

SUMMARY
A typical pathogen presents a combination of Toll-like receptor (TLR) ligands during infection. Although individual TLR pathways have been well characterized, the nature of this “combinatorial code” in pathogen sensing remains unclear. Here, we conducted a comprehensive transcriptomic analysis of primary macrophages stimulated with all possible pairwise combinations of four different TLR ligands to understand the requirements, kinetics, and outcome of combined pathway engagement. We find that signal integration between TLR pathways leads to non-additive responses for a subset of immune mediators with sustained expression (>6 hr) properties and T cell polarizing function. To identify the underlying regulators, we conducted a focused RNAi screen and identified four genes—Helz2, Phf11d, Sertad3, and Zscan12—which preferentially affect the late phase response of TLR-induced immune effector expression. This study reveals key molecular details of how contemporaneous signaling through multiple TLRs, as would often be the case with pathogen infection, produce biological outcomes distinct from the single ligands typically used to characterize TLR pathways.
In Brief
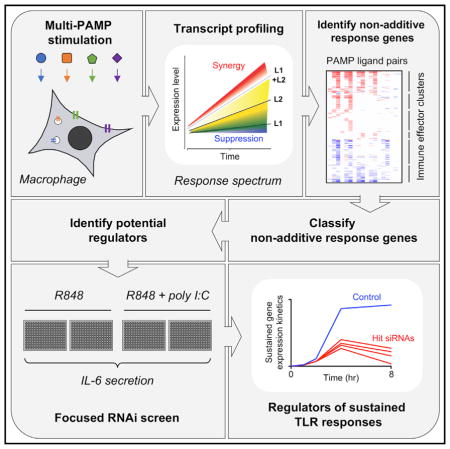
Lin et al. conducted a systematic transcriptomic analysis of dual TLR-ligand-stimulated mouse macrophages stimulated with single and paired TLR ligands to understand the requirements, kinetics, and outcome of combined TLR pathway engagement. RNAi screening revealed previously unreported regulators of sustained TLR gene activation.
INTRODUCTION
Toll-like receptors (TLRs) recognize conserved pathogen-associated molecular patterns (PAMPs) to initiate innate immune responses through the induction of numerous effectors, including many cytokines (Takeuchi and Akira, 2010). After nearly two decades of intensive investigation, the TLR signaling response to individual PAMPs has been well characterized (Alexopoulou et al., 2001; Chen and Chen, 2013; Garber et al., 2012; Hemmi et al., 2002; Hoebe et al., 2003; Horng et al., 2002; Kagan and Medzhitov, 2006; Kagan et al., 2008; Kawai et al., 1999; Lin et al., 2010; Smale, 2010; Takeuchi and Akira, 2010; Takeuchi et al., 2001; Yamamoto et al., 2003a, 2003b). However, immune cells are unlikely to encounter single PAMPs in real infection scenarios, as each pathogen invariably presents multiple PAMPs to the host cell. Indeed, multi-TLR cooperation is important for combating viral and bacterial infections (Tabeta et al., 2004; Weiss et al., 2004), and multi-PAMP challenge has been shown to lead to synergistic induction of many cytokines (Bagchi et al., 2007; Liu et al., 2015; Napolitani et al., 2005; Suet Ting Tan et al., 2013). The nature of this “combinatorial code” in pathogen sensing remains unclear, with previous reports suggesting contributions by signaling adaptor use, cellular localization, or pathogen origination (Bagchi et al., 2007; Liu et al., 2015; Napolitani et al., 2005; Suet Ting Tan et al., 2013). Nevertheless, since previous studies have either examined only selective TLR-ligand combinations, or have limited their analysis to a few outputs, a comprehensive analysis is needed to reveal the intrinsic factor(s) mediating TLR signaling crosstalk.
The complex gene program induced by TLR pathway activation is classified into early primary, late primary, and secondary response genes (Escoubet-Lozach et al., 2011; Hargreaves et al., 2009; Ramirez-Carrozzi et al., 2009; Ramsey et al., 2008; Tong et al., 2016). This classification is based primarily on transcriptional initiation, and the majority of published studies have focused on the first 2 hr after TLR activation to address this issue. However, genes with the same initial kinetics can behave differently at later time points. For example, both Tnf and Nfkbiz have an early peak at 1 hr post Lipid A stimulation, but Tnf shows a second wave of sustained expression at 4–12 hr, while Nfkbiz does not (Figure S1). Although some studies have extended to later time points (Escoubet-Lozach et al., 2011; Kawagoe et al., 2008; Litvak et al., 2009; Qiao et al., 2013; Yamamoto et al., 2004), the mechanisms that can sustain TLR-induced gene activation for up to 12 hr are not well characterized. Many key immune mediators induced by TLR activation, such as Tnf, Il1a, and Il6, exhibit peak expression >6 hr after ligand challenge (Figure S1). Since extended mRNA transcription may be required for these critical cytokines to accumulate to an effective expression level, it is important to understand specific mechanisms that support sustained gene activation beyond 6 hr.
The mechanisms regulating TLR pathway crosstalk and sustained gene expression may be related, as multi-PAMP activation often leads to synergistic enhancement of late-peaking T cell polarizing cytokines such as interleukin-6 (IL-6). To better understand the consequences of TLR signaling crosstalk, and the mechanisms regulating sustained TLR-induced gene expression, we first conducted a comprehensive microarray analysis with four single and six pairwise TLR ligands across a time course to understand the requirements, kinetics, and outcome of combined TLR signaling pathway engagement in primary mouse macrophages. We identify a clear and selective pattern of MyD88/TRIF pathway interaction that leads to synergistic and sustained expression (>6 hr) of a subset of TLR-induced genes. We then conducted a further microarray analysis with a higher resolution time course and used transcriptional kinetics to implicate a set of 218 candidate regulators. Using IL-6 secretion as readout, we perturbed each of these candidate regulators in a focused RNAi screen. Screen hits were validated against a larger panel of immune effectors and regulators. We identify several genes required to support TLR-induced sustained immune effector expression (>6 hr), which could be future targets for clinical regulation of TLR-mediated inflammatory conditions.
RESULTS
Dual TLR-Ligand-Induced Pathway Crosstalk Enhances Cytokine Secretion beyond Maximal Single Ligand Responses
TLR ligation can lead to the activation of different signaling pathways through the MyD88 and/or TRIF adaptor proteins, and prior studies have shown that MyD88/TRIF pathway interactions can lead to both suppressive (Amit et al., 2009; Hacker et al., 2006) and synergistic responses (Bagchi et al., 2007; Napolitani et al., 2005; Suet Ting Tan et al., 2013). In addition to their differences in adaptor usage, the TLR receptor family also varies with respect to their cellular localization and ligand origination (Figure 1A) (Takeuchi and Akira, 2010), and these factors are also proposed to affect TLR pathway crosstalk (Napolitani et al., 2005). To investigate systematically the characteristics of pathway crosstalk among TLRs with varied features, we selected a panel of four TLR ligands: R848, poly(I:C), Pam3CSK4, and LPS (Figure 1A). Among these PAMPs, R848, and poly(I:C) are recognized in the endosome (by TLR7/8 and TLR3) as mimics of single- and double-stranded RNA, respectively (Alexopoulou et al., 2001; Hemmi et al., 2002), Pam3CSK4 is recognized at the cell membrane by TLR2/1 as a mimic of bacterial lipopeptide (Takeuchi et al., 2001), while LPS derived from a Gram-negative bacterium is recognized by TLR4 sequentially at the cell membrane and the endosome (Kagan et al., 2008; Takeuchi and Akira, 2010). R848 (R) and Pam3CSK4 (P) signal through the adaptor MyD88 (Hemmi et al., 2002; Takeuchi et al., 2001; Yamamoto et al., 2003a), poly(I:C) (I) signals through TRIF (Yamamoto et al., 2003a), while LPS (L) signals initially through MyD88 at the cell membrane, then also TRIF as it traffics to the endosome (Kagan et al., 2008; Kawai et al., 1999; Yamamoto et al., 2003a).
Figure 1. TLR Pathway Crosstalk Enhances Cytokine Secretion beyond Maximal Single Ligand Responses.

(A) Features of four representative TLR ligands selected for analysis of pathway crosstalk.
(B) Mouse BMDMs were stimulated with increasing concentrations of single or dual TLR-ligand combinations of poly(I:C) and R848, as indicated in (C), for 24 hr, and cytokine secretion was measured by ELISA. Data are presented as mean ± SD of three independent experiments. Significant differences between the observed dual ligand-stimulated cytokine level and the calculated additive output of the combined single ligand values are shown. ****p < 0.0001 (two-tailed t test).
(C) TLR-ligand doses used for evaluation of IL-6 and IL-12 p40 responses (Figure S2). The doses shown in red were selected for subsequent experiments.
First, we tested whether the previously described synergy in the TLR pathways (Bagchi et al., 2007; Napolitani et al., 2005; Suet Ting Tan et al., 2013) could be simply due to an exponential dose response that could also occur with single TLR ligands. To address this question, we tested a comprehensive stimulation matrix of four doses of each TLR ligand, either singly or in all possible pairwise combinations, and measured cytokine secretion from mouse bone-marrow-derived macrophages (BMDM). We observed dual ligand-dependent synergy across a wide TLR-ligand dose range in the secretion of IL-6 and IL-12p40 (Figures S2A and S2B). Closer examination of the effects of dual exposure to a MyD88-specific (R848) and a TRIF-specific (poly(I:C)) ligand pair revealed IL-6 and IL-12p40 responses that far exceeded those seen at saturating amounts of the single ligands (Figure 1B). This suggests that the interaction of the MyD88 and TRIF pathways leads to a level of macrophage activation that is not achievable through increased activation of either single pathway. To focus our further analysis on characteristics of synergy that occur beyond saturated single ligand dose responses, we chose saturation or near saturation TLR-ligand doses for subsequent experiments (Figures S2C, S2D, and 1C).
Transcriptional Analysis of TLR-Ligand Combinations in Primary Macrophages Identifies Inter- and Intra-TLR MyD88/TRIF Crosstalk
To investigate how different TLR-ligand combinations regulate the macrophage transcriptional response, we employed micro-array analysis using RNA from BMDMs activated for 1, 4, and 8 hr with either single or pairwise combinations of the four TLR ligands. This permitted the assessment of both the early and late phases of TLR-induced transcription. We measured the mRNA induction from each single ligand and, using a variation of a previously described model (Zhu et al., 2006), we calculated the predicted additive gene expression for each TLR-ligand pair (see STAR Methods). To investigate the global pattern of dual TLR-activated transcription, we compared observed and predicted additive gene expression for each TLR-ligand pair for 9,134 genes induced in at least 3 of the 30 tested conditions (see STAR Methods). This analysis identified several different response patterns dictated by the profile of MyD88/TRIF engagement by the two ligands (Figure S3; Data S1). A pattern of near additive response was observed for the combinations of the MyD88- and TRIF-specific ligands, P3C + PIC and R848 + PIC, suggesting the relative independence of the MyD88 and TRIF pathways (Figure S3A). In contrast, in all other pairs, where both ligands could activate a common adapter, a broadly less than additive response was shown, especially for the MyD88-only combination of R848 + P3C (Figures S3B–S3D). Only a small subset of genes showed a clearly greater than additive response (Figure S3, points above red diagonal), and such synergistic responses were most evident in the combinations of MyD88- and TRIF-specific ligands (Figure S3A).
To examine the non-additive transcript responses more closely, we measured the difference in expression (DIF value) between the predicted gene response and the actual expression level measured from dual TLR-stimulated cells, and defined criteria for genes showing saturated, additive, synergistic, and antagonistic dual ligand-induced transcription (Figure 2A; STAR Methods). Particularly, we define antagonism as a dual ligand response lower than the highest single ligand response, in order to separate the effect of saturation from active antagonism of expression (Figure 2A). Among the 9,134 induced transcripts, only 282 genes showing synergy or antagonism in at least one of the six ligand combinations, with 188 showing synergy, 127 showing antagonism, and 33 showing both synergy and antagonism at different time points (Figure 2B; Data S2). Among the 188 synergized transcripts, 80 have a DIF >4, and a further 33 have a DIF >8, indicating that a substantial proportion of these transcripts are robustly synergized. The predominantly additive/saturation response observed for most genes in dual TLR-stimulated macrophages suggests that simultaneously activated TLR pathways can act independently for induction of most genes, and that only a selective subset of genes are regulated by pathway crosstalk.
Figure 2. Transcriptional Analysis of TLR-Ligand Combinations in Primary Macrophages Identifies Inter- and Intra-pathway MyD88 and TRIF Interactions.

(A) Schematic for calculation of non-additivity (DIF value) in dual TLR-ligand transcriptional responses. Fold-inductions relative to the non-treated sample are calculated for single ligand-stimulated samples (PAMP A or PAMP B) and dual ligand-stimulated samples, with the basal expression in non-treated sample taken as 1-fold. Synergy (red) is defined as a dual ligand response more than the simple additive level of the two single ligands. Antagonism (blue) is defined as a dual ligand response less than the highest single ligand response.
(B) Left: clustering of 282 transcripts with a DIF ≥2 or DIF ≤ −2 under 6 pairwise PAMP combinations at 3 time points. Antagonized/synergized fold DIF values (converted to log2 scale), are indicated by the color gradient of blue/red respectively, while additive or saturated outcomes are white. Right: percent mRNA expression normalized to the maximum level (100%) for each gene, under basal conditions, after treatment with a MyD88 ligand alone, a TRIF ligand alone, or with LPS or dual ligand. For the MyD88 alone (either R848 or Pam3CSK4) and dual ligand conditions (six pairwise PAMP combinations), the highest expression value is shown. Dual ligand combinations are shown in one-letter abbreviations as follows: I, poly(I:C); R, R848; P, Pam3CSK4; L, LPS.
(C) Number of gene data points (across the 8 hr time course) showing synergy or antagonism under different dual ligand treatments. One pairwise PAMP combination at one time point is counted as one data point.
(D) BMDMs from WT or the indicated strains of knockout mice were stimulated with single or dual TLR ligands (25 ng/mL R848, 10 μg/mL PIC, 10 ng/mL P3C, 5 nM Lipid A) for 24 hr, and IL-6 secretion was measured by ELISA (mean + SD).
(E) qPCR validation of a subset of synergized genes showing internal synergy between the MyD88 and TRIF branches of the TLR4-LPS pathway. Left: antagonized/synergized fold DIF values are indicated by the color gradient of blue/red, respectively, while additive or saturated outcomes are white. Right: percent mRNA expression in either Myd88−/− cells or Trif−/− cells normalized to the maximum Lipid A response of WT cells.
Data were derived from duplicate microarrays (A–C) (see STAR Methods; Figure S4; Data S2), duplicate experiments from two separate mice (D), **p < 0.01, ***p < 0.001, ****p < 0.0001 (two-tailed t test), and from duplicate qPCR experiments from two separate mice (E) (see Data S3). Ligand abbreviations: R (R848); I or PIC (poly(I:C)); P or P3C (Pam3CSK4); L (LPS).
Hierarchical clustering of the 282 non-additive transcripts revealed that non-additive dual TLR responses are driven primarily by crosstalk between the TRIF and MyD88 pathways, and less related to other features of the TLRs such as cellular location and ligand origin (Figure 2B). This is evident by the fact that the TRIF-specific ligand poly(I:C), in combination with either of the MyD88-specific ligands (Pam3CSK4 or R848), induces the highest number of both synergized (169) and antagonized transcripts (89) compared with other ligand combinations (Figures 2B and 2C, IR and IP combinations). In contrast, the combination of only MyD88-specific ligands (R848 + Pam3CSK4) led to an almost exclusively saturated response, with only two transcripts slightly synergized and 11 antagonized (Figures 2B and 2C and Data S2). The synergistic response to poly(I:C) combined with either R848 or Pam3CSK4 was confirmed to be mediated by TRIF and not the MAVS-dependent RIG-I or MDA5 pathways, as BMDM from Trif−/−, but not Mavs−/− mice showed loss of synergy for IL-6 (Figure 2D).
To determine whether the pattern of MyD88-TRIF crosstalk also exists within the TLR4-LPS pathway (which activates both the MyD88 and TRIF branches), we measured mRNA expression by qPCR of a subset of synergized genes in Myd88−/−, Trif−/−, and wild-type (WT) BMDMs stimulated with Lipid A (the activating component of LPS) for 1, 2, 4, 8, 12, and 20 hr (Figure 2E; Data S3). Synergy level was determined as the DIF between WT cells compared with the sum of expression from Myd88−/− and Trif−/− cells (Figure 2E, left panel). All genes showed a synergy pattern of minimal expression driven by either the MyD88 or TRIF pathway alone, but strong induction with Lipid A activation of both pathways in WT cells (Figure 2E, right panel). We also noted that, in Trif−/− macrophages, the Lipid A-activated MyD88 pathway alone induced no substantial IL-6 secretion, while the MyD88-specific ligands, R848 and Pam3CSK4, were able to induce IL-6 at a low level (Figure 2D). This might be explained by TLR4 trafficking and the transition from MyD88 to TRIF in LPS/Lipid A responses (Kagan et al., 2008), where a shorter-term engagement of TLR4 with MyD88 is insufficient to induce IL-6 in the absence of TRIF responses. These data also support an important role for synergism between the MyD88 and TRIF pathways to facilitate a more robust activation of the innate immune system in response to LPS.
This inherent internal MyD88-TRIF crosstalk in the TLR4-LPS pathway is further supported by our observations in dual TLR-stimulated macrophages. As shown in Figure 2B, ligand combinations with LPS are complicated by the internal crosstalk (Hoebe et al., 2003; Kawai et al., 1999; Yamamoto et al., 2003a), such that the outcome of adding another ligand depends on whether the internal synergy in the LPS response has been saturated. At our employed LPS ligand dose of 10 ng/mL, we observed only 23 transcripts synergized in the LPS + poly(I:C) (LI) combination (Figures 2B and 2C), suggesting near saturation of the TRIF branch at this LPS dose. In contrast, LPS + R848 or Pam3CSK4 (LR/P) combinations retained 64 out of the 169 transcripts synergized in pure MyD88 + TRIF combinations (Figures 2B and 2C), suggesting less saturation of the MyD88 branch by LPS. As suggested above, the transition from MyD88 to TRIF activation in the LPS response may lead to less-sustained MyD88 activation than can be achieved by a dedicated MyD88-activating ligand.
TLR Signaling Crosstalk Simultaneously Synergizes and Antagonizes Different Gene Subsets
We noted that all of the 188 synergized transcripts showed synergy in either the poly(I:C) + R848/Pam3CSK4 (IR/P) or LPS + R848/Pam3CSK4 (LR/P) combinations, and there were no transcripts selectively synergized in either the LPS + poly(I:C) (LI) or R848 + Pam3CSK4 (RP) combinations. We classified the 188 synergized genes into 4 primary clusters, based on their behavior in the four major synergy combinations poly(I:C) + R848 (IR), poly(I:C) + Pam3CSK4 (IP), LPS + R848 (LR), and LPS + Pam3CSK4 (LP) (Figure 2B, left panel; Data S2). We also include a heatmap of comparative expression of each transcript in response to a MyD88 ligand alone, a TRIF ligand alone, LPS, and the maximum response to dual ligand to observe the typical expression characteristics of the gene clusters (Figure 2B, right panel; Data S2). Cluster 1 is composed of genes synergized in all the IR, IP, LR, and LP ligand combinations, and includes Il1a, Il27, Il6, Il10, Edn1, Nos2, Lcn2, and Cd40. Genes in this cluster generally have higher synergy than other clusters due to low induction by either MyD88 or TRIF alone (Figure 2B, right panel). Cluster 2 contains genes, including Socs3, Il1b, and Il18, synergized in both IR and IP combinations but not synergized in LR and LP combinations, likely due to internal synergy inside the LPS pathway already maximizing their expression. Cluster 3 genes are also selectively synergized by the pure Myd88 + TRIF IR and IP combinations, but are noteworthy as they separate into IR- and IP-specific subgroups (Figure 2B, left panel; Data S2). It is possible that the ligand specificity of these genes relates to receptor location and specific aspects of pathway crosstalk that require either TLR7/TLR3 co-localization in the endosome for the IR-synergized genes, or separate activation of TLR2 and TLR3 (at the cell surface and endosome, respectively) for the IP-synergized genes. Cluster 4 are genes synergized in LR/P conditions but not in IR/P conditions. These genes are induced to a lesser extent by TLR activation than genes in clusters 1 to 3, and their LR/P-specific synergy is primarily due to a slightly stronger response to LPS than the other single ligands (Figure S4).
In addition to the 188 synergized transcripts, we observed 127 antagonized transcripts with a DIF of ≤2-fold in at least one sample (Figure 2B; Data S2). We find that the antagonized transcripts separate into two clusters (5 and 6) of genes that are highly induced by either a TRIF-activating ligand (cluster 5) or a MyD88-activating ligand alone (cluster 6), and that they typify genes involved in interferon and inflammatory gene programs, respectively (Figure 2B). While mechanisms of antagonistic effects between certain inflammatory and interferon transcriptional programs induced by MyD88 and TRIF have been proposed (Amit et al., 2009; Hacker et al., 2006), the synergistic interaction between the two pathways is poorly understood. Moreover, it is unclear how such differential non-additivity can emerge for these different classes of genes in response to the same stimuli.
Transcription Factor Motif and Pathway Analysis Identifies Selective Characteristics of TLR Non-additive Response Genes
To further interrogate the properties of the synergized and antagonized gene clusters, we conducted motif analysis to determine whether similarities or differences in predicted regulatory transcription factors might relate to the gene cluster groupings. We used DiRE to identify shared regulatory elements in gene sets both within and outside of proximal promoter regions (Gotea and Ovcharenko, 2008). Interestingly, the four synergized clusters (1–4) showed a pattern of shared nuclear factor κB (NF-κB) enrichment at both promoter and distal regulatory sites, but with selective requirements for additional transcription factors (TFs) from the AP1/ATF, IRF, STAT, and ETS families (Figure 3A). The most strongly synergized clusters 1 and 2 were the only group enriched for AP1/ATF, suggesting an important role for MAPK signaling for these gene subsets, while clusters 3 and 4 showed a selective requirement for ETS family members. The antagonized gene clusters lacked an NF-κB enrichment in their distal regulatory regions, and showed no AP1 or ETS enrichment (Figure 3B).
Figure 3. Transcription Factor Motif and Pathway Analysis of TLR Non-additive Response Genes.

(A and B) DiRE analysis of enriched transcriptional regulatory motifs at the promoter and promoter + distal regions of (A) synergized and (B) antagonized gene clusters as identified in Figure 2B. Members of recurrent NF-kB, AP1, IRF, STAT, and ETS transcription factor families are highlighted.
(C) Ingenuity pathway analysis (IPA) functional annotation of genes within the synergized and antagonized gene clusters as identified in Figure 2B.
Both synergized and antagonized gene clusters showed enrichment for IRF and STAT TFs that would not be directly activated by the primary TLR pathway response. This may suggest an important role for autocrine/paracrine type I interferon and other cytokine responses which could feedback on the TLR-activated macrophage to modulate the non-additive immune effector responses promoted by combined TLR activation (Xue et al., 2015). Such mediators likely also perform a key function in innate/adaptive immune cell communication through leukocyte recruitment and activation, as demonstrated by the selective functional annotation of the synergized gene clusters derived from ingenuity pathway analysis (IPA) (Figure 3C).
Synergized Transcripts Are Enriched for Sustained Immune Effector Genes
Of the 188 synergized transcripts, 181 are induced by at least one single ligand, with only 7 of them dual ligand specific (Data S2). All 181 transcripts induced by a single ligand are inducible by LPS, 16 of them are LPS specific, 34 are MyD88 specific (including Lcn2, Il19, Serpinb2, Il12a, Il6), 18 are TRIF specific (including Ifnb1, Ifit3, Mx1, Oasl1, Stat3, Irf1), and 113 are inducible by either pathway alone (Figure S5A; Data S2). While some transcripts are only induced by one pathway, they are further enhanced by the “non-inducing” pathway.
A closer examination of the synergized genes reveals that they are highly enriched for immune effectors rather than signaling mediators (Figure S5B). Functional annotation in IPA also reveals that synergized genes are strongly and specifically enriched for roles in leukocyte activation, proliferation, and accumulation (Figure 3C). Specifically, these genes are either cytokines (Il1a, Il27, Il19, Il1b, Ccl12, Cxcl2, Cxcl9), secondary immune effectors (Lcn2, Edn1, Mmp13), or immune suppressors with key roles in resolution of inflammation and tissue repair (Socs3, Dusp2, Il10, AA467197 [miR-147]) (Figure S5B). This suggests that selective transcriptional synergy in response to combined TLR stimuli plays a central role not only in triggering an inflammatory immune response, but also in promoting the subsequent process of immune attenuation and resolution. A case in point is the highly synergistic induction of Il1a and Il1b, along with the IL-1 receptor antagonist (Il1rn), possibly to facilitate a timely shut down of the inflammatory effects of IL-1 signaling.
As many of the immune effectors are sustained for up to 12 hr, we wanted to determine whether this sustaining expression is a general feature for the synergized transcripts. For this purpose, we carried out a microarray analysis of poly(I:C) + R848 (single and dual ligand) stimulated BMDM across a more detailed time course of 0.5, 1, 2, 4, 8, and 12 hr (Data S4). We identified 86 induced transcripts with a DIF of ≥2-fold in at least one condition (Data S5). Similar to our initial transcript analysis (Figure 2B), the genes synergized at the later time points are enriched for immune effectors, including Il6, Il1a, Il1b, Cxcl1, Cxcl2, and Lcn2 (Figure 4A). A closer examination of the expression kinetics found that among the 86 poly (I:C) + R848 synergized transcripts, 38% peaked at 8 hr and 23% peaked at 12 hr under R848 stimulation (Figure 4B, middle panel). Dual ligand treatment further enhanced this trend, as the proportion of transcripts peaking at 12 hr increased to 43% (Figure 4B, bottom panel). Calculation of the average expression across the time course indicated an intrinsic trend of late and sustained expression (>6 hr) for the synergized transcripts (Figure 4C). However, while most of these genes tend to have a later onset of expression, some of them (e.g., Cxcl1 and Cxcl2) are induced early at 1 hr (Data S5).
Figure 4. Time-Resolved Transcriptional Analysis Reveals Properties and Kinetics of Synergy.

(A) Predicted and observed mRNA expression levels in BMDM stimulated with 10 μg/mL poly(I:C) + 25 ng/mL R848 for 4, 8, and 12 hr. Predicted mRNA expression was calculated by summing the net fold induction of the two single ligand stimulations for 7,342 transcripts induced in at least 2 of the 18 tested single or dual TLR-ligand conditions. Each dot represents one upregulated gene, and the predicted versus observed mRNA expression values are plotted. Red lines are the diagonal reference for additive expression if the predicted and observed mRNA levels are the same, while green lines are fitted to the observed data points.
(B) mRNA expression kinetics of 86 synergized transcripts under dual and single ligand treatment. Percent mRNA expression normalized to the maximum level (100%) for each gene across all time points of the same treatment is shown in a color gradient from blue (lowest) to red (highest) expression.
(C) Percent gene expression (mean ± SEM) of all 86 synergized transcripts (averaged) over a 12 hr time course of single and dual ligand treatment.
(D) Distribution of dual ligand-induced gene expression synergy (DIF value) for the 86 synergized transcripts over a 12 hr time course of poly(I:C) + R848 stimulation.
(E) Kinetics of synergy emergence among the 86 synergized transcripts over a 12 hr time course of poly(I:C) + R848 stimulation, as in (D).
Data were derived from duplicate microarrays; see STAR Methods; Data S4 for (A) and Data S5 for (B–E). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Two-tailed t test (C) and Wilcoxon signed rank test (D).
We also analyzed the kinetics of synergy for 86 transcripts over the higher resolution time course (Figures 4D and 4E). The number of transcripts synergized and the degree of synergy steadily increased from 0.5 to 12 hr, with more than 80% of the 86 transcripts showing synergy at 8 and 12 hr. Although synergy peaks late, it initiates early between 0.5 and 1 hr, with the proportion of transcripts synergized increasing by more than 2-fold from 0.5 to 1 hr (9.3%–25.6%).
A Focused siRNA Screen for Regulators of Dual TLR-Ligand-Induced IL-6 Secretion
While the transcriptional regulatory motif analysis of synergized genes predicted co-regulation by numerous TF families (Figure 3A), it did not identify a common regulatory factor unique to all synergized genes. To further investigate candidate regulators for the sustained TLR response, we interrogated our microarray data for genes showing an expression change of at least 1.5-fold at 0.5 or 1 hr after ligand treatment, when synergy just initiates, as the regulators for sustaining the response would likely be enriched in the synergy context (Figure 5A). Based on these and related criteria, we identified a total of 218 candidate genes for primary RNAi screening (Data S6).
Figure 5. A focused siRNA Screen for Regulators of Dual TLR-ligand-Induced IL-6 Secretion.

(A) Selection criteria for putative regulators for the primary RNAi screen (see STAR Methods; Data S6).
(B) Workflow for the RNAi screen employing three siRNA sequences per gene distributed in separate regions of duplicate 384-well plates (red circles show example of siRNA locations for a single gene). Blue region of plate, gene-specific siRNA; orange region, control siRNA. At 48 hr after reverse transfection of siRNA, mouse RAW G9 cells in duplicate plates were treated for a further 24 hr with either single or dual TLR ligands, and IL-6 secretion was measured by ELISA (see STAR Methods; Data S7).
(C) Secondary RNAi screen of 24 putative hits from the primary screen. IL-6 secretion levels are shown normalized to the dual ligand response of non-target control siRNA-transfected cells. The range of expression variation for non-target control siRNA data points are shown in the blue and pink shaded regions for single and dual ligand treated cells, respectively. The data points for the three siRNAs per gene are shown as stars, circles, and triangles, with single- and dual-ligand response data point in blue and red, respectively. Representative data from one of three independent screening experiments is shown.
Using previously established methods for robust small interfering RNA (siRNA)-based gene knockdown in RAW264.7 G9 macrophages (RAW G9) (Li et al., 2015), each gene was targeted by three independent siRNAs in separate wells (Figure 5B). Plates were run in duplicate to permit stimulation with either R848 alone or poly(I:C)+R848 (Figure 5B). As poly(I:C) alone does not induce any IL-6 in RAW G9 cells under the screen conditions, a separate plate was not required for this ligand. Supernatants were collected and subjected to ELISA for IL-6. After data normalization, the effect of the three siRNAs per gene were evaluated for both single and dual ligand-induced IL-6 levels (see STAR Methods). The primary screen analysis implicated 24 genes as regulators of TLR-induced IL-6 (Data S7), including known positive and negative regulatory genes, such as Irak2 and Tnfaip3 (Figure S6A). These 24 genes were subjected to secondary screening with a modified protocol that combined the R848 alone and poly(I:C) + R848-treated wells in the same screening plate to reduce variability (Figure S6B). A high validation rate was observed in the secondary screen, with 22 of 24 genes replicating the primary screen phenotype (Figure 5C; Data S7).
In further investigating the putative screen hits, pathway analysis suggested that the 22 hits separated into 3 subnetworks. The best-characterized genes, such as Junb, Nfkbiz, and Tank, are incorporated into a heavily interconnected network around Il6, providing an important validation of our screen (Figure 6A). Two histone hits form a second network around the NF-κB complex (Figure S7A). A third network, centered around the known transcriptional regulators Pparg, Sp1, and Hnf4a, contains many of the poorly characterized hits identified in our screen, but these putative regulators are distributed on the edge of the network, often with single connections to known components, consistent with the relative novelty of the hits (Figure 6B).
Figure 6. Pathway Analysis of Network Interactions among Screen Hits.

(A and B) Network analysis of screen hits using IPA identifies (A) a network of hits with direct and indirect associations with IL-6 regulation, and (B) an interconnected network around the transcriptional regulators PPARG, SP1, and HNF4A. Screen hits are indicated by gray shading, genes connected to the hits are white. Hits selected for follow up are highlighted. Arrows indicate a known functional link between the genes.
(C) Protein domain analysis of the screen hits HELZ2, PHF11D, ZSCAN12, and SERTAD3, conducted using SMART (http://smart.embl-heidelberg.de).
Newly Identified Regulators Preferentially Regulate TLR-Induced Sustained Gene Expression
While our screen provides important supporting data linking numerous well-studied genes to regulation of IL-6, and possibly to the broader TLR-induced gene program, we focused our further investigation on four genes not previously associated with regulation of inflammatory mediator expression downstream of TLR activation, Helz2, Phf11d, Sertad3, and Zscan12. Protein domain-scanning analysis identified several different domains in the four hits (Figure 6C), but with a unifying theme that indicates possible roles in transcriptional regulation. For example, ZSCAN12 contains multiple repeats of the C2H2 type zinc finger DNA-binding domain, and PHF11D contains the zinc-finger-like PHD domain, suggesting these two hits might serve as TFs. Although no known DNA-binding motifs are found in SERTAD3 and HELZ2, they contain additional conserved domains and have been reported to function as transcriptional co-activators (Cho et al., 2000; Surapureddi et al., 2002; Tomaru et al., 2006).
To determine if the effects of these four hits extend beyond IL-6, we used microfluidic qPCR to interrogate a broad panel of immune genes induced by TLR activation. We targeted the four screen hits with the two most potent siRNAs from our screen, using an optimized protocol for efficient siRNA-based knockdown in primary macrophages (see STAR Methods; Figure S7B). The transfected BMDMs were treated with 5 nM Lipid A across an 8 hr time course, and mRNA expression was measured for 41 genes from the previously identified “synergized” group (Figure 2B) plus 22 prominent immune regulators (Data S3). We find that all four hits preferentially support late gene expression, particularly at the 8 hr time point. None of the hits have an effect on basal expression, with only a few genes affected at 1 or 2 hr (Figure 7A; Data S3). IPA analysis of the genes affected by the screen hits shows enrichment for cytokines that mediate innate/adaptive immune cell communication and for polarization of T helper cell subsets (Figure 7B). The important immune effectors Mmp13, Socs3, Spic, and Il12b are enhanced by all four hits, while Il27, Il1a, Il6, Nos2, and Lcn2 are enhanced by at least two of the hits. Many of the genes modulated by the hits are cytokines, which often cross-regulate each other, so the gene set we measured here could be direct or indirect targets of the four screen hits. We also find that the number of genes affected by Phf11d and Zscan12 is greater than those affected by Helz2 or Sertad3. Seventeen genes are not affected by any of the four hits, suggesting further selectivity in the regulation of TLR-induced immune effector genes. This pattern of selective regulation is also validated at the protein level (Figures 7C–7F). Discovery of these hits with selective activity on late gene expression provides new insight to regulation of TLR-mediated sustained gene expression.
Figure 7. Screen Hits Preferentially regulate sustained TLR-Induced Transcription of Immune Effector Genes.

(A) Left panel: effect of Helz2, Phf11d, Zscan12, and Sertad3 screen hit knockdown on Lipid A-induced mRNA expression of 63 immune related genes in mouse BMDM. Each hit was knocked down by two independent siRNAs in separate wells, then the BMDMs were treated with 5 nM Lipid A for 0, 1, 2, and 8 hr. mRNA levels were assayed by Fluidigm microfluidic qPCR (see STAR Methods; Data S3). Genes with consistent and significant reduction of TLR-induced expression (assessed by a two-way ANOVA analysis) by both siRNAs, in each of two replicate experiments are shown as cyan. Right panel: induction kinetics of each gene. Percent mRNA expression in 0, 1, 2, and 8 hr relative to the maximum expression (100%) is shown in a color gradient from blue (low) to red (high).
(B) IPA of genes selectively affected by screen hit knockdown shows enrichment for innate/adaptive immune system communication and T cell polarization.
(C–F) BMDM transfected with control or gene-specific siRNA were stimulated with Lipid A (5 nM) for 24 hr, and the indicated cytokine secretion levels were measured by ELISA. Data are mean + SD of two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA).
DISCUSSION
The numerous studies on the architecture of TLR signaling pathways since their discovery reflects the importance of these pathways to the immune response; however, the comparatively limited analysis of how TLR pathways interact when challenged with a multi-PAMP microbe leaves us with many questions on how simultaneously engaged pathways cooperate to mount an optimal innate immune response. Here we describe a systematic approach to this issue that combines a comprehensive transcriptomic analysis with a focused RNAi screen to identify both the characteristics and putative modulators of dual TLR-induced responses in macrophages. Our data clearly support a model where TLR pathway crosstalk is mediated through combined activation of the two branches of TLR signaling downstream of the MyD88 and TRIF TIR-domain adaptor proteins.
A key observation in this regard is the unique characteristic of the TLR4 agonist LPS, which can essentially mimic the robust outcome of combined MyD88 and TRIF activation with a single ligand. This suggests a host requirement to mount a more robust transcriptional response to LPS over other TLR ligands, possibly due to the inherent dangers from exposure to this stimulus. While the LPS-activated TRIF pathway induces what is often considered an antiviral gene program, its activation by a bacterial ligand suggests an important function in the response to Gram-negative infection. One possibility is that it might function as an amplifier of late/sustained TLR-induced gene expression. Indeed, multi-pathway cooperation is a common strategy for stringent control of late gene expression intensity and kinetics in immune cells, such as the interferon gamma and CD40 ligand cooperation in enhancing inflammatory gene expression in dendritic cells and macrophages (Abdi et al., 2012; Napolitani et al., 2005; Qiao et al., 2013; Schulz et al., 2000). In this context, activation through a single pathway is not sufficient to induce robust and sustained expression (>6 hr). Moreover, as initial signaling usually generates secondary events, such multi-pathway synergy takes longer than single pathway signaling, and this, in part, can determine the later onset kinetics of synergized immune effectors. This stringent regulation of both expression levels and kinetics is likely an important factor in proper temporal regulation of immune responses. The MyD88-TRIF cooperation inside the TLR4-LPS pathway (Figure 2C) (Hoebe et al., 2003; Kawai et al., 1999; Ramsey et al., 2008; Yamamoto et al., 2003a) fits such a model, as knockout of either adapter pathway has dramatic effects on many synergized late/sustained effector genes (Kawai et al., 1999; Yamamoto et al., 2003a). Above all, the recruitment of both MyD88 and TRIF signaling branches by the TLR4 pathway is likely a mechanism to employ multi-pathway cooperation to deal with a dangerous infectious stimulus.
Since we observe that productive inter-TLR pathway crosstalk essentially parallels the MyD88/TRIF crosstalk inside the TLR4-LPS pathway (Figure 2D), synergistic responses to combined TLR ligands could be a means to mimic the robust innate response to endotoxin, and could represent a metric for the host to assess the danger level of an infection (Chen and Nunez, 2010; Gallucci et al., 1999; Matzinger, 2002; Shi et al., 2003). In this scenario, the co-presence of signals activating both pathway branches may indicate a dangerous situation that requires MyD88-TRIF cooperation to sustain the transcriptional response for immune effector gene expression. It is also possible that the MyD88 and TRIF branch could represent different classes of danger/infection signals. While the likelihood of synergistic MyD88-TRIF activation would be increased by co-infection with multiple pathogens, we do not find any evidence in our transcriptional analysis that suggests there are selective responses to ligand pairs that could mimic co-infection.
Regulation of the sustained TLR-induced gene program is poorly understood compared with the well-characterized early activation events (Smale, 2010; Takeuchi and Akira, 2010; Tong et al., 2016). Since these sustained genes, such as IL-6 and IL-12, play important roles in the later adaptive immune response, it is important to understand how their expression levels are maintained at high levels in innate immune cells encountering combined microbial ligands. Here, we carried out a targeted RNAi screen and identified numerous regulators of sustained gene expression. Our success in identifying regulators by selecting de novo transcribed candidates highlights the importance of de novo protein synthesis for late gene regulation. We focused on genes induced at 0.5 to 1 hr when the synergy initiates, assuming that regulators would be enriched in this time frame. However, considering the fact that the greatest synergy is observed at later time points, it is likely that additional regulators are induced after 1 hr, and we expect further investigation will reveal additional regulators and provide insights to the mechanism of late gene regulation.
In summary, we have carried out a comprehensive analysis of TLR pathway crosstalk in macrophages to gain insight to the function and regulation of TLR-induced gene expression. Our systematic analysis reveals a pattern of inter- and intra-TLR pathway MyD88-TRIF cooperation facilitating immune gene expression, especially for late response genes. This pathway structure may be designed to optimally sense dangerous infectious signals, either from a single TLR4 ligand in the case of sepsis-inducing Gram-negative bacterial infections, or from a combination of different classes of infection/danger signals separately inducing the MyD88 and TRIF pathways. We have further identified several genes that selectively regulate TLR-induced sustained immune effector expression, which could be viable targets for clinical regulation of TLR-driven inflammatory conditions.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS (Salmonella minnesota R595 TLRgrade) | Alexis Biochemicals, San Diego, CA | ALX-581-008-L002 |
| Pam3CSK4 | InvivoGen, San Diego, CA | tlrl-pms |
| R848 | InvivoGen, San Diego, CA | tlrl-r848 |
| poly(I:C) (low molecular weight) | InvivoGen, San Diego, CA | tlrl-picw |
| lipid A | Avanti Polar Lipids, Alabaster, AL | 699500P |
| M-CSF | R&D, Minneapolis, MN | 416-ML-050 |
| Deposited Data | ||
| Microarray data | This paper | GSE89988 |
| Experimental Models: Cell Lines | ||
| RAW264.7 G9 cells | (Li et al., 2015) | |
| Experimental Models: Organisms/Strains | ||
| Mus musculus/C57BL/6J, male | Jax (https://www.jax.org/jax-mice-and-services) | Jax000664 | Black 6 |
| Mus musculus/C57BL/6J-Ticam1Lps2/J, male | Jax (https://www.jax.org/jax-mice-and-services) | Jax005037 | TrifLps2 |
| Mus musculus/B6.129P2(SJL)-Myd88tm1.1Defr/J, male | Jax (https://www.jax.org/jax-mice-and-services) | Jax009088 | Myd88 null |
| Mus musculus/B6;129-Mavstm1Zjc/J, male | Jax (https://www.jax.org/jax-mice-and-services) | Jax008634 | Mavs- |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Dr. Iain D.C. Fraser (fraseri@niaid.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Line
RAW264.7 G9 cells were derived from an authenticated batch of RAW264.7 cells used by the Alliance for Cell Signaling consortium (Li et al., 2015). RAW264.7 G9 cells were maintained in complete DMEM media, comprising DMEM with 4.5 g/L glucose without L-gluta-mine (Lonza, Walkersville, MD, cat# 12-614F/12), 10% FBS (Gemini Bio-Products, West Sacramento, CA), 2 mM Glutamine (Lonza) and 20 mM HEPES (Lonza).
Mice and Primary Cell Cultures
C57BL/6J, Myd88−/− (B6.129P2(SJL)-Myd88tm1.1Defr/J), Trif−/− (C57BL/6J-Ticam1Lps2/J) (Hoebe et al., 2003) and Mavs−/− (B6;129-Mavstm1Zjc/J) (Sun et al., 2006) mice were purchased from Jax (www.jax.org/jax-mice-and-services) and housed in a specific-pathogen-free mice facility for one week before use for bone-marrow cell harvesting. BMDM were prepared from 6–8 week old C57BL/6J male mice by the following procedure. Bone marrow was flushed out with complete DMEM, plated at a density of 0.3 million cells/ml in non-tissue culture treated flasks (BD bioscience, Chicago, IL), and cells were cultured in complete DMEM supplemented with 60 ng/ml of recombinant mouse M-CSF (R&D, Minneapolis, MN, cat# 416-ML-050) for 6 days. Mice were maintained in specific-pathogen-free conditions and all procedures were approved by the NIAID Animal Care and Use Committee (NIH).
METHOD DETAILS
TLR Ligand Stimulation
TLR ligand stocks were prepared as suggested by the manufacturer and stored at −80°C. Working stocks were diluted into complete DMEM at 10X concentration before use. TLR ligand sources: LPS was from Alexis Biochemicals, San Diego, CA, Salmonella minnesota R595 TLRgrade, cat# ALX-581-008-L002; Pam3CSK4 was from InvivoGen, San Diego, CA cat# tlrl-pms; R848 was from InvivoGen, cat# tlrl-r848; poly(I:C) (low molecular weight) was from Invivogen, cat# tlrl-picw; lipid A was from Avanti Polar Lipids, Alabaster, AL, cat# 699500P.
Microarray and Calculation of Non-Additivity
BMDM were plated at a density of 0.22 x 106 cells/ml in a 24-well dish (BD Falcon) in a total volume of 0.9 mL. 24 hr after plating, the cells were treated with single or dual TLR ligands diluted in 0.1 mL of complete DMEM, for time periods as indicated. Cells from 4 wells were pooled for total RNA extraction with RNeasy Mini Kit (Qiagen), and each condition was represented by two biological replicates. cRNA amplification and labeling were performed using the Illumina TotalPrep RNA Amplification Kit (Ambion), microarray hybridization and scanning protocols followed standard Illumina protocols. Signal data was extracted from the image files with the Gene Expression module (v. 1.9.0) of the GenomeStudio software (v. 2011.1), and Log2 signal intensity were determined. Microarray replicates for each condition routinely showed a high correlation of r > 0.99. For the four TLR ligand study (LPS, P3C, R848 and poly I:C), 9,134 genes were selected which showed a significant change in at least 3 of the 30 single and dual ligand conditions tested (Data S1). For the two ligand extended time course study (R848 and Poly I:C), 7,342 genes were selected which showed a significant change in at least 2 of the 18 single and dual ligand conditions tested (Data S4).
Non-additive differential expression values (DIF) in dual ligand transcriptional responses were calculated as previously described (Zhu et al., 2006) (see also Figure 2A). Briefly, microarray data points (log2-(treated/control)) without significant changes were set to 0. Log2 ratios were transformed into fold change by 2^(log2-ratio), and only transcripts showing >2-fold induction in at least two time points of one ligand treatment were selected for further analysis. Then the net fold change above or below 1-fold was calculated for each single/dual ligand. The sum of the net fold change of two ligands was taken as the predicted additive response. The DIF value was then calculated as the net fold change of the observed dual ligand response minus the predicted additive response (Figure 2A). As an extension to the previous model, we defined antagonism as a dual ligand response lower than the highest single ligand response, in order to separate a saturation outcome from active antagonism of expression (Figure 2A). Thus, fold antagonism was calculated by net fold change of dual ligand treatment minus net fold change of the highest single ligand response. Genes with DIF >= 2 were considered synergized, <= −2 were antagonized and −2 < DIF < 2 were considered as additive/saturated. Among the 9,134 induced transcripts, only 282 genes showing synergy or antagonism in at least one of the six ligand combinations, with 188 showing synergy, 127 showing antagonism, and 33 showing both synergy and antagonism at different time points (Figure 2B, Data S2). Among the 188 synergized transcripts, 126 showed synergy in at least 2 ligand combinations. Moreover, 80 transcripts exhibited a DIF > 4, and 33 a DIF > 8, suggesting a robust and selective synergy response. Hierarchichal clustering and heatmap generation was done with GENE-E (http://www.broadinstitute.org/cancer/software/GENE-E/index.html), and log2 (DIF) values were used for heat map plotting to better distinguish samples across the heatmap.
Cytokine ELISA
BMDM or RAW G9 cells were treated with single or dual TLR ligands for 24 hr and supernatants were collected for cytokine quantification by ELISA (Mouse IL-6 and IL-12p40 (BD biosciences) and mouse CXCL10, LCN2, and IL-10 ELISA duoset (R&D)). Capture antibodies were coated on Nunc MaxiSorp 384 well plates (cat# 464718) following the manufactors instructions, and plates were washed using a Biotek EL406 washer/dispenser. Each sample was assayed in triplicate, and the median of the three ELISA replicates was calculated.
Candidate Selection for RNAi Screening
Since predicted candidate regulators are newly synthesized and synergy is evident at 2 hr after dual TLR activation, we first selected all genes changed +/− 1.5-fold by 0.5 and/or 1 hr. If a gene was induced by both MyD88 and TRIF, we also required this gene to be synergized. By this criterial, a total of 218 genes were selected for RNAi screening (Data S6).
RNAi Screening
RNAi screening was carried out using the RAW G9 cell clone as described previously (Li et al., 2015). Briefly, 2 μl of siRNAs (2.5 μM) were spotted in 384-well plates (BD Falcon), and 0.2 μl of TransIT-TKO transfection reagent pre-mixed with 7.8 μl of Dulbecco’s phosphate buffered saline (DPBS) was added to each well. After incubation for 10 min at room temperature, 5,000 cells in 40 μl of complete DMEM were added per well for a final siRNA concentration of 100 nM. Cells were incubated at 37°C in a humidified atmosphere with 5% CO2 for 48 hr, media was replaced, and cells were stimulated with single or dual TLR ligands as indicated. Culture supernatants were harvested at 24 hr post ligand stimulation for cytokine quantification by ELISA. Each gene was targeted by 3 independent siRNAs from Thermofisher (Data S6), and the RNAi screen was repeated 3 times. The screen data were normalized to the non-targeting negative control siRNA (NTC5; Dharmacon), and the IL-6 levels in NTC5-transfected wells was used to establish the range of variation of the single ligand R848 response (R cloud; R848 median ± R848 MAD (median absolute deviation)), and the dual ligand PIC+R848 response (IR cloud; PIC+R848 median ± PIC+R848 MAD; see Figure S6B). 36 and 48 NTC5-transfected control wells were used for R cloud and IR cloud determination, respectively (Figure S6B). The large number of control wells used here ensures that we get an accurate estimate of the data variation in the control siRNA condition, and we can more reliably define a hit by its deviation from the variation range in the control (R and IR clouds). If both R and IR responses were below their respective control variation clouds for a given gene in the screen, this would be considered as a possible positive regulator hit, while if both R and IR responses were above their respective clouds, this would be considered as a possible negative regulator hit. A single siRNA was required to show a consistent deviation from the cloud in at least two of the three replicate screens to be considered a reproducible hit. Since we targeted each gene with three independent siRNAs, we also required a hit gene to have two of the three tested siRNAs showing the same effect to be considered a hit (which greatly reduces the likelihood of an off-target effect), and this effect (positive or negative regulator) was defined as the phenotypic effect of this gene. The hits in both the primary and secondary screens were ranked in this fashion by the total number of active siRNAs, the total number of data points showing a phenotypic effect, and the sum of the deviation from the respective cloud (see Data S7).
BMDM RNAi
BMDMs were reverse transfected with siRNAs against screen hit genes using Viromer Green (Lipocalyx, cat# VG-01LB-00). 5 μl of siRNAs (0.5 μM) were spotted in 96-well plates (BD Falcon, cat# 353219), and 0.1 μl of Viromer Green transfection reagent pre-mixed with 4.9 μl of Viromer Green Buffer was added to each well. Plates were tapped to mix the transfection reagents and centrifuged at 400 g for 1 s. After incubation for 30 min at room temperature, 40,000 BMDMs in 90 μl of complete DMEM were seeded per well for a final siRNA concentration of 25 nM. Plates were incubated at room temperature for 10 min to allow the cells to settle, then at 37°C in a humidified atmosphere with 5% CO2 for 48 hr. Cells were stimulated with 5 nM lipid A (Avanti Polar Lipids, Alabaster, AL, cat# 699500P) for time periods as indicated. Cells from 24 wells were pooled for total RNA extraction with RNeasy Mini Kit (Qiagen), then reverse transcribed with iScript cDNA synthesize kit (Biorad, cat# 1708891). For measurement of secreted protein level, supernatants were collected and subject to ELISA as described above. Selected siRNAs for the 4 hit genes were all from Thermofisher with the following siRNA IDs: s106096 and 298393 for Helz2; s104443 and s104445 for Phf11d, s100717 and 163480 for Sertad3, s76360 and s76361 for Zscan12.
Fluidigm Quantitative PCR
Quantitative PCR was carried out according to the manufacturers instructions using the BioMark HD system (Fluidigm), with DELTAgene primer sets designed by the manufacturer.
QUANTIFICATION AND STATISTICAL ANALYSIS
DiRE Transcription Factor Binding Motif Analysis
The six clusters of non-additive genes described in Figure 2B were each analyzed for potential regulatory elements using DiRE (Gotea and Ovcharenko, 2008) (https://dire.dcode.org). Analysis was performed using default DiRE settings for either promoter only or promoter+distal regions using a background of 5000 random genes.
Pathway Analysis
Entrez gene IDs of select gene sets were imported into Ingenuity Pathway Analysis software (Qiagen), and core analysis using the Ingenuity Knowledge Base was carried out, considering both direct and indirect relationships. For analysis of the 29 screen hit set as shown in Figure 6A, Il6 was included to identify the relationship between the hits and Il6.
Analysis of Fluidigm Quantitative PCR Data
Ct values were automatically calculated and exported from the BioMark HD system, then normalized to housekeeping gene Hprt. For TLR4-LPS pathway internal synergy experiment, fold-change relative to the wild-type 0 hr samples were calculated, and the DIF values were calculated as described above. For RNAi knocking down of the 4 hits experiment, mRNA expression levels relative to the non-targeting negative control siRNA NC#1 (Silencer Select Negative Control No. 1 siRNA, Thermofisher, cat# 4390843) transfected samples were calculated using Hprt as the normalization housekeeping gene. 2-way ANOVA analyses were done with Graphpad Prism 7, using Dunnett’s multiple comparisons test to compare each siRNA treatment to the respective NC#1 treatment at the same time point, with adjusted p value <0.05 considered significant. A gene was considered to be affected by a screen hit if perturbation of the hit by 2 different siRNAs led to a consistent and significant effect on that gene. Heatmaps of the siRNA effect on target genes were generated using GENE-E (http://www.broadinstitute.org/cancer/software/GENE-E/index.html).
DATA AND SOFTWARE AVAILABILITY
Microarray data has been deposited in the Gene Expression Omnibus database (GEO: GSE89988).
Supplementary Material
Highlights.
TLR crosstalk is driven by adaptor usage (MyD88/TRIF) of each TLR pathway
TLR crosstalk simultaneously synergizes and antagonizes different gene subsets
TLR synergized transcripts are enriched for sustained immune effectors
Regulators of sustained TLR-induced immune effector expression are identified
Acknowledgments
We thank Ron Germain, Rachel Gottschalk, and colleagues in the Laboratory of Systems Biology for helpful discussions and critical reading of the manuscript, and we thank Tim Myers for assistance with microarray data analysis and GEO data deposition. This work was generously supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases. B.D. is currently an employee of AstraZeneca Inc.
Footnotes
Supplemental Information includes seven figures and seven data files and can be found with this article online at http://dx.doi.org/10.1016/j.cels.2017.06.014.
AUTHOR CONTRIBUTIONS
B.L. and I.D.C.F. designed the study and wrote the paper. B.L. conducted the experiments. B.L. and B.D. conducted the microarray and RNAi screen data analysis.
References
- Abdi K, Singh NJ, Matzinger P. Lipopolysaccharide-activated dendritic cells: “exhausted” or alert and waiting? J Immunol. 2012;188:5981–5989. doi: 10.4049/jimmunol.1102868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, Hellman J. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol. 2007;178:1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- Chen J, Chen ZJ. Regulation of NF-kappaB by ubiquitination. Curr Opin Immunol. 2013;25:4–12. doi: 10.1016/j.coi.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JM, Song DJ, Bergeron J, Benlimame N, Wold MS, Alaoui-Jamali MA. RBT1, a novel transcriptional co-activator, binds the second subunit of replication protein A. Nucleic Acids Res. 2000;28:3478–3485. doi: 10.1093/nar/28.18.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoubet-Lozach L, Benner C, Kaikkonen MU, Lozach J, Heinz S, Spann NJ, Crotti A, Stender J, Ghisletti S, Reichart D, et al. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet. 2011;7:e1002401. doi: 10.1371/journal.pgen.1002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell. 2012;47:810–822. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotea V, Ovcharenko I. DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic Acids Res. 2008;36:W133–W139. doi: 10.1093/nar/gkn300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of inter-feron-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, Akira S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–691. doi: 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Li N, Sun J, Benet ZL, Wang Z, Al-Khodor S, John SP, Lin B, Sung MH, Fraser ID. Development of a cell system for siRNA screening of pathogen responses in human and mouse macrophages. Sci Rep. 2015;5:9559. doi: 10.1038/srep09559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich I, Aderem A. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat Immunol. 2009;10:437–443. doi: 10.1038/ni.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhu Y, Yong WK, Sze NS, Tan NS, Ding JL. Cutting edge: synchronization of IRF1, JunB, and C/EBPbeta activities during TLR3-TLR7 cross-talk orchestrates timely cytokine synergy in the proinflammatory response. J Immunol. 2015;195:801–805. doi: 10.4049/jimmunol.1402358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Giannopoulou EG, Chan CH, Park SH, Gong S, Chen J, Hu X, Elemento O, Ivashkiv LB. Synergistic activation of inflammatory cytokine genes by interferon-gamma-induced chromatin remodeling and toll-like receptor signaling. Immunity. 2013;39:454–469. doi: 10.1016/j.immuni.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138:114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey SA, Klemm SL, Zak DE, Kennedy KA, Thorsson V, Li B, Gilchrist M, Gold ES, Johnson CD, Litvak V, et al. Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics. PLoS Comput Biol. 2008;4:e1000021. doi: 10.1371/journal.pcbi.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S, Sher A, Reis e Sousa C. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13:453–462. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010;140:833–844. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suet Ting Tan R, Lin B, Liu Q, Tucker-Kellogg L, Ho B, Leung BP, Ling Ding J. The synergy in cytokine production through MyD88-TRIF pathways is co-ordinated with ERK phosphorylation in macrophages. Immunol Cell Biol. 2013;91:377–387. doi: 10.1038/icb.2013.13. [DOI] [PubMed] [Google Scholar]
- Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Surapureddi S, Yu S, Bu H, Hashimoto T, Yeldandi AV, Kashireddy P, Cherkaoui-Malki M, Qi C, Zhu YJ, Rao MS, et al. Identification of a transcriptionally active peroxisome proliferator-activated receptor alpha -interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc Natl Acad Sci USA. 2002;99:11836–11841. doi: 10.1073/pnas.182426699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalo-virus infection. Proc Natl Acad Sci USA. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- Tomaru T, Satoh T, Yoshino S, Ishizuka T, Hashimoto K, Monden T, Yamada M, Mori M. Isolation and characterization of a transcriptional cofactor and its novel isoform that bind the deoxyribonucleic acid-binding domain of peroxisome proliferator-activated receptor-gamma. Endocrinology. 2006;147:377–388. doi: 10.1210/en.2005-0450. [DOI] [PubMed] [Google Scholar]
- Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, Smale ST. A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell. 2016;165:165–179. doi: 10.1016/j.cell.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DS, Raupach B, Takeda K, Akira S, Zychlinsky A. Toll-like receptors are temporally involved in host defense. J Immunol. 2004;172:4463–4469. doi: 10.4049/jimmunol.172.7.4463. [DOI] [PubMed] [Google Scholar]
- Xue Q, Lu Y, Eisele MR, Sulistijo ES, Khan N, Fan R, Miller-Jensen K. Analysis of single-cell cytokine secretion reveals a role for paracrine signaling in coordinating macrophage responses to TLR4 stimulation. Sci Signal. 2015;8:ra59. doi: 10.1126/scisignal.aaa2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003a;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003b;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature. 2004;430:218–222. doi: 10.1038/nature02738. [DOI] [PubMed] [Google Scholar]
- Zhu X, Chang MS, Hsueh RC, Taussig R, Smith KD, Simon MI, Choi S. Dual ligand stimulation of RAW 264.7 cells uncovers feedback mechanisms that regulate TLR-mediated gene expression. J Immunol. 2006;177:4299–4310. doi: 10.4049/jimmunol.177.7.4299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.