

Abstract
In previous structure–activity relationship (SAR) studies, (3R)-7-hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl]-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic, 3) was identified as the first potent and selective κ-opioid receptor antagonist from the trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine class of opioid antagonists. In the present study, we report the synthesis of analogues 8a–p of 3 and present their in vitro opioid receptor functional antagonism using a [35S]GTPγS binding assay. Compounds 8a–p are analogues of 3 containing one, two, or three methyl groups connected to the JDTic structure at five different positions. All the analogues with one and two added methyl groups with the exception of 8k had subnanomolar Ke values at the κ receptor. The three most potent analogues were the monomethylated (3R)-7-hydroxy-N-[(1S,2S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidine-1-yl]methyl}-2-methylbutyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8a) and (3R)-7-hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-(2-methylpropyl)]-3-methyl-1,2,3, 4-tetrahydroisoquinoline-3-carboxamide (8e) with Ke values of 0.03 nM at the κ receptor and (3R)-7-hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylpropyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8d) with Ke=0.037 nM at the κ receptor. All three compounds were selective for the κ receptor relative to the μ and δ receptors. Overall, the results from this study highlight those areas that are tolerant to substitution on 3.
Introduction
Stress can induce despair and increase the risk of clinical depression and drug abuse.1,2 Dynorphin, the endogenous ligand for the κ-opioid receptor, is a stress-related neuropeptide in the brain that may mediate these responses.3 Activation of the κ-opioid receptor causes place aversion in rodents and dysphoria in humans.4,5 The dynorphin/κ-opioid receptor system has been reported to be critical for stress-induced depression-like behaviors and reinstatement to drug-seeking behavior.4,6–10 The results from these studies have led to an increased interest in selective κ-opioid receptor antagonists.
The first nonpeptide, highly selective antagonists of the κ-opioid receptor were nor-BNI11 (1, Figure 1) andGNTI12 (2, Figure 1), which were derived from the nonselective opioid receptor antagonist naltrexone. More recently, JDTic (3, Figure 1) was discovered as the first highly potent and selective κ-opioid receptor antagonist from the N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine (4, Figure 1) class of antagonist,13,14 and arodyn (5, Figure 1) was developed from dynorphin.15 Studies with these compounds have shown that this system is intimately involved in brain processes that relate to stress, fear, and anxiety as well as reward-seeking behavior.16 Studies have shown that 3 and 1 dose-dependently reduce fear and stress-induced responses in multiple behavioral paradigms with rodents (immobility in the forced-swim assay,8,10 reduction of exploratory behavior in the elevated plusmaze, fear-potentiated startle).17 Furthermore, selective κ antagonists have been shown to reduce stress-induced reinstatement of cocaine self-administration in rats,8 block the stress-induced potentiation of cocaine place preference conditioning,7,9,18 decrease dependence-induced ethanol self-administration,19 diminish deprivation-induced eating in rats,20 and prevent prepulse inhibition mediated by the κ agonist U50,488.21 These observations regarding the behavioral consequences of receptor blockade in several animal tests suggest that κ antagonists might be useful for the treatment of anxiety, depression, schizophrenia, addiction, and eating disorders.
Figure 1.

In vivo, 3 has been shown to be more potent at blocking κ-opioid agonist-induced activity than other κ-opioid antagonist. 22 Compound 3 was also shown to have oral activity in antagonizing the antinociceptive activity of the κ agonist enadoline in mice22 and preventing stress-induced cocaine reinstatement of self-administration in rats.8 To our knowledge 3 remains the only orally active κ-opioid receptor antagonist.
In a recent structure–activity relationship study, it was reported that 8a, which has an extra methyl group on the (1S)-isopropyl group of 3, had a Ke value of 0.03 nM at the κ-opioid receptor, relative to 0.02 nMfor 3, and retained 100- and 800-fold κ selectivity relative to the μ and δ opioid receptors, respectively.23 It was also reported that the methyl ether 8b was a highly potent antagonist with a Ke value of 0.06 nM at the κ-opioid receptor, making it only 3-fold less potent than 3. Compound 8b was 857- and 1970-fold selective for the κ receptor relative to the μ and δ receptors, respectively. The synthesis of the N-methyl analogue 8c has also been reported; however, this analogue had not been evaluated for inhibition of agonist-stimulated [35S]GTPγSa binding at cloned μ-, δ-, and κ-opioid receptors in our laboratory.14 In this study, we report the synthesis of a series of methylated analogues of 3 (8d–p, see Table 1 for structures) and report results on their ability to inhibit agonist-stimulated [35S]GTPγS binding in cells expressing cloned μ-, δ-, and κ-opioid receptors. Even though 3 has drug-like properties and has performed well in several animal behavioral tests,8,17,22 we reasoned that analogues 8a–d could have better pharmacokinetic properties and ability to penetrate the brain. All 16 analogues (8a–p) had calculated logBB values24 that suggested they would possess better brain penetration than 3. All the mono- and dimethylated 3 analogues with the exception of 8k had subnanomolar Ke values at the κ-opioid receptor. The most potent new analogue was 8e with a Ke value of 0.03 nM for the κ-opioid receptor and 120- and 28000-fold selective relative to the μ- and δ-opioid receptors. However, analogues 8d and previously reported 8a and 8b were also potent and selective κ antagonists.
Table 1.
Comparison of Inhibition of Agonist Stimulated [35S]GTPγS Binding in Cloned Human μ, δ, and κ-opioid Receptors for Compounds 8a–d to 3 and 1a
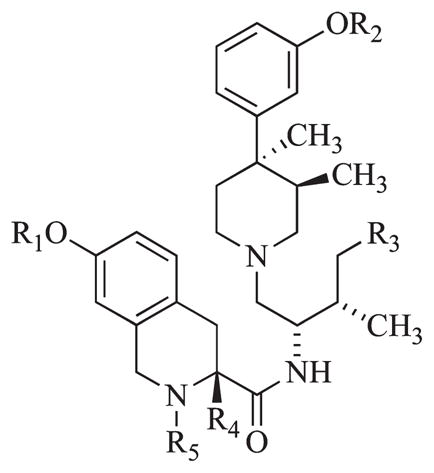
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| compd | R1 | R2 | R3 | R4 | R5 | μ, DAMGO Ke (nM) | δ, DPDPE Ke (nM) | κ, U69,593 Ke (nM) | μ/κ | δ/κ |
| 1 | 26±7 | 29±8 | 0.05±0.02 | 520 | 580 | |||||
| 3 | H | H | H | H | H | 25.1±3.5b | 76.4±2.7b | 0.02±0.01b | 1255 | 3830 |
| 8a | H | H | CH3 | H | H | 3±1c | 24±4c | 0.03±0.02c | 100 | 800 |
| 8b | CH3 | H | H | H | H | 51.4±15c | 118±45c | 0.06±0.01c | 857 | 1970 |
| 8c | H | H | H | H | CH3 | 210±60 | 491±120 | 0.16±0.06 | 1313 | 3070 |
| 8d | H | CH3 | H | H | H | 24±8 | 21.2±5 | 0.037±0.003 | 649 | 573 |
| 8e | H | H | H | CH3 | H | 3.6±1 | 854±210 | 0.03±0.008 | 120 | 28500 |
| 8f | H | CH3 | CH3 | H | H | 5.1±2 | 1170±400 | 0.96±0.4 | 5 | 1220 |
| 8g | CH3 | H | H | CH3 | H | 3.8±1.1 | 36.8±6.9 | 0.93±0.05 | 4 | 40 |
| 8h | H | CH3 | H | CH3 | H | 123±30 | 2200±900 | 0.26±0.1 | 473 | 8500 |
| 8i | H | H | CH3 | CH3 | H | 8.7±2.7 | 149±13 | 0.11±0.01 | 79 | 1350 |
| 8j | H | H | CH3 | H | CH3 | 7.2±1.8 | 132±24 | 0.11±0.03 | 66 | 1200 |
| 8k | H | CH3 | H | H | CH3 | 880±220 | 2300±900 | 4.3±2.7 | 204 | 535 |
| 8l | CH3 | CH3 | H | CH3 | H | 1450±490 | IAd | 15.2±3.7 | 95 | |
| 8m | CH3 | H | CH3 | CH3 | H | 17.5±3.6 | 18.7±1.5 | 3.5±0.8 | 5 | 5 |
| 8n | H | CH3 | CH3 | CH3 | H | 59.1±16 | 2100±600 | 0.52±0.2 | 114 | 4040 |
| 8o | CH3 | H | H | CH3 | CH3 | 7.0±1.4 | 117±30 | 2.2±0.6 | 3 | 53 |
| 8p | H | CH3 | CH3 | H | CH3 | 360±120 | IAd | 2.03±0.03 | 180 | |
Chemistry
The structure of 3 was modified to introduce methyl groups at five different sites of the molecule (see Table 1 for structure): at the phenolmoieties (R1, R2), on the linker of the phenylpiperidine to the tetrahydroisoquinoline carboxamide fragments (R3), at the position α to the carboxamide moiety (R4), and at the isoquinoline nitrogen (R5). Analogues 8a–c were synthesized as previously reported.14,23 The synthesis of the new analogues 8d–p is shown in Scheme 1. Coupling of the appropriate 1,2,3,4-tetrahydroisoquinoline carboxylic acids 6a–e with 7a–d using benzotriazole-1-yloxy-tris-(dimethylamino)phosphonium hexafluorophosphate (BOP) in tetrahydrofuran (THF) or O-(benzotriazol-1-yl)-N, N, N′, N′-tetramethyluronium hexafluorophosphate (HBTU) in acetonitrile (followed by removal of the Boc-protecting group with trifluoroacetic acid in methylene chloride when 6a and 6c were used) yielded 8d–p.
Scheme 1a.

a Reagents: (a) BOP, THF, Et3N (for 8d, 8f–g, 8i–l, 8n–p), or HBTU, CH3CN, Et3N, (for 8e, 8h, 8m) for coupling with 6a and 6c; (b) CF3CO2H, CH2Cl2; (c) Raney Ni, H2, HCHO, CH3OH.
The tetrahydroisoquinoline carboxylic acids 6a–d needed for the synthesis of 8e, 8g, 8h, 8i, 8l, 8m, 8n, and 8o were prepared following the transformations outlined in Scheme 2. D-Alanine (9) was converted to the sodium salt using sodium hydroxide in ethanol, followed by conversion to the chiral oxazolidinone 10 by condensation with benzaldehyde under azeotropic distillation conditions and benzoylation using benzoyl chloride.25 Alkylation of 10 with 4-methoxybenzyl bromide using lithium hexamethyldisilazide as the base at −78 °C proceeded with high diastereomeric selectivity to give the p-methoxybenzylated intermediate 11.26 Acid hydrolysis of the chiral intermediate 11 gave the amino acid 12. Formation of the tetrahydroisoquinoline ring system was achieved via the Pictet–Spengler reaction. This was carried out by bromination of 12 to give 13 to protect the ortho positions of the methoxy group, followed by treatment with hydrobromic acid and formaldehyde at 80 °C to give 14. Compound 14 was converted to 6a by treatment with concentrated hydrobromic acid to demethylate the 7-methoxy to a phenol, followed by catalytic debromination using palladium on carbon under hydrogen, and finally treatment with di-tert-butyl dicarbonate in dimethylformamide containing triethylamine to give 6a. The N-methyl analogue 6b was obtained by treating 6a with trifluoroacetic acid to give the free amine followed by reductive methylation using Raney nickel catalyst, hydrogen, and formaldehyde in methanol. Compounds 6c and 6d were obtained from 14 by protection as the tert-butoxycarbonyl ester using di-tert-butyl dicarbonate and then debromination using palladium on carbon as catalyst under hydrogen to give 6c. Removal of the Boc-protecting group from6c using hydrochloric acid followed by reductive methylation using the same conditions as for 6b gave the N-methyl analogue 6d.
Scheme 2a.

a Reagents: (a) NaOH, C6H5CHO; (b) C6H5COCl; (c) LiHMDS, THF, −78 °C; CH3OC6H4CH2Br; (d) conc HCl; (e) Br2, HCl; (f) HCHO, HBr, H2O, CF3CO2H; (g) conc HBr, reflux; (h) H2, Pd/C, CH3OH; (i) (Boc)2O, DMF, Et3N; (j) CF3CO2H, CH2Cl2; (k) Raney Ni, H2, CH3OH, HCHO; (l) 12 M HCl, THF.
Compound 7b was synthesized by coupling N-Boc-L-valine with (3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidine (16a)27 using BOP in tetrahydrofuran followed by reduction with diborane in tetrahydrofuran (Scheme 3). Coupling of 16b and 16a with N-Boc-L-isoleucine using HBTU in acetonitrile followed by reduction with diborane gave 7c and 7d, respectively. Compound 7a was synthesized as previously reported.28
Scheme 3a.

a Reagents: (a) N-Boc-L-valine, BOP, THF; (b) N-Boc-L-isoleucine, HBTU, CH3CN, Et3N, THF; (c) B2H6, THF.
Pharmacology
Compounds 1, 3, and 8a–p were first evaluated at 10 μM for intrinsic activity in the [35S]GTPγS binding assay at all three opioid receptors. As none of the compounds displayed measurable intrinsic activity at this concentration, they and the reference compound 1 were evaluated for functional antagonism and selectivity at the opioid receptors. These data were obtained by monitoring the ability of test compounds to inhibit stimulated [35S]GTPγS binding produced by the selective agonists DAMGO (μ), DPDPE (δ), or U69,593 (κ) using cloned human opioid receptors expressed in CHO cells.29 Agonist dose response curves were run in the presence or absence of a single concentration of test compound. Test compound assay concentrations ranged from 1–5000 nM, depending on their activity. The Ke values were calculated using the formula: Ke=[L]/DR − 1, where [L] is the concentration of test compound and DR is the ratio of agonist EC50 value in the presence or absence of test compound, respectively. At least two different concentrations of test compound were used to calculate the Ke, and the concentrations were chosen such that the agonist EC50 exhibited at least a 4-fold shift to the right and there was a clear upper asymptote to the agonist + compound concentration response curve. The Ke values along with those for the reference compound 1 are shown in Table 1.
The calculated logP, tPSA, and logBB values for compounds 1, 3, and 8a–p are given in Table 2. The logBB values were calculated using eq 6 (the Clark equation) given in ref 24. Topological polar surface areas (tPSA) and logP values were calculated using ChemAxon’s Instant JChem version 5.03 software.
Table 2.
Calculated logP, tPSA, and logBB for 1, 3, and 8a–pa
| compd | logP | tPSA | logBB |
|---|---|---|---|
| 1 | 1.57 | 121.65 | −1.42 |
| 3 | 3.75 | 84.83 | −0.55 |
| 8a | 4.09 | 84.83 | −0.49 |
| 8b | 4.11 | 73.83 | −0.33 |
| 8c | 4.12 | 76.04 | −0.36 |
| 8d | 3.83 | 73.83 | −0.37 |
| 8e | 3.98 | 84.83 | −0.51 |
| 8f | 4.18 | 73.83 | −0.32 |
| 8g | 4.33 | 73.83 | −0.30 |
| 8h | 4.06 | 73.83 | −0.34 |
| 8i | 4.32 | 84.83 | −0.46 |
| 8j | 4.46 | 76.04 | −0.31 |
| 8k | 4.20 | 65.04 | −0.19 |
| 8l | 4.74 | 62.83 | −0.07 |
| 8m | 4.71 | 73.83 | −0.24 |
| 8n | 4.41 | 73.83 | −0.28 |
| 8o | 4.69 | 65.04 | −0.11 |
| 8p | 4.55 | 65.04 | −0.13 |
logBB was calculated using eq 6 in ref 24.
Results and Discussion
Even though 3 (Ke = 0.02 nm) was more potent as a κ-opioid receptor antagonist than any of the methylated analogues studied, many of the analogues were potent and selective κ antagonists. All of the monomethylated analogues 8a–8e, the dimethylated analogues 8f–8j, and the trimethylated analogue 8m retained subnanomolar potency at the κ-opioid receptor. All of the monomethylated compounds 8a–8e, the dimethylated compounds 8h and 8k, and the trimethylated compound 8n retained greater than 100-fold κ selectivity relative to the μ and δ receptors. The two most potent analogues were 8a (R3=CH3) and 8e (R4=CH3), both with Ke values of 0.03 nM at the κ-opioid receptor. Both compounds had 100-fold or greater selectivity for the κ receptor relative to the μ receptor. The κ selectivity for 8a and 8e relative to the δ receptor was 800 and 28500, respectively. The N-methyl compound 8c (R5=CH3) with a Ke value of 0.16 nM at the κ-opioid receptor and 1313- and 3070-fold selectivity for the κ receptor relative to the μ and δ receptors was the most κ selective analogue of this new series. Compound 8b (R2=CH3), with a Ke value of 0.06 nM, was 3 times less potent than 3, and with μ/κ and μ/δ ratios of 857 and 1970, it was also highly κ selective.
Compound 8d, with a methyl substituted at the 3-hydroxyl in the phenylpiperidine fragment, had only a 2-fold decrease in potency for the κ receptor (Ke = 0.037 nM) relative to 3.
Methylation of the alkyl side chain on the linker between the phenylpiperidine and tetrahydroisoquinoline carboxamide fragments (R3) produced compounds 8a, 8f, 8i, 8j, and 8m that had increased potency at μ receptors compared to 3. This effect was most notable in the monomethyl substituted compound 8a, which had an 8-fold increase in potency at μ receptors compared to 3. These observations mirror those seen in previous studies where large substituents at this position increased μ receptor potency.28
N-Methylation at the tetrahydroisoquinoline nitrogen to give the N-methyl 3 analogue 8c resulted in a reduction in potency at all receptor subtypes. At κ receptors, this modification consistently gave decreases in potency for all analogues 8j, 8k, 8o, and 8p. Nevertheless, analogues 8c and 8j, with Ke values of 0.16 and 0.11 nM, respectively, were still highly potent κ antagonists.
In general, it was observed that introduction of multiple methyl groups into the structure of 3 was detrimental for potency and selectivity at κ receptors. Compounds 8i and 8j with Ke values of 0.11 nM each at the κ receptor were the two most potent analogues with multiple methyl groups. The effect was more noticeable for compounds with three methyl substitutions. The most potent analogue containing three methyl groups was 8n, which had a Ke value of 0.52 nM at the κ receptor.
The calculated logP, tPSA, and logBB values for 1, 3, and 8a–p are given in Table 2.24 In contrast to standard compound 1 (calculated logBB=−1.42), the calculated logBB for 3 and 8a–p (−0.07 to −0.55) are above the threshold proposed by Clark to indicate low blood–brain barrier penetration. The calculated logBB values24 show that all 16 methylated analogues would be expected to show enhanced brain penetration relative to 3. In the case of 8b for instance, monomethylation shifts the calculated logBB value positively by 0.22 log units (calculated logBBs for 3 and 8b are −0.55 and −0.33, respectively). This change in the relative concentration of drugs implies an approximately 66% increase in the concentration of the drug in the brain.
Conclusions
In summary, 16 analogues of 3 with methyl substituents at five different positions on the 3 structure were synthesized. Eleven of the analogues had subnanomolar Ke values at the κ opioid receptor. The monomethylated analogues 8a, 8b, 8d, and 8e with Ke values of 0.03–0.06 nM were the most potent compounds. Even though the efficacy at the κ opioid receptor is not as good as that for 3, the calculated logBB values suggest that these analogues may have activity comparable to that of 3 in vivo. While beyond the scope of this investigation, a pharmacokinetic (PK) study could suggest further development of one or more of these analogues.
Experimental Section
1H NMR spectra were determined on a Bruker 300 spectrometer using tetramethylsilane as an internal standard. Mass spectral data were obtained using a Finnegan LCQ electrospray mass spectrometer in positive ion mode at atmospheric pressure. Medium-pressure flash column chromatography was done on a CombiFlash Companion system using Teledyne Isco prepacked silica gel columns or using EM Science silica gel 60 Å (230–400 mesh). All reactions were followed by thin-layer chromatography using Whatman silica gel 60 TLC plates and were visualized by UV. Optical rotations were measured on an Auto Pol III polarimeter. All solvents were reagent grade. HCl in dry diethyl ether was purchased from Aldrich Chemical Co. and used while fresh before discoloration. CMA-80 is a mixture of 80% chloroform, 18% methanol, and 2% concentrated ammonium hydroxide. Purity of compounds (>95%) was established by elemental analysis. Elemental analyses were performed by Atlantic Microlab, Inc., Atlanta, GA. Care should be used when using BOP in coupling reactions as it yields the carcinogenic byproduct HMPA.
(2S,4R)-4-(4-Methoxybenzyl)-4-methyl-2-phenyl-3-(phenylcarbonyl)-1,3-oxazolidin-5-one (11)
Compound 1025 (6.35 g, 0.023 mol) in 50 mL of THF at −78 °C was added over 20 min to a solution of LiHMDS in THF(25 mL of 1 M solution in THF). After 10 min, 1.1 equiv of 4-methoxybenzyl bromide (25 mmol, 5 mL) was added in one portion. The mixture was stirred at −78 °C for 3 h and then at room temperature overnight. Saturated NH4Cl solution was added, the THF was removed in vacuo, Et2O (100 mL) was added, and the phases were separated. The organic layer was washed with 50 mL of NaHCO3 solution and brine. After drying (Na2SO4), filtration, and removal of the solvent, the residue was purified by chromatography using a silica gel Isco column with 9% EtOAc in hexanes as eluent. Concentration of the product fractions gave 7.4 g (82%) of 11 as a white solid: mp 128–129 °C; [α]25D=−260 (c 0.8, MeOH). 1H NMR (CDCl3) δ 7.27 (2H, d, J=8 Hz), 7.19–7.14 (2H, m), 7.09–7.05 (4H, m), 6.94 (d, 2H, J=8 Hz), 6.76–6.72 (m, 4H), 5.68 (s, 1H), 3.88 (d, 1H, J=12 Hz), 3.86 (s, 3H), 3.27 (d, 1H, J=12 Hz), 2.14 (s, 3H). 13CNMR 175.1, 169.4, 159.6, 136.7, 131.5, 130.1, 130.0, 128.8, 128.7, 128.2, 127.2, 126.3, 114.6, 90.7, 65.9, 55.8, 40.5, 24.6. ESIMS: m/z 402 (M + 1, 100).
O,α-Dimethyl-D-tyrosine (12)
Compound 11 (2.2 g, 0.0055 mol) was suspended in 20 mL of concentrated HCl solution. After nitrogen flush, the mixture was heated under reflux for 3 h. After filtration and removal of the HCl solution, the white precipitate was dried. 1HNMR(CD3OD) δ 7.24 (d, 2H, J=6 Hz), 6.91 (d, 2H, J=6 Hz), 3.77 (3H, s), 3.26 (d, 1H, J=14 Hz), 3.13 (d, 1H, J=14 Hz), 1.66 (s, 3H). 13C NMR 173.8, 161.3, 132.9, 115.9, 62.4, 56.4, 43.5, 23.2. MS (ESI) 210 (M + 1). The product was used in the next step without purification.
3,5-Dibromo-O,α-dimethyl-D-tyrosine (13)
To a solution of compound 12 from above in distilled water (20 mL), 12 M HCl (4 mL) was added. The reaction mixture was cooled to 5 °C, and bromine (2.1 mL, 41 mmol) was injected into the stirred solution. After 15 min, N2 gas was passed through the reaction mixture until the product precipitated. APCIMS: m/z 366 (M + 1, 100). The product was used in the next step without purification.
(3R)-6,8-Dibromo-7-methoxy-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid (14)
Compound 13 from above (assumed to be 4.8 mmol) was added to trifluoroacetic acid (5 mL). HBr (33% in acetic acid, 0.9 mL, 4.8 mmol) was added dropwise to the reaction mixture under a nitrogen atmosphere. After the addition of the acid, formaldehyde (8.64 mmol, 260 mg, 0.7 mL) was added dropwise and the mixture stirred at 70–80 °C for 17 h. The reaction mixture was cooled, dried, and concentrated. APCIMS: m/z 378 (M + 1). The product was used in the next step without purification.
(3R)-6,8-Dibromo-2-(tert-butoxycarbonyl)-7-methoxy-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid (15)
The crude compound 14 reported above (assumed to be 4.8 mmol) was dissolved in DMF (7 mL) and water (2 mL). Triethylamine (1.01 g, 0.01 mol) was added, followed by di-tert-butyl dicarbonate (1.57 g, 0.007 mol). The reaction mixture was stirred at room temperature for 4 h and then concentrated to dryness. The resulting residue was treated with water (30 mL) and EtOAc (30 mL). KHSO4 (2 g) was added to the mixture (pH=2), and the organic layer was separated, dried, and concentrated. The product was purified by chromatography on silica gel (Isco column) using 35% EtOAc in hexanes as eluent to afford 500 mg of 15 (22% from 13) as a syrup. 1H NMR (CD3OD) δ 7.55 (s, 1H), 4.84 (d, 1H, J = 16 Hz), 4.54 (d, 1H, J = 16 Hz), 3.85 (s, 3H), 3.19 (d, 1H, J = 16 Hz), 2.92 (d, 1H, J = 16 Hz), 1.47 (s, 9H), 1.42 (s, 3H). 13C NMR 177.7, 154.7, 138.1, 135.4, 132.9, 118.6, 117.9, 62.5, 62.0, 46.1, 41.7, 29.1, 28.3, 23.9. ESIMS: m/z 478 (M + 1).
(3R)-2-(tert-Butoxycarbonyl)-7-hydroxy-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid (6a)
A suspension of 14 (5.00 g, 0.012 mol) in 75 mL of 48% aqueous HBr was heated to reflux for 5 h. The solution was then evaporated to dryness under reduced pressure and dissolved in 30 mL of MeOH and 7.00 mL of Et3N (0.05 mol). This solution was added to 300 mg of 10%Pd on carbon and shaken for 12 h in a Parr hydrogenator under 60 psig H2. The suspension was filtered and the solvents removed under reduced pressure to leave a solid product (containing the product and triethylammonium salts) with a mass of 9.77 g. This solid was dissolved in 15 mL of H2O, 40 mL of DMF, and 4.78 mL (34.29 mmol) of Et3N. Into this solution, di-tert-butyl dicarbonate (2.1 mL, 22.26 mmol) was introduced and the mixture stirred for 10 h. The solution was reduced to 1/10 of its volume under reduced pressure and partitioned between 30 mL of H2O and 30 mL of EtOAc. The water layer was extracted with EtOAc (3 × 15 mL). The pooled organic extracts were washed once each with 10 mL of H2O, 10 mL of brine, dried over MgSO4, filtered, and concentrated to dryness to yield 3.42 g of 6a as a foam that was pure byNMR.1HNMR(CDCl3) δ 7.17 (d, 1H, J = 8.1 Hz), 6.73 (m, 2H), 4.60–4.41 (2d, 2H), 3.12 (d, 1H, J = 14.7 Hz), 2.78 (d, 1H, J = 14.7 Hz), 1.56–1.24 (2s, 12H). ESIMS: m/z 207 (M + 1-Boc).
(R)-7-Hydroxy-2,3-dimethyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid (6b) Triethylammonium Salt
The Boc-protected isoquinoline 6a (534 mg, 1.74 mmol) was dissolved in 5 mL of a 1:1 mixture of CF3CO2H/CH2Cl2 and stirred overnight. The solvents were removed under reduced pressure and the residue suspended in 2 mL of water. The pH of the solution was adjusted to 7 by addition of saturated NaHCO3. To this solution was added 300 mg of Raney Ni slurry in MeOH using a spatula along with 1 mL of a 37% solution of formaldehyde in water (13.4 mmol), and the resulting suspension was stirred under 1 atm of H2 overnight. The suspension was filtered, and the solvents were removed under reduced pressure to yield a residue that was subjected to silica gel flash-column chromatography. Elution with CHCl3/MeOH/NH4OH (60:30:10) afforded 384 mg of the ammonium salt of 6b after removal of solvents. The triethylammonium salt of 6b was prepared by addition of 5 mL of Et3Nto a solution of the compound in 2 mL of MeOH, followed by removal of the volatiles:mp >220 °C. 1H NMR (CD3OD) δ 7.07 (d, 1H, J=8 Hz), 7.77 (d, 1H), 6.58 (s, 1H), 4.43 (bd, 1H), 4.31 (bd, 1H), 3.37 (d, 1H), 3.20 (m, 9H), 2.95 (d, 1H, J=14.7 Hz), 1.52 (s, 3H), 1.25 (t, 9H), 2.88 (m, 1H), 2.75 (m, 1H). ESIMS: m/z 222 (M + 1, 100).
(3R)-2-(tert-Butoxycarbonyl)-7-methoxy-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid (6c)
Triethylamine (3 mmol, 0.42 mL) and 10% Pd/C (20 mg) were added to 15 (337 mg, 1.05 mmol) in MeOH (5 mL). This mixture was shaken for 90 min under 40 psig of H2 in a Parr apparatus. The mixture was then filtered and concentrated under reduced pressure to give 6c in quantitative yield. An analytical sample was prepared by recrystallization from EtOAc-hexanes: mp 191 °C dec. 1H NMR (CD3OD) δ 7.10 (d, 1H, J = 8 Hz), 6.82 (m, 3H), 4.69 (d, 1H), 4.40 (d, 1H), 3.18 (d, 1H, J=14.7 Hz), 2.79 (d, 1H, J=14.7 Hz), 1.46 (s, 9H), 1.39 (s, 3H). ESIMS: m/z 322 (M+ 1, 100).
(3R)-7-Methoxy-2,3-dimethyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid (6d) Triethylammonium Salt
At 0 °C, 6c (266 mg, 1.13 mmol) was dissolved in 5 mL of THF and 2 mL of 12 M HCl. After this solution was strirred for 4 h, the solvents were removed under reduced pressure. The residue was dissolved in 5 mL of MeOH. Into this solution were added 0.12 mL of Et3N, 0.5 mL of 37% formaldehyde in H2O, and 0.3 mL of Raney Ni slurry in MeOH. The mixture was stirred overnight under an atmosphere of H2, filtered, and the solvents removed under reduced pressure to yield a residue that contained the title compound and Et3N · HCl. This residue was used without further purification. 1H NMR (CD3OD) δ 7.13 (d, 1H), 6.88 (d, 1H), 6.75 (s, 1H), 4.57 (d, 1H), 4.32 (d, 1H) 3.77 (s, 3H), 3.41 (d, 1H, J=14.7 Hz), 3.21 (q, 6H), 2.99 (d, 1H), 2.90 (s, 3H), 1.53 (s, 3H), 1.31 (t, 9H). ESIMS: m/z 322 (M + 1, 100).
(3R)-7-Hydroxy-2-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid (6e) Hydrochloride
Compound 6f30 (1.087 g, 0.0034 mol) was suspended in 15 mL of CH2Cl2, and the mixture was cooled to 0 °C. Into this solution was added 7 mL of CF3COOH, and the mixture was stirred for 6 h. The solvents were removed under reduced pressure, and the residue was suspended in 100 mL of MeOH and 5 mL of formalin. Into this suspension was added 1 g of a slurry of Raney Ni in MeOH using a spatula. The mixture was stirred under an atmosphere of H2 for 5 h and was filtered through celite. To the filtered solution was added 10 mL of a 2 M solution of HCl in ethanol. The solvents were removed under reduced pressure, and the residue was recrystallized from MeOH to give 879 mg (64%) of 6e · HCl as a white powder: mp >220 °C. 1HNMR(DMSO-d6) δ 9.59 (s, 1H), 7.09 (d, 1H, J=8.4 Hz), 6.73 (m, 1H), 6.59 (s, 1H), 4.53 (b, 1H), 4.38 (b, 2H), 3.31 (dd, 1H, J=5.7 Hz), 3.16–3.07 (m, 1H), 2.91 (s, 3H). ESIMS: m/z 208 (M + 1, 100).
(2S)-1-[(3R,4R)-4-(3-Methoxyphenyl)-3,4-dimethylpiperidin-1-yl]-3-methylbutan-2-amine (7b)
To a heterogeneous solution of (3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidine27 (41.1 g, 0.161 mol), N-Boc-L-valine (34.9 g, 0.161 mol), BOP reagent (71.0 g, 0.161 mol) in THF (450 mL) was added triethylamine (51.9 g, 0.513 mol) in THF (50 mL). The reaction mixture became homogeneous within 5 min after addition of Et3N. The reaction was stirred for 4 h at room temperature and then added to ether (500 mL)/H2O (300 mL). The organic layer was separated, washed with saturated NaHCO3 and then brine, and separated. The extracts were dried (Na2SO4) and concentrated in vacuo to afford an off-white solid. This material was purified by silica gel column chromatography, eluting with 70% hexanes in EtOAc to yield 63.6 g (94%) of a white amorphous solid.
Diborane (260 mL, 1.0 M in THF, 0.260 mol) was added to the material described above (54.6 g, 0.130 mol) in THF (350 mL). The reaction mixture was stirred underN2 at reflux for 2 h. The slightly heterogeneous reaction mixture was cooled to room temperature, and 6 N HCl was added (initially cautiously). After stirring at reflux for 2 h, the mixture was concentrated in vacuo and diluted with water. The reaction mixture was made basic with solid Na2CO3 and extracted with CH2Cl2. The organic layer was separated, dried (Na2SO4), and concentrated in vacuo to afford 44 g (100%) of a thick oil. 1H NMR (CDCl3) δ 7.21 (t, 1H), 6.88 (d, 1H, J=8.1 Hz), 6.83 (s, 1H), 6.71 (d, 1H), 3.81 (s, 3H), 2.77 (m, 1H), 2.63–2.13 (m, 8H), 2.10 (bm, 1H), 2.00 (m, 1H), 1.60 (m, 1H), 1.41 (s, 3H), 0.91 (m, 7H), 0.76 (d, 3H, J = 7.2 Hz). ESIMS: m/z 305 (M + H+, 100). Anal. (C19H32N2O) C, H, N.
3-{(3R,4R)-1-[(2S,3S)-2-Amino-3-methylpentyl]-3,4-dimethylpiperidin-4-yl}phenol (7c)23
3-[(3R,4R)-3,4-Dimethylpiperidin-4-yl]phenol (2.42 g, 11.79 mmol) and L-Boc-Ile (2.73 g, 11.79 mmol) were stirred in 30 mL of CH3CN and the solution cooled to 0 °C. Into this solution, HBTU (4.47 g, 11.79 mmol) was added followed by Et3N(3.3 mL, 23.57 mmol). The solution was stirred for 2 h and was then partitioned between 60 mL of EtOAc and 20 mL of H2O. The organic layer was washed with saturated NaHCO3 (10 mL × 3) and brine (10 mL). The solvent was dried over Na2SO4, filtered, and removed under reduced pressure. Flash column chromatography on silica gel eluting with a solvent gradient (80% hexanes in EtOAc to 66% hexanes in EtOAc) gave fractions that contained 3.39 g of pure amide. The amide (3.37 g, 8.33 mmol) was dissolved in 20 mL of dry THF, and 16.67 mL of a 1 M solution of BH3 in THF was added. The solution was heated at reflux for 3 h, cooled to ambient temperature, and carefully added to 3 mL of H2O. Then 7 mL of conc HCl was added. The mixture was heated at reflux for 2 h, and the volume of the reaction was reduced to one-third under reduced pressure. The remaining mixture was made basic by addition of solid NaHCO3 and extracted thoroughly with a 4:1 mixture ofCH2Cl2/THF. The pooled extracts were washed once with 20 mL of H2O, dried over MgSO4, filtered, and concentrated to give 2.50 g (70%) of a clear oil that slowly crystallized. An analytical sample was prepared by recrystallization from EtOAc: mp 150–153 °C. 1H NMR (CDCl3) δ 7.13 (t, 1H), 6.80 (m, 1H), 6.71 (s, 1H), 6.64 (d, 1H), 2.82–2.78 (m, 3H), 2.74–2.52 (m, 4H), 2.42–2.26 (m, 6H), 1.97 (m, 1H), 1.54 (m, 2H), 1.49–1.38 (m, 1H), 1.31 (s, 3H), 1.27–1.19 (m, 1H), 0.89 (t, 6H), 0.77 (d, 3H, J=6.9 Hz). ESIMS: m/z 305 (M + H+, 100). Anal. (C19H32N2O) C, H, N.
(2S,3S)-1-[(3R,4R)-4-(3-Methoxyphenyl)-3,4-dimethylpiperidin-1-yl]-3-methylpentan-2-amine (7d)
(3R,4R)-4-(3-Methoxyphenyl)-3,4-dimethylpiperidine27 (533 mg, 2.43 mmol) and Boc-L-Ile (562 mg, 2.43 mmol) were stirred in 20 mL of CH3CN, and the solution was cooled to 0 °C. Into this solution was added HBTU (922 mg, 2.43 mmol), followed by Et3N (0.7 mL, 4.87 mmol). The solution was stirred for 2 h and was then partitioned between 30 mL of EtOAc and 10 mL of H2O. The organic layer was washed with saturated NaHCO3 (7 mL × 3) and brine (5 mL) solutions. The solvent was dried over Na2SO4, filtered, and removed under reduced pressure. Flash column chromatography on silica gel eluting with 83% hexanes in EtOAc gave fractions that after removal of solvent yielded 680 mg of pure amide. The amide (675 mg, 1.56 mmol) was dissolved in 20 mL of dry THF, and 3.12 mL of a 1Msolution of BH3 in THF was added. The solution was heated at reflux for 3 h, cooled to room temperature, and then 1 mL of H2O was added carefully, followed by 3mL of conc HCl. The mixture was heated at reflux for 2 h, and the volume of the reaction was reduced to one-third under reduced pressure. The remaining mixture was made basic by addition of NaHCO3 and extracted thoroughly with CH2Cl2. The pooled extracts were washed once with 10mL of H2O, dried over MgSO4, filtered, and the solvents removed to give 540 mg (70%) of a clear oil. 1H NMR (CD3OD) δ 7.20 (t, 1H, ArH), 6.89 (m, 1H, ArH), 6.82 (s, 1H, ArH), 6.72 (m, 1H, ArH), 3.77 (s, 3H, CH3OAr), 2.88–2.25 (m, 9H), 2.03 (m, 1H), 2.57–2.40 (m, 3H), 1.30 (d, 3H, CH3, J = 6.6 Hz).), 1.28–1.10 (m, 1H), 0.96–0.89 (m, 7H), 1.60–1.70 (dd, 3H, CH3). EIMS: m/z 319 (M + H+, 100). Anal. (C20H34N2O) C, H, N.
General Procedures for the Preparation of Compounds 8d–p
a. BOP Coupling Procedure
A phenylpiperidine 7 (1 equiv) was dissolved along with a tetrahydroisoquinoline 6 (1.05 equiv) in 10 mL of dry THF and cooled to 0 °C. Into this flask was introduced BOP (1.05 equiv) dissolved in 5 mL of dry THF. Immediately afterward, Et3N (1.05 equiv) was added and the solution was warmed to room temperature and allowed to stir for 3 h. The solution was added to 30 mL of saturated NaHCO3. The resulting mixture was extracted 3× with 10 mL of EtOAc. The pooled organic solvents were washed once with 5 mL of water and dried over MgSO4. The mixture was then separated by flash chromatography on silica gel. For the reactions employing Boc-protected tetrahydroisoquinolines, and the crude coupling mixture was dissolved in 10 mL of a 20% CF3CO2H solution in CH2Cl2 and stirred overnight. The solvents were removed and the crude product stirred in 10 mL of saturated NaHCO3 and 10 mL of EtOAc. The layers were separated, and the aqueous layer was extracted 2× with 5 mL of EtOAc. The pooled EtOAc extracts were washed once with 3 mL of brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield a crude residue. When needed, the impure compound was purified by preparative thick layer chromatography. The dihydrochloride salts were formed by dissolving the freebase in 5 mL of EtOH, followed by addition of 5 mL of 2M HCl in EtOH and evaporation of the solution under reduced pressure.
b. HBTU Coupling Procedure
A phenylpiperidine 7 (1 equiv) was dissolved along with a tetrahydroisoquinoline 6 (1.05 equiv) in 15 mL of a 50% solution of THF in CH3CN and cooled to 0 °C. Into this flask was introduced HBTU (1.05 equiv) dissolved in 10 mL of CH3CN. Immediately afterward, Et3N (1.05 equiv) was added and the solution was warmed to room temperature and allowed to stir for 3 h. To the reaction solution was added 30 mL of saturated NaHCO3. The resulting mixture was extracted three times with 10 mL of EtOAc. The pooled organic solvents were washed once with 5 mL of water and dried over MgSO4. The mixture was then separated by chromatography. For the reactions employing Boc-protected tetrahydroisoquinolines, the crude coupling mixture was dissolved in 10 mL of a 20% CF3CO2H solution in CH2Cl2 and stirred overnight. The solvents were removed and the crude product stirred in 10 mL of saturated NaHCO3 and 10 mL of EtOAc. The layers were separated, and the aqueous layer was extracted 2× with 5 mL EtOAc. The pooled EtOAc extracts were washed once with 3 mL of brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield a crude residue. When needed, the impure compound was purified by preparative thick layer chromatography. The dihydrochloride salts were formed by dissolving the freebase in 5 mL of EtOH, followed by addition of 5 mL of 2 M HCl in EtOH and evaporation of the solvents under reduced pressure.
(3R)-7-Hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylpropyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8d) Dihydrochloride
General procedure (a) was employed using 100 mg (0.328 mmol) of 7b and 112 mg (0.382 mmol) of 6f to afford 65 mg (35%) of the freebase. 1HNMR(CD3OD) δ 7.26 (t, 1H, J=8.1 Hz), 7.00 (d, 1H, J = 8.1 Hz), 6.91 (d, 1H, J = 8.1 Hz), 6.86–6.75 (m, 2H), 6.63 (s, 1H), 4.30–4.37 (m, 3H), 3.50 (m, 2H), 3.15–3.11 (m, 1H), 2.80–2.71 (m, 1H), 2.45 (m, 1H), 1.93–1.87 (m, 1H), 1.49 (s, 3H), 1.29–1.13 (m, 1H), 1.07 (2d, 6H), 0.85 (d, 3H). The hydrochloride salt synthesized by the general procedure had mp >220 °C dec [α]D25+69° (c 0.35, MeOH). 1HNMR(CD3OD) δ 7.31 (t, 1H, J = 9 Hz), 7.12 (d, 1H, J = 9 Hz), 6.95 (d, 1H), 6.86–6.73 (m, 3H), 6.63 (s, 1H), 4.40 (d, 1H), 4.34 (d, 1H), 4.27 (m, 2H), 3.81 (s, 3H), 3.63 (d, 1H), 3.60–3.24 (m, 6H), 3.20 (d, 1H), 2.63 (dt, 1H), 2.43 (m, 1H), 1.95 (m, 1H), 1.48 (s, 3H), 1.03–0.87 (m, 3H), 0.83 (d, 3H, J=9 Hz), 0.80–0.68 (m, 6H). ESIMS: m/z 480 (M + 1, 50). Anal. (C29H43Cl2N3O3 · 2H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S)-1-[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-(2-methylpropyl]-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8e) Dihydrochloride
General procedure (b) was employed using 100 mg (0.344 mmol) of 7a and 111 mg (0.361 mmol) of 6a to afford 65 mg (35%) of the freebase after separation by preparative TLC eluting with 1:1 CMA-80/CH2Cl2. 1H NMR (CD3OD) δ 7.09 (t, 1H, J=8.4 Hz), 6.87 (d, 1H, J=8.4 Hz), 6.70 (m, 2H), 6.55 (m, 2H), 6.47 (s, 1H), 4.02 (d, 1H), 3.77 (m, 2H), 3.16 (d, 1H), 2.74–2.33 (m, 7H), 2.14 (dt, 1H), 1.89–1.75 (m, 2H), 1.47 (d, 1H), 1.38 (d, 3H), 1.26–1.17 (m, 7H), 0.82 (t, 6H), 0.58 (d, 3H). The hydrochloride salt synthesized by the general procedure had mp >220 °C dec [α]D25+47.2° (c 1, MeOH). 1HNMR (CD3OD) δ 7.18 (m, 2H), 6.75 (m, 3H), 6.62 (m, 1H), 4.42 (d, 1H), 4.27 (d, 1H), 4.25 (m, 1H), 3.67–3.30 (m, 6H), 3.18 (d, 1H), 2.63 (dt, 1H), 2.39 (m, 1H), 1.90 (d, 1H), 1.85–1.60 (m, 1H), 1.79 (s, 3H), 0.85 (d, 3H, J = 9 Hz), 0.68 (2d, 6H). ESIMS: m/z 480 (M + 1, 50). Anal. (C29H41Cl2N3O3 · 2H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S,2S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylbutanyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8f) Dihydrochloride
General procedure (a) was employed using 120 mg (0.377 mmol) of 7d and 116 mg (0.396 mmol) of 6f to afford 50 mg (27%) of the freebase after separation by preparative TLC eluting with 1:1 CMA-80/Et2O. 1H NMR (CD3OD) δ 7.19 (t, 1H), 6.87–6.81 (m, 2H), 6.80 (s, 1H), 6.69 (ds, 1H), 6.58 (ds, 1H), 6.47 (s, 1H), 4.06 (m, 1H), 3.77 (dd, 2H), 3.75 (s, 3H), 3.50 (dd, 1H), 2.82 (dd, 1H), 2.78–2.70 (m, 2H), 2.65–2.39 (m, 5H), 2.27 (dt, 1H), 1.99 (m, 1H), 1.70–1.50 (m, 4H), 1.40 (m, 5H), 1.22–1.03 (m, 2H), 0.8 (m, 9H), 0.69 (d, 3H). The hydrochloride salt synthesized by the general procedure had mp >220 °C dec [α]D25 +95.9° (c 0.71, MeOH). 1HNMR(CD3OD) δ 7.30 (t, 1H, J=9 Hz), 7.12 (d, 1H, J = 9 Hz), 6.91 (d, 1H, J = 9 Hz), 6.89–6.73 (m, 2H), 6.62 (s, 1H), 4.40–4.20 (m, 3H), 3.89 (d, 1H), 3.81 (s, 3H), 3.67–3.23 (m, 7H), 3.12 (m, 1H), 2.82 (dt, 1H), 2.45 (m, 1H), 1.92 (d, 1H), 1.70 (m, 1H), 1.55 (m, 1H), 1.50 (s, 3H), 1.42 (m, 1H), 1.31 (m, 1H), 1.20 (m, 1H), 1.10–0.89 (m, 9H). ESIMS:m/z 494 (M + 1, 80). Anal. (C30H45Cl2N3O3 ·H2O) C, H, N.
(3R)-7-Methoxy-N-[(1S)-1-[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-(2-methylpropyl]-3-methyl-1,2,3, 4-tetrahydroisoquinoline-3-carboxamide (8g) Dihydrochloride
General procedure (b) was employed using 172 mg (0.593 mmol) of 7a and 200 mg (0.622 mmol) of 6c to afford 250 mg of the freebase after isolation (68% yield): mp 210–212 °C. 1H NMR (CD3OD) δ 7.09 (t, 1H, J=8.1 Hz), 6.73–6.57 (m, 2H), 4.13 (d, 1H), 3.95–3.87 (m, 2H), 3.64 (s, 3H), 3.25 (d, 2H), 2.68 (m, 1H), 2.65–2.50 (m, 2H), 2.40 (m, 4H), 2.18 (dt, 1H), 1.82 (m, 2H), 1.52 (d, 1H), 1.37 (s, 3H), 1.24 (s, 3H), 0.85 (2d), 0.47 (d, 3H). The hydrochloride salt synthesized by the general procedure had mp 210–212 °C dec [α]D25 +15° (c 1.2, MeOH). 1H NMR (CD3OD) δ 7.17–6.74 (m, 5H), 6.61 (m, 1H), 4.44 (d, 1H), 4.25 (d, 1H), 4.23 (m, 1H), 3.79 (s, 3H), 3.65–3.29 (m, 6H), 3.17 (d, 1H), 2.63 (dt, 1H), 2.40 (m, 1H), 1.91 (d, 1H), 1.84–1.59 (m, 1H), 1.79 (s, 3H), 0.84 (d, 3H, J=9 Hz), 0.67 (2d, 6H). ESIMS: m/z 494 (M + 1, 100). Anal. (C30H45Cl2N3O3 ·H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylpropyl]-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8h) Dihydrochloride
General procedure (b) was employed using 100 mg (0.328 mmol) of 7b and 120 mg (0.390 mmol) of 6a to afford 27 mg (17%) of the freebase after separation by preparative TLC eluting with 75:1 EtOAc/Et3N. 1H NMR (CD3OD) δ 7.17 (t, 1H), 6.79–6.90 (m, 2H), 6.70 (m, 1H), 6.56 (m, 1H), 6.48 (s, 1H), 4.00 (d, 1H), 3.87 (m, 2H), 3.78 (s, 3H), 3.17 (d, 1H), 2.72–2.28 (m, 7H), 2.15 (dt, 1H), 1.89 (m, 1H), 1.79 (sextet, 1H), 1.50 (d, 1H), 1.38–1.20 (m, 7H), 1.10–0.77 (m, 7H), 0.56 (d, 3H). The hydrochloride salt synthesized by the general procedure had mp >220 °C dec [α]D25 +49.8° (c 0.45, MeOH). 1H NMR(CD3OD) δ 7.30 (t, 1H, J=9 Hz), 7.13 (d, 1H, J=9 Hz), 6.94 (d, 1H, J=9 Hz), 6.85–6.74 (m, 3H), 6.63 (s, 1H), 4.41 (d, 1H), 4.35 (d, 1H), 4.27 (m, 1H), 3.81 (s, 3H), 3.63 (d, 1H), 3.60–3.25 (m, 6H), 3.20 (d, 1H), 2.63 (dt, 1H), 2.45 (m, 1H), 1.95 (m, 1H), 1.78 (s, 3H), 1.48 (s, 3H), 1.05–0.89 (m, 3H), 0.83 (d, 3H, J=9 Hz), 0.80–0.68 (m, 6H). ESIMS:m/z 494 (M+1, 80). Anal. (C30H45Cl2N3O3 ·H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S,2S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylbutanyl]-3-methyl-1, 2,3,4-tetrahydroisoquinoline-3-carboxamide (8i) Dihydrochloride
General procedure (a) was employed using 104 mg of 7d (0.341 mmol) and 110 mg (0.358 mmol) of 6a to afford 29 mg of the freebase after separation by preparative TLC eluting with 1:1 CMA-80/CH2Cl2 (17% yield). 1HNMR(CD3OD) δ 7.19 (t, 1H), 6.79 (d, 1H, J=8.1 Hz), 6.77–6.66 (m, 2H), 6.58 (m, 2H), 6.48 (s, 1H), 4.09 (q, 1H), 4.02–3.95 (d, 1H), 3.93–3.8 (m, 2H), 3.15 (d, 2H), 2.70–2.50 (m, 3H), 2.49–2.32 (m, 3H), 2.15 (dt, 1H), 1.88 (m, 1H), 1.62–1.3 (m, 11H), 0.91–0.79 (m, 9H), 0.58 (d, 3H, CH3). The hydrochloride salt synthesized by the general procedure had mp >220 °C dec [α]D25+42.2° (c 0.51, MeOH). 1HNMR (CD3OD) δ 7.20 (t, 1H, J=9 Hz), 7.13 (d, 1H, J=9 Hz), 6.80–6.75 (m, 2H), 6.69–6.64 (m, 2H), 4.41 (d, 1H), 4.30 (d, 1H), 4.28 (m, 1H), 3.64–3.32 (m, 7H), 3.17 (d, 1H), 2.63 (dt, 1H), 2.40 (m, 1H), 1.92 (d, 1H), 1.76 (s, 3H), 1.47 (m, 4H), 1.20 (m, 2H), 0.92–0.71 (m, 9H). ESIMS: m/z 494 (M +1, 80). Anal. (C30H45Cl2N3O3 ·H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S,2S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylbutanyl]-2-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8j) Dihydrochloride
General procedure (a) was employed using 130 mg (0.427 mmol) of 7c and 109 mg (0.448 mmol) of 6e to afford 55 mg (25%) of the freebase after separation by preparative TLC eluting with 2:1 CMA-80/CH2Cl2. 1H NMR (CD3OD) δ 7.19 (t, 1H, J=8.1 Hz), 6.90 (d, 1H, J=8.1 Hz), 6.73 (m, 2H), 6.59 (m, 2H), 6.51 (s, 1H), 4.09 (q, 1H), 3.98 (m, 1H), 3.84 (d, 1H), 3.50 (d, 1H), 3.13 (t, 1H), 2.99 (m, 1H), 2.88 (m, 1H), 2.75 (m, 1H), 2.55 (m, 2H), 2.45 (s, 3H), 2.37 (m, 2H), 2.23 (m, 1H) 1.94 (m, 2H), 1.63 (m, 1H), 1.50 (m, 2H), 1.62–1.3 (m, 11H), 0.80–1.0 (m, 9H), 0.7 (d, 3H, CH3). The hydrochloride salt synthesized by the general procedure had mp 180 °C dec [α]D25 +84.3° (c 0.6, MeOH). 1HNMR(CD3OD) δ 7.19–7.11 (m, 2H), 6.89–6.65 (m, 4H), 4.50 (m, 2H), 4.32 (m, 1H), 3.73 (d, 1H), 3.55–3.16 (m, 8H), 3.08 (s, 3H), 2.78 (dt, 1H), 2.39 (m, 1H), 1.86 (d, 1H, J = 15 Hz), 1.68 (m, 1H), 1.51–1.40 (m, 4H), 1.21 (m, 2H), 1.02 (d, 3H, J=6 Hz), 0.97–0.80 (m, 6H). ESIMS:m/z 494 (M + 1, 100). Anal. (C30H45Cl2N3O3 ·H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylpropyl]-2-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8k) Dihydrochloride
General procedure (a) was employed using 120 mg (0.394 mmol) of 7b 86 mg (0.414 mmol) of 6e to afford 67 mg (35%) of the freebase after separation by preparative TLC eluting with 2:1:1 CMA-80/EtOAc/hexanes. 1H NMR (CD3-OD) δ 7.20 (t, 1H, J=8.1 Hz), 6.91 (d, 1H, J=8.1 Hz), 6.86 (d, 1H, J=8.1 Hz), 6.81 (s, 1H), 6.71 (dd, 1H), 6.59 (dd, 1H), 6.51 (d, 1H), 4.09 (q, 1H), 3.92 (m, 1H), 3.84 (dd, 1H), 3.77 (s, 1H), 3.50 (d, 1H), 3.13 (dd, 1H), 3.05 (dd, 1H), 2.96 (dd, 1H), 2.73 (m, 1H), 2.53 (dd, 1H), 2.50–2.40 (m, 4H), 2.40–2.37 (m, 2H), 2.22 (dt, 1H) 1.98 (m, 2H), 1.84 (m, 1H), 1.57 (t, 1H), 1.33 (m, 5H), 0.94–0.84 (m, 8H), 0.84–075 (dd, 1H), 0.73–0.68 (m, 3H). The hydrochloride salt synthesized by the general procedure had mp 210–215 °C dec [α]D25+75.7° (c 1, MeOH). 1HNMR(CD3OD) δ 7.29 (t, 1H, J = 9 Hz), 7.12 (d, 1H, J = 9 Hz), 6.91 (d, 1H), 6.87–6.61 (m, 3H), 4.48 (d, 1H), 4.35 (d, 1H), 4.30 (m, 1H), 3.81 (s, 3H), 3.78 (d, 1H), 3.63–3.15 (m, 6H), 3.09 (s, 3H), 3.07 (m, 1H), 2.80 (dt, 1H), 2.45 (m, 1H), 1.91 (m, 2H), 1.49 (s, 3H), 1.08–0.90 (m, 7H), 0.86 (d, 3H, J = 9 Hz). ESIMS: m/z 494 (M + 1, 100). Anal. (C30H45Cl2N3O3 ·H2O) C, H, N.
(3R)-7-Methoxy-N-[(1S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylpropyl]-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8l) Dihydrochloride
General procedure (a) was employed using 126 mg (0.414 mmol) of 7b 140 mg (0.435 mmol) of 6c to afford 51 mg (23%) of the freebase after separation by preparative TLC eluting with 2:1 CHCl3/CMA-80. 1H NMR (CD3OD) δ 7.39–7.19 (m, 2H), 7.92–6.78 (m, 5H), 4.5–4.32 (q, 2H), 4.25 (m, 1H), 3.70 (d, 6H), 3.6–3.30 (m), 3.20 (d, 1H), 2.67 (dt, 1H), 2.45 (m, 1H), 1.92 (bd, 1H), 1.78 (s, 3H), 1.75–1.60 (m, 1H), 1.47 (s, 3H), 0.86 (d, 3H), 0.70 (t, 6H). The hydrochloride salt synthesized by the general procedure had mp >220 °C dec [α]D25 +49.8° (c 1, MeOH). 1H NMR (CD3OD) δ 7.30 (t, 1H), 7.26 (d, 1H, J=6 Hz), 6.92–6.80 (m, 2H), 6.84–6.80 (m, 2H), 4.48 (d, 1H), 4.36 (d, 1H), 4.38 (m, 1H), 3.81 (s, 3H), 3.79 (s, 3H), 3.58 (d, 1H), 3.44–3.25 (m, 6H), 3.23 (d, 1H), 2.64 (dt, 1H), 2.46 (m, 1H), 1.95 (d, 1H), 1.78 (s, 3H), 1.80 (m, 1H), 0.83 (d, 3H, J=7.5 Hz), 0.79 (m, 6H). ESIMS: m/z 508 (M + 1, 100). Anal. (C31H47Cl2N3O3 ·H2O) C, H, N.
(3R)-7-Methoxy-N-[(1S,2S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]}-2-methylbutanyl]-3-methyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8m) Dihydrochloride
General procedure (b) was employed using 65 mg (0.213 mmol) of 7c and 46 mg (0.224 mmol) of 6c to afford 25 mg (25%) of the freebase after separation by preparative TLC eluting with 1:1 CMA-80/CH2Cl2. 1H NMR (CDCl3) δ 7.41 (d, 1H), 7.12 (t, 1H), 6.96 (d, 1H), 6.82–6.52 (m, 5H), 4.18–3.77 (m, 4H), 3.73 (s, 3H), 3.11 (d, 1H), 2.82–2.57 (m, 4H), 2.48–2.28 (m, 4H), 2.17–2.02 (m, 2H), 1.91–1.54 (m, 3H), 1.55–1.22 (m, 9H), 0.98–083 (m, 6H), 0.48 (d, 3H). 1.47 (d, 1H), 1.38 (d, 3H), 1.26–1.17 (m, 7H), 0.82 (t, 6H), 0.58 (d, 3H). The hydrochloride salt synthesized by the general procedure had mp >220 °C dec [α]D25 +44.7° (c 0.45, MeOH). 1H NMR (CD3OD) δ 7.24 (d, 1H, J=9 Hz), 7.18 (t, 1H, J=9 Hz), 6.92 (m, 1H), 6.80 (s, 1H), 6.76 (m, 1H), 6.68 (m, 1H), 4.48 (d, 1H), 4.38 (d, 1H), 4.30 (m, 1H), 3.79 (s, 3H), 3.70 (d, 1H, J=15.9 Hz), 3.60–3.31 (m, 5H), 3.20 (d, 1H, J = 15.9 Hz), 2.67 (dt, 1H), 2.40 (m, 1H), 1.91 (d, 1H), 1.79 (s, 3H), 1.46 (bs, 4H), 1.13 (m, 1H), 0.85 (d, 3H), 0.80–0.69 (m, 6H). ESIMS: m/z 508 (M + 1, 100). Anal. (C31H47Cl2N3O3 · 2H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S,2S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylbutanyl)-3-methyl-1, 2,3,4-tetrahydroisoquinoline-3-carboxamide (8n) Dihydrochloride
General procedure (a) was employed using 145 mg (0.455 mmol) of 7d and 146 mg (0.478 mmol) of 6a to afford 60 mg (26%) of the freebase after separation by preparative TLC eluting with 3:1 CHCl3/CMA-80. 1H NMR (CD3OD) δ 7.18 (t, 1H, J=8.1 Hz), 6.88 (d, 1H, J=8.1 Hz), 6.82 (d, 1H), 6.78 (t, 1H), 6.89 (dd, 1H, J2 = 5.7 Hz, J1 = 2.1 Hz), 6.53 (dd, 1H J2=5.7 Hz, J1=2.1 Hz), 6.46 (d, 1H, J=2.4 Hz), 3.96 (d, 1H), 3.92 (m, 1H), 3.76 (s, 3H), 3.30 (m, 1H), 3.15 (d, 1H, J = 15.9 Hz), 2.69 (m, 1H), 2.64 (d, 1H), 2.54 (b, 1H), 2.47–2.36 (m, 5H), 2.17 (dt, 1H), 1.91 (m, 1H), 1.52–1.35 (m, 5H), 1.32 (s, 3H), 1.26 (m, 4H), 1.15 (d, 1H), 1.06–0.9 (m, 2H), 0.86 (t, 3H, J=7.2 Hz), 0.81 (d, 3H, J = 6.9 Hz), 0.57 (d, 3H, J = 6.9 Hz). The hydrochloride salt synthesized by the general procedure had mp 210 °C dec [α]D25 +39.7° (c 0.41, MeOH). 1HNMR(CD3OD) δ 7.27 (t, 1H, J = 9 Hz), 7.11 (d, 1H, J = 9 Hz), 6.90–6.70 (m, 3H), 6.61 (s, 1H), 4.38 (d, 1H), 4.29 (m, 1H), 3.65 (d, 1H), 3.60–3.25 (m, 5H), 3.15 (d, 1H), 2.64 (dt, 1H), 2.42 (m, 1H), 1.92 (d, 1H), 1.76 (s, 3H), 1.46 (bs, 4H), 1.12 (m, 1H), 0.82 (d, 3H, J= 9 Hz), 0.78–0.71 (m, 6H). ESIMS: m/z 508 (M + 1, 100). Anal. (C31H47Cl2N3O3 · 2H2O) C, H, N.
(3R)-7-Methoxy-N-[(1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl}-2-methylpropyl]-2,3-dimethyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8o) Dihydrochloride
General procedure (a) was employed using 104 mg (0.358 mmol) of 7a and 88 mg (0.376 mmol) of 6d to afford 80 mg (44%) of the freebase after separation by preparative TLC eluting with 3:1 CHCl3/CMA-80. 1H NMR (CD3OD) δ 7.07 (t, 1H), 6.93 (d, 1H), 6.73–6.52 (m, 5H), 4.86 (s, 3H), 4.07 (d, 1H, J = 16.5 Hz), 3.89 (m, 1H), 3.80 (d, 1H, J = 16.5 Hz), 3.67 (s, 3H), 3.14 (d, 1H, J=16.5 Hz), 2.72 (m, 1H), 2.62–2.57 (m, 2H), 2.50–2.37 (m, 5H), 2.31 (dd, 1H), 2.18 (dt, 1H), 1.90 (m, 1H), 1.89–1.75 (m, 1H), 1.51 (bd, 1H), 1.37–1.25 (m, 7H), 1.01–0.84 (m, 8H), 0.54 (d, 3H, J = 6.9 Hz). The hydrochloride salt synthesized by the general procedure had mp 199 °C dec [α]D25 +50.2° (c 0.55, MeOH). 1H NMR (CD3OD) δ 7.29 (d, 1H, J= 9 Hz), 7.18 (t, 1H, J=9 Hz), 6.95 (d, 1H, J=9 Hz), 6.83–6.68 (m, 2H), 6.67 (d, 1H), 4.70–4.48 (bm, 2H), 4.33 (bm, 1H), 3.80 (s, 3H), 3.67–3.30 (m, 7H), 2.99 (s, 1H), 2.68 (dt, 1H), 2.42 (m, 1H), 1.92 (d, 1H), 1.48 (s, 3H), 0.99–0.50 (s, 9H). ESIMS: m/z 508 (M + 1, 100). Anal. (C31H47Cl2N3O3 · 2H2O) C, H, N.
(3R)-7-Hydroxy-N-[(1S,2S)-1-{[(3R,4R)-4-(3-methoxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl]-2-methylbutanyl]-2-methyl-1, 2,3,4-tetrahydroisoquinoline-3-carboxamide (8p) Dihydrochloride
General procedure (a) was employed using 100 mg (0.328 mmol) of 7d and 71 mg (0.345 mmol) of 6e to afford 40 mg (34%) of the freebase after separation by preparative TLC eluting with 1:1 CMA-80/Et2O. 1H NMR (CD3OD) δ 7.19 (t, 1H, J =7.8 Hz), 6.92–6.84 (m, 2H), 6.82 (s, 1H), 6.71 (d, 1H), 6.59 (m, 1H), 6.21 (m, 1H), 4.09 (q, 1H), 3.98 (m, 1H), 3.87 (m, 1H), 3.77 (s, 3H), 3.49 (d, 1H, J = 15 Hz), 3.14–2.82 (m, 4H), 2.79–2.60 (m, 2H), 2.60–2.28 (m, 9H), 2.23 (dt, 1H) 1.97 (m, 1H), 1.68–1.35 (m, 4H), 1.29 (d, 3H), 1.23 (t, 3H), 0.93 (t, 5H), 0.90–0.77 (m, 2H), 0.70 (t, 3H). The hydrochloride salt, synthesized by the general procedure, had mp 180 °C dec [α]D25+66.2° (c 0.5, MeOH). 1HNMR(CD3OD) δ 7.29 (t, 1H, J=9 Hz), 7.13 (d, 1H, J = 9 Hz), 6.93–6.78 (m, 3H), 6.67 (d, 1H), 6.85–6.74 (m, 3H), 6.67 (d, 1H), 4.48–4.28 (m, 3H), 3.81 (s, 3H), 3.63 (d, 1H), 3.60–3.25 (m, 6H), 3.09–3.01 (m, 4H), 2.78 (dt, 1H), 2.46 (m, 1H), 1.92 (m, 1H), 1.78 (s, 3H), 1.48 (bs, 4H), 1.1–0.8 (m, 10H). ESIMS: m/z 508 (M + 1, 100). Anal. (C31H47Cl2-N3O3 ·H2O) C, H, N.
Supplementary Material
Acknowledgments
This research was supported by the National Institute on Drug Abuse, grant DA 09045.
Footnotes
We are pleased to have our study published in this special issue of the Journal of Medicinal Chemistry that acknowledges the 100th anniversary of the Division of Medicinal Chemistry.
Abbreviations: GPCRs, G-protein-coupled receptors; cDNAs, cDNA; SAR, structure–activity relationship; [35S]GTPγS, sulfur-35 guanosine-5′-O-(3-thio)triphosphate; DAMGO, [D-Ala2, MePhe4, Glyol 5]enkephalin; DPDPE, [D-Pen 2, D-Pen5]enkephalin; U69,593, (5α, 7α,8β)-(−)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]benzeneacetamide; CHO, Chinese hamster ovary; GDP, guanosine diphosphate; BOP, benzotriazole-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate; HBTU, O-(benzotriazol-1-yl)-N, N, N′, N′-tetramethyluronium hexafluorophosphate; Tic, tetrahydroisoquinolinecarboxylic acid; tPSA, topological polar surface area.
Supporting Information Available: Elemental analysis data for compounds 7c,d, 8d–p. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Volkow ND, Li TK. Drug addiction: the neurobiology of behaviour gone awry. Nat Rev Neurosci. 2004;5:963–970. doi: 10.1038/nrn1539. [DOI] [PubMed] [Google Scholar]
- 2.Koob G, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- 6.Carlezon WA, Jr, Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- 7.Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berlin) 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 11.Portoghese PS, Nagase H, Lipkowski AW, Larson DL, Takemori AE. Binaltorphimine-related bivalent ligands and their kappa opioid receptor antagonist selectivity. J Med Chem. 1988;31:836–841. doi: 10.1021/jm00399a026. published erratum appears in J. Med. Chem, 1988, 31, 2056. [DOI] [PubMed] [Google Scholar]
- 12.Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. Mutational evidence for a common kappa antagonist binding pocket in the wild-type kappa and mutant mu[K303E] opioid receptors. J Med Chem. 1998;41:4911–4914. doi: 10.1021/jm9805182. [DOI] [PubMed] [Google Scholar]
- 13.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J Med Chem. 2001;44:2687–2690. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 14.Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, Mascarella SW, Rothman RB, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Identification of (3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J Med Chem. 2003;46:3127–3137. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- 15.Bennett MA, Murray TF, Aldrich JV. Identification of arodyn, a novel acetylated dynorphin A-(1–11) analogue, as a kappa opioid receptor antagonist. J Med Chem. 2002;45:5617–5619. doi: 10.1021/jm025575g. [DOI] [PubMed] [Google Scholar]
- 16.Metcalf MD, Coop A. Kappa opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-Like Effects of κ-Opioid Receptor Antagonists in Models of Unlearned and Learned Fear in Rats. J Pharmacol Exp Ther. 2007;323:838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- 18.Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berlin) 2008;200:59–70. doi: 10.1007/s00213-008-1122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker BM, Koob GF. Pharmacological evidence for a motivational role of κ-opioid systems in ethanol dependence. Neuropsychopharmacology. 2007:1–10. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodnar RJ, Glass MJ, Ragnauth A, Cooper ML. General, mu and kappa opioid antagonists in the nucleus accumbens alter food intake under deprivation, glucoprivic and palatable conditions. Brain Res. 1995;700:205–212. doi: 10.1016/0006-8993(95)00957-r. [DOI] [PubMed] [Google Scholar]
- 21.Bortolato M, Aru GN, Frau R, Orru M, Fa M, Manunta M, Puddu M, Mereu G, Gessa GL. Kappa opioid receptor activation disrupts prepulse inhibition of the acoustic startle in rats. Biol Psychiatry. 2005;57:1550–1558. doi: 10.1016/j.biopsych.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 22.Carroll FI, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Pharmacological properties of JDTic: a novel κ-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Cai TB, Zou Z, Thomas JB, Brieaddy L, Navarro HA, Carroll FI. Synthesis and in vitro opioid receptor functional antagonism of analogues of the selective kappa opioid receptor antagonist (3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic) J Med Chem. 2008;51:1849–1860. doi: 10.1021/jm701344b. [DOI] [PubMed] [Google Scholar]
- 24.Clark DE. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood–brain barrier penetration. J Pharm Sci. 1999;88:815–821. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 25.Hsiao Y, Hegedus LS. Synthesis of optically active imidazolines, azapenams, dioxocyclams, and bis-dioxocyclams. J Org Chem. 1997;62:3586–3591. [Google Scholar]
- 26.Ma D, Ma Z, Kozikowski AP, Pshenichkin S, Wroblewski JT. Synthesis and biological evaluation of two analogues of (S)-alpha-methyl-3-carboxyphenylalanine. Bioorg Med Chem Lett. 1998;8:2447–2450. doi: 10.1016/s0960-894x(98)00409-0. [DOI] [PubMed] [Google Scholar]
- 27.Zimmerman DM, Leander JD, Cantrell BE, Reel JK, Snoddy J, Mendelsohn LG, Johnson BG, Mitch CH. Structure–activity relationships of the trans-3,4-dimethyl-4-(3-hydroxyphenyl) piperidine antagonists for μ and κ opioid receptors. J Med Chem. 1993;36:2833–2841. doi: 10.1021/jm00072a001. [DOI] [PubMed] [Google Scholar]
- 28.Thomas JB, Fall MJ, Cooper JB, Rothman RB, Mascarella SW, Xu H, Partilla JS, Dersch CM, McCullough KB, Cantrell BE, Zimmerman DM, Carroll FI. Identification of an opioid κ receptor subtype-selective N-substituent for (+)-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine. J Med Chem. 1998;41:5188–5197. doi: 10.1021/jm980511k. [DOI] [PubMed] [Google Scholar]
- 29.Carroll FI, Chaudhari S, Thomas JB, Mascarella SW, Gigstad KM, Deschamps J, Navarro HA. N-Substituted cis-4a-(3-hydroxyphenyl)-8a-methyloctahydroisoquinolines are opioid receptor pure antagonists. J Med Chem. 2005;48:8182–8193. doi: 10.1021/jm058261c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu C, Thomas JB, Brieaddy L, Berrang B, Carroll FI. An improved synthesis of (3R)-2-(tert-butoxycarbonyl-D-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid. Synthesis. 2008:856–858. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.