

Abstract
Background
Tazemetostat (EPZ-6438) is a selective inhibitor of the histone methyltransferase EZH2, currently in clinical development for non-Hodgkin lymphoma and genetically defined tumors.
Procedures
Tazemetostat was tested against the PPTP solid tumor xenografts using a dose of 400 mg/kg administered twice-daily by oral gavage for 28 days. H3K27me3:H3 ratios were determined in control and treated tumors.
Results
Tazemetostat induced significant differences in event free survival (EFS) distribution compared to control in 9 of 30 (30%) of the xenografts studied. Significant differences in EFS distribution were observed in 5 of 7 (71%) rhabdoid tumor xenograft lines compared to 4 of 23 (17%) non-rhabdoid xenograft lines [chi square (χ2) test p=0.006]. Tazemetostat induced tumor growth inhibition meeting criteria for intermediate and high EFS treated to control (T/C) activity in 2 of 25 (8%) and 1 of 25 (4%) xenografts, respectively. Intermediate and high activity for the EFS T/C metric was observed exclusively among rhabdoid tumor xenografts (3 of 5 rhabdoid tumor versus 0 of 22 non-rhabdoid tumors (χ2 test p<0.001). One rhabdoid tumor xenograft (G401) showed stable disease. For one rhabdoid tumor (G401), delayed tumor regression to tazemetostat was noted following 1 week of tumor growth. Tazemetostat induced significant reduction of H3K27me3 levels in the majority of tumors compared to controls.
Conclusions
Tazemetostat demonstrated significant antitumor activity in rhabdoid tumor models, but showed no consistent activity against any other histology. Tazemetostat reduced H3K27me3 levels irrespective of tumor response. Further preclinical testing to evaluate tazemetostat in combination with other anticancer agents is warranted.
Keywords: Preclinical Testing, Developmental Therapeutics, EZH2 inhibitor
INTRODUCTION
EZH2 (Enhancer of Zeste Homolog 2) is the functional enzymatic component of the multiprotein histone methyltransferase complex known as Polycomb Repressive Complex 2 (PRC2) and is responsible for the H3K27 trimethylation activity of the complex [1]. An oncogenic role for EZH2 has been demonstrated for several cancer types [2]. For example, heterozygous EZH2 mutations within the catalytic SET domain have been observed in approximately 20% of cases of germinal center B-cell (GCB)-like diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL) [7]. These activating mutations in EZH2 result in massive increases in H3K27 trimethylation, leading to abnormal repression of PRC2 targets and to lymphoma development [8,9]. These findings provided the basis for EZH2 inhibition as a specific rational therapy for germinal center–derived B-cell NHL.
Regulation of stem cell differentiation by polycomb group proteins is mediated via repression of gene transcription [10]. In differentiated myogenic tissues, EZH2 is either very low or not detectable [11]. For RD embryonal rhabdomyosarcoma cells, forced expression of EZH2 inhibited differentiation, whereas genetic or pharmacologic inhibition of EZH2 induced differentiation [11]. In PAX3-FOXO1 expressing alveolar rhabdomyosarcoma cells, suppression of EZH2 led to apoptosis [12]. Indirect inhibition of EZH2 by 3-deazaneplanocin A (an inhibitor of S-adenosylhomocysteine hydrolase) and inhibition by an EZH2 catalytic inhibitor slowed tumor growth for an alveolar rhabdomyosarcoma xenograft [12]. Multiple cell lines and animal models have demonstrated sensitivity to EZH2 inhibition, including tumor models of diffuse large B-cell lymphoma [13,14], rhabdoid tumor [15], mesothelioma [16], and ependymoma [17]. It has been proposed that tumors such as malignant rhabdoid tumors (MRT) and atypical teratoid rhabdoid tumors (ATRT) that are deficient in the SWI/SNF complex by virtue of SMARCB1 (also known as INI1) deletion have a synthetic lethal dependency on EZH2 [18].
Tazemetostat is one of several potent and selective small molecule EZH2 inhibitors which have been recently reported [13–15,19,20]. These inhibitors compete with the methyl-group donor S-adenosyl methionine (SAM) to suppress the enzymatic activity of EZH2. Tazemetostat inhibits wild type and mutant EZH2 and is highly selective against EZH2 over other histone methyltransferases. Tazemetostat is among the most potent EZH2 inhibitors described to date and demonstrates superior pharmacokinetic properties, including good oral bioavailability in animals [14,21]. Tazemetostat showed impressive in vitro and in vivo activity against a rhabdoid tumor model (G401) [21]. It is currently under clinical investigation for diffuse large cell B-lymphoma and follicular lymphoma as well as SMARCB1-deficient and SMARCA4-deficient solid tumors. Here we report the antitumor activity of tazemetostat against seven panels of pediatric solid tumor xenograft models including an expanded panel of MRT/ATRT.
MATERIALS AND METHODS
In vivo tumor growth inhibition studies
CB17SC scid−/− female mice (Taconic Farms, Germantown, NY), were used to propagate subcutaneously implanted kidney/rhabdoid tumors, sarcomas (Ewing, osteosarcoma, rhabdomyosarcoma), neuroblastoma, and brain tumors. Female mice were used irrespective of the patient gender from which the original tumor was derived. All mice were maintained under barrier conditions and experiments were conducted using protocols and conditions approved by the institutional animal care and use committee at the Research Institute, Nationwide Children’s Hospital. Ten mice were used in each control or treatment group. Tumor volumes (cm3) were determined and responses were calculated using three activity measures as previously described [22]. An in-depth description of the analysis methods is included in the Supplemental Appendix S1. Response Definitions section.
Statistical methods
The exact log-rank test, as implemented using Proc StatXact for SAS®, was used to compare EFS distributions between treatment and control groups. P-values were two-sided and were not adjusted for multiple comparisons given the exploratory nature of the studies.
Drugs and formulation
Tazemetostat was provided to the Pediatric Preclinical Testing Program by Epizyme Inc., through the Cancer Therapy Evaluation Program (NCI). Tazemetostat (HBr salt) was formulated as a 40 mg/ml suspension in sodium carboxymethylcellulose (0.5%) and Tween-80 (0.1%), and sonicated and vortexed until a clear solution was prepared. Solutions were prepared from powder each morning and stored at 4°C until the second daily dose. Solutions were brought to room temperature with stirring (30 min) then administered. Tazemetostat, with a volume of 0.1 m per 10 grams of body weight, was administered by oral gavage twice daily for 28 days at a dose of 400 mg/kg (350 mg/kg calculated as free base). Tazemetostat was provided in coded vials for blinded testing.
Pharmacodynamic studies
Tumors were harvested during treatment when they evented (achieved 4-fold increase in volume relative to the starting tumor volume). This ranged from day 7 (EW-5) to day 22 (KT-14), and tumors were harvested two hours after the final dose of tazemetostat. For three models, OS-1, NB-1382 and RBD-1, tumors were excised 1, 7 and 14 days after completion of tazemetostat treatment. Tumors were rapidly excised, snapfrozen in liquid N2, and stored at −80°C prior to acid extraction of histones and determination of H3K27me3 and total H3 by ELISA. Briefly, frozen samples (30–60 mg) were lysed in 1 mL lysis buffer using a Precellys 24 homogenizer (2.8 mm Beads Kit tube, 15 sec. 5000 rpm), transferred to a fresh microfuge tube (1.5 mL) and incubated on ice (5 min). Samples were centrifuged (4°C, 600 × g, 5 min.) and the supernatant discarded. The pellet was washed once in PBS. After removal of the supernatant the pellet was resuspended in 0.4N H2SO4 (150 μL). The sample was vortexed (5 sec) and incubated on ice for 60 min with vortexing every 15 min. The samples were re-centrifuged (10,000 × g, 10 min, 4°C), and supernatant transferred to a microfuge tube to which 1 mL ice-cold acetone was added and sample incubated for at least 2 hr at −20°C. The samples were briefly vortexed, centrifuged (10,000 × g, 10 min, 4°C), supernatant discarded and the samples allowed to air dry for 5 minprior to resuspension in deionized water (150μL) with mixing. After one hour at room temperature, samples were centrifuged (10,000 × g, 5 min) to remove debris and transferred to a fresh microfuge tube. Samples were stored at −80°C. For the ELISA control (DMSO) and tazemetostat-treated acid extracted histones from WSU-DLCL2 lymphoma cells (EZH2 Y641 mutant) were used as negative and positive controls, respectively. A standard curve for H3K27me3 and total H3 was constructed using acid extracted histones from DMSO treated WSU-DLCL2 cells in coating buffer to establish the linear range of the assay. Protein concentration of the acid extracted histones from the tumor samples was determined and diluted to 2–4 ng/ul in PBS+0.05% BSA. Samples and controls (100 μL/well) were plated in triplicate. The 96 well plate was sealed and incubated at 4°C overnight. Plates were blocked with PBS+2%BSA+0.05%Tween20 (PBST) for 120 min at room temperature. Plates were washed three times and primary antibodies were prepared (rabbit α-H3K27me3 [catalog #9733] at a 1:1,000 dilution and rabbit α-H3 [catalog # 4620] at a 1:10,000 dilution in PBST. Samples were incubated with antibody (100 μL/well) at room temperature for 90 min. Wells were washed 3 times with PBST. Secondary antibody rabbit α-IgG-HRP (catalog #7074) at 1:2000 dilution (H3K27Me3) or 1:6000 (total H3) were diluted in PBST, and samples incubated at room temperature for 90 min. All antibodies were purchased from Cell Signaling Technologies (Malvern, MA). Wells were washed 4 times (PBST) and 100 μL/well BioFx Supersensitive TMB substrate was added. Samples were developed for 5 min at room temperature. The reaction was terminated (100 μL/well 1N H2SO4), and absorbance was read at 450 nm (Envision plate reader).
Expression analysis
Agilent Sureprint 3 data was collected, the foreground signal of each feature was extracted and the effect of hybridization differences was removed by quantile normalization across 350 arrays, running through Partek Genomics Suit. The biological replicates per tumor line (n=4) were collapsed by computing the mean expression level for all features by Perl script. The expression of SMARCB1 gene was extracted and aligned (by LibreOffice Calc) to create the histogram presented.
RESULTS
Tazemetostat was tested against the PPTP panels of solid tumor xenografts using a dose of 400 mg/kg (350 mg/kg active drug) administered twice-daily by oral gavage for 28 days. The total planned treatment and observation period was 6 weeks. Tazemetostat was generally well tolerated, with a 3.1% (9 of 291) mortality rate in the treated groups, which is a non-significant increase compared to control animals (3 of 286; 1%). Complete details of testing are elucidated in Supplemental Table S1 including total numbers of mice, number of mice that died (or were otherwise excluded), numbers of mice with tumor events and average times to event, tumor growth delay, as well as numbers of responses and T/C values.
Thirty of 30 tested xenograft models were considered evaluable for efficacy. Tazemetostat induced significant differences in EFS distribution compared to control in 9 of 30 (30%) of the evaluable xenografts studied (Table I). Significant differences in EFS distribution were observed in 5 of 7 (71%) rhabdoid tumor xenograft lines compared to 4 of 23 (17%) non-rhabdoid xenograft lines (χ2 test p=0.006). For these analyses, renal MRT xenografts and the CNS ATRT xenograft are grouped together, referred to as rhabdoid tumors and characterized by loss of SMARCB1. For those xenografts with a statistically significant difference in EFS distribution between treated and control groups, the EFS T/C activity measure additionally requires an EFS T/C value of > 2.0 for intermediate activity and indicates a substantial agent effect in slowing tumor growth. High activity further requires a reduction in final tumor volume compared to the starting tumor volume. Tazemetostat induced tumor growth inhibition meeting criteria for intermediate and high EFS T/C activity in 2 of 27 (8%) and 1 of 27 (4%) models, respectively. Intermediate and high activity for the EFS T/C metric was observed exclusively among rhabdoid tumor xenografts (3 of 5 rhabdoid tumor versus 0 of 22 non-rhabdoid tumor, χ2 test p<0.001). As expected, the expression level of SMARCB1 in rhabdoid tumor models available to the PPTP (as shown in Supplemental Figure 1), including four models tested here, is undetectable.
Table 1.
In vivo activity of tazemetostat (EPZ-6438) against PPTP solid tumor lines.
| Line | Tumor Type | Estimate of Median Time to Event | P-value | EFS T/C | Median RTV/CD45 at End of Study | Tumor Volume T/C | Median Group Response | EFS Activity |
|---|---|---|---|---|---|---|---|---|
| KT-16 | Rhabdoid | > EP | <0.001 | > 5.6 | 3.7 | 0.48 | PD2 | Int |
| KT-14 | Rhabdoid | > EP | 0.008 | > 2.1 | 2.6 | 0.84 | PD2 | Int |
| KT-12 | Rhabdoid | > EP | 1 | . | 2.3 | 0.91 | NE | NE |
| NCH-RBD1 | Rhabdoid | > EP | 0.002 | > 1.4 | 3.4 | 0.82 | PD2 | NE |
| NCH-RBD2 | Rhabdoid | 10.4 | 0.486 | 0.9 | >4 | 1.17 | PD1 | Low |
| BT-16 | Rhabdoid (CNS) | 28.5 | 0.005 | 1.9 | >4 | 0.67 | PD2 | Low |
| G401 | Rhabdoid | > EP | <0.001 | > 4.4 | 1 | 0.7 | SD | High |
| KT-10 | Wilms | 14.9 | 0.203 | 1.1 | >4 | 0.85 | PD1 | Low |
| KT-13 | Wilms | 13.9 | 0.003 | 1.5 | >4 | 0.7 | PD2 | Low |
| EW5 | Ewing | 12.3 | 0.432 | 1 | >4 | 0.95 | PD1 | Low |
| EW8 | Ewing | 11.4 | 0.057 | 1.2 | >4 | 0.87 | PD1 | Low |
| TC-71 | Ewing | 14 | 0.104 | 1.1 | >4 | 0.78 | PD1 | Low |
| CHLA258 | Ewing | 12.5 | 0.12 | 1.1 | >4 | 0.95 | PD1 | Low |
| Rh10 | Alveolar RMS | 14.9 | 0.374 | 1.1 | >4 | 0.93 | PD1 | Low |
| Rh30 | Alveolar RMS | 12.7 | 0.131 | 1.1 | >4 | 0.81 | PD1 | Low |
| Rh41 | Alveolar RMS | 9.8 | 0.839 | 1 | >4 | 0.95 | PD1 | Low |
| Rh65 | Alveolar RMS | 29.1 | 0.024 | 0.8 | >4 | 0.77 | PD1 | Low |
| BT-50 | Medulloblastoma | > EP | 0.025 | > 1.6 | 3.5 | 0.9 | PD2 | NE |
| BT-36 | Ependymoma | 26.4 | 0.615 | 1 | >4 | 1.1 | PD1 | Low |
| BT-35 | Astrocytoma | 18.1 | 0.108 | 1.2 | >4 | 0.92 | PD1 | Low |
| BT-39 | Glioblastoma | 11.1 | 0.872 | 1.1 | >4 | 0.92 | PD1 | Low |
| BT-56 | Glioblastoma | 12 | 0.93 | 1 | >4 | 0.86 | PD1 | Low |
| NB-1691 | Neuroblastoma | 13.3 | 0.918 | 1.3 | >4 | 0.91 | PD1 | Low |
| NB-EBc1 | Neuroblastoma | 6.8 | 0.598 | 1 | >4 | 1.02 | PD1 | Low |
| NB-1643 | Neuroblastoma | 6.9 | 0.961 | 1 | >4 | 1 | PD1 | Low |
| NB-1382 | Neuroblastoma | 32.4 | 0.843 | 1.1 | >4 | 0.87 | PD1 | Low |
| OS-1 | Osteosarcoma | 23.8 | 0.036 | 1.2 | >4 | 0.87 | PD1 | Low |
| OS-2 | Osteosarcoma | 25.7 | <0.001 | 2 | >4 | 0.46 | PD2 | Low |
| OS-17 | Osteosarcoma | – | – | – | – | – | PD1 | Low |
| OS-9 | Osteosarcoma | – | – | – | – | – | PD1 | Low |
Tumor Volume T/C value: Relative tumor volumes (RTV) for control (C) and treatment (T) mice were calculated at day 21 or when all mice in the control and treated groups still had measurable tumor volumes (if less than 21 days). The T/C value is the mean RTV for the treatment group divided by the mean RTV for the control group. High activity = T/C ≤ 0.15; Intermediate activity = T/C ≤ 0.45 but > 0.15; and Low activity = T/C > 0.45.
EFS T/C value is defined by the ratio of the median time to event of the treatment group and the median time to event of the respective control group. High activity requires: a) an EFS T/C > 2; b) a significant difference in EFS distributions, and c) a net reduction in median tumor volume for animals in the treated group at the end of treatment as compared to at treatment initiation. Intermediate activity demonstrates criteria a) and b) above, but not having a net reduction in median tumor volume for treated animals at the end of the study. Low activity demonstrates EFS T/C < 2.
Objective response measures are described in detail in the Supplemental Response Definitions. PD1 = progressive disease with EFS T/C ≤ 1.5, and PD2 = progressive disease with EFS T/C > 1.5. Bold font, P-value <0.05.
For the objective response metric, one MRT xenograft (G401) showed stable disease (SD). Progressive Disease 2 (PD2) response, defined by progressive disease with growth delay (EFS T/C >1.5), was observed in an additional three MRT xenografts (Figure 1). Tazemetostat also produced PD2 responses against BT-16, derived from a CNS ATRT, as well as three non-rhabdoid tumor xenografts [medulloblastoma (BT-50), Wilms tumor (KT-13), and osteosarcoma (OS-2); (Table 1 and Figure 2) evaluable for this measure. The objective response results for both solid tumor models are represented in Figure 3 using a ‘COMPARE’ and “heatmap” formats. In Figure 3, xenografts with PD2 are indicated by a score of −3, and xenografts with regression (PR or CR) are indicated by bars to the right of the midpoint line in the COMPARE graph. Red bars indicate xenografts with significant differences in EFS distribution between the treated and control groups.
Figure 1.
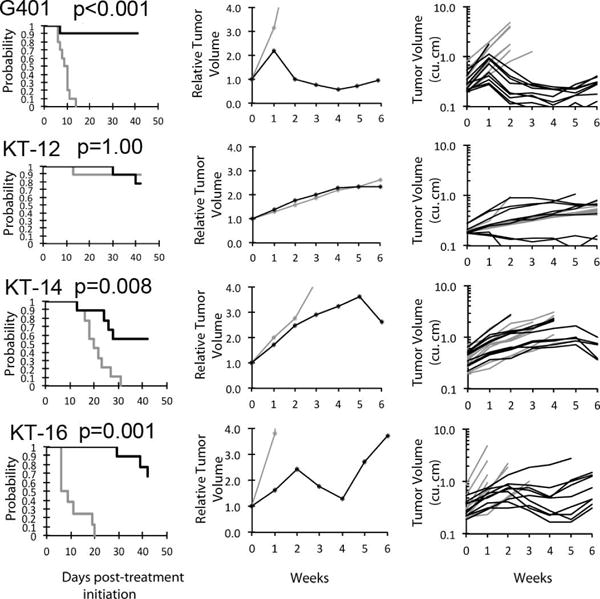
Tazemetostat (EPZ-6438) in vivo objective response activity kidney rhabdoid tumor models. A. Kaplan-Meier curves with statistical significance indicated by p-values for EFS of the control (gray) and treated (black) groups. B, median relative tumor volume graphs (center), and C. individual tumor volume graphs (right) are shown for selected lines.
Figure 2.
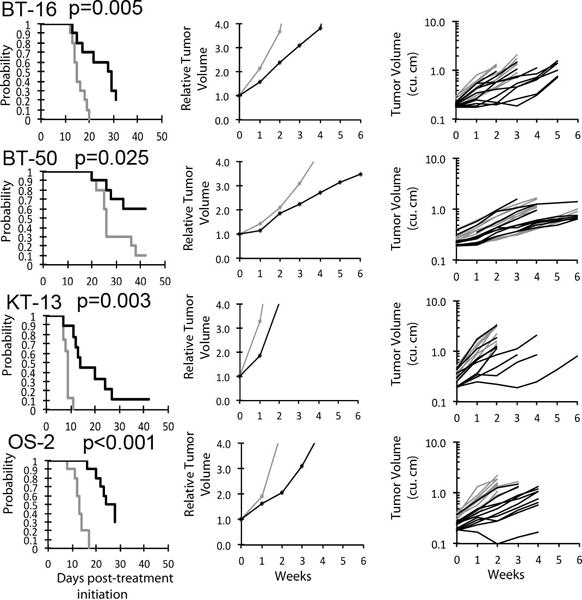
Tazemetostat (EPZ-6438) in vivo objective response activity in one CNS rhabdoid xenograft model (BT-16) and non-rhabdoid tumor models (BT-50, KT-13, and OS-2). A. Kaplan-Meier curves for EFS, B. median relative tumor volume graphs, and C. individual tumor volume graphs (right) are shown for selected lines. Control groups are indicated in gray, and treated groups shown in black. Statistical significance (p-values) of the difference between treated and control groups are included in the Kaplan-Meier plots.
Figure 3.
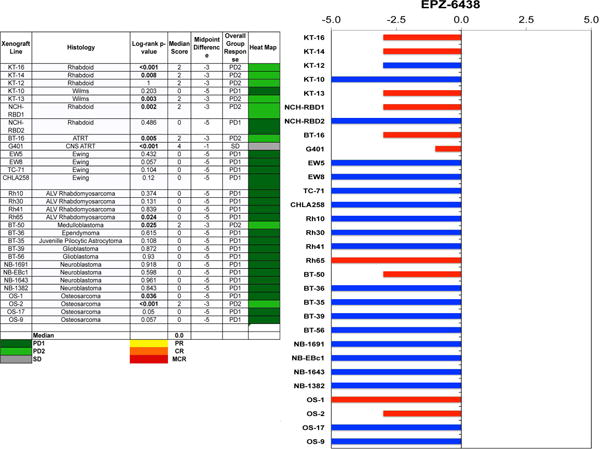
A. The heat map depicts group response scores. A high level of activity with tumor regression is indicated by a “Median Score” of 6 or more, intermediate activity by a score of ≥2 but <6, and low activity by a score of <2. The “Median Score” ranges from 0 for PD1 responses to 10 for MCR objective responses, with the “Midpoint Difference” reflecting the difference between the Median Score and the Midpoint Response (5 for stable disease). The log-rank P-value is for the difference in EFS distribution for treated and control groups.
B. representation of tumor sensitivity based on the difference of individual tumor lines from the midpoint response (stable disease). Bars to the right of the median represent tumor regression, and to the left are tumor models that progress. Red bars indicate xenograft models with a significant difference in EFS distribution between treatment and control groups, while blue bars indicate lines for which the EFS distributions were not significantly different.
To determine pharmacodynamic effects of tazemetostat, tumors were harvested at time of event on treatment, or at various time periods after completion of treatment when tumor bearing mice completed the 28 day course of treatment. Histones were extracted using the procedures of Daigle et al. [23], and assayed for total Histone H3 and H3K27me3 using an ELISA as described by Knutson et al. [14]. As shown in Figure 4A, tazemetostat reduced H3K27me3 levels in most tumors that were harvested during drug treatment, the exceptions being the KT-10 (Wilms tumor) and TC-71 (Ewing sarcoma) xenograft lines. Of note H3K27me3 levels were still significantly suppressed in RBD-1 rhabdoid tumors 14 days after stopping tazemetostat treatment (Figure 4A). The highest H3K27me3:H3 ratios in control tumors were in the rhabdoid models (KT-14, RBD-1, RBD-2), although the level of EZH2 gene expression was generally lower than the mean across the PPTP tumor panel (Figure 4B). The Ewing sarcoma CHLA-258 also exhibited high H3K27me3 levels, but also had high EZH2 expression at the mRNA level (Figure 4B).
Figure 4.
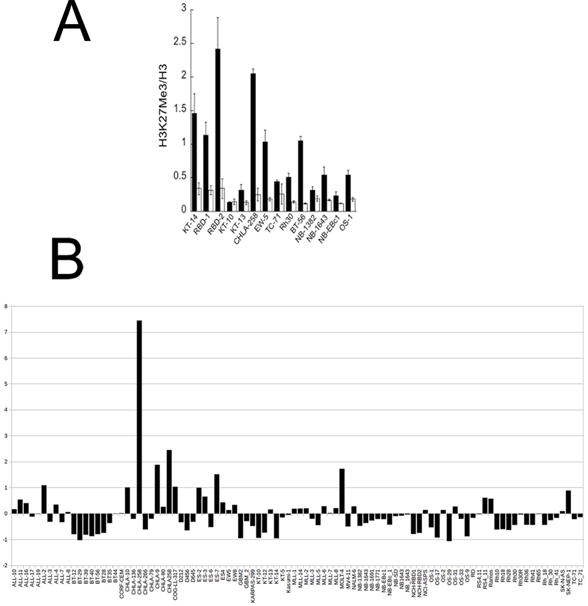
A. Tazemetostat induced changes in H3K27me3:H3 ratios in treated xenografts as measured by ELISA; controls (solid bars), treated (open bars). B. Relative expression levels of EZH2 in xenograft and cell line models. Data are expressed around the mean expression level (0) for the complete datset. The results show lower than mean levels of expression for rhabdoid tumors KT-12, KT-14, NCH-RBD1, NCH-RBD2, BT-29, marked with arrows (Agilent SurePrint G3 arrays).
DISCUSSION
Mono-, di- and tri-methylation at the H3K27 site is uniquely mediated by PRC2, being specified by the EZH2 SET domain [24]. Mice lacking Ezh2 are not viable and show severe developmental and proliferative defects [25]. Through H3K27me3 EZH2 suppresses differentiation of mesenchymal stem cells and potentially cancer stem cells [26]. Point mutations that increase the catalytic activity of EZH2 promote cell transformation in subsets of B-cell neoplasms, thus making EZH2 a compelling target in this genetically defined patient population [27]. EZH2 inhibitors were shown to have strong antitumor effects against human B-cell lymphoma xenograft models [14,20]. MRTs and ATRTs are extremely aggressive pediatric cancers that are characterized by the homozygous loss of SMARCB1, a core component of the SWI/SNF chromatin remodeling complex [28]. Reciprocal expression has been reported between EZH2 and SMARCB1, and EZH2 has been shown to be required for the development of SMARCB1-deficient tumors such as MRTs and ATRTs [18,28,29]. Tazemethostat showed in vitro and in vivo activity against a SMARCB1-mutant MRT model [21]. SMARCA4-deleted ovarian cancer, such as small cell cancer of the ovary of the hypercalcemic type (SCCOHT), has also been shown to be responsive to EZH2 inhibition [30]. Clinical trials are underway to evaluate tazemetostat against these SWI/SNF mutant cancers.
We tested 30 xenograft models from the PPTP solid tumor panels. Tazemetostat induced high or intermediate responses in two MRT xenograft lines established by direct transplantation of tumor into immunocompromised mice (KT-14 and KT-16) and also in one rhabdoid tumor cell line-derived xenograft model (G401). The G401 rhabdoid tumor xenograft showed stable disease with a delayed response to tazemetostat treatment, with the tumor starting to regress during week two of treatment (Figure 1). This is in agreement with previously reported results for the G401 xenograft in which tumor regression was observed with a delay of 5–7 days after starting treatment [21]. Tumor re-growth after cessation of dosing was observed in all three MRT xenograft lines with intermediate and high objective responses. It has been reported previously that G401 xenograft re-grew at lower dosage (125mg/kg) but showed complete tumor regression without regrowth following high-dose tazemetostat treatment (250–500mg/kg, BID) [14]. Tazemetostat was administered to G401 xenograft line in our study at 400 mg/kg (BID), however, it still demonstrated tumor re-growth at weeks 4–6.
Measurement of H3K27me3 in tumor tissues showed that tazemetostat significantly inhibited global EZH2 marking (H3K27me3). This was particularly evident in rhabdoid tumors for which the ratio of H3K27me3 to Histone H3 was higher than in the other tumors examined and was significantly suppressed by treatment. Interestingly, expression of EZH2 (Agilent surePrint G3 array) was lower in rhabdoid tumors than the mean for all xenograft models, with generally higher expression in Ewing sarcoma models.
In contrast to the SMARCB1-deficient rhabdoid tumor models, none of the non-rhabdoid tumors showed intermediate or high response activity for the EFS T/C metric. These included Ewing tumors, rhabdomyosarcoma, brain tumors, neuroblastoma and osteosarcoma. Some of these indications contain mutations that have been hypothesized to sensitize cells to EZH2 inhibition. For instance, mutations in histone H3 cause reduced EZH2 histone methyltransferase activity leading to reprogramming of H3K27 and H3K36 methylation in certain malignant pediatric brain tumors [31–33]. The BT-35 astrocytoma cell line has a mutation in histone H3.3 (H3F3A K27M), but was unresponsive to tazemetostat. It currently is unclear whether these tumor types are not sensitive to EZH2 inhibition, have acquired resistance to tazemetostat, or only represent a small subset of a heterogenous indication which otherwise might respond. Resistance to tazemetostat and other EZH2 inhibitors has been suggested to be caused by increased expression of ABCB1 and ABCG2 drug transporters [34]. Tazemetostat is a substrate for ABCB1 (P-glycoprotein)-mediated efflux, and we noted that the expression of this pump is greater than the panel mean level in two xenografts (NB-EBc1 and NB-1691 xenografts (data not shown). However, H3K27me3 levels were reduced to a similar level in the NB-EBc1 xenograft as other xenograft models, suggesting that this xenograft model is not preventing tazemetostat target engagement, but rather is not sensitive to EZH2 inhibition.
Clinical results for tazemetostat support the importance of loss of SWI/SNF function as one predictor for sensitivity to the agent [35]. In a phase 1 trial for adults with cancer, dose levels from 100 to 1600 mg twice-daily were evaluated with expansion cohorts at 800 and 1600 mg BID [35]. Among 30 solid tumor patients, 8 had SMARCB1-negative tumors [5 with MRTs and 3 with epithelioid sarcomas (ES)] and 3 had SMARCA4-negative tumors [2 with small cell carcinoma of the ovary hypercalcemic type (SCCOHT) and one with thoracic sarcoma]. Two of 5 MRT patients achieved objective responses, with one having a CR that was maintained for more than one year. Both SCCOHT patients remained on-therapy for more than 6 months, with one achieving a PR. Two of 3 ES patients remained on-therapy for more than 6 months, with one achieving a PR. Tazemetostat demonstrated no clinical activity in the remaining 19 solid tumor patients, including three patients with synovial sarcoma. For patients with relapsed or refractory B-cell NHL, tazemetostat demonstrated substantial clinical activity. Nine of 15 evaluable patients had an objective response, including two patients with CRs maintained for more than one year [36].
In summary, pediatric preclinical testing of tazemetostat, a novel, potent and selective EZH2 inhibitor, demonstrated significant anti-tumor activity in rhabdoid tumor xenograft models, however, limited activity was found in the panel of solid tumors studied as a whole. The phenomenon of delayed tumor response should be further investigated and may need to be considered when developing clinical trials for this class of agents. Our results suggest that there is heterogeneity in response to EZH2 inhibitors and that the heterogeneity in response is unlikely to be the result of differential reduction in H3K27me3. These results additionally provide impetus for further studying this heterogeneity. The data also provide a cautionary note against expectations for uniform high level clinical activity for EZH2 inhibitors against rhabdoid tumors.
EZH2 inhibitors have been reported to sensitize mutant non-small cell lung carcinoma cells to topoisomerase II poisons [37], enhance the activity of EGFR inhibition in colon cancer cells [38,39], synergize with glucocorticoid receptor agonists in models of germinal center non-Hodgkin lymphoma, and enhance doxorubicin and paclitaxel activity [40,41]. Further evaluation of tazemetostat in preclinical models with mutations considered to predispose to drug sensitivity (e.g., SMARCB1, SMARCA4) in combination with conventional chemotherapeutics agents may be a valuable approach to develop for this class of epigenetic modifier.
Supplementary Material
Supplemental Appendix S1. Response Definitions.
Supplemental Figure S1. Expression of SMARCB1 in PPTP models.
Supplemental Table S1. Efficacy of tazemetostat (EPZ-6438) against PPTP Xenograft Models.
Acknowledgments
This work was supported by NO1-CM-42216, CA21765, and CA108786 from the National Cancer Institute and used tazemetostat (EPZ-6438) supplied the Epizyme, Inc., Cambridge, MA. In addition to the authors this paper represents work contributed by the following: Sherry Ansher, Catherine Billups, Hulyun Wu, and Jian Zhang.
Abbreviations
- PPTP
Pediatric Preclinical Testing Program
- EFS T/C
Ratio of Event Free Survival Treated/Control
- Cmax
Maximum plasma concentration
- (NOD)/scid−/−
Non-Obese diabetic/severe combined immune deficient
- EPZ-6438
Tazemetostat
- EZH2
Enhancer of Zeste Homolog 2
- MRT/ATRT
Malignant rhabdoid tumor/atupical teratoid-rhabdoid tumor
- H3K27me3
Histone H3 lysine 27 trimethylation
Footnotes
Conflict of interest statement: Dr. Cosmopoulos, and C. Klaus are employees of Epizyme Inc. Dr. Keilhack is an employee of Ribon Therapeutics, Inc.
The other authors consider that there are no actual or perceived conflicts of interest.
References
- 1.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17(9):2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 3.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, Saksena G, Stransky N, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Hernandez-Lemus E, Schwarz-Cruz y Celis A, Imaz-Rosshandler I, Ojesina AI, Jung J, Pedamallu CS, Lander ES, Habermann TM, Cerhan JR, Shipp MA, Getz G, Golub TR. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodor C, O’Riain C, Wrench D, Matthews J, Iyengar S, Tayyib H, Calaminici M, Clear A, Iqbal S, Quentmeier H, Drexler HG, Montoto S, Lister AT, Gribben JG, Matolcsy A, Fitzgibbon J. EZH2 Y641 mutations in follicular lymphoma. Leukemia. 2011;25(4):726–729. doi: 10.1038/leu.2010.311. [DOI] [PubMed] [Google Scholar]
- 7.Lunning MA, Green MR. Mutation of chromatin modifiers; an emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J. 2015;5:e361. doi: 10.1038/bcj.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107(49):20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, Gascoyne RD, Humphries RK, Arrowsmith CH, Morin GB, Aparicio SA. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibaja V, Shen F, Harari J, Korn J, Ruddy D, Saenz-Vash V, Zhai H, Rejtar T, Paris CG, Yu Z, Lira M, King D, Qi W, Keen N, Hassan AQ, Chan HM. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2015 doi: 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchesi I, Fiorentino FP, Rizzolio F, Giordano A, Bagella L. The ablation of EZH2 uncovers its crucial role in rhabdomyosarcoma formation. Cell Cycle. 2012;11(20):3828–3836. doi: 10.4161/cc.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciarapica R, De Salvo M, Carcarino E, Bracaglia G, Adesso L, Leoncini PP, Dall’Agnese A, Walters ZS, Verginelli F, De Sio L, Boldrini R, Inserra A, Bisogno G, Rosolen A, Alaggio R, Ferrari A, Collini P, Locatelli M, Stifani S, Screpanti I, Rutella S, Yu Q, Marquez VE, Shipley J, Valente S, Mai A, Miele L, Puri PL, Locatelli F, Palacios D, Rota R. The Polycomb group (PcG) protein EZH2 supports the survival of PAX3-FOXO1 alveolar rhabdomyosarcoma by repressing FBXO32 (Atrogin1/MAFbx) Oncogene. 2014;33(32):4173–4184. doi: 10.1038/onc.2013.471. [DOI] [PubMed] [Google Scholar]
- 13.Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, Balasubramanian S, Campbell R, Zhao F, Bergeron L, Audia JE, Albrecht BK, Harmange JC, Cummings R, Trojer P. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20(11):1329–1339. doi: 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13(4):842–854. doi: 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- 15.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang JH, Yan X, Fang J, Mi Y, Zhang M, Zhou T, Feng G, Chen Z, Li G, Yang T, Zhao K, Liu X, Yu Z, Lu CX, Atadja P, Li E. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109(52):21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T, Konstantinoff K, Micol JB, Durham B, Knutson SK, Campbell JE, Blum G, Shi X, Doud EH, Krivtsov AV, Chung YR, Khodos I, de Stanchina E, Ouerfelli O, Adusumilli PS, Thomas PM, Kelleher NL, Luo M, Keilhack H, Abdel-Wahab O, Melnick A, Armstrong SA, Levine RL. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21(11):1344–1349. doi: 10.1038/nm.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jager N, Jones DT, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJ, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, Hovestadt V, Tzaridis T, Dubuc AM, Northcott PA, Peacock J, Bertrand KC, Agnihotri S, Cavalli FM, Clarke I, Nethery-Brokx K, Creasy CL, Verma SK, Koster J, Wu X, Yao Y, Milde T, Sin-Chan P, Zuccaro J, Lau L, Pereira S, Castelo-Branco P, Hirst M, Marra MA, Roberts SS, Fults D, Massimi L, Cho YJ, Van Meter T, Grajkowska W, Lach B, Kulozik AE, von Deimling A, Witt O, Scherer SW, Fan X, Muraszko KM, Kool M, Pomeroy SL, Gupta N, Phillips J, Huang A, Tabori U, Hawkins C, Malkin D, Kongkham PN, Weiss WA, Jabado N, Rutka JT, Bouffet E, Korbel JO, Lupien M, Aldape KD, Bader GD, Eils R, Lichter P, Dirks PB, Pfister SM, Korshunov A, Taylor MD. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–450. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 20.Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, Kawano S, Minoshima Y, Choi HW, Porter Scott M, Waters NJ, Smith JJ, Chesworth R, Moyer MP, Copeland RA. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity. ACS Med Chem Lett. 2015;6(5):491–495. doi: 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R, Carol H, Tajbakhsh M, Reynolds CP, Maris JM, Courtright J, Keir ST, Friedman HS, Stopford C, Zeidner J, Wu J, Liu T, Billups CA, Khan J, Ansher S, Zhang J, Smith MA. The pediatric preclinical testing program: Description of models and early testing results. Pediatr Blood Cancer. 2006 doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- 23.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keilhack H, Smith JJ. Small molecule inhibitors of EZH2: the emerging translational landscape. Epigenomics. 2015;7(3):337–341. doi: 10.2217/epi.15.14. [DOI] [PubMed] [Google Scholar]
- 25.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23(20):4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Momparler RL, Cote S. Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin Investig Drugs. 2015;24(8):1031–1043. doi: 10.1517/13543784.2015.1051220. [DOI] [PubMed] [Google Scholar]
- 27.Shen C, Vakoc CR. Gain-of-function mutation of chromatin regulators as a tumorigenic mechanism and an opportunity for therapeutic intervention. Curr Opin Oncol. 2015;27(1):57–63. doi: 10.1097/CCO.0000000000000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh M. Mutation spectra of histone methyltransferases with canonical SET domains and EZH2-targeted therapy. Epigenomics. 2015 doi: 10.2217/epi.15.89. [DOI] [PubMed] [Google Scholar]
- 29.Kakkar A, Biswas A, Goyal N, Suri V, Sharma MC, Gupta D, Julka PK, Sarkar C. The expression of Cyclin D1, VEGF, EZH2, and H3K27me3 in Atypical Teratoid/Rhabdoid Tumors of the CNS: A Possible Role in Targeted Therapy. Appl Immunohistochem Mol Morphol. 2015 doi: 10.1097/PAI.0000000000000247. [DOI] [PubMed] [Google Scholar]
- 30.Knutson SK, Drew AE, Plescia C, Copeland RA, Smith JJ, Keilhack H, Ribich S. EZH2 inhibition leads to decreased proliferation in SMARCA4-deleted ovarian cancer cell lines. Boston, MA. Philadelphia (PA): AACR; 2015. p Abstr #C87. [Google Scholar]
- 31.Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24(5):567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124(3):439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 34.Zhang P, de Gooijer MC, Buil LC, Beijnen JH, Li G, van Tellingen O. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-Inhibitors. Int J Cancer. 2015;137(8):2007–2018. doi: 10.1002/ijc.29566. [DOI] [PubMed] [Google Scholar]
- 35.Italiano A, Keilhack H, Toulmonde M, Coindre JM, Michot JM, Massard C, Ottesen LH, Reyderman L, Blakemore SJ, Kraljevic S, Thomson B, McDonald A, Ho PT, Ribrag V. A phase 1 study of EPZ-6438 (E7438), an Enhancer of Zeste-Homolog 2 (EZH2) inhibitor: Preliminary activity in INI1-negative tumors. Eur J Cancer. 2015;51(Supplement S3):S54–S55. [Google Scholar]
- 36.Ribrag V, Soria J, Reyderman L, Kraljevic S, Chen R, Salazar P, Kuznetsov G, Keilhack H, Waters NJ, Hedrick E, Ottesen LH, Italiano A. Phase 1 study of E7438 (EPZ-6438), an enhancer of Zeste homolog 2 (EZH2) inhibitor: dose determinaton and preliminary activity in non-Hodgkin lymphoma. Hematol Oncol. 2015;33:179. Abstr #145. [Google Scholar]
- 37.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, Zhang H, Marquez VE, Hammerman PS, Wong KK, Kim CF. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520(7546):239–242. doi: 10.1038/nature14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katona BW, Liu Y, Ma A, Jin J, Hua X. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol Ther. 2014;15(12):1677–1687. doi: 10.4161/15384047.2014.972776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A, Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014;9(12):e111840. doi: 10.1371/journal.pone.0111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gharpure KM, Chu KS, Bowerman CJ, Miyake T, Pradeep S, Mangala SL, Han HD, Rupaimoole R, Armaiz-Pena GN, Rahhal TB, Wu SY, Luft JC, Napier ME, Lopez-Berestein G, DeSimone JM, Sood AK. Metronomic docetaxel in PRINT nanoparticles and EZH2 silencing have synergistic antitumor effect in ovarian cancer. Mol Cancer Ther. 2014;13(7):1750–1757. doi: 10.1158/1535-7163.MCT-13-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai J, Ma M, Cai M, Xu F, Chen J, Wang G, Shuai X, Tao K. Inhibition enhancer of zeste homologue 2 promotes senescence and apoptosis induced by doxorubicin in p53 mutant gastric cancer cells. Cell Prolif. 2014;47(3):211–218. doi: 10.1111/cpr.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Appendix S1. Response Definitions.
Supplemental Figure S1. Expression of SMARCB1 in PPTP models.
Supplemental Table S1. Efficacy of tazemetostat (EPZ-6438) against PPTP Xenograft Models.