kcross to form dimers (D). The amount of dimers formed allows us to determine which FcγR play the major roles in signaling for phagocytosis. Quantified binding affinities were used as parameters in the model of phagocytosis.
Figure 2.

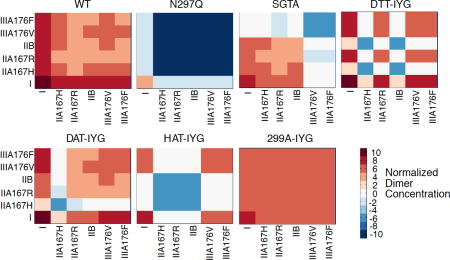
Mathematical Model of Phagocytosis. (a) Diagram of phagocytosis where the red dotted outline represents the contact volume between tumor cell (top) and macrophage (bottom). Antibody “Ab” can bind to the antigen “Ag” with an equilibrium constant of KAb to become bound antibody “B”. Free FcγR “Rf” can diffuse into the contact volume (red dotted line) to become Rcv, and only FcγR in the contact volume “Rcv” can bind to bound antibody “B” with an equilibrium constant of KR to form complexes “C”. Only complexes “C” can dimerize with an equilibrium constant of Kcross to form dimers “D”. (b) Heat maps of normalized dimer concentrations based on the mathematical model. Heat maps are plotted on log10 scale. Red signals an increase in concentration and blue a decrease in concentration. Labels represent each different human Fcγ receptor and allelic variants.
Normalized dimer concentrations were plotted as heat maps for each aglycosylated construct (Fig. 2b). The wild-type glycosylated antibody has an increase in all possible dimer formations. The FcγRI:FcγRI is by far the highest concentration, and then the remaining FcγRI:FcγRIIA, FcγRI:FcγRIIB, and FcγRI:FcγRIIIA concentrations are roughly equivalent. From the heat map, it appears that FcγRIIB dimers with other FcγRs are of lower concentration and therefore probably play a lesser role in driving biological activity.
The N297Q construct has no dimerization of the low affinity FcγRs and very low dimerization levels of FcγRI. This agrees with the fact that N297Q is aglycosylated and loses all binding to FcγR. The SGTA construct was engineered to only regain binding to FcγRIIA and FcγRIIB and not FcγRIIIA. Therefore, the majority of its dimer concentrations are of FcγRI in complex with FcγRI, FcγRIIA, or FcγRIIB. The FcγRIIA and FcγRIIB dimers with each other are at lower levels. The DTT-IYG aglycosylated construct also mainly forms FcγRI:FcγRI dimers in addition to slightly lower levels of FcγRI:FcγRIIA167R and FcγRI:FcγRIIIA dimers, along with a lower concentration of FcγRIIIA:FcγRIIIA dimers. There are very few FcγRIIA167H and FcγRIIB dimers, since the DTT-IYG construct has no detectable binding to those two receptors.
The DAT-IYG construct is also mostly driven by FcγRI dimers, but also shows some FcγRIIIA dimers. It has very few FcγRIIA167H dimers, since it also had no measureable binding to that receptor.
The HAT-IYG construct mainly has FcγRI:FcγRI dimers and a lower level of FcγRI:FcγRIIIA and FcγRIIIA:FcγRIIIA dimers. HAT-IYG has no detectable binding to both the activating and inhibitory FcγRII. The 299A-IYG construct is unique in that it has restored binding to all of the low affinity FcγR as shown in the heat map of all the low affinity receptors. When observing the FcγRI:FcγRI dimer concentration level, it is lower than that of the wild-type glycosylated antibody and the DTT-IYG and DAT-IYG constructs.
Aglycosylated variants promote phagocytosis of tumor cells
In order to determine if these aglycosylated variants displayed biological activity along with their restored binding to the various human FcγR, a phagocytosis assay was conducted. Macrophages, which express all of the FcγR, [5,24] were differentiated using GM-CSF from monocytes isolated from human peripheral blood, were combined with B16F10 cells that were pre-coated with or without antibodies. Macrophages were identified by staining with an anti-CD14 antibody and analyzed on flow cytometry. Phagocytosis results indicate that besides the wild-type glycosylated antibody, only the DTT-IYG and DAT-IYG constructs promoted significant levels of phagocytosis (Fig. 3a).
Figure 3.

Phagocytosis of tumor cells driven by aglycosylated antibodies. The negative control of “cells only” combine monocytes/macrophages with B16F10 tumor cells, which leads to a basal level of phagocytosis. Wild-type glycosylated antibody serves as the positive control. N297Q aglycosylated antibody serves as a second negative control. Macrophages are stained as CD14+ cells and B16F10 tumor cells as CFSE+. (a) Phagocytosis assay conducted from macrophages that were derived from monocytes harvested from whole human blood and matured with human GM-CSF. *** p-val < 0.001, **** p-val <0.0001 as compared to negative control of cells only (b) Microscopy images of phagocytosis of CFSE+ B16F10 cells (green) by CD14+ macrophages (red) at 20×. Arrows point to phagocytosis events. Scale bars at 50 µm.
Phagocytosis was further confirmed using microscopy. Cells only and N297Q treated cells show very little phagocytosis, whereas cells treated with wild-type antibody or DTT-IYG have higher levels of phagocytosis (Fig. 3b, Supplementary Fig. S5a). Aglycosylated variants SGTA, DAT-IYG, HAT-IYG and 299A-IYG show intermediate levels of phagocytosis (Supplementary Fig. S5a) in agreement with the flow cytometry data. Higher magnification at 60× visually confirms engulfment of tumor cells in macrophages on an individual cell level for samples treated with wild-type, SGTA, DTT-IYG, DAT-IYG, HAT-IYG, and 299-IYG antibodies (Supplementary Fig. S5b). Phagocytosis assays confirm restoration of biological activity in the new aglycosylated variants that initially lost their ADCP activity with loss of glycosylation as represented by the N297Q variant.
Aglycosylated variants promote tumor killing in vivo
Aglycosylated variants were evaluated for proper in vivo characteristics and stability through pharmacokinetic (PK) studies. The PK of each antibody through intraperitoneal dosing was measured in triplicate (Fig. 4a, Supplementary Fig. S6). The wild-type glycosylated antibody has the longest beta half-life of approximately 108 hours, whereas the aglycosylated antibodies N297Q, SGTA, DTT-IYG, DAT-IYG and 299A-IYG have similar beta half-lives ranging from approximately 70–90 hours. The aglycosylated variant HAT-IYG has a shorter beta half-life of 54 hours, which suggests that in vivo it is slightly less stable than the other antibodies. The PK studies suggest that the aglycosylated antibodies are slightly less stable than the wildtype glycosylated antibody, but still have fairly long beta half-lives that would allow them to be viable therapeutics.
Figure 4.

In vivo pharmacokinetic characterization and lung metastasis reduction with aglycosylated variants. (a) Table of alpha half-lives and beta half-lives of each aglycosylated antibody based on pharmacokinetic curves. Readings are fit to: C(t) = Ae−αt + Be−βt. Each study was done in triplicate. (b) Aglycosylated antibodies were used to treat B16F10 lung metastases. Wild-type (WT) glycosylated IgG1 serves as the positive control and PBS serves as the negative vehicle control. ** p-val < 0.01 as compared to negative control of PBS treatment. Other treatments were not significantly different from PBS. DTT-IYG was not significantly different from WT treatment. Five to 10 mice were used per group.
In vivo tumor control studies were conducted with aglycosylated variants in FcγR-humanized mice, in which all human FcγR family members are expressed as BAC transgenes on a murine FcγR-deficient background [25]. Antibody efficacy was assessed in the B16F10 tumor lung metastasis model, by counting the number of metastatic foci in the lungs (Fig. 4b). Wild-type antibody has decreased numbers of foci as compared to the PBS control. The aglycosylated mutant DTT-IYG demonstrated the best tumor control and was also significantly different from PBS, but not significantly different from WT treatment. There was no statistical decrease in tumor foci in the DAT-IYG, HAT-IYG and 299A-IYG treated mice as compared to PBS.
Hydrogen deuterium exchange characterization of DTT-IYG
Since aglycosylated mutant DTT-IYG demonstrated the best in vivo tumor clearance data, we monitored differences in deuterium exchange between a glycosylated wild-type (WT) IgG1 and the N297Q and DTT-IYG variants of the aglycosylated mutated IgG1’s to assess the impact of the amino acid substitutions in the IgG1 conformation. Hydrogen/deuterium exchange (H/DX) MS data were obtained for the entire antibody, but we mainly focused on the results from the antibody Fc region, given that all mutations were localized to the antibody CH2 and CH3 domains. In total 58 IgG1 Fc peptic peptides were identified and compared. Because the mutations modified the amino acid sequences of the IgG1, different peptides were produced in the region of the mutations. Comparing data from such different peptides is challenging. We only report here the comparison of those peptides that were identical in sequence (i.e. residues 242–252 vs. residues 242–252 etc.).
Comparing the glycosylated WT IgG1 with the aglycosylated mutants, as well as mutant to mutant comparisons, revealed several areas of significant difference within the IgG1-CH2 domain (Fig. 5) including residues 318–332, 278–295, and 243–252. Most of the protein showed no differences in deuterium incorporation between WT and mutant proteins (e.g., residues 369–378, Fig. 5d; remaining data not shown). In both aglycosylated IgG1 mutants, residues 243–252 incorporated more deuterium relative to the glycosylated WT IgG1 (Fig. 5a). These results were expected as removal of the IgG1 N-linked glycan is known to cause increased deuteration in this region [26,27], most likely as a result of decreased H-bonds between the glycan and the protein amide backbone. In addition to changes caused by removal of the glycosylation, two regions were found to be different in the mutants: residues 318–332 of the FG-loop and residues 278–295 of the C’E-loop. For residues 318–332 (Fig. 5b) (this identical peptide was not detected for the WT IgG1), comparison between N297Q and the DTT-IYG mutant showed that while exchange was similar for shorter time points, it was quite different at longer times (1min – 4 hrs). Such H/DX results suggest that the proteins differed in structural dynamics [28]. Conversely, in residues 278–295 (Fig. 5c) (again this peptide was not detected for the WT IgG1) deuterium incorporation of DTT-IYG was less than N297Q at early time points, suggesting that rapidly exchanging backbone amide hydrogens usually found on the surface of proteins [29] were more protected from hydrogen exchange in DTT-IYG relative to N297Q. This difference disappeared as the exchange time increases to 10 minutes through 4 hours indicating that their long-term dynamics were similar.
Figure 5.

Comparison of deuterium levels in glycosylated WT IgG1 versus aglycosylated IgG1 mutants N297Q and N297D/S298T/K326I/A327Y/L328G. (a–d) The relative percent deuteration is shown for four heavy chain peptides (a) 243–252, (b) 318–332, (c) 278–295 and, (d) 369–378. For each measurement the error was ± 0.15 Da (see the Material and Methods). (e) The structure of the IgG1 Fc region (PDB ID 1HZH10); glycans are colored cyan and peptides with differences in exchange are colored red (318–332), blue (278–295) and green. (f) DSC thermograms of two IgG1 variants. The glycosylated WT IgG1 trace is shown in solid black whereas the mutated DTT-IYG is shown in red.
DSC data
The thermogram of the glycosylated WT IgG1 (Fig. 5f, black trace) exhibits 3 main peaks with Tm values of about 60 °C, 72 °C and 84 °C. The main peak at 60 °C contains a large fronting profile and is likely composed of 2 peaks, corresponding to the variable and CH2 domains, whereas the 72 °C and 84 °C peaks correspond to the CH1 and CH3 domains, respectively. The DTT-IYG mutant (Fig. 5f, red trace) displayed a significant shift of the 60 °C peak to a lower temperature, losing about 3 °C to a Tm of 57 °C. The profile of the peak was more symmetrical, indicating a more uniform and stable conformational distribution. Additionally in DTT-IYG, the 72 °C peak appeared to gain stability with an increase in Tm to 74 °C, indicative of a more stable conformation. The slight loss in stability observed in the first peak (WT 60 °C vs. DTT-IYG 57 °C) correlates well with the increase in H/D exchange observed in the upper CH2 domain (residues 318–332).
Discussion
In the present study, we demonstrate that aglycosylated IgG variants can be engineered to engage FcγRIIIA at wild-type or improved levels, and that some of these variants can bind to all of the human low-affinity FcγRs. In engineering these aglycosylated variants, we chose a modular design strategy, based upon the hypothesis that properties imparted by altered contact loops will be additive. By combining previously identified mutant aglycosylated C’/E loops with an altered F/G loop isolated for improved FcγRIIIA176F binding, we have generated a series of aglycosylated Fc variants capable of binding FcγRIIIA whose relative receptor binding properties mirror those of the C’/E loop variants alone.
Previous work has shown reduced stability of deglycosylated wild-type hIgG1 [30] as well as the reduced stability of a glycosylated Fc variant engineered for improved FcγR binding [31]. Although we determined the biophysical properties and serum half-life of these IgGs to be comparable to wild-type, it was observed that some of the aglycosylated antibodies tended to aggregate over time, which suggested slight instability.
The engineered panel of aglycosylated variants suggests there may be overlapping roles between the activating FcγRs (FcγRI, FcγRIIA, and FcγRIIIA) that can compensate for each other in in vitro and in vivo efficacy. Engagement with the inhibitory receptor FcγRIIB might decrease efficacy results as shown with DAT-IYG. Interestingly, the HAT-IYG variant demonstrated that sole engagement of FcγRIIIA was not able to induce phagocytosis or demonstrate tumor control, which seems counter to prior works demonstrating that FcγRIIIA engagement is necessary and sufficient for tumor clearance [10]. It is noted that WT IgG1 was insufficient to enable Fc effector function [10], which suggests the need to increase the binding affinity of HAT-IYG to FcγRIIIA for efficacy. Also, 299A-IYG restored binding to all of the low affinity FcγRs and was expected to behave most similarly to wild-type glycosylated antibody. In our studies, 299A-IYG also showed less activity compared to WT IgG1, which could also suggest a potential role for FcγRI in the phagocytosis assay.
We identified an aglycosylated variant, DTT-IYG, which engaged all of the activating receptors without engaging the inhibitory receptor and demonstrates a maximum activating to inhibitory (A/I) ratio. In biological activity assays, this variant also behaved the most similar to the WT IgG1 and demonstrated near WT levels of phagocytosis and decreased lung metastatic nodules. This further confirms the importance in engaging the activating FcγRs for tumor control [10,25].
We further investigated the DTT-IYG to determine if it had any special structural properties that accounted for its restored binding to the FcγRs. The use of mass spectrometry (MS) to measure backbone amide hydrogen exchange (H/DX) in proteins has been described previously [32]. The folded conformation of a protein can influence the exchange rate at backbone amide positions, particularly in cases where hydrogen bonds are created or destroyed. By measuring H/DX for the intact protein or for the peptides created after the labelling reaction is quenched, information about protein conformation can be obtained. Anything that perturbs the conformation of a protein, such as post-translational modifications, protein activation, complex formation, etc., may perturb some of the amide hydrogen exchange rates allowing one to observe the location and magnitude of conformational changes. We previously demonstrated that H/DX MS could be used to interrogate the conformational properties of an intact IgG1 antibody [26,27]. The H/DX data from this study suggested that at residues 318–332, which includes the IYG mutations, the DTT-IYG aglycosylated variant had increased deuterium incorporation over longer periods of time and differed from N297Q in structural dynamics. This increased incorporation could suggest more accessibility of the region to enable binding to FcγRIIIA. Interestingly, peptide 278–295 showed less incorporation of deuterium at short periods of time, which suggested less exposure of the backbone amide hydrogens initially. This peptide stretch might have been impacted by the N297D/S298T (DTT) mutation downstream of the 278–295 peptide. The differences between H/DX between N297Q and DTT-IYG suggest the aglycosylated DTT-IYG has less protection from deuterium exchange at a peptide region of the FG loop, which could explain the restoration of FcγR binding.
Murine models and clinical data suggest a strong correlation between therapeutic activity and the presence of the low-affinity FcγRs [4] – FcγRIIA, FcγRIIB, FcγRIIIA. Thus the variants described here have provided a panel to test the importance of various combinations of FcγR engagement in in vivo efficacy; however, the panel is unable to demonstrate whether a single activating receptor is sufficient for effector function. Higher affinity binders to individual FcγRs tested in single receptor expressing transgenic mice would most likely be able to demonstrate this sufficiency. Our study suggests that FcγRI may play an additional role, though not necessarily an essential role, in in vitro and in vivo studies. Further confirmation with a mathematical model that suggests FcγRI:FcγRI and FcγRI:FcγRIIIA dimers might be a driving force of phagocytosis.
We have found that the aglycosylated variant DTT-IYG may effectively substitute in these models for wild-type Fc. In this study, we have engineered various aglycosylated IgG1 mutants that can theoretically be produced in any expression system without the need for glycosylation and effectively engage with FcγRs, which expands the potential for antibody variants for clinical therapeutics.
Experimental Procedures
Cell lines
HEK293F cells (Invitrogen) were cultured in Freestyle 293 expression medium (Invitrogen) in suspension. The murine melanoma cell line B16F10 (ATCC) was cultured in DMEM (ATCC) with 10% heat inactivated FBS (Invitrogen) and 100 U/ml penicillin and 100 µg/ml streptomycin (Corning Cellgro). All cell lines were maintained at 37°C with 5% CO2. Cells were subcultured every 2–3 days and when necessary detached using 0.25% trypsin and 1mM EDTA (Invitrogen).
Proteins
DNA for transfection was obtained using a PureLink HiPure Plasmid FP Maxiprep Kit (Invitrogen). Proteins were obtained through transient transfection of HEK293F cells with DNA and 2 mg of polyethyleneimine per liter of culture per manufacturer’s protocol. Supernatants were harvested and sterile filtered with a 0.2 µm filter one week after transfection. All antibodies were purified using Protein A resin (Genscript) per manufacturer’s protocol. Proteins are all buffer exchanged and stored in 1× PBS.
Protein characterization
Proteins were run on a TSKgel G3000SWxl (Tosoh Bioscience) SEC-HPLC column in 50mM sodium phosophate, 150 mM NaCl pH 7.2 buffer. Dynamic light scattering (DLS) analysis was performed on a DynaPro NanoStar (Wyatt Technology). Readings were taken with UVette disposable cuvettes (Eppendorf).
Biacore characterization
Readings were taken on a Biacore T200 instrument (GE Healthcare Life Sciences). Antitetra HIS antibody was immobilized to a CM5 series S chip using amine chemistry to a level of 14,000 RU. HIS-tagged human FcγR were diluted in HBS-EP+ binding buffer (GE Healthcare Life Sciences) and captured on the chip. Aglycosylated and wild-type IgG1 antibodies with TA99 variable regions were buffer exchanged into the HBS-EP+ buffer and injected into the instrument in serial dilutions at various concentrations to measure the binding. A hIgG1 was used as a control to confirm proper binding. Chip surfaces were regenerated with 10 mM glycine pH 1.5. Binding affinities to FcγRI were determined using kon and koff rates determined by global fits with the Langmuir model. The remaining equilibrium binding constants to the low affinity FcγR were determined by plotting steady state binding affinities.
Mathematical model
The mathematical model was modified from the one developed by Jung et al. [19] to model the phagocytosis of B16F10 melanoma cells by macrophages. In this system, free FcγR (Rf) can diffuse into the contact volume to become Rcv, where free FcγR is equivalent to the total FcγR (RT) minus Rcv. Antibody (Ab) can bind to the antigen (Ag) with an equilibrium constant of KAb to become bound antibody (B). Only FcγR in the contact volume (Rcv) can bind to bound antibody (B) with an equilibrium constant of KR to form complexes (C). Only complexes (C) can dimerize with an equilibrium constant of Kcross to form dimers (D).
Using the system of equations at steady state as follows:
Several of the parameters, given in Supplementary Table 1, were used from Jung et al. including tumor cell diameter, macrophage diameter, and the method of calculating contact area and Kdiff. Antibody concentration used in the model was the same used in the phagocytosis assays of 100 nM. The antigen expression number of TRP-1 on B16F10s was measured using Quantum Simply Cellular anti-mouse beads (Bangs Laboratories). The total FcγR level was taken from literature [33]. Kcross was calculated by using the dimer cross-linking rate constant konx and the dimer dissociation rate constant koffx. The koffx was taken from Wofsy et al. to be 1×10−5 s−1 and the konx was calculated to be kx1Ro/Ro, where the Ro was the initial cell surface FcγR expression [33]. This system of equations was expanded to include all combinations of FcγR dimers and was solved in MATLAB using fsolve to obtain the concentrations of the dimers. The data was normalized to the highest dimer value, wild-type glycosylated antibody FcγRI:FcγRI dimer. In order to plot the data as heat maps, the log10 was taken of the matrices and 11 was added to the data to center the range around 0 with +/−11. This allows all of the data to be compared under the same scale.
Phagocytosis assay
Monocytes were isolated from whole human blood using RosetteSep™ Human Monocyte Enrichment Cocktail (Stemcell Technologies). For differentiation into macrophages, monocytes were seeded into a 96-well tissue culture plate at 1×105 cells per well and treated with 50 ng/ml of recombinant human GM-CSF (Biolegend) in RPMI (Corning Cellgro) with 10% heat-inactivated fetal bovine serum (Invitrogen) and 1× penicillin-streptomycin solution on days 0, 2 and 5 after harvest. Cells were ready to use on day 7 after harvest and treatment with GM-CSF.
B16F10s were detached with 0.25% trypsin and 1mM EDTA, and washed and resuspended with 1×PBS and 0.1% Bovine serum albumin (PBSA) at 5×106 cells/ml. Cells were labeled with 5µM carboxyfluorescein succinimidyl ester (CFSE) for 10 minutes at room temperature in the dark. Cells were then quenched with excess PBSA and washed two more times with serum free DMEM. Cell lines were then incubated with 100 nM of TA99 hIgG1 WT, N297Q, SGTA, DTT-IYG, DAT-IYG, HAT-IYG, or 299A-IYG for 30 minutes at room temperature in the dark. Cells incubated in protein were then transferred to the 96-well plate the macrophages were seeded in previously. Plates were incubated at 37°C with 5% CO2 for at least 3 hours to allow for phagocytosis.
After phagocytosis, plates were pelleted, supernatants removed, and incubated with Versene (Invitrogen) for 15 min at 37°C with 5% CO2. Cells were transferred to a new plate and washed with excess PBSA before staining for 1 hour at 4°C with an anti-human CD14 Alexa Fluor 647 (Biolegend) antibody as a macrophage marker. Plates were analyzed on a FACSCalibur HTS instrument.
Microscopy
Imaging was performed on an Applied Precision DeltaVision (GE healthcare) deconvolution microscope. The phagocytosis assay was prepared as stated above with 3×105 macrophages per well in an 8 well Lab-Tek Chambered Coverglass (Thermo Scientific). 1×105 CFSE labeled B16F10 cells were incubated with the proteins and then added to the macrophages for at least 3 hours before imaging. Samples were stained with anti-human CD14 Alexa Fluor 647 antibody before imaging on the DeltaVision microscope. Images were recorded on the 20× lens and 60× oil lens.
Pharmacokinetic study
Animal work was conducted in accordance with federal, state, and local guidelines under the approval of the Massachusetts Institute of Technology Division of Comparative Medicine. Animals were maintained under pathogen-free conditions. C57BL/6 mice were injected intraperitoneally with 100 µg of antibody labeled with IRDye® 800CW (LI-COR Biosciences). Time points are measured by taking samples of blood at the tip of the tail immediately after injection at 0 hour and then subsequent hours after at 1, 3, 5, 8, 12, 24, 25, 27,29, 32, 48, 72, 96, and 120 hrs after injection. Fluorescent readings are read on the Odyssey scanner (LI-COR Biosciences) and taken on serum only. All readings are normalized to the first reading and fit to: C(t) = Ae−αt + Be−βt. All PK studies were conducted in triplicate with three separate mice. Because with an IP injection it takes a while for the antibody to distribute into the blood, the first 2 time points were ignored for the curve fit for SGTA, DAT-IYG, HAT-IYG, and 299A-IYG. For WT, N297Q, and DTT-IYG the first 3 time points were ignored for the curve fit.
Lung metastasis model
All mice were maintained under specific pathogen-free conditions and studies were performed under the approval of the Rockefeller University Institutional Animal Care and Use Committee. The B16F10 cells (1×106 cells) were injected intravenously through tail vein into FcγR-humanized C57BL/6 mice [10,25]. TA99 aglycosylated variants were injected I.P. on days 0, 2, 4, 7, 9, and 11 at 100 µg per mouse. After two weeks, the mice were euthanized and the lungs were harvested and fixed in Fekete’s solution before lung metastasis nodules were manually counted.
Statistical Analysis
One-way ANOVA with Dunnett’s multiple comparisons test were performed for the phagocytosis assay and the lung metastasis study. The phagocytosis percentages (%CD14+CFSE+) were compared to the negative control “cells only” and the lung metastatic foci compared to the PBS control with the various antibody treatments. Statistical significance was determined for p-values below 0.05.
Hydrogen/deuterium exchange mass spectrometry
Hydrogen/deuterium exchange was performed as previously described [26]. Briefly, 1.0 µL aliquots of IgG1 (~ 20 µM) were diluted into 19 µL of deuterated buffer (50 mM sodium phosphate, 100 mM NaCl, pD 6.0) and incubated for various amounts of time (10 seconds, 1, 10, 60 and 240 minutes) before quenching with a 1:1 dilution of 200 mM sodium phosphate, 0.5 M TCEP and 4 M guanidine HCl, pH 2.3. The quench dilution reduced the pH to 2.6 while simultaneously unfolding the protein and reducing disulfide bonds [27]. Quenched samples were digested online with pepsin, desalted, and separated using a Waters UPLC system designed for H/D exchange [34]. An 8 minute linear acetonitrile gradient (5–55%) was used to separate the peptides. Eluate was directed into a Waters QTof Premier mass spectrometer with electrospray ionization and lock-mass correction (using Glu-fibrinogen peptide). Mass spectra were acquired from 255 to 1800 m/z.
Pepsin fragments were identified using a combination of exact mass and MSE, aided by Waters IdentityE software [35]. Peptide deuterium levels were determined as described by Weis et al. using the Excel based program HX-Express [36] and other custom macros. No adjustment was made for deuterium back-exchange during analysis and therefore all results were reported as relative deuterium level [32]. The peptic peptides identified and followed covered 97.3 % of the IgG1 sequence. Each analysis was repeated twice. Based on our observations using historical data along with repeat injections, the error of deuterium level determination was ±0.15 Da for each data point, similar to that reported elsewhere [27,37]. Therefore, we concluded that reproducible changes in deuterium content greater than 0.5 Da were meaningful.
Differential scanning calorimetry
DSC measurements were performed using a MicroCal capillary VP-DSC system (Northampton, MA, USA) with Origin VPViewer2000 version 2.0.64 controlling software. Sample concentrations for DSC measurements were 0.5 mg/mL in IgG1 buffer. The thermograms were generated by scanning the temperature from 25 to 100 °C at a rate of 2 °C / minute. All data was processed using Origin 7SR2 software.
Supplementary Material
Highlights.
Previous Fc engineered aglycosylated antibodies restored binding to FcγRI or FcγRII
We have aglycosylated variants that restore binding to all low-affinity FcγR
In vitro phagocytosis and in vivo tumor control confirm activity of Fc variants
Mathematical modeling and H/DX were used to characterize the Fc variants
We have an aglycosylated variant that may effectively substitute for wild-type Fc
Acknowledgments
This work was supported by a Sanofi-Aventis Biomedical Innovation Award to K.D.W., the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute to K.D.W., and the National Cancer Institute Awards R01CA080757 and R35CA196620 to J.V.R. The H/DX MS work was partially supported by a research collaboration with the Waters Corporation. We thank the Koch Institute Swanson Biotechnology Center, Flow Cytometry Core, and Microscopy Core for their technical support. The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-0070319) is gratefully acknowledged. We also thank Dr. Eric F. Zhu for critical review of the manuscript.
J.B. and H.Q. are employees of Sanofi Genzyme. D.H. is currently an employee of Codiak Biosciences. S.L.S. is currently an employee of Jounce Therapeutics. T.F.C is currently an employee of Rubius Therapeutics.
Abbreviations used
- hFcγR
human Fc gamma receptor
- WT
wild-type
- SGTA
S298G/T299A
- DTT
N297D/S298T
- DAT
N297D/S298A
- HAT
N297H/S298A
- IYG
K326I/A327Y/L328G
- DLS
dynamic light scattering
- GM-CSF
Granulocyte macrophage colony-stimulating factor
- PBS
Phosphate-Buffered Saline
- H/DX MS
Hydrogen deuterium exchange mass spectrometry
- DSC
Differential scanning calorimetry
- ADCP
antibody dependent cellular phagocytosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All other authors declare no competing financial interests.
Author Contributions
T.F.C., S.L.S., K.D.W. designed research, coordinated the studies, performed experiments, analyzed data, and wrote the manuscript. D.H. and J.R.E. designed, performed, analyzed, and wrote up the experiments shown in Figure 5. J.B. and H.Q. designed, performed, and analyzed experiment in Table 1 and Figure S4. D.J.D. and J.V.R. provided animals and performed the experiment in Figure 4b. K.K.L. and G.T.G. provided technical assistance and contributed to the preparation of the figures.
References
- 1.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 2.Reichert JM, Valge-Archer VE. Development trends for monoclonal antibody cancer therapeutics. Nat. Rev. Drug Discov. 2007;6:349–356. doi: 10.1038/nrd2241. [DOI] [PubMed] [Google Scholar]
- 3.Carter P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer. 2001;1:118–129. doi: 10.1038/35101072. [DOI] [PubMed] [Google Scholar]
- 4.Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007;19:239–245. doi: 10.1016/j.coi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 6.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99:754–758. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 7.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, Neri TM, Ardizzoni A. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008;26:1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 8.Weng W-K, Czerwinski D, Timmerman J, Hsu FJ, Levy R. Clinical outcome of lymphoma patients after idiotype vaccination is correlated with humoral immune response and immunoglobulin G Fc receptor genotype. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004;22:4717–4724. doi: 10.1200/JCO.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Weng W-K, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003;21:3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 10.DiLillo DJ, Ravetch JV. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell. 2015;161:1035–1045. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jefferis R, Lund J. Interaction sites on human IgG-Fc for FcgammaR: current models. Immunol. Lett. 2002;82:57–65. doi: 10.1016/s0165-2478(02)00019-6. [DOI] [PubMed] [Google Scholar]
- 12.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, Fox JA, Presta LG. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 13.Simmons LC, Reilly D, Klimowski L, Raju TS, Meng G, Sims P, Hong K, Shields RL, Damico LA, Rancatore P, Yansura DG. Expression of full-length immunoglobulins in Escherichia coli: rapid and efficient production of aglycosylated antibodies. J. Immunol. Methods. 2002;263:133–147. doi: 10.1016/s0022-1759(02)00036-4. [DOI] [PubMed] [Google Scholar]
- 14.Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. Baltim. Md 1950. 1989;143:2595–2601. [PubMed] [Google Scholar]
- 15.Mimura Y, Sondermann P, Ghirlando R, Lund J, Young SP, Goodall M, Jefferis R. Role of oligosaccharide residues of IgG1-Fc in Fc gamma RIIb binding. J. Biol. Chem. 2001;276:45539–45547. doi: 10.1074/jbc.M107478200. [DOI] [PubMed] [Google Scholar]
- 16.Walker MR, Lund J, Thompson KM, Jefferis R. Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing Fc gamma RI and/or Fc gamma RII receptors. Biochem. J. 1989;259:347–353. doi: 10.1042/bj2590347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. U. S. A. 2008;105:20167–20172. doi: 10.1073/pnas.0809257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung ST, Reddy ST, Kang TH, Borrok MJ, Sandlie I, Tucker PW, Georgiou G. Aglycosylated IgG variants expressed in bacteria that selectively bind FcgammaRI potentiate tumor cell killing by monocyte-dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 2010;107:604–609. doi: 10.1073/pnas.0908590107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung ST, Kelton W, Kang TH, Ng DTW, Andersen JT, Sandlie I, Sarkar CA, Georgiou G. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcγRIIa affinity and selectivity. ACS Chem. Biol. 2013;8:368–375. doi: 10.1021/cb300455f. [DOI] [PubMed] [Google Scholar]
- 20.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SHA, Presta LG. Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human FcγRIII and Antibody-dependent Cellular Toxicity. J. Biol. Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 21.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-Inflammatory Activity of Immunoglobulin G Resulting from Fc Sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 22.Bethea D, Wu S-J, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O’Neil KT, Gilliland GL, Feng Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng. Des. Sel. PEDS. 2012;25:531–537. doi: 10.1093/protein/gzs047. [DOI] [PubMed] [Google Scholar]
- 23.Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY. Optimizing engagement of the immune system by anti-tumor antibodies: an engineer’s perspective. Drug Discov. Today. 2007;12:898–910. doi: 10.1016/j.drudis.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Griggs J, Zinkewich-Peotti K. The state of the art: immune-mediated mechanisms of monoclonal antibodies in cancer therapy. Br. J. Cancer. 2009;101:1807–1812. doi: 10.1038/sj.bjc.6605349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl. Acad. Sci. 2012;109:6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 2009;81:2644–2651. doi: 10.1021/ac802575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol. Cell. Proteomics MCP. 2010;9:1716–1728. doi: 10.1074/mcp.M900540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgan CR, Engen JR. Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr. Protoc. Protein Sci. Editor. Board John E Coligan Al. 2009 doi: 10.1002/0471140864.ps1706s58. Chapter 17 Unit 17.6.1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dharmasiri K, Smith DL. Mass spectrometric determination of isotopic exchange rates of amide hydrogens located on the surfaces of proteins. Anal. Chem. 1996;68:2340–2344. doi: 10.1021/ac9601526. [DOI] [PubMed] [Google Scholar]
- 30.Mimura Y, Church S, Ghirlando R, Ashton PR, Dong S, Goodall M, Lund J, Jefferis R. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: properties of a series of truncated glycoforms. Mol. Immunol. 2000;37:697–706. doi: 10.1016/s0161-5890(00)00105-x. [DOI] [PubMed] [Google Scholar]
- 31.Oganesyan V, Damschroder MM, Leach W, Wu H, Dall’Acqua WF. Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol. Immunol. 2008;45:1872–1882. doi: 10.1016/j.molimm.2007.10.042. [DOI] [PubMed] [Google Scholar]
- 32.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 33.Wofsy C, Torigoe C, Kent UM, Metzger H, Goldstein B. Exploiting the difference between intrinsic and extrinsic kinases: implications for regulation of signaling by immunoreceptors. J. Immunol. Baltim. Md 1950. 1997;159:5984–5992. [PubMed] [Google Scholar]
- 34.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva JC, Gorenstein MV, Li G-Z, Vissers JPC, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics MCP. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Weis DD, Engen JR, Kass IJ. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 37.Burkitt W, O’Connor G. Assessment of the repeatability and reproducibility of hydrogen/deuterium exchange mass spectrometry measurements. Rapid Commun. Mass Spectrom. RCM. 2008;22:3893–3901. doi: 10.1002/rcm.3794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.