

Abstract
The molecular mechanisms underlying the multiresistant phenotype of leukemic and other cancer cells are incompletely understood. We used expression arrays to reveal differences in the gene expression profiles of an apoptosis-resistant T cell leukemia clone (A4) and normally apoptosis-sensitive parental Jurkat cells. CD73 (ecto-5′-nucleotidase) was the most up-regulated gene in the resistant A4 cell clone. A4 cells displayed CD73 surface expression and significant ecto-5′-nucleotidase activity. The role of CD73 was confirmed by transfection of wild-type CD73 into native Jurkat cells, which led to specific resistance against TRAIL-induced apoptosis, but not other types of apoptosis. The protective role of CD73 was further confirmed by small interfering RNA-mediated down-regulation of CD73, restoring TRAIL sensitivity. CD73-mediated resistance was independent of enzymatic activity of CD73, but was reliant on the anchoring of the protein to the membrane via GPI. We suggest that the inhibition of TRAIL signaling works through interaction of CD73 with death receptor 5, as CD73 and death receptor 5 could be coimmunoprecipitated and were shown to be colocalized in the plasma membrane by confocal microscopy. We propose that CD73 is a component of multiresistance machinery, the transcription of which is activated under selective pressure of the immune system.
The resistance of tumor cells against immune surveillance and/or anticancer therapy is one of the major reasons for failure of anticancer treatment. Even when chemotherapy is initially successful, many neoplastic cells are able to develop resistance to further treatment. Deciphering the molecular events responsible for the development of drug resistance in cancer cells has been a highly active endeavor in cancer research during the past decades. It is now established that a primary cause for drug resistance in tumor cells is reduced sensitivity to apoptotic stimuli reviewed in Ref. (1), either by modulation or rerouting of apoptotic signaling pathways and/or by elevated expression of antiapoptotic regulator proteins. Identification of critical hubs in the apoptotic regulatory machinery, whose functions have been altered in resistant cells, is a way to identify new therapeutic opportunities when conventional cancer therapies fail (2).
Several regulatory systems have the capacity to promote resistance against apoptosis. For example, activation of survival pathways, such as those including protein kinase B (3), PI3K (4), NF-κB (5), and ERK (6, 7) (for review, see Ref. 8), can override many different types of death-inducing stimuli, especially in combination with defects in the p53/p21 system. These signaling pathways are often activated in highly resistant tumor cells and provide major routes to escape DNA damage-mediated cell cycle arrest and apoptosis (9). ATP-binding cassette transporters (MDR1/pgp, MDR-related protein, lung resistance-related protein, breast cancer resistance protein, etc.) rescue cancer cells preferentially from drug-induced apoptosis (10). Overexpression or misbalance among the intrinsic antiapoptotic regulators, such as Bcl-2, Bcl-xL, and inhibitors of apoptosis, have been identified as major causes for protection of tumor cells and is usually associated with poor clinical prognosis (11). The protection may also be targeted at the extrinsic apoptosis inducers, as inhibition of apoptotic signaling from the transmembrane death receptors and their adaptor proteins is commonly observed in many different cancer cell types (12).
Judging from the above-listed wide range of different antiapoptotic mechanisms activated in tumor cells, it seems obvious that there is no single universal source of apoptotic resistance. Instead, tumor cells display complex patterns of resistance programs, with several resistance mechanisms engaged at the same time. It is plausible to expect that the tumor would display an increasingly complex apoptosis resistance, often initially based on single-resistance determinants but with escalating complexity in the resistance program under the selective pressure of the immune system and/or various cancer therapies.
The human leukemic T cell line Jurkat is a useful model for studying the development of apoptotic resistance. These cells have a high proliferation rate, are moderately sensitive to most anticancer drugs and ionizing radiation (13), and do not express the p53 protein (14). These factors lead to reduced genetic stability and clonal divergence under the pressure of apoptosis-inducing factors (15).
In a previous study, we observed that pulse treatment of Jurkat cells with gradually elevated concentrations of agonistic Abs against the death receptor Fas (FasR/CD95/APO-1) led to a selection of cells that lacked functional FasR on their surface (16). Several groups have obtained similar results and have reported different causes for the gained resistance. Depletion of FasR has been observed in a Jurkat clone, which was still sensitive to anticancer drugs (17). In another study, the resistant Jurkat clone JB-6 showed presence of FasR, but was lacking caspase-8 (18). A similar approach led to the development of a cell line defective in caspase-3 (19), but with retained sensitivity to drug-induced apoptosis. When we followed the above-described approach to produce Fas-insensitive cell lines, we obtained a clone, Jurkat/A4 (referred to as A4), which displayed, apart from a Fas-negative phenotype, to our surprise, partial or complete insensitivity to many other apoptogenic stimuli, including UV- and x-ray irradiation, anticancer drugs (etoposide, doxorubicin, and cycloplatam), oxidative stress (H2O2 and menadione), the general kinase inhibitor staurosporin, and also TRAIL (despite the presence of the TRAIL receptor, DR5). Growth, morphology, and ploidy of these cells remained similar to the parent line (16).
Because A4 cells acquired resistance to several prominent apoptosis-inducing pathways during the course of desensitization to FasR, we chose to use this clone as a starting point for identifying and characterizing individual molecular determinants of a multiresistant phenotype. To identify potential candidate genes and their products that could explain the observed phenotype, we used a large scale gene expression profiling approach. Among the topranking genes differentially expressed between parental Jurkat and A4 cells, we report here that CD73 has a defined and TRAIL-targeted role in the apoptosis resistance machinery. These results help explain previous observations regarding elevated expression of CD73 in various types of malignant cells and the reported correlation between CD73 expression and poor prognosis (20, 21).
Materials and Methods
Cells and treatments
The Jurkat cell line was obtained from American Type Culture Collection (clone EG-1). A multiresistant cell line (A4) was prepared from this Jurkat cell line by consecutive treatment with stepwise increasing concentrations (50–1000 ng/ml) of agonistic Abs against the FasR (16). Phenotype is stable for at least 2 years of cultivation without selective pressure of agonistic Abs. Both lines were maintained in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% FCS without antibiotics in an humidified atmosphere of 5% CO2 in air at 37°C. The cells were maintained in the log phase by routine passage every 2–3 days. The cell number was kept at 0.5–1.0 × 106 cells/ml.
The transfected Jurkat cell lines B-NT5.1, B-TM15.1, β92.20, and NC-1 that express human GPI-anchored, human transmembrane-anchored, human enzyme-inactive GPI-anchored, and murine GPI-anchored CD73, respectively, were described previously (22, 23). Apoptosis was induced by treatments with rFasL (Alexis) at 50 ng/ml for 4 h, with TRAIL (Alexis) cross-linked with Abs against FLAG epitope (M2; Sigma-Aldrich) at 50–100 ng/ml (or SuperKillerTRAIL at various concentrations) for 4 h, and with etoposide (Alexis) at 10 μM for 20 h.
The ecto-5′-nucleotidase-positive endothelial cell line HEC, equivalent to EaHy-926 (24), was maintained in Iscove’s DMEM medium (Sigma-Aldrich) supplemented with 10% FCS without antibiotics in a humidified atmosphere of 5% CO2 in air at 37°C.
Oligonucleotide array analyses
Total RNA was isolated from ~80 × 106 resting parental Jurkat and multiresistant A4 cells in two independent biological replicates with interval of 3 mo between replicates, and 5 μg of total RNA was used as a starting material for the sample preparation according to the protocol provided by the manufacturer (Affymetrix). Fifteen micrograms of biotin-labeled cRNA was fragmented and hybridized to human genome U133 A oligonucleotide arrays comprised of >22,000 probe sets and enabling simultaneous analysis of the expression level of 18,400 transcripts and variants, including 14,500 well-characterized human genes (Affymetrix; performed in duplicate with two biological repeats). Arrays were scanned and results were normalized by the Microarray Suite software.
Ecto-5′-nucleotidase assays
Ecto-5′-nucleotidase activity was analyzed by a thin-layer chromatography (TLC)3 assay with [2-3H]AMP (specific activity 18.6 Ci/mM; Amersham) as described previously (25). Briefly, cell suspensions (1–2 × 105 cells) were incubated at 37°C for 45 min in a final volume of 120 μl of RPMI 1640 containing 5 mM β-glycerophosphate and 300 μM AMP with tracer [3H]AMP (~4 × 105 dpm). In some experiments, the cells were preincubated for 30 min at 37°C with recombinant phosphatidylinositol-specific phospholipase C (PI-PLC; 2.5 U/ml) from Bacillus cereus, and then washed with RPMI 1640. Catalytic reactions were terminated by applying aliquots of the mixture to Alugram SIL G/UV254 sheets (Macherey-Nagel). [3H]AMP and its dephosphorylated nucleoside derivatives were separated by TLC and quantified by scintillation counting with a Wallac-1409 β-spectrometer. Ecto-5′-nucleotidase activity was expressed as nanomoles of AMP hydrolyzed by 106 cells in 1 h.
Abs and Western blotting analyses
Ecto-5′-nucleotidase was detected by using a rabbit polyclonal Ab (gift of Dr. J. Spychala, University of North Carolina, Chapel Hill, NC) raised from immunization with a peptide corresponding to residues 155ETPFL-SNPGTNLVFGD of the human ecto-5′-nucleotidase and normal rabbit serum (1:100) as a negative control, as described previously (26). A polyclonal Ab against PP2A was the gift of D. Brautigan (University of Virginia, Charlottesville, VA) and a mAb against caspase-8 was the gift of Dr. P. Krammer (German Cancer Research Center, Heidelberg, Germany). Commercial Abs against p53, Bcl-2, FLAG tag (M2) (Sigma-Aldrich), Bcl-xL, Bax, caspase-3 (BD Pharmingen), Fas-associated death domain (FADD; 610399; BD Transduction Laboratories), FLIP (NF6; Axxora), DR4 (1167; ψProSci), DR5 (45B872.1; Abcam), Raf-1, MKK3, JNK, phospho-JNK, phospho-c-Jun, ERK, phospho-ERK (New England Biolabs), and heat shock protein (HSP)-70, heat shock cognate (HSC)-70, HSP-90 (StressGen Biotechnologies) were also used. A 15-mer peptide PVLAPQWEGYDELQT from a sequence neighboring the caspase-8 cleavage site of human BH3-interacting domain death agonist (BID) was used to produce mAbs. Resulting Abs recognize specifically full-length BID as well as the cleaved 6.6-kDa fragment. Western blotting was performed after lysing cells in Laemmli sample buffer and then resolving the proteins on a SDS-PAGE (10%). The separated proteins were transferred to a polyvinylidene fluoride filter (Amersham). Filters were blocked with 5% BSA in buffer solution (125 mM NaCl, 25 mM MOPS, 12.5 mM KOH, 0.1% Tween 20). Membranes were treated overnight with working dilutions of primary Abs in 5% BSA in buffer solution with 0.1% NaN3, followed by reaction with appropriate HRP-conjugated secondary Abs. Detection was conducted with the ECL+ system (Amersham); quantitation was performed with help of MCID Analysis Software Version 7.0 (InterFocus Imaging).
Apoptosis and immunofluorescence analyses
CD73 expression was assessed by flow cytometry using Cy2-conjugated 4G4 mAb (24) (Amersham). TRAIL receptors for flow cytometry were stained with the following Abs against DR5 (DJR2-1), DR4 (DJR1), DcR1 (DJR3; Abcam), DcR2 (DJR4-2; Abcam). Cells were washed with cold PBS and then incubated with diluted Ab on ice for 30 min. Apoptosis was detected by flow cytometry on the basis of phosphatidylserine exposure and membrane integrity. Cells were washed once with cold PBS and stained using the Vybrant Apoptosis Assay kit 9 with allophycocyanin, annexin V, and Sytox Green stain (Molecular Probes) according to the manufacturer’s instructions, and then analyzed with a LSRII flow cytometer (BD Biosciences). Data acquisition was performed by FACSDiva software with enforced autocompensation. Cells with low staining with both fluorophores were considered to be surviving cells and were located at lower left quadrant of the two-dimensional graphs. Three to five independent experiments were performed in each experimental setting.
For confocal microscopy, cells were labeled in growth medium with Cy2-conjugated Abs against CD73 (4G4 for human CD73 or TY/11.8 for mouse CD73 (27)) and Cy3B-conjugated Abs against DR5 (DJR2-1) for 1 h at +20°C. Cells were then washed three times, fixed with 3% paraformaldehyde, and observed with an inverted confocal microscope (Zeiss LSM 510) on glass-bottom 24-well plates. As a control, we used a Cy3B conjugate of an isotype-matched Ab against nodularin (28), which is known not to react with any proteins on Jurkat cells or human sera. Pixel correlations were calculated with Zeiss LSM imaging software.
Activity of caspases-8 and -3 was measured by LANCE Caspase-3 and -8 kits (Wallac/PerkinElmer) in 384-well plates according to the manufacturer’s instructions.
Small interfering RNA (siRNA) treatment
HEC cells were seeded into 100-mm petri dishes at low density. Treatment was initiated when cell cultures reached 10% confluence. Three different synthetic 21-mer siRNAs (5′-ACAGCAGCAUUCCUGAAGATT-3′, 5′- CGCAACAAUGGCACAAUUATT-3′ and 5′-CCUGGAGACAGAGUAGUCATT-3′; MWG Biotech) and scrambled control RNA were transfected by jetSI (Polyplus-Transfection) at a concentration of 4.8 μg per dish according to the protocol supplied by the manufacturer. Alternatively, B-NT5.1 cells were repeatedly transfected with 25 nM of a 1:1:1 mixture of the above-described siRNAs in suspension by electroporation (400 v/cm, 1 mF, two pulses) for 3 consecutive days before additional experiments.
Results
Immunochemical screening reveals only minor differences between parental Jurkat and clone A4
In a previous study (16), we assessed the apoptotic sensitivity of the apoptosis-insensitive Jurkat A4 clone and observed that it is highly resistant to a number of different apoptotic stimuli (results reviewed in Table I). In this study, we found that the A4 cells express only traces of FasR on the cell surface. To evaluate the possible role of other apoptosis-regulating proteins for the insensitivity, whole cell lysates of resting parental Jurkat and multiresistant A4 cells were analyzed by Western blotting for the levels of candidate apoptosis-regulating proteins that could be involved in the multiresistant phenotype. The levels of the following proteins were analyzed: p53, Bcl-2, Bcl-xL, Bax, FADD, FLIP, DR4, DR5, DcR1, DcR2, Cas-8, Cas-3, Raf-1, MKK3, JNK, phospho-JNK, phospho-c-Jun, ERK, phospho-ERK, HSP-70, HSC-70, HSP-90, PP2A. Of all these proteins, only the amount of HSP-90 was significantly higher in A4 cells, whereas the levels of Bcl-2 and Bcl-xL were unexpectedly lower in the multiresistant cells (Fig. 1A, Table II, and data not shown).
Table I.
Survival of parental Jurkat cells and multiresistant A4 cells subjected to different apoptosis-inducing treatmenta
| Treatment | Survival %
|
Reference | |
|---|---|---|---|
| Jurkat | A4 | ||
| Agonistic Abs against FasR, 100 ng/ml, 6 h | 10 ± 5 | 95 ± 3 | 16 |
| TRAIL, 50 ng/ml, 4 h | 45 ± 11 | 90 ± 2 | |
| Doxorubicin, 0.5 μg/ml, 46 h | 54 ± 7 | 95 ± 3 | 16 |
| Etoposide, 10 μg/ml, 22 h | 10 ± 2 | 71 ± 6 | 16 |
| Staurosporine, 0.46 μM, 46 h | 25 ± 8 | 75 ± 11 | |
| H2O2, 10−5 M, 22 h | 58 ± 5 | 92 ± 6 | |
| Menadion, 10 μM, 46 h | 53 ± 6 | 90 ± 3 | |
| X-ray, 20 Gy, 48 h after treatment | 38 ± 7 | 80 ± 5 | |
| UV irradiation, 254 nm, 8 min | 50 ± 9 | 84 ± 4 | |
The presented data are from biological triplicates, original treatment in five biological repeats; table shows mean values ± SD.
FIGURE 1.
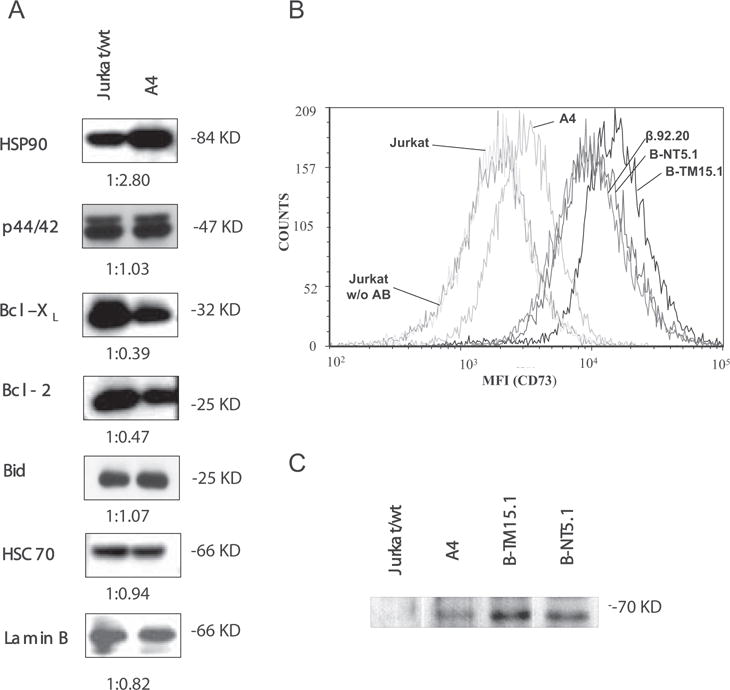
A, Immunochemical screening of antiapoptotic proteins. Lysates of parental Jurkat and A4 cells were subjected to Western blotting with Abs against HSP-90, ERK (p44/42), Bcl-2, Bcl-xL, and BID. Reactions were developed with HRP conjugates of corresponding secondary Abs and visualized by chemiluminescence. HSC70 and Lamin B were used as loading controls. Densitometry ratios are shown below the individual graphs. The densitometric value obtained with Jurkat cells equals 1. B, Analysis of CD73 expression by flow cytometry. The elevated CD73 expression in A4 cells was confirmed by FACS analysis. Resting Jurkat, A4 and different CD73 transfectant cells were labeled with anti-CD73 Ab 4G4, triple washed and visualized with secondary Abs conjugated to Alexa 647. The cells were washed once with PBS and data from 105 cells were collected on a FACScan (BD Biosciences) flow cytometer and analyzed with FCSExpress3 software. The following mean fluorescence intensity values were obtained from the FACS analyses: Jurkat wild-type labeled with nonspecific Ab, 2209; Jurkat wt, 2239; A4, 3672; β.92.20, 12294; B-NT5.1, 13345; B-TM15.1, 18289. C, Confirmation of CD73 expression on A4 cells by Western blotting. Lysates of 105 resting Jurkat and A4 cells equivalent to those shown in A were subjected to Western blotting with Abs against CD73. Lysates of 2 × 104 Jurkat cells stably transfected with CD73 (B-NT5.1 and B-TM15.1) were used as positive controls.
Table II.
Distribution of TRAIL receptors and TRAIL-binding sites on the surface of the cell lineda
| Cells
|
|||||
|---|---|---|---|---|---|
| Jurkat | β.92.20 | B-TM15.1 | B-NT5.1 | A4 | |
| Treatments | |||||
| Secondary Ab | 1 | 0.97 ± 0.04 | 0.95 ± 0.04 | 0.98 ± 0.08 | 1.15 ± 0.05 |
| DR5 | 19.30 ± 2.11 | 21.30 ± 0.93 | 19.33 ± 1.10 | 23.00 ± 1.33 | 23.16 ± 0.64 |
| Dr4 | 1.65 ± 0.14 | 1.90 ± 0.35 | 1.71 ± 0.29 | 1.74 ± 0.10 | 1.80 ± 0.34 |
| DcR1 | 1.27 ± 0.20 | 1.64 ± 0.22 | 1.58 ± 0.06 | 1.59 ± 0.13 | 1.42 ± 0.09 |
| DcR2 | 1.35 ± 0.17 | 1.39 ± 0.19 | 1.14 ± 0.13 | 1.29 ± 0.38 | 1.80 ± 0.29 |
| TRAIL binding | 8.18 ± 0.45 | 8.96 ± 1.17 | 8.87 ± 0.48 | 9.40 ± 0.64 | 9.32 ± 1.54 |
Life cells were treated in suspension with primary Abs against DR5 (clone DJR2-1), Dr4 (clone DJR1), DcR1 (clone DJR3), DcR2 (clone DJR4-2) for 1 h at +37°C in culture media. Cells were washed three times in PBS and treated with secondary Abs against mouse IgG conjugated with Alexa 546 for 30 min at room temperature. In parallel, other aliquots of cells were treated with rFlag-tagged TRAIL (Axxora) at 4°C with addition of mouse anti-Flag mAb M2 was obtained from Sigma-Aldrich for 30 min. Cells were washed three times in cold PBS and treated with secondary Abs against mouse IgG conjugated with Alexa 546 for 30 min at +4°C. Some cells were treated with secondary Abs only. All cells were washed with cold PBS and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature, diluted 1/10 with cold PBS and subjected for FACS analysis. Data in secondary Ab raw are relative ratios to mean fluorescence for Jurkat cells. All measurements were done in triplicates. Table shows mean values ± SD. Data in columns are relative ratios to mean fluorescence for secondary Abs for the designated cell line.
Large-scale gene expression profiling of the multiresistant cell line
As the approach of “educated guesses” did not yield any answer to the question of why clone A4 had become so deficient in its execution of apoptosis, we conducted a large-scale gene expression profiling to identify possible candidates responsible for the acquired phenotype. Transcription profiles of parental-sensitive (Jurkat) and apoptosis-resistant (A4) cells were compared. RNA was isolated from resting Jurkat and multiresistant A4 cells, converted to cDNA, and was hybridized with oligonucleotide DNA microarrays (Affymetrix 133A; performed in duplicate with samples from two independent experiments). Arrays were scanned and results were normalized by the Microarray Suite software. Comparison of the signals showed that only a rather limited number of genes on the array were up- or down-regulated more than four (22) times. Moreover, quite surprisingly, the list of up- or down-regulated genes contained neither genes involved in the initiation or execution of apoptosis, nor genes encoding for proteins among major survival signaling pathways (Table III, complete data submitted into the MIAMExpress Database, accession number E-MEXP-530). In the current study, we focused on the most significantly altered gene in the multiresistant A4 line, CD73 (elevated 362 (28.5) times), because CD73 has been associated with multidrug-resistant tumor phenotypes in previous reports (20, 21). It is noteworthy that we did not detect substantial changes in expression of other components of the purinergic signaling cascade between the parental Jurkat cells and the multiresistant A4 line, including various adenosine receptors, another nucleotide-hydrolyzing ectoenzyme, NTPDase1, and other purine-converting enzymes (Table III).
Table III.
DNA-microarray analysis of gene expression in resistant A4 cells as compared to the parental Jurkat cell linea
| Reference No. | Signal from A4 Cells | Signal Ratios A4 vs Jurkat (Binary Log Ratio, Folds in Parentheses) | Change p Value | Description of the Gene |
|---|---|---|---|---|
| Top up-regulated genes | ||||
| 203939_at | 252 257 |
8.5 (362) 3.5 (11) |
0.00002 0.021224 |
5′-nucleotidase (CD73) (NT5) |
| 218559_s_at | 217.5 | 6.1 (68) | 0.000189 | Kreisler maf-related leucine zipper homolog (KRML) |
| Signal lower threshold | ||||
| 201218_at | 201.5 248.9 |
5.9 (59.7) 7.1 (137) |
0.00002 0.000023 |
C-terminal binding protein 2 |
| 214254_at | 275.8 53.2 |
4 (16) 3.9 (14.9) |
0.00002 0.000035 |
Melanoma Ag, family A |
| 204379_s_at | 212.2 75 |
3.7 (12.9) 3.7 (12.9) |
0.000078 0.004925 |
Fibroblast growth factor receptor 3 |
| 205691_at | 144.4 93.9 |
2.9 (7.46) 1.7 (3.25) |
0.000618 0.030967 |
Synaptogyrin 3 |
| 203471_s_at | 398.9 64.9 |
2.8 (6.96) 0 (1) |
0.00002 0.330589 |
Pleckstrin |
| 210554_s_at | 346.8 556.1 |
2.8 (6.96) 4.4(21.1) |
0.00003 0.000023 |
C-terminal binding protein 2, clone MGC:1563 |
| 205768_s_at | 201.7 154.2 |
2.7 (6.49) 2.3 (4.92) |
0.00002 0.000241 |
Fatty-acid-coenzyme A ligase, very long-chain 1 (FACVL1) |
| 201220_x_at | 389.7 815.5 |
2.6 (6.06) 3.5 (11.3) |
0.00002 0.00002 |
C-terminal binding protein 2 (CTBP2), transcript variant 1 |
| 201215_at | 134.5 59.5 |
2.6 (6.06) 1.3 (2.46) |
0.00002 0.000068 |
Plastin 3 (T isoform) |
| 218718_at | 1263.8 1212.2 |
2.5 (5.66) 2.4 (5.28) |
0.00002 0.00002 |
Platelet-derived growth factor C |
| 209589_s_at | 142.5 66.7 |
2.5 (5.66) 2.9 (7.46) |
0.00002 0.00225 |
Protein-tyrosine kinase EPHB2v |
| 200704_at | 109.4 99.9 |
2.4 (5.28) 1.8 (3.48) |
0.000023 0.00002 |
Integral membrane protein of lysosomelate endosome, LPS-induced TNF-α factor |
| 205488_at | 1018 184.5 |
2.4 (5.28) 1.2(2.297) |
0.00002 0.000052 |
Granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine esterase 3) |
| 210835_s_at | 361.9 485.2 |
2.4 (5.28) 2.8 (6.96) |
0.000023 0.00002 |
Ribeye/C-terminal-binding protein 2 |
| Top down-regulated genes | ||||
| 205347_s_at | 28.5 23.5 |
−4.9 (0.0334) −2.1 (0.2332) |
0.999759 0.996645 |
Thymosin, β, identified in neuroblastoma cells (TMSNB) |
| 202149_at | 2 10.2 |
−4.6 (0.0412) −2.8 (0.1436) |
0.99998 0.999948 |
Consensus includes 3 part of the gene For enhancer of filamentation (HEF1) |
| 209209_s_at | 4.2 2.3 |
−4.5 (0.0442) −5.3 (0.0254) |
0.999973 0.999448 |
Mitogen-inducible 2 |
| 201670_s_at | 4 19 |
−4.1 (0.0583) −1.7 (0.3077) |
0.99987 0.995927 |
Myristoylated alanine-rich protein kinase C substrate (MARCKS, 80K-L) |
| 201170_s_at | 6.5 17.4 |
−3.6 (0.0825) −2.3 (0.203) |
0.999853 0.977068 |
Basic helix-loop-helix domain, class B, 2 (BHLHB2) |
| 209210_s_at | 29.7 32 |
−2.9 (0.1339) −3 (0.125) |
0.99998 0.99998 |
Mitogen-inducible gene mig-2, complete CDS |
| 210432_s_at | 34.8 120.5 |
−2.8 (0.1436) −0.7 (0.6155) |
0.99998 0.995519 |
Voltage-gated sodium channel α subunit splice variant SCN3A-s |
| 214464_at | 27.8 48.5 |
−2.6 (0.1649) −0.8 (0.574) |
0.99998 0.99996 |
Ser-Thr protein kinase related to the myotonic dystrophy protein kinase (PK428) |
| 215784_at | 265 130.5 |
−2.5 (0.1767) −2.8 (0.1436) |
0.99998 0.99998 |
CD1E Ag, e polypeptide |
| Death receptors | ||||
| 209295_at | 242.7 185.6 |
0.2 (1.148) 0.4 (1.319) |
0.026698 0.213188 |
TRAIL receptor 2 |
| 202535_at | 283.1 463.6 |
0.1 (1.07171) −0.3 (0.812) |
0.5 0.952736 |
Fas (TNFRSF6)-associated via death domain (FADD) |
| 210654_at | 10.8 11.7 |
0.2 (1.148) 1.1 (2.143) |
0.467656 0.318909 |
TRAIL-R4-B (TRAIL-R4) mRNA |
| 203508_at | 22.3 28.1 |
2.3 (4.924) 2.4 (5.278) |
0.099852 0.354442 |
TNFR2 (75 kDa) TNFR superfamily, member 1B |
| Caspases | ||||
| 209790_s_at | 152.9 204.3 |
−0.7 (0.6155) 0.6 (1.515) |
0.99994 0.354442 |
Caspase 6, apoptosis-related cysteine protease |
| 202763_at | 148 197.5 |
−0.4 (0.757) −0.1 (0.933) |
0.998514 0.725952 |
Caspase 3, apoptosis-related cysteine protease (CASP3) |
| 203984_s_at | 152 276.5 |
−0.1 (0.933) 0.3 (1.231) |
0.5 0.083826 |
Protease Mch6, caspase 9, apoptosis-related cysteine protease |
| 207181_s_at | 166.7 105.8 |
0 (1) 0 (1) |
0.5 0.5 |
Caspase 7, apoptosis-related cysteine protease (CASP7) |
| 207686_s_at | 81.9 45 |
0 (1) −1.2(0.435) |
0.681091 0.558077 |
Caspase 8, apoptosis-related cysteine protease (CASP8) |
| Apoptosis-related genes | ||||
| 201746_at | 84.1 104.2 |
0.5 (1.414) 0.7 (1.6245) |
0.161038 0.050553 |
Tumor protein p53 |
| 205385_at | 29.9 24.5 |
1.3 (2.462) −0.4 (0.7578) |
0.005409 0.330589 |
Human p53-associated mRNA, complete CD |
| 207004_at | 27.6 9.6 |
0.8 (1.741) −2.7 (0.1538) |
0.041201 0.284967 |
Homo sapiens B cell CLL lymphoma 2 (BCL2), nuclear gene encoding mitochondrial protein, transcript variant β |
| 215037_s_at | 102.9 68.5 |
0.1 (1.07177) −0.9 (0.5358) |
0.5 0.905721 |
Human Bcl-x β (bcl-x) gene |
| 219350_s_at | 556.6 713.4 |
0.1 (1.07177) −0.1 (0.933) |
0.5 0.60871 |
Second mitochondria-derived activator of caspase (SMAC) |
| 210563_x_at | 148.2 180.2 |
0.3 (1.231) 0.6 (1.515) |
0.161038 0.001077 |
FLICE-like inhibitory protein short, CASP8 and FADD-like apoptosis regulator |
| 208478_s_at | 32 63.7 |
0.1 (1.07177) 0.4 (1.319) |
0.5 0.330589 |
BCL2-associated X protein (BAX) |
| 204493_at | 297.2 220.9 |
0.3 (1.231) 0.2 (1.1486) |
0.111714 0.274048 |
BH3-interacting domain death agonist (BID) |
| Heat shock proteins | ||||
| 117_at | 28.9 7.4 |
0 (1) −2.8 (0.1435) |
0.5 0.603256 |
Human heat-shock protein HSP70B gene |
| 211015_s_at | 487 324.8 |
−0.1 (0.933) −0.2 (0.8705) |
0.5 0.961585 |
Human heat shock protein 70 (hsp70) mRNA |
| 200064_at | 3516.6 3904.4 |
0 (1) 0.6 (1.1557) |
0.5 0.000023 |
Chaperone protein HSP90 β (HSP90β) mRNA |
| Kinases and phosphatases | ||||
| 212046_x_at | 136 150 |
0.4 (1.3195) −0.1 (0.933) |
0.570859 0.583567 |
ERK1 mRNA for protein serine threonine kinase |
| 214693_x_at | 301 380.7 |
0 (1) 0.2 (1.1486) |
0.5 0.124552) |
Phosphatase type 2A |
| 211500_at | 9.2 7.3 |
0.6 (1.1557) 0.3 (1.231) |
0.005409 0.000966 |
p38β MAPK mRNA, complete CD |
| 208351_s_at | 138.1 111.8 |
0.4 (1.3195) −0.5 (0.7071) |
0.004481 0.964215 |
Mitogen-activated protein kinase 1 (MAPK1) |
| 201461_s_at | 77 57.6 |
0 (1) −0.1 (0.933) |
0.545234 0.5 |
Mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2) |
| 210570_x_at | 78.7 207.6 |
0 (1) −0.4 (0.7578) |
0.5 0.861614 |
JNK2β l protein kinase (JNK2B1) |
| 206369_s_at | 7.2 9.4 |
−2 (0.25) −0.2 (0.8705) |
0.992212 0.715033 |
Phosphoinositide-3-kinase γ catalytic subunit (PI3KCG) |
| Adenosine receptors and purine-metabolizing enzymes | ||||
| 205481_at | 50.7 82.8 |
0 (1) 1 (2) |
0.493524 0.558077 |
Adenosine A1 receptor (ADORA1) |
| 205013_s_at | 47.7 101.1 |
0.3 (1.231) 0.1 (1.072) |
0.022932 0.037598 |
Adenosine A2a receptor (ADORA2A) |
| 205891_at | 91.1 215.3 |
−0.2 (0.8705) 0.8 (1.741) |
0.930187 0.023926 |
Adenosine A2b receptor (ADORA2B) |
| 206171_at | 50.2 49.7 |
0.1 (1.072) 0 (1) |
0.5 0.246094 |
Adenosine A3 receptor (ADORA3) |
| 207691_x_at | 34.1 16.7 |
0.5 (1.414) −0.4 (0.7578.) |
0.5 0.219482 |
Ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, CD39) |
| 202588_at | 27.7 22.9 |
0.9 (1.866) 0.8 (1.741) |
0.242585 0.753906 |
Adenylate kinase 1 (AK1) |
| 205851_at | 84.2 122.9 |
−0.2 (0.8705) 0.1 (1.072) |
0.838962 0.01416 |
Nucleoside diphosphate kinase |
| 204639_at | 4056.9 3819.5 |
0.2 (1.1486) 0.5 (1.414) |
0.232549 0.000244 |
Adenosine deaminase (ADA) |
| 201695_s_at | 684.9 422.9 |
0.5 (1.414) −0.3 (0.812) |
0.000552 0.000244 |
Purine nucleoside phosphorylase |
The microarray analysis reveals CD73 as the prominent candidate for further investigation.
Confirmation of the presence of CD73 protein
To verify the presence of CD73 on the surface of the multiresistant A4 line (the parent Jurkat cell line is known to be CD73 negative (23, 26, 29)), we measured the level of CD73 by flow cytometry with the 4G4 mAb specific for CD73. The analysis revealed the presence of CD73 Ag on the surface of A4 cells (Fig. 1B). The presence of CD73 in A4 cells was further established by Western blotting with polyclonal CD73 Abs. A4 cell lysates showed a single band at a molecular mass of ~70 kDa at the same position as the positive control lysates, i.e., Jurkat cells stably transfected with CD73 (Fig. 1C) (22). These results show that Jurkat cells, normally known to be CD73 negative (26), started to express full-length CD73 protein on their surface during acquisition of the multiresistant phenotype after prolonged culture with agonistic anti-Fas Abs.
Enzymatic activity of 5′-nucleotidase (CD73)
Because CD73 is an ectoenzyme, it was important not only to verify the expression at the protein level, but also to demonstrate that the enzyme is properly folded and catalytically active. Direct radio-TLC analysis further confirmed that A4 cells were able to hydrolyze [3H]AMP into [3H]adenosine (although not as efficiently as the stably CD73-transfected cell lines that have much higher expression levels of CD73 on their surface), whereas no detectable 5′-nucleotidase activity was observed in experiments with the native Jurkat T cells (Table IV). Thus, not only is CD73 expressed in the resistant A4 cells, but it is also fully functional catalytically.
Table IV.
Ecto-5′-nucleotidase activitya
| Cells | Jurkat | A4 | A4 | A4 | B-NT5.1 | B-NT5.1 | B-NT5.1 | B-NT5.1 | B-TM15.1 | B-TM15.1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Control | Control | AMPCP | PI-PLC | control | AMPCP | PI-PLC | control | AMPCP | PI-PLC |
| Ecto-5′-nucleotidase (nmol/106 cells/h) | Not detected | 2.75 ± 1.33 | 0.11 ± 0.20 | 0.43 ± 0.29 | 45.0 ± 9.35 | 0.97 ± 0.66 | 15.2 ± 6.05 | 154 ± 19.0 | 3.5 ± 2.24 | 131.2 ± 40 |
Activity was measured in parental Jurkat cells, multirésistant A4 cells, and Jurkat cell clones B-NT5.1 and B-TM15.1, stably transfected with GPI-anchored and transmembrane CD73, respectively (mean ± SD; n = 3). Some cells were preincubated for 30 min at 37°C with rPI-PLC (2.5 U/ml) from Bacillus cereus, or treated with AMPCP (1 mM).
CD73 transfectants and apoptotic resistance
To isolate the CD73 pathway from the complex mechanisms of multiple resistance in A4 cells and to determine whether CD73 per se is able to bring any apoptotic resistance, we treated Jurkat-derived cell lines stably transfected with either human (clone B-NT5.1) or murine (line NC-1) CD73 with a panel of apoptosis-inducing agents. In contrast to the results with A4 cells, the two transfectants expressing GPI-anchored CD73 showed significantly increased resistance specifically against TRAIL-induced apoptosis (Fig. 2A). The presence of the GPI-anchored wild-type CD73 also suppressed TRAIL-mediated activation of both caspases-3 and -8 (Fig. 3C, Table V), indicating that the apoptosis inhibition takes place at the level of the receptor.
FIGURE 2.
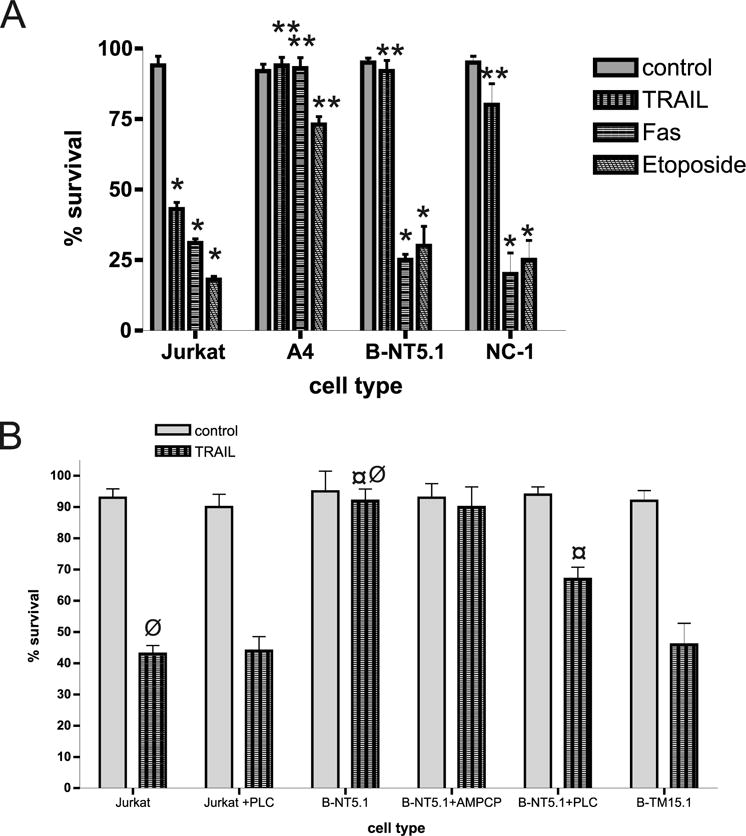
A, Jurkat cells transfected with GPI-anchored CD73 are resistant to TRAIL-induced apoptosis. Jurkat, A4, B-NT5.1 (stable transfectants with GPI-anchored human CD73), and NC-1 (stable transfectants with GPI-anchored murine CD73) cells were treated with rFasL for 4 h, 50 ng/ml TRAIL for 4 h, or 10 μM etoposide for 20 h to induce apoptosis. Cells were then stained with allophycocyanin-annexin V and Sytox Green and evaluated for apoptosis as described in Materials and Methods. Bars (mean values + SD values from five repeats, 104 cells counted) labeled with one asterisk (*) represent statistically significant difference (p < 0.01) from the untreated control; bars labeled with two asterisks (**) represent statistically significant difference (p < 0.01) from untransfected Jurkat cells having the same treatment. B, The GPI anchor, but not the enzyme activity of ecto-5′-nucleotidase is required for resistance against TRAIL-induced apoptosis. Jurkat (±pretreatment with 2.5 U/ml PI-PLC), B-NT5.1 cells (±pretreatment with 1 mM AMPCP or with 2.5 U/ml PI-PLC), and B-TM15.1 cells were exposed to 50 ng/ml TRAIL for 4 h to induce apoptosis. Cells were then stained with allophycocyanin-annexin V and Sytox Green and evaluated for apoptosis as described in Materials and Methods (bars represent mean values + SD values from five repeats, 104 cells counted; *, p < 0.01 between values marked with ¤ and ø).
FIGURE 3.
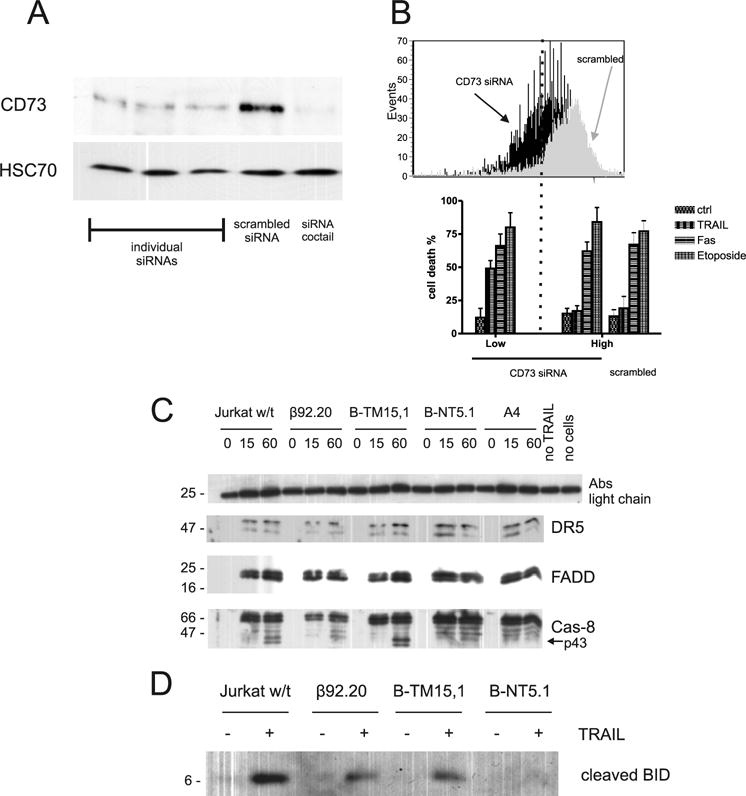
The roles of CD73 in TRAIL resistance. A, siRNAs against CD73 efficiently down-regulate its endogenous expression. HEC cells were transiently transfected with three different CD73 siRNAs, scrambled synthetic siRNA, and a mixture of 3 siRNAs (1:1:1) and CD73 expression was evaluated after 44 h by Western blotting. Western blotting was also performed with Abs to HSC70 (lower lane) to demonstrate equivalent protein loading. B, siRNAs against CD73 down-regulate its expression in B-NT5.1 cells and resensitize cells to TRAIL-induced apoptosis. B-NT5.1 cells were transiently triple transfected with a mixture of three CD73 siRNAs (1:1:1) or with scrambled siRNA. After 20 h, they were incubated with 50 ng/ml TRAIL, 50 ng/ml rFasL, for 4 h or 10 μM etoposide for 16 h and then stained with Cy2 anti-CD73 and allophycocyanin-annexin V. Cells with low and high surface expression of CD73 were analyzed separately for annexin labeling as were cells transfected with scrambled siRNA. Bars indicate mean values + SD from three repeats. C, DISC analysis reveals no caspase-8 processing in B-NT5.1 cells. Cells were exposed to 200 ng/ml FLAG-tagged rTRAIL and cross-linking M2 Abs for designated time. Then cells were spinned down for 30 s at 5,000 × g + 4C and cell pellet were lysed with Pierce M-prot lysis buffer. Lysate was clarified by centrifugation 2 min at 12,000 × g + 4C and supernatant exposed to protein G Sepharose for 15 min. Sepharose slurry washed with lysing buffer and boiled with Laemmli buffer and subjected to Western blotting. D, BID is not cleaved in B-NT5.1 cells. The cell lysates for 0 and 60 min TRAIL stimulation described above were subjected to Western blot and developed with primary mAbs against epitope of BID neighboring to caspase-8 cleavage site.
Table V.
Treatment of resistant A4 cells or CD73-transfected cells with TRAIL does not activate caspases 3 and 8a
| Cells and Treatments
|
||||||
|---|---|---|---|---|---|---|
| Jurkat | Jurkat + TRAIL | B-NT5.1 | B-NT5.1 + TRAIL | A4 | A4 + TRAIL | |
| RFU per 104 cells | ||||||
| Cas-8 | 83 ± 27 | 1160 ± 201 | 105 ± 33 | 147 ± 64 | 77 ± 30 | 93 ± 16 |
| Cas-3 | 144 ± 41 | 1303 ± 170 | 150 ± 49 | 152 ± 40 | 155 ± 34 | 178 ± 26 |
Activity of initiator (8) and effector (3) caspases does not change significantly after TRAIL treatment (50 ng/ml, 4 h) in resistant (A4) cells and cells stably transfected with CD73 (B-NT5.1) as compared to the corresponding caspase activities in TRAIL-stimulated Jurkat cells. Measurements are relative fluorescent units (RFU) done in triplicate. Mean values ± SD are shown.
To determine whether CD73 must be anchored into the membrane to confer TRAIL resistance, B-NT5.1 cells were treated with 2.5 U/ml PI-PLC. This induced efficient cleavage of CD73 (as confirmed by the effect on the enzymatic activity, Table IV) and also partially reversed the antiapoptotic effect of CD73 against TRAIL (Fig. 2B). Lower doses (1 U/ml and less, as used in Ref. 22) did not have any significant effect on the resistance of B-NT5.1 to TRAIL-induced apoptosis.
GPI-anchoring rather than -enzymatic activity of 5′-nucleotidase affects resistance to TRAIL-induced apoptosis
It has been previously demonstrated that the enzymatic activity of CD73 may not be important for the signaling of this surface molecule (23). To estimate the role of the enzymatic activity of CD73 for TRAIL resistance, we treated B-NT5.1 cells with α,β-methylene adenosine 5′-diphosphate (AMPCP; 1 mM), a specific competitive inhibitor of ecto-5′-nucleotidase activity (25, 29), followed by subsequent treatment with TRAIL (Fig. 2B). The ability of AMPCP to inhibit ecto-5′-nucleotidase activity was directly confirmed using an enzymatic assay (Table IV). Even though the enzyme activity was potently inhibited by AMPCP by >95%, the sensitivity of the CD73-transfected lymphocytes to TRAIL-induced apoptosis remained unchanged (Fig. 3A). It is important to note that even at relatively high concentrations, AMPCP itself displayed no obvious signs of cellular toxicity and did not significantly change the percentage of apoptotic cells in control experiments (data not shown).
To determine whether the GPI anchor of CD73 is needed for TRAIL resistance, a Jurkat transfectant (B-TM15.1) that expresses a form of the enzyme that is also fully catalytically active, but is anchored into the membrane with a conventional transmembrane domain, was studied (22). The level of CD73 expression is similar to that on B-NT5.1 as assessed by staining with anti-CD73 mAbs (Fig. 1B). This clone showed a sensitivity to apoptosis induced by TRAIL (Fig. 2B) and etoposide and FasL that was similar to that of parental Jurkat cells. These data suggest that the GPI anchor of CD73 is needed for its apoptosis-inhibiting activity.
siRNA-mediated down-regulation of CD73 expression restores sensitivity to TRAIL
To conclusively confirm the involvement of CD73 in TRAIL resistance, we used the siRNA technique to test the effect of CD73 down-regulation on TRAIL sensitivity in B-NT5.1 cells with high levels of CD73 and an assumed CD73-specific protection against TRAIL-induced apoptosis. To this end, we first tested the efficacy of different siRNAs on HEC cells with high levels of CD73 (Fig. 3A; Ref. 24). Individual siRNAs against CD73 were able to downregulate CD73 expression up to 80% 72 h after transfection (Fig. 3A). When mixed together, siRNA inhibition reached 92% (Fig. 3A). Transfection of B-NT5.1 cells with the mixture of siRNAs yielded a population heterogeneous in CD73 surface expression, as measured by FACS analysis (Fig. 3B). When cells with low CD73 expression were gated, their degree of apoptosis (as measured by annexin-allophycocyanin as a marker for apoptosis) after TRAIL stimulation was significantly higher than in the population gated for high CD73 expression or in B-NT5.1 cells transfected with scrambled siRNA (Fig. 3B).
CD73 interacts with DR5
The above-listed observations, including the fact that TRAIL-induced activation of caspases-3 and -8 was inhibited in the presence of CD73, implied that the action of CD73 is likely to be at the level of the TRAIL receptor, possibly through molecular interactions with DR5. In a death receptor signaling complex (DISC) assay, we showed that despite the fact that DISC complex is assembling properly in all tested lines, the processing of caspase-8 into the p43 form does not take place in B-NT5.1 and A4 cells after TRAIL stimulation (Fig. 3C). To test the idea of interaction between DR5 and CD73 further, we immunolabeled CD73 Jurkat transfectants with the CD73-specific Ab 4G4 and with an Ab specific to DR5 under resting conditions and where capping could occur. Confocal microscopy showed that in both B-NT5.1 and NC-1 cells, DR5 occurred in clusters that colocalized to a high degree with the immunofluorescence pattern of CD73 (Fig. 4A). Half an hour treatment of B-NT5.1 cells with 2.5 U/ml PI-PLC partially reversed the distribution of DR5 on the cell surface to the pattern typical for untransfected Jurkat cells (Fig. 4A). The colocalization of CD73 and DR5 on B-NT5.1 and NC-1 cells was more apparent when the length of time for capping of CD73 was increased to 3 h and a cross-linking anti-rat Ig Ab was added (Fig. 4B). In contrast, cells with transmembrane CD73 (B-TM15.1) showed little to no colocalization of CD73 and DR5. We also studied a Jurkat clone (β92.20) that expresses a mutant form of CD73 that is totally lacking in ecto-5′-nucleotidase enzyme activity due to mutation of histidine 92 to alanine (23). Somewhat to our surprise, this clone was sensitive to TRAIL-induced apoptosis (data not shown). However, when incubated with a mixture of CD73- and DR5-specific Abs and examined by confocal microscopy, CD73 and DR5 were colocalized to a lower extent (Fig. 4A), suggesting that the mutation in CD73 may have changed the conformation of CD73 such that it weakened the interaction with DR5. Areas of diffuse staining of DR5 also coexist in the same areas of cell surface as another marker of lipid rich domains, caveolin-1 (Fig. 5A), but an image with more magnification shows little actual colocalization between DR5 and caveolin-1 (Fig. 5B).
FIGURE 4.
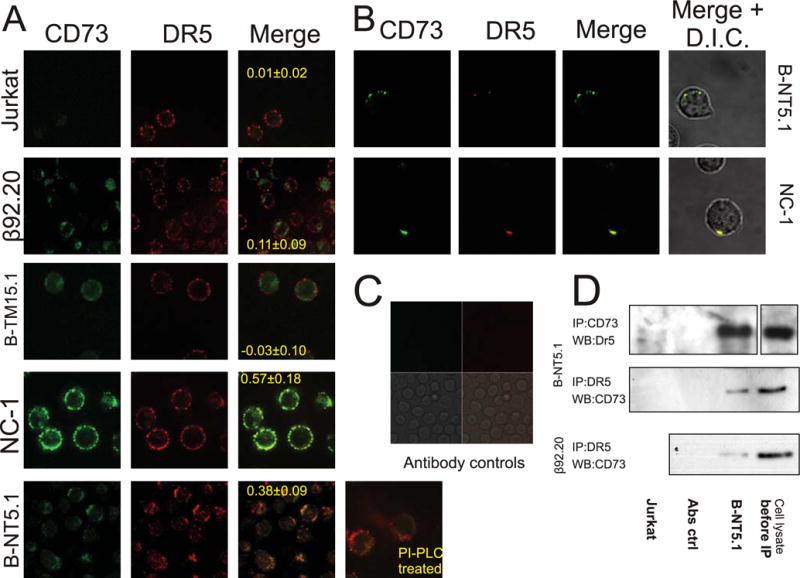
Confocal microscopy reveals colocalization of TRAIL-receptor DR5 and CD73 in TRAIL-resistant transfectants. A, Jurkat, B-NT5.1, NC-1, B-TM15.1, and β92.20 cells were labeled with Cy2-conjugated anti-CD73 (green) plus Cy3B-conjugated anti-DR5 (red) for 1 h at 20°C and processed as described in Materials and Methods. In a separate experiment, B-NT5.1 was pretreated with 2.5 U/ml PI-PLC. Separate, as well as merged images are shown, with the pixel correlation shown over the merged images (mean values ± SD from 10 ratio measurements over 10 individual cells). B, B-NT5.1 and NC-1 cells were incubated with Cy2-conjugated rat anti-mouse CD73 for 3 h at 37°C, followed by 1 h with goat anti-rat Ig to allow cross-linking and capping of CD73. The cells were then washed, fixed with 3% formaldehyde, washed again and stained with Cy3B-conjugated anti-DR5 or Cy3B-conjugated isotype control Ab (data not shown) in the presence of 10% normal goat serum. Individual images as well as merged images are shown. C, Ab controls: Jurkat cells were stained with a mixture of Cy2-conjugated CD73-specific Abs (upper left, green) and Cy3B-conjugated isotype-matched mAb against nodularin (upper right, red). Phase contrast image showing cells (lower left); merged image (lower right). D, TRAIL-receptor DR5 coimmunoprecipitates with CD73. Resting B-NT5.1, β92.20, and Jurkat cells were treated with Abs against either DR5 or CD73 or species/class matching control Abs, washed, lysed with hypotonic buffer and Ab complexes extracted with protein G Sepharose. Beads were boiled with sample Laemmli buffer and the supernatant subjected to SDS-PAGE. The figure shows Western blots developed with Abs against CD73 and DR5, respectively.
FIGURE 5.
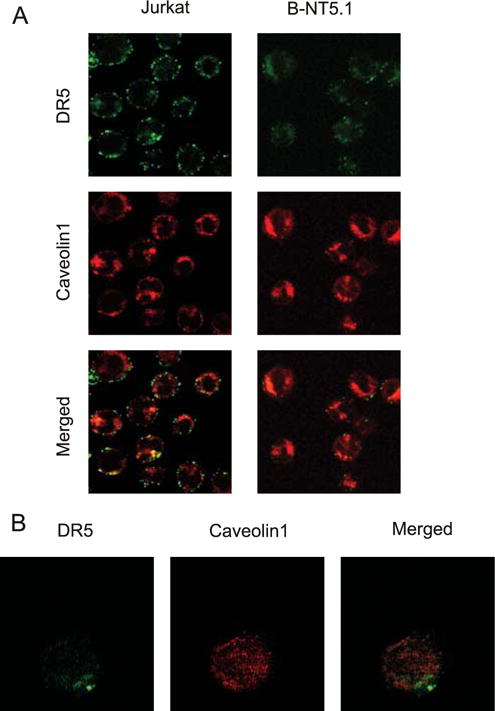
Colocalization of DR5 with caveolin 1-rich regions. Viable B-NT5.1 and Jurkat cells were stained with primary Abs against DR5 (DJR2-1) and caveolin 1 (ab2910) in suspension, then washed three times with PBS and developed with corresponding (Alexa 405 goat anti-mouse IgG and Alexa 547 goat anti-rabbit IgG) secondary Abs. Stained cells were washed three times with PBS, fixed with 4% paraformaldehyde, and subjected to confocal microscopy. A, Images were taken at objective magnification ×40 with a metadetector registering emission wavelengths 411–507 nm (laser emission 405 nm) represented as the green channel, and 550–753 nm (laser emission 543 nm) for the red channel. B, B-NT5.1 cell stained as described above at objective magnification ×100. Total Z-stack projection from the optical slice of maximal diameter to the top of the cell.
The colocalization of CD73 and DR5 in Jurkat transfectants exhibiting TRAIL resistance suggested a molecular interaction between the two proteins, either directly or through one or more additional proteins that interact with both CD73 and DR5. This conclusion was supported by immunoprecipitation experiments that demonstrated that the two wild-type proteins could be coprecipitated with each other, but not with Fas (data not shown), while mutant CD73 was pulled down at lower efficiency (6.3 times lower OD, mean from three measurements, Fig. 4C). This interaction between CD73 and DR5 provides a highly plausible molecular mechanism by which CD73 can modulate DR5 signaling. We also observed that the effect was not associated with the tyrosine kinase activity that has previously been associated with CD73 (24), as neither specific (p2) nor nonspecific (genistein) tyrosine kinase inhibitors could modulate the apoptosis-inhibiting effect of CD73 (data not shown). Therefore, the protective mechanism is likely to be a consequence of some kind of direct or indirect interaction between CD73 and DR5.
Discussion
Continuous treatment of target cells with agonistic FasR Abs mimics the real situation of a chronic immune response against a developing tumor, during which the tumor cells (through the action of the immune system) are constantly being exposed to high levels of both soluble and membrane-bound FasL and subjected to continuous FasR stimulation. As a result, cells that are able to escape the immune surveillance are selected on the basis of their Fas insensitivity, thereby acquiring resistance to FasR stimulation. Our results indicate that during this process cells may not only be desensitized to stimulation of this particular death receptor, but can, in some circumstances, also acquire a multiresistant phenotype. An analogous situation could occur when young patients with strong immune systems develop highly malignant tumors that are not only resistant to immune surveillance but also resistant to chemotherapy (30).
When we examined at the protein level a set of possible genes that could explain the insensitive phenotype, we could not identify any candidates whose elevated/decreased expression could explain the observed resistant phenotype. FasR itself was down-regulated in the resistant cell line A4, but the loss of the FasR could obviously not account for the resistance toward TRAIL receptor stimulation. In fact, some survival proteins (Bcl-2, Bcl-xL) showed decreased expression, contrary to what the default assumption would have been. Interestingly, the proteins that were observed to be down-regulated (FasR, Bcl-2, Bcl-xL) were insignificantly affected at the transcriptional level. When we tried to provide an explanation for the observed apoptosis insensitivity by gene screening, the DNA microarrays did not reveal changes in the expression of any known components of proapoptotic or prosurvival pathways (Table III). This evidence indicates that screening of the RNA expression profile for known inhibitors of apoptosis or known activators of survival pathways is not a sufficient approach to identify candidate genes for acquired apoptosis resistance in tumor cells.
Therefore, we adopted the approach of looking for individual candidate genes from the DNA microarray assay that could account for the apoptotic resistance observed in A4 cells. From the literature (15) and from our current data, it does not seem likely that a single component would be responsible for all manifestations of multiresistance. Instead, it is possible that cells have a single evolutionarily conserved “multiresistance program” on the genetic level, which can be activated by multiple stimuli, but leads to resistance against a variety of different death-threatening stimuli. Therefore, we started deciphering the molecular basis of apoptotic resistance to multiple stimuli by investigating the role of one of the most altered genes: CD73. Because A4 cells likely underwent several genetic alterations during acquisition of their multiresistant phenotype, we used Jurkat cells transfected with CD73 to dissect out the specific consequences of CD73 expression.
Elevated activity of CD73 has been found in breast carcinoma (31), gastric cancer, pancreatic cancer (32), chronic myelogenous leukemia, cutaneous T cell lymphoma, glioblastoma, and in Walker 256 carcinoma (33), indicating that its presence is likely to be beneficial for the survival of tumor cells. Further support for this assumption comes from the reported increase in CD73 expression in several multidrug-resistant cell lines (34). In these earlier studies, the effects of CD73 were dependent on its enzymatic activity and proposed to be connected with the ATP-binding cassette transporter superfamily (33). In certain resistant cell lines, it is assumed that CD73 would serve as a required accessory molecule that would, through the production of growth-sustaining nucleosides, provide an ATP-dependent purine salvage pathway (33). Although CD73 expression on tumors has obvious deleterious consequences to cancer patients, it is also important for normal homeostasis. In this respect, it is interesting to note that recent data with ecto-5′-nucleotidase/CD73-deficient mice confirmed the important beneficial role of CD73-generated adenosine in the maintenance of pulmonary integrity and lung function (35, 36), renal and cardiac protection during hypoxia (37, 38), thromboregulation, and vascular inflammatory responses (39).
In our study, when CD73 was introduced into the parental-sensitive cells, it actively protected them against apoptosis induced by stimulation of the TRAIL receptor, DR5. Because Jurkat T cells have been shown to maintain a micromolar “ATP halo” constitutively in their immediate vicinity (29), it might be anticipated that up-regulation of CD73 will activate a nucleotide-degrading pathway with concomitant generation of extracellular adenosine and activation of adenosine receptors. In contrast, the high specificity of its action is not explained by its enzymatic activity, as this is absolutely dispensable for the apoptosis-inhibiting effect. Most likely, inhibition of TRAIL-induced apoptosis in our experiments cannot be an indirect consequence of the adenosine produced by CD73, but is rather a consequence of the presence of the molecule itself. Our data with transfectants of enzyme-inactive CD73 showing very limited effects on TRAIL resistance suggest that the native conformation of CD73 is essential for its interaction with DR5, or that this interaction requires the ability of CD73 to bind AMP (or NMP). Furthermore, CD73 must have a GPI anchor and presumably be localized in lipid rafts, as a highly enzymatically active transmembrane version of CD73 did not confer any apoptotic resistance. Finally, as no elevated caspase-8 or -3 activity was registered in protected cells, the protection provided by CD73 was likely to be targeted above the initiation of caspase activity.
It has been previously shown that some mAbs raised against CD73 can serve as agonistic ligands for TCR signaling (23, 40). However, in our experiments, these Abs failed both to increase already acquired resistance against TRAIL-induced apoptosis or to promote apoptosis in all tested cell models (data not shown), which indicates that the effects of CD73 on TRAIL resistance are not linked to the costimulatory activities of CD73. Until now, there are no known natural ligands for CD73. Our results suggest that DR5 (or another protein that links DR5 to CD73) could be such a ligand and those interactions between CD73 and DR5 on the same cell alter the ability of cells to respond to TRAIL.
The above-listed observations imply that the action of CD73 is based at the level of the death receptor, perhaps by molecular interactions with DR5. This hypothesis is supported by the finding that colocalization of CD73 with DR5 correlates with resistance against TRAIL stimulation in our panel of CD73 transfectants. Our assertion that there is a molecular interaction between the proteins is supported by immunoprecipitation experiments that demonstrated that the two proteins could be coprecipitated with each other, but not with FasR (data not shown). The colocalization of CD73 and DR5, either direct or indirect (through some CD73 and DR5-interacting protein or proteins), suggests a molecular mechanism by which CD73 could modulate DR5 signaling. We have recently shown the presence of distinct lipid patches on the surface of nonstimulated Jurkat cells (29). However, the role of these microdomains in the described resistance still remains unclear. There are several reports where mobilization of DR5 into lipid rafts could change the sensitivity of the cells to TRAIL stimulation (41). The fact that incubation with TRAIL fails to activate caspase-3 or -8 in B-NT5.1 cells suggests that the interaction of CD73 with DR5 uncouples DR5 from downstream signaling components. This phenomenon could be highly physiologically relevant in the regulation of cancer cell survival, as we have shown in a separate study that clustering of death receptors can promote survival rather than apoptosis (42). The importance of survival signaling from death receptors has been established during the past few years (for review, see Ref. 8) and we have observed that tumor cells have the capacity to steer death receptor signaling into survival-promoting pathways, such as that mediated by ERK (6). Therefore, it is an attractive idea that CD73 can reorganize DR5 into a survival-promoting mode. Testing this hypothesis will be a goal for future studies.
In summary, the multiresistant phenotype is likely to be the consequence of alterations in multiple individual genes, each with high specificity toward individual stimuli. Our results suggest that CD73 is one component of a complex nonspecific antiapoptotic program, which was in our case up-regulated during selection for Fas resistance. Within this program, CD73 has its own specific responsibility and is clearly not a solitary source for the acquired multiresistance. Our results strongly suggest that CD73 specifically inhibits TRAIL-mediated signaling upstream of caspases by molecular interaction with DR5 (Fig. 6). The molecular details and physiological implications of this mechanism are currently under investigation.
FIGURE 6.
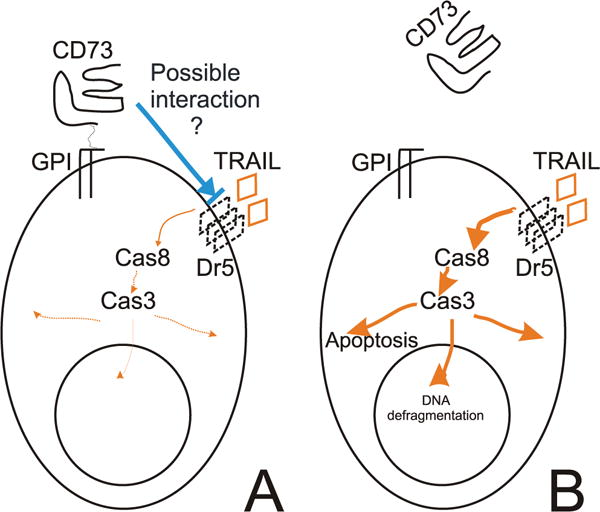
Model for the role of CD73 in TRAIL resistance. A, Ectopic or acquired expression of CD73 linked via GPI to the surface of cells protects them against TRAIL-induced apoptosis by interaction with molecules such as DR5. B, Although the protection is independent of ecto-5′-nucleotidase enzymatic activity, it is dependent on the GPI-anchoring of CD73 to the cell membrane, as cleaved CD73 loses its protective property.
Acknowledgments
We thank Catherine Angénieux, Josef Spychala, Peter Krammer, and David Brautigan for the Abs they kindly provided. We also thank Helena Saarento, Miina Miller, and the Finnish DNA Microarray Centre (Turku Centre for Biotechnology, Turku, Finland) for great technical help. We are grateful to the members of our laboratory for critical comments on the manuscript and technical help during the course of this study. An explanation of author contributions follows: A. Mikhailov designed the research, performed the experiments, and wrote the manuscript; A. Sokolovskaya performed the experiments; G. G. Yegutkin performed the experiments and wrote sections in the manuscript; H. Amdahl performed the experiments; A. West analyzed data; H. Yagita contributed vital new reagents and know how; R. Lahesmaa analyzed data; L. F. Thompson performed the experiments and edited the manuscript; S. Jalkanen edited the manuscript; D. Blokhin designed the experiments; and J. E. Eriksson edited the manuscript and supervised the consortium.
Footnotes
This work was supported by the Academy of Finland and the National Technology Agency, and National Institutes of Health Grant AI18220 (to L.F.T.). L.F.T. holds the Putnam City Schools Distinguished Chair in Cancer Research.
Abbreviations used in this paper: TLC, thin-layer chromatography; PI-PLC, phosphatidylinositol-specific phospholipase C; FADD, Fas-associated death domain; HSP, heat shock protein; HSC, heat shock cognate; AMPCP, α,β-methylene adenosine 5′-diphosphate; DISC, death receptor signaling complex; siRNA, short interfering RNA; BID, BH3-interacting domain death agonist.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Ng C, Bonavida B. A new challenge for successful immunotherapy by tumors that are resistant to apoptosis: two complementary signals to overcome cross-resistance. Adv Cancer Res. 2002;85:145–174. doi: 10.1016/s0065-230x(02)85005-9. [DOI] [PubMed] [Google Scholar]
- 2.Piche A, Rancourt C. Gene therapy to overcome drug resistance in cancer: targeting key regulators of the apoptotic pathway. Curr Gene Ther. 2001;1:317–324. doi: 10.2174/1566523013348382. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson K, Anderson N. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 4.Stambolic V, Mak T, Woodgett J. Modulation of cellular apoptotic potential: contributions to oncogenesis. Oncogene. 1999;18:6094–6103. doi: 10.1038/sj.onc.1203126. [DOI] [PubMed] [Google Scholar]
- 5.Leong K, Karsan A. Signaling pathways mediated by tumor necrosis factor α. Histol Histopathol. 2000;15:1303–1325. doi: 10.14670/HH-15.1303. [DOI] [PubMed] [Google Scholar]
- 6.Holmstrom TH, Tran SE, Johnson VL, Ahn NG, Chow SC, Eriksson JE. Inhibition of mitogen-activated kinase signaling sensitizes HeLa cells to Fas receptor-mediated apoptosis. Mol Cell Biol. 1999;19:5991–6002. doi: 10.1128/mcb.19.9.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran SEF, Holmstrom TH, Ahonen M, Kahari VM, Eriksson JE. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J Biol Chem. 2001;216:16484–16490. doi: 10.1074/jbc.M010384200. [DOI] [PubMed] [Google Scholar]
- 8.Tran SE, Meinander A, Eriksson JE. Instant decisions: transcription-independent control of death-receptor-mediated apoptosis. Trends Biochem Sci. 2004;29:601–608. doi: 10.1016/j.tibs.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Weller M. Predicting response to cancer chemotherapy: the role of p53. Cell Tissue Res. 1998;292:435–445. doi: 10.1007/s004410051072. [DOI] [PubMed] [Google Scholar]
- 10.Ross D. Novel mechanisms of drug resistance in leukemia. Leukemia. 2000;14:467–473. doi: 10.1038/sj.leu.2401694. [DOI] [PubMed] [Google Scholar]
- 11.Hersey P, Zhang X. Overcoming resistance of cancer cells to apoptosis. J Cell Physiol. 2003;196:9–18. doi: 10.1002/jcp.10256. [DOI] [PubMed] [Google Scholar]
- 12.Igney F, Krammer P. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 13.Wang GQ, Gastman BR, Wieckowski E, Goldstein LA, Rabinovitz A, Yin XM, Rabinowich H. Apoptosis-resistant mitochondria in T cells selected for resistance to Fas signaling. J Biol Chem. 2001;276:3610–3619. doi: 10.1074/jbc.M006222200. [DOI] [PubMed] [Google Scholar]
- 14.Vigorito E, Plaza S, Mir L, Mongay L, Vinas O, Serra-Pages C, Vives J. Contributions of p53 and PMA to γ-irradiation induced apoptosis in Jurkat cells. Hematol Cell Ther. 1999;41:153–161. doi: 10.1007/s00282-999-0153-0. [DOI] [PubMed] [Google Scholar]
- 15.Boesen-de Cock JGR, Tepper AD, de Vries E, van Blitterswijk WJ, Borst J. Common regulation of apoptosis signaling induced by CD95 and the DNA-damaging stimuli etoposide and γ-radiation downstream from caspase-8 activation. J Biol Chem. 1999;274:14255–14261. doi: 10.1074/jbc.274.20.14255. [DOI] [PubMed] [Google Scholar]
- 16.Sokolovskaya AA, Zabotina TN, Blokhin DY, Inshakov AN, Mikhailov AD, Kadagidze ZG, Baryshnikov AY. CD95-deficient cells Jurkat/A4 subline are resistant to drug-induced apoptosis. Exp Oncol. 2001;23:174–180. [Google Scholar]
- 17.Friesen C, Herr I, Krammer P, Debatin K. Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med. 1996;2:574–577. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- 18.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Lorenzo M, Gamen S, Etxeberria J, Lasierra P, Larrad L, Pineiro A, Anel A, Naval J, Alava M. Resistance to apoptosis correlates with a highly proliferative phenotype and loss of Fas and CPP32 (caspase-3) expression in human leukemia cells. Int J Cancer. 1998;75:473–481. doi: 10.1002/(sici)1097-0215(19980130)75:3<473::aid-ijc23>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 20.Lee H, Lin EC, Liu L, Smith JW. Gene expression profiling of tumor xenografts: in vivo analysis of organ-specific metastasis. Int J Cancer. 2003;107:528–534. doi: 10.1002/ijc.11428. [DOI] [PubMed] [Google Scholar]
- 21.Ludwig HC, Rausch S, Schallock K, Markakis E. Expression of CD73 (ecto-5′-nucleotidase) in 165 glioblastomas by immunohistochemistry and electronmicroscopic histochemistry. Anticancer Res. 1999;19:1747–1752. [PubMed] [Google Scholar]
- 22.Resta R, Hooker S, Laurent A, Shuck J, Misumi Y, Ikehara Y, Koretzky G, Thompson L. Glycosyl phosphatidylinositol membrane anchor is not required for T cell activation through CD73. J Immunol. 1994;153:1046–1053. [PubMed] [Google Scholar]
- 23.Gutensohn W, Resta R, Misumi Y, Ikehara Y, Thompson L. Ecto-5′-nucleotidase activity is not required for T cell activation through CD73. Cell Immunol. 1995;161:213–217. doi: 10.1006/cimm.1995.1029. [DOI] [PubMed] [Google Scholar]
- 24.Airas L, Niemela J, Salmi M, Puurunen T, Smith DJ, Jalkanen S. Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J Cell Biol. 1997;136:421–431. doi: 10.1083/jcb.136.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yegutkin GG, Henttinen T, Jalkanen S. Extracellular ATP formation on vascular endothelial cells is mediated by ecto-nucleotide kinase activities via phosphotransfer reactions. FASEB J. 2001;15:251–260. doi: 10.1096/fj.00-0268com. [DOI] [PubMed] [Google Scholar]
- 26.Yegutkin G, Henttinen T, Samburski S, Spychala J, Jalkanen S. The evidence for two opposite, ATP-generating and ATP-consuming, extracellular pathways on endothelial and lymphoid cells. Biochem J. 2002;367:121–128. doi: 10.1042/BJ20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita Y, Hooker SW, Jiang H, Laurent AB, Resta R, Khare K, Coe A, Kincade PW, Thompson LF. CD73 expression and fyn-dependent signaling on murine lymphocytes. Eur J Immunol. 1998;28:2981–2990. doi: 10.1002/(SICI)1521-4141(199810)28:10<2981::AID-IMMU2981>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 28.Mikhailov A, Harmala-Brasken AS, Polosukhina E, Hanski A, Wahlsten M, Sivonen K, Eriksson JE. Production and specificity of monoclonal antibodies against nodularin conjugated through N-methyldehydrobutyrine. Toxicon. 2001;39:1453–1459. doi: 10.1016/s0041-0101(01)00104-0. [DOI] [PubMed] [Google Scholar]
- 29.Yegutkin GG, Mikhailov A, Samburski SS, Jalkanen S. The detection of micromolar pericellular ATP pool on lymphocyte surface by using lymphoid ecto-adenylate kinase as intrinsic ATP sensor. Mol Biol Cell. 2006;17:3378–3385. doi: 10.1091/mbc.E05-10-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knutson K, Schiffman K, Rinn K, Disis M. Immunotherapeutic approaches for the treatment of breast cancer. J Mammary Gland Biol Neoplasia. 1999;4:353–365. doi: 10.1023/a:1018714300217. [DOI] [PubMed] [Google Scholar]
- 31.Kruger K, Thompson L, Kaufmann M, Moller P. Expression of ecto-5′-nucleotidase (CD73) in normal mammary gland and in breast carcinoma. Br J Cancer. 1991;63:114–118. doi: 10.1038/bjc.1991.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ujhazy P, Berleth E, Pietkiewicz J, Kitano H, Skaar J, Ehrke M, Mihich E. Evidence for the involvement of ecto-5′-nucleotidase (CD73) in drug resistance. Int J Cancer. 1996;68:493–500. doi: 10.1002/(SICI)1097-0215(19961115)68:4<493::AID-IJC15>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 33.Ujhazy P, Klobusicka M, Babusikova O, Strausbauch P, Mihich E, Ehrke M. Ecto-5′-nucleotidase (CD73) in multidrug-resistant cell lines generated by doxorubicin. Int J Cancer. 1994;59:83–93. doi: 10.1002/ijc.2910590117. [DOI] [PubMed] [Google Scholar]
- 34.Garlanda C, Dejana E. Heterogeneity of endothelial cells: specific markers. Arterioscler Thromb Vasc Biol. 1997;17:1193–1202. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- 35.Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, Rosenberger P, Eltzschig HK. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–8137. doi: 10.4049/jimmunol.178.12.8127. [DOI] [PubMed] [Google Scholar]
- 36.Kiss J, Yegutkin GG, Koskinen K, Savunen T, Jalkanen S, Salmi M. IFN-β protects from vascular leakage via up-regulation of CD73. Eur J Immunol. 2007;37:3334–3338. doi: 10.1002/eji.200737793. [DOI] [PubMed] [Google Scholar]
- 37.Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann S, Deng C, Briggs J, Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′-nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 39.Koszalka P, Ozuyaman B, Huo Y, Zernecke A, Flogel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sevigny J, et al. Targeted disruption of CD73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. 2004;95:814–821. doi: 10.1161/01.RES.0000144796.82787.6f. [DOI] [PubMed] [Google Scholar]
- 40.Massaia M, Perrin L, Bianchi A, Ruedi J, Attisano C, Altieri D, Rijkers GT, Thompson LF. Human T cell activation: synergy between CD73 (ecto-5′-nucleotidase) and signals delivered through CD3 and CD2 molecules. J Immunol. 1990;145:1664–1674. [PubMed] [Google Scholar]
- 41.Gajate C, Mollinedo F. Cytoskeleton-mediated death receptor and ligand concentration in lipid rafts forms apoptosis-promoting clusters in cancer chemotherapy. J Biol Chem. 2005;280:11641–11647. doi: 10.1074/jbc.M411781200. [DOI] [PubMed] [Google Scholar]
- 42.Soderstrom TS, Nyberg SD, Eriksson JE. CD95 capping is ROCK-dependent and dispensable for apoptosis. J Cell Sci. 2005;118:2211–2223. doi: 10.1242/jcs.02343. [DOI] [PubMed] [Google Scholar]